

Abstract
Recent advances in canine genomics are changing the landscape of veterinary biology, and by default, veterinary medicine. No longer are clinicians locked into traditional methods of diagnoses and therapy. Rather major advances in canine genetics and genomics from the past five years are now changing the way the veterinarian of the 21st century practices medicine.
First, the availability of a dense genome map gives canine genetics a much needed foothold in comparative medicine, allowing advances made in human and mouse genetics to be applied to companion animals. Second, the recently released 7.5x whole genome sequence of the dog is facilitating the identification of hereditary disease genes. Finally, development of genetic tools for rapid screening of families and populations at risk for inherited disease means that the cost of identifying and testing for disease loci will significantly decrease in coming years.
Out of these advances will come major changes in companion animal diagnostics and therapy. Clinicians will be able to offer their clients genetic testing and counseling for a myriad of disorders. Such advances are certain to generate healthier and more long lived dogs, improving quality of life for owner and pet alike. The clinician of the 21st century, therefore, faces incredible opportunities as well as challenges in the management of genetic disease. In this review we summarize recent findings in canine genomics and discuss their application to the study of canine cardiac health.
Keywords: inherited disease, genome, canine, cardiac, heart
Introduction
Veterinary cardiologists have appreciated the importance of genetics in the development of cardiac diseases since the early 1970s 1–5. Although the true prevalence of heart disease in dogs is unknown, two independent studies completed 30 years apart suggest that approximately 11% of canine patients presenting for evaluation at large veterinary hospitals have cardiac disease 6,7. Many of the most common congenital and adult onset cardiovascular diseases are familial, in at least some breeds (Table 1), including myxomatous valve disease in the Cavalier King Charles Spaniel 8 and Dachshunds 9 and dilated cardiomyopathy in the Doberman Pinscher, Great Dane and Portuguese Water Dog 10–12. In many other cases, a subset of breeds have been suggested to have an elevated relative risk compared to the general canine population indicating a familial nature of the disease though genetic studies are required to confirm this (Figure 1).
Table 1.
Mode of Inheritance for Common Canine Cardiac Disorders
| Mode of Inheritance and Disease | Breed and Ref |
|---|---|
| X-linked recessive | |
| Dilated cardiomyopathy | Great Dane 42 |
|
| |
| Autosomal dominant | |
| Tricuspid dysplasia | Labrador 76 |
| Dilated cardiomyopathy | Doberman Pinscher 10 |
| Arrhythmogenic right ventricular cardiomyopathy | Boxer 77 |
|
| |
| Autosomal recessive | |
| Dilated cardiomyopathy | Portuguese water dog 12 |
|
| |
| Polygenic | |
| Patent Ductus Arteriosus | Poodle 1,4 |
| Subvalvular Aortic Stenosis | Newfoundland 3 |
| Pulmonic Stenosis | Beagle 5 |
| Conotruncal defect | Keeshond 43,61 |
| Myxomatous valve disease | Cavalier King Charles Spaniel 8 and Dachshund 9 |
Figure 1.

Association of canine heart disorders and breeds. Increased incidence of disease was assigned according to the Canine Inherited Disorders database, the Cambridge Inherited Diseases in Dogs database and The Textbook of Canine and Feline Cardiology. The breeds and diseases reported are based on observation at veterinary hospitals and publications in veterinary and medical journals. We fully acknowledge the limitations of such data and present this chart as a starting point for further investigation into the breed specific incidence of disease. The authors strongly encourage input from other researchers to supplement this data.
1. Reported by Canine Inherited Disorders database 78 (http://www.upei.ca/~cidd/intro.htm).
2. Reported by Cambridge Inherited Diseases in Dogs database41 (http://www.upei.ca/~cidd/intro.htm).
3. Reported by Buchanan et al. in The Textbook of Canine and Feline Cardiology7.
Canine genetics and genomics is in the midst of an information explosion 13,14. Recent studies have brought advances in understanding the genetics of breed development 15 and the availability of ever higher resolution maps 16–21. These have led to the mapping of disease loci for a variety of diseases including metabolic disorders 22,23, blindness 24–28, cancer 29,30 neurologic disorders 31,32, hip dysplasia 33 epilepsy 34, as well as several morphologic traits 35,36. In addition, the advancement of a framework for how to study complex canine traits 37 has led to deepening of our knowledge about the organization of the canine genome 38,39 and how it relates to morphological variation between breeds.
Given these advances, what has slowed progress in understanding the molecular nature of familial canine cardiovascular disease? In part, the lack of tools for studying what are arguably some of the most complex diseases in companion animal medicine has been problematic. For simple Mendelian traits, mapping studies have moved forward with great success. Cardiac diseases, however, are typically more complex and may involve multiple genes, age dependent penetrance, and contributions from environmental factors. The frequent late onset of disease is particularly problematic as individuals often reproduce before the disease is manifest. While progress has certainly been made in some areas, as reviewed below, the field of canine cardiac genetics, in general, has struggled to keep pace with the rest of the canine genetics community. We believe this is about to change.
In this review we discuss, first, how to utilize information from pedigrees to understand issues related to mode of inheritance. This sets the stage for future genetic evaluation. We next discuss both candidate gene and linkage-based approaches for mapping cardiac diseases. We summarize recent advances related to the canine genome sequence and understanding genome organization. Finally, we conclude with a brief discussion of genomic tools under development that will influence the way canine cardiac genetic studies are performed, highlighting the roles of the owner, clinician, and research scientist in conducting a successful inherited disease study.
Mode of Inheritance
The identification of mode of inheritance for a familial disease is an important first step for any genetic study (Figure 2). This may best be determined by performing outcross breedings, but such matings are costly and may be unpopular as they require intentional breeding of carrier and/or affected animals. In many cases, careful evaluation of pedigree information and clinical records can provide the needed information to correctly assess mode of inheritance.
Figure 2.

Flow chart for designing a mapping study. The flow chart shows the basic steps for designing a mapping study using either families or cases and controls. Affected individuals are designated by the blue ice pack on the head. The unaffected individuals have no head gear. Arrows indicate order of tasks. Alleles at each hypothetical locus are represented by uppercase and lowercase versions of the same letter.
The most common inheritance patterns reported in the dog are autosomal dominant, autosomal recessive and X- linked recessive 40,41. Autosomal dominant traits are classically defined by the presence of an essentially equal number of affected males and females. Every affected individual will typically have one carrier parent, and all dogs with one copy of the mutant allele can manifest the disease, however if the variant allele is not fully penetrant the number of affected offspring will fall below the theoretical 50%. Modifier genes can also affect allele penetrance by working to either enhance or suppress the function of the mutant allele. Also, exposure to particular environmental factors can alter the appearance of the disease. Carriers will transmit the mutant allele, and the potential to develop the disease, to 50% of their offspring.
Autosomal recessive traits require two copies of a mutant allele in order for the trait to appear in the dog. The mating of carriers, each of which has one variant allele would typically produce a 3:1 ratio of unaffected to affected dogs. This, as discussed above, assumes the disease allele is fully penetrant. A juvenile form of familial dilated cardiomyopathy in the Portuguese Water Dog has been reported to be consistent with an autosomal recessive mode of inheritance12.
X-linked traits are almost always recessive and may be defined by the presence of only affected males. Carrier females have a 50:50 chance of passing the trait on to male offspring while there is no male to male (father to son) inheritance. Affected females are typically rare, but can result from a cross between an affected male and a female carrier who passes on her mutant allele. When considering X-linked traits the frequency of such affected females will depend wholly on the allele frequency of the deleterious allele in the general population. Adult onset familial dilated cardiomyopathy in the Great Dane appears to be an X- linked recessive trait 42.
As always, manifestation of any disease may be complicated by age dependent penetrance. This is certainly the case with dilated cardiomyopathy or myxomatous valve disease, where the disease usually appears in older dogs making the mode of inheritance more difficult to trace.
In many cases, the defect or disease cannot be explained by a single gene mutation with a Mendelian mode of inheritance. Such diseases are often caused by the interplay of multiple genes and are referred to as polygenic. Extensive breeding studies performed on a family of poodles segregating Patent ductus arteriosus suggest that, at least in this breed, it is inherited as a polygenic defect1,4. Molecular studies have suggested that multiple genes contribute to the development of myxomatous valve disease in the Cavalier King Charles Spaniels and Dachshunds 8,9 and the conotruncal heart defect in the Keeshond dog 43.
In order to ascertain the mode of inheritance, a complex segregation analysis (CSA) is often done. CSA tests the hypothesis that a locus inherited in a Mendelian fashion substantially contributes to the risk of a disease. It can provide information about the likely mode of inheritance and penetrance of underlying susceptibility genes. These analyses can also identify subsets of families whose disease is most likely due to mutations in inherited susceptibility genes. CSA can not determine the number of susceptibility loci, nor can it provide information about the location of loci. Instead, it provides an estimate of the overall frequency of mutant alleles with a specific set of characteristics. This type of data can be extremely useful for developing statistical models that are, in turn, used for linkage analysis, as discussed below.
Candidate Gene Approaches
To find a disease gene, many scientists initially use a “candidate gene” approach. Genes are selected for detailed examination based on 1) molecular information from a similar disease in a unique species such as human or mouse; 2) the prevalence of the candidate gene in a tissue of interest (myocardium, valve, etc); or 3) the specific function of the gene (i.e. involved in channel regulation, etc). For instance, causative mutations have been observed in six different genes for human arrhythmogenic right ventricular cardiomyopathy (ARVC) 44,45. The canine homology of each of these six genes is therefore a candidate for molecular analysis in the Boxer dog with ARVC. Each gene is, or has been, evaluated by direct DNA sequencing of exons, introns, and promoters to hopefully identify mutations that would affect transcription or translation of the resultant protein. Northern blot analysis or expression arrays can be used to ascertain differences in gene expression between affected and unaffected dogs. Southern blots can be used to identify large genomic rearrangements. Finally, in situ antibody staining can determine if the encoded protein localizes correctly and is stable. In all likelihood only one or two genes will be causative for a given disease within a single breed. But when all affected breeds are considered, many candidates originally identified from studies in humans could result in some form of related canine disease.
This candidate gene approach has been used to evaluate several genes for familial dilated cardiomyopathy in the Doberman Pinscher 46. However, the initial results have all been negative. Evaluation of the desmin and delta sarcoglycan did not support a role for these genes in this disease 47,48. In addition, the promoter and coding regions of the phospholamban gene and coding region of the actin gene have been excluded by comparison of direct sequencing results in affected and unaffected dogs 49,50. However as more human disease variants are identified, the candidate gene approach will become increasingly fruitful for companion animal geneticists. Currently, a primary disadvantage of this approach is that a significant amount of time and resources can be spent chasing what are frequently negative results. Therefore, we advocate using candidate gene studies as the first approach only when there are a small number of genes of large effect that have been found to cause the same or a similar disease in another species. Additionally, the candidate gene approach may be a reasonable starting point when the collection of extended families for linkage analysis has proven difficult. One example of such is X-linked dilated cardiomyopathy in the Great Dane. This is an adult onset disease that is rarely diagnosed early. By the time the proband is identified, properly phenotyped and samples collected, the previous generations have likely passed away.
Mapping Disease Causing Loci
In the absence of obvious candidate genes or successful candidate gene studies, investigators use linkage-based approaches to find loci of interest. Linkage analysis involves tracking alleles of markers through multiple generations in a set of families to determine if there are regions of the genome co-inherited in affected versus unaffected dogs. This approach requires well-characterized, multi-generation families with high quality phenotypes. Larger number of families and families with the greatest number of affected individuals offer the best opportunity for finding evidence of true linkage.
Generally, 300–500 markers spanning the genome at a 5–10 centimorgan (cM) density are used for an initial genome wide scan 19,20,51. Genetic markers are places in the genome where there is variation, or polymorphism, between alleles. Millions of markers have been identified for most mammalian genomes. The most informative markers to use are those that are highly polymorphic and tend to have a large number of alleles in the population. After the genotyping is completed, the data set is checked for errors and examples of non-paternity/maternity 52–54. The latter is often anxiety provoking to veterinary clients, who worry that events occurring several generations prior to the current one will be discovered and thus devalue their pedigrees. Scientists typically include a statement of informed consent that assures clients that identification of any such event is highly unlikely, and furthermore if found would not be disclosed. In a linkage study, each individual marker as well as set of adjacent markers are evaluated using several statistical methods to consider the likelihood that the marker is linked or co-segregates with the disease in question 55–59.
Data can be analyzed in a variety of ways, using both parametric (model-driven) or non-parametric (parameter free) approaches. The Lod score method is used to assign confidence levels to the hypothesis that linkage exists between genetic markers at known locations and a putative disease locus. This analysis will also provide a maximum likelihood estimate of the recombination fraction (theta) between each marker and the putative gene. This in turn provides an estimate of the distance between the marker and the disease locus 55–57.
In the case of parametric analysis, a marker that has a Lod score of > 3.3 is considered “linked” to the disease locus 58 and indicates odds of greater then 1000:1 that the disease locus has been correctly positioned. In the case of non-parametric analysis, no assumptions are made about the methods of disease inheritance; only that affected individuals carry a germline change that unaffected individuals do not. If excess sharing of a particular allele or set of alleles from adjacent markers is observed among affecteds, as indicated by a significant p-value, then the region surrounding the marker(s) is investigated further.
By way of example, genetic linkage analysis has been used to study familial tricuspid dysplasia in the Labrador Retriever 60. Investigators identified three purebred Labrador Retriever kindreds that were enriched for tricuspid valve dysplasia and carried out a 15cM genome scan. They found linkage on canine chromosome 9 (CFA9) with a statistically significant maximum multipoint Lod score of 3.33. Two markers define a region of about 4.3 cM in which the disease gene is presumed to lie. Interestingly, to find the disease locus the investigators had to assume that reduced penetrance was associated with the variant allele. The linkage was only detected when investigators assumed that about 70% of dogs carrying the mutation would go on to get the disease at some point in their life.
By comparison, use of the same technique in the Keeshond with conotruncal defect revealed linkage to three different canine chromosomes 43. This suggests a polygenic mode of inheritance 43,61. The next steps for these investigators will be challenging as they do yet know how the three loci interact with one another.
Once a marker is found to be linked to a disease locus, additional markers are genotyped and, if possible, additional samples are tested in order to verify the initial findings. If the chromosome region associated with the disease is large, more families will be needed to identify critical recombinants that reduce the region of linkage. In the case the Andelfinger et al. study, the original region of linkage was nearly 11cM in size. Identification of dogs in which a meiotic recombination event had occurred on the chromosome carrying the mutation reduced the region to a much more manageable 4.3 cM 60.
In this “fine mapping” stage, researchers often turn to population studies to look for a haplotype or pattern of alleles on one chromosome that is shared by affected but not unaffected individuals. If the findings of the initial linkage analysis are validated, and the chromosomal region is modest in size, obvious candidate genes are tested. For example, in the case of the Labrador Retriever with triscuspid dysplasia, the region of interest on chromosome nine could be evaluated for genes involved in valvular development. If such genes are found, the germline sequence of exons associated with each gene should be compared between affected and unaffected dogs in the hope of finding a disease causing mutation. Additionally, expression studies would be appropriate to determine if any given candidate gene is present in the appropriate tissues and if so, is expression altered in animals with the disease. If a change in gene sequence is identified that segregates faithfully with the disease, the variant can be typed in dogs from both the breed in question as well other breeds to determine the population frequency of the mutant allele. Such information is often of enormous use to the practicing vet, who can use the data to estimate the likelihood that any dog of an “at risk” breed will eventually get the disease in question.
Using Molecular Information for Genetic Screening and Therapeutic Development
Once a disease locus has been identified, the information can be used in two ways. First, breeders are often anxious to obtain access to genetic tests that will allow them to improve their breeding programs. Tests based on linkage results alone are less desirable then those based on an identified variant. Rare recombination events occurring between the critical marker and disease variant itself are always possible and can distort findings.
The development of a gene-based test is therefore optimal. Gene-based tests directly screen for mutations in the disease gene. Such tests are most economical when only one variant in the gene segregates with the disease. More often, however, multiple mutations can occur in a single gene, especially when several breeds have the disease. Then, the entire gene must be screened in every dog undergoing the test.
Genetic testing is available for a host of canine diseases, including several associated with blindness, metabolic defects, cancer, narcolepsy, and urinary dysfunction. Breeders and veterinarians need to be cautious, however, about how such information is used. In breeds with small gene pools derived from limited numbers of founders, or breeds that have experienced population bottlenecks, strict screening and removal programs can be detrimental to the breed, as other recessive diseases will appear when the gene pool is reduced. For instance, the Irish wolfhound is a fairly small breed with less than 1000 new puppies registered by the AKC each year. Contributing to its’ small gene pool is the fact that the breed suffered a severe bottleneck in the mid 1800’s, such that Irish Wolfhound were considered by many to be extinct. The breed is at high risk for dilated cardiomyopathy, with the disorder possibly affecting as much as one-third of the population (www.iwclubofamerica.com) 62. Once the disease gene is identified, a strict screening and removal policy that would restrict dogs carrying even one disease allele from producing registered dogs would likely destabilize the gene pool, resulting in many new recessive disorders “popping-up” in the breed. For recessive disorders in particular, the ideal breeding program allows carriers to remain in the gene pool, but limits their matings to primarily non-carriers, thus diluting the deleterious alleles out of the population slowly.
While the above information is most useful to the breeder, the research scientist is often more interested in what information the disease gene reveals in terms of mechanism of disease. The identification of the hypocretin 2 receptor as an important component of sleep biology 63 by Emmanuel Mignot and colleagues offers a clear example. Linkage analysis and eventual cloning of the disease gene using pedigrees of Doberman pinschers affected with narcolepsy 31 led to significant advances in understanding the biology of sleep, and development of new programs in neuroscience focused on the molecular biology of sleep. As genes for canine heart disease are found, similar success stories will likely emerge.
Canine Genome Maps and the Canine Genome Sequence
Many advances in canine genomics over the past two years can be traced to the recent release of a high quality draft sequence of the dog genome, produced from a female Boxer 64. The sequence assembly validated much of what was already hypothesized from comparative genome map studies. The genome is approximately 2.4 billion bases and is comprised of approximately 250 conserved segments when compared to the human genome 16,17,21,65–67. The assembled sequence is estimated to cover 98% of the dog genome with the majority of genes containing no sequence gaps. On average, two segments of continuous sequence cover each of the dog’s 38 chromosomes. The current gene count is listed as ~19,000 with about 75% representing 1-1-1 orthologs between dog, human and mouse. The full genome sequence can be accessed through: http://www.genome.ucsc.edu; http://www.ncbi.nih.gov; http://www.ensembl.org.
In addition to the Boxer sequence, a low density (1.5x) sequence of the Standard Poodle is also available 68. These resources, together with one million sequence reads completed on each of nine dogs representing all of the AKC breed groups have resulted in the generation of more then 2 million single nucleotide polymorphisms (SNPs). SNPs are bi-allelic markers that can be easily automated for use in linkage studies, and have thus recently come into great favor. A SNP occurs about once every 1000 bases in dogs. The public databases currently contain information on about 2.1 million canine SNPs. A subset of these is currently being verified for development of a canine specific “SNP chip” that, once released, will be used for whole genome association studies in dogs. Such studies make use of populations of affected dogs that are not themselves closely related, but because they are of the same breed likely share a common ancestral mutation for any given disease. Comparison of allele frequencies for adjacent SNPs between affected cases and unaffected controls is another useful way to localize disease loci. Such studies, when they encompass markers spanning all chromosomes, are referred to as whole genome association studies.
Linkage Disequilibrium Facilitates Disease Gene Mapping
With the availability of a full coverage genome sequence, investigators have been able to improve the way they design mapping studies in the dog. Integral in these changes have been studies of the extent of linkage disequilibrium (LD) in the dog 64,69–71. LD refers to the nonrandom association of two or more usually adjacent loci that segregate together through several generations (Figure 3). LD thus is a measure of the distance that separates two markers or loci before they can be considered independent. The most informative studies have been those of Sutter et al., 71 and Lindblad-Toh et al. 64, the latter performed as part of the dog genome sequencing effort.
Figure 3.
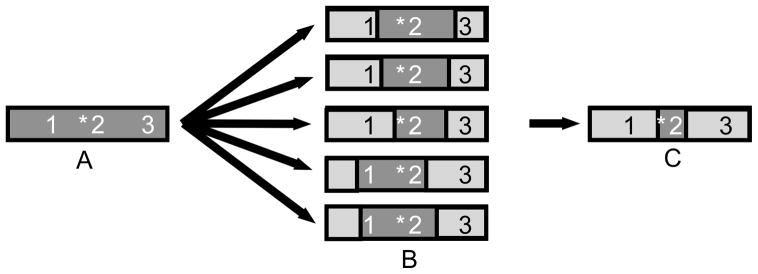
Extensive linkage disequilibrium maintains the association between marker and mutation over multiple generations. A) A mutation (*) arises on an ancestral chromosome carrying markers 1, 2, and 3. B) Generations of recombination disrupt the haplotype across the chromosome. On chromosomes where the mutation (*) remains, markers in close proximity (2) also remain unchanged. C) Modern chromosomes retain the mutation and the markers within the span of linkage disequilibrium (LD).
In an initial series of studies, Sutter and colleagues examined the extent of LD in five breeds with distinct breed histories; the Akita, Golden Retriever, Labrador Retriever, Bernese Mountain Dog and Pekingese. The average length of LD in these five breeds is approximately 2 Mb. This is 40–100 times further than LD typically extends in the human genome. Thus while a typical whole genome association study in humans requires about 500,000 SNPs72, the increased level of LD in the dog means that only about 10–30,000 markers would be required for a comparable study. For diseases found in both humans and dogs, such as many cardiac diseases, it will thus be far easier to do the initial mapping study in dogs instead of humans, a fact not overlooked by those applying for funding to do such studies.
These investigators also found that the extent of LD varied over a near ten-fold range between breed of dog (0.4–3.2 Mb) 71. Thus, selection of a particular breed for a mapping study over other available breeds could dramatically reduce the work load and expense associated with the study, and judicious selection of breeds for studies of shared disorders becomes very important. Finally, this study demonstrated that haplotypes or blocks of alleles are frequently shared between dog breeds, a result that was not surprising given the recent evolution of the dog from the wolf 73,74. This means that a single map and a single SNP chip will be useful for mapping studies in any breed of dog. These results were corroborated and greatly extended in a much larger study by Lindblad-Toh et al., using 10 breeds and nearly 1300 SNPs 64. Indeed, their data predicts that as little as 10,000 informative SNPs will be needed for most whole genome association studies in the dog.
Canine Breed Clusters Facilitate Mapping Efforts
Given the information above, one important way to improve power for mapping studies, especially once a locus is known and researchers are trying to reduce the region of linkage, is to combine data across breeds. To determine the ancestral relationship between breeds, Parker et al. have analyzed data from 96 microsatellite markers genotyped on five unrelated dogs from each of 85 breeds. They then performed an unsupervised clustering analysis using the computer program structure 15. The 85 breeds were ordered into four clusters, generating a new canine classification system for dog breeds based on similar patterns of alleles, presumably from a shared ancestral pool 15. For instance, Cluster 1 comprised dogs of Asian and African origin. Cluster 2 included largely mastiff type dogs with big, boxy heads and large, sturdy bodies. The 3rd and 4th clusters split a group of herding dogs and sight hounds away from the general population of modern hunting dogs including terriers, hounds and gun dogs. Studies are underway to expand this research, with the goal of including all AKC recognized breeds.
The Parker clusters 15, together with the findings of Sutter et al., and Lindblad-Toh et al. offered the first look at relationships between breeds, and in doing so suggest study designs for trait mapping 64,71. The latter two studies both demonstrate high haplotype sharing between breeds and low haplotype diversity within breeds. Thus, disease alleles will be most easily identified by comparison of haplotypes that are identical by descent (IBD) in affected individuals from two or more breeds localizing to the same Parker cluster, as they likely share a common ancestral mutation. Data from additional breeds can then be used for fine resolution mapping to reduce the region of linkage.
In the example provided (Figure 4) the four color graph shows the population distribution of genomic patterns in 414 dogs representing 85 breeds 15. Each vertical line represents a single dog. Each line is divided into four colored segments that correspond to one of four breed clusters. The length of the colored segment relates to the proportion of the genome that matches the indicated cluster. In this specific example, nine breeds demonstrating an increased incidence of dilated cardiomyopathy, arguably one of the most common adult onset cardiovascular diseases in the dog, are shown. In this case we observe that several of the most commonly reported affected breeds, including the Doberman Pinscher, English Cocker Spaniel and Great Dane fall into Cluster 4. We can reasonably hypothesize that one ancestral mutation is responsible for this particular disease in these breeds. Of note, genetic studies such as these will be successful only if they are supported by strong clinical studies that precisely define disease presentation, detail appropriate methods of diagnosis, and track response to treatment. Indeed, such clinical studies may suggest that only a subset of affected breeds within a cluster should be considered together. In that case, separate genome scans would be set up for the different groups of breeds with the hope that all susceptibility loci would eventually be identified.
Figure 4.
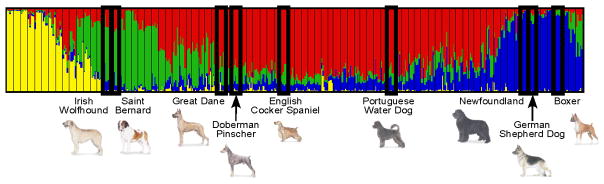
Genomic classification of nine dog breeds displaying an increased incidence of cardiomyopathy. The four color graph shows the population distribution of genomic patterns in 414 dogs representing 85 breeds 15. Each vertical line represents a single dog. Each line is divided into four colored segments corresponding to a different breed cluster. The length of the colored segment is relative to the proportion of the genome that matches the indicated cluster. Nine breeds that show an increased incidence of cardiomyopathy are outlined in bold and names and pictures are given below the graph. Increased incidence of disease was assigned according to the Canine Inherited Disorders database and the Cambridge Inherited Diseases in Dogs database41,78.
Conclusions
Canine cardiac diseases are common, complex, and devastating to owners. They are often silent killers, leaving owners and breeders wondering what could have been done to prevent the loss of their treasured pet. The advances of the past three years by the canine genomics community offer hope to dog owners and breeders alike, as the tools and resources are now available to understand the genetic susceptibility for many cardiac diseases.
Key to the success of such endeavors will be the partnership between the owner, veterinarian, and research scientist. Every genetic study must start with a sampling of individuals affected with the disease under scrutiny. In order to achieve success, owners must be willing to provide accurate medical histories for their dogs as well as blood and, where appropriate, tissue samples. Even with unlimited samples, a genetic study is only as good as the pathology and clinical data that it is built upon. Veterinarians must be willing to abstract important information from owners regarding disease presentation, family history, and past treatment responses, and to encourage the completion of tests needed to provide unambiguous diagnosis. Finally, the geneticist must communicate clearly what samples and information are required in order for the study to take place and should facilitate the collection of both by sharing experimental plans and information with clinicians and owners alike.
The genome community has met many of the goals it set for itself 15 years ago when the canine genome project began 75. What remains now is for owners and veterinarians to join with motivated researchers to embrace new paradigms in canine genetic health. New ways of diagnosing disease based on molecular tests, genetic testing and counseling, and development of breeding strategies that optimize health over conformation are all topics of deep discussion in the dog world today. The veterinarian of the 21st century must be sufficiently well informed to not only take part in, but to lead, this debate.
Acknowledgments
We thank our many colleagues who read the article during preparation and made excellent suggestions, including Dr. Nathan Sutter. We also thank Dr. Sutter for Figure 3. We acknowledge the long term support of the AKC-CHF (EAO and KMM). Ongoing work (EAO) is supported in part by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health. Finally, we thank the many dog owners and breeders who continue to support our efforts.
Glossary of Terms
- allele
One copy of a genetic sequence, haplotype, or locus that has more than one form. This can refer to a marker, a gene, or a single base-pair
- allele frequency
The proportion of a sample of chromosomes that carries a particular allele
- centimorgan
A unit of genetic distance based on recombination frequency. One centimorgan is equal to a 1% chance that a marker at one genetic locus will be separated from a marker at a second locus due to crossing over in a single generation. In dogs, one centimorgan is equivalent, on average, to 800 kilobases
- complex segregation analysis (CSA)
A test to establish mode of inheritance and penetrance of a disease-associated allele by examining the segregation of the allele through generations of affected and unaffected individuals
- conserved segments
A region of the genome containing two or more contiguous, homologous genes in at least two separate species
- deleterious allele
A genetic sequence that is disease-associated
- direct sequencing
Sequencing a gene or region of interest directly from affected individuals in order to look for variations from wild-type
- expression arrays
An array of gene specific DNA sequences, usually affixed to a chip, that can be probed with RNA extracted from specific tissues in order to identify the genes that are expressed in that tissue
- haplotype
“Haploid genotype”, marker alleles that are aligned along a single chromosome
- identical by descent
Two or more individuals having the same pattern of alleles across a region of the chromosome because the alleles were inherited from a common ancestor
- linkage
The proximity of two or more markers on a chromosome; the closer the markers, the lower the probability that they will be separated during meiosis and hence the greater the probability that they will be inherited together and therefore linked
- linkage disequilibrium
Alleles at different markers occur together more often than can be accounted for by chance. This usually indicates that the two markers are physically close on a piece of DNA. The extent of linkage disequilibrium (distance between correlated markers) varies by breed and is increased by forces that reduce effective population size
- Lod score
Logarithm of the ratio of the odds, a Lod of 3.0 means that such a result is obtained only one time per 1000 by chance
- markers
A portion of DNA correlated to a specific chromosomal location. Polymorphic markers can be used to trace the inheritance of a chromosome through generations of families
- mutation
Any change in the reference DNA sequence. Such changes may be, but are not necessarily, deleterious
- Northern blot
A method for determining the amount and size of transcribed RNA in a cell. Total mRNA is extracted from tissue and then separated by gel electrophoresis and transferred to a membrane. Individual reads specific to a gene of interest are identified by the adhesion of a complementary piece of DNA used as a probe. Northern blot analysis can identify alternate splicing events as well as overall gene expression levels and patterns
- ortholog
A gene found in at least two different species that evolved from a common ancestral gene by speciation. Normally, orthologs retain the same function
- penetrance
The probability of a gene or genetic trait being expressed. “Complete” penetrance means that every individual that carries the gene that causes a trait will express that trait. “Incomplete” penetrance means the genetic trait is expressed in only part of the population carrying the gene
- polygenic
Genetic disorder resulting from the combined action of alleles of more than one gene (e.g. most heart disease, diabetes, and some cancers)
- polymorphic
Having more than one allel
- population bottleneck
An extreme reduction in population size, retained for multiple generations, which in turn leads to a reduction in genetic variation
- recombination
The process by which progeny derive a combination of genes different from that of either parent. This occurs through cross-over events that take place during meiosis
- recombination fraction
The fraction of chromosomes examined, in a linkage study, in which a recombination event took place between the two loci of interest
- sequence (DNA or genomic)
The relative order of base pairs
- single nucleotide polymorphism (SNP)
Variation that occurs when a single nucleotide (A, T, C, or G) in the genome sequence is altered - sometimes referred to as a point mutation
- Southern blot
Genomic DNA is fragmented, separated by gel electrophoresis and transferred to membrane filters for detection of specific base sequences by labeled complementary probes. Used to identify deletions, insertions or other changes that create or destroy restriction enzyme sites
- susceptibility locus (loci)
Location on a chromosome that is linked to, or associated with, disease susceptibility
Contributor Information
Heidi G. Parker, Email: hgparker@mail.nih.gov.
Kathryn M. Meurs, Email: meurs@vetmed.wsu.edu.
Elaine A. Ostrander, Email: eostrand@mail.nih.gov.
References
- 1.Patterson DF, Pyle RL, Buchanan JW, et al. Hereditary patent ductus arteriosus and its sequelae in the dog. Circ Res. 1971;29:1–13. doi: 10.1161/01.res.29.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Patterson DF, Pyle RL, Van Mierop L, et al. Hereditary defects of the conotruncal septum in Keeshond dogs: pathologic and genetic studies. Am J Cardiol. 1974;34:187–205. doi: 10.1016/0002-9149(74)90198-2. [DOI] [PubMed] [Google Scholar]
- 3.Pyle RL, Patterson DF, Chacko S. The genetics and pathology of discrete subaortic stenosis in the Newfoundland dog. Am Heart J. 1976;92:324–34. doi: 10.1016/s0002-8703(76)80113-5. [DOI] [PubMed] [Google Scholar]
- 4.Patterson DF. Congenital defects of the cardiovascular system of dogs: studies in comparative cardiology. Adv Vet Sci Comp Med. 1976;20:1–37. [PubMed] [Google Scholar]
- 5.Patterson DF, Haskins ME, Schnarr WR. Hereditary dysplasia of the pulmonary valve in beagle dogs. Pathologic and genetic studies. Am J Cardiol. 1981;47:631–41. doi: 10.1016/0002-9149(81)90548-8. [DOI] [PubMed] [Google Scholar]
- 6.Detweiler DK, Patterson DF. The prevalence and types of cardiovascular disease in dogs. Ann N Y Acad Sci. 1965;127:481–516. doi: 10.1111/j.1749-6632.1965.tb49421.x. [DOI] [PubMed] [Google Scholar]
- 7.Buchanan JW. Prevalence of cardiovascular disorders. In: Fox P, Sisson D, Moise NS, editors. Textbook of canine and feline cardiology. Principles and clinical practice. 2. Philadelpia: WB Saunders; 1999. pp. 457–70. [Google Scholar]
- 8.Haggstrom J, Kvart C, Hansson K. Heart sounds and murmurs: changes related to severity of chronic valvular disease in the Cavalier King Charles spaniel. J Vet Intern Med. 1995;9:75–85. doi: 10.1111/j.1939-1676.1995.tb03276.x. [DOI] [PubMed] [Google Scholar]
- 9.Olsen LH, Fredholm M, Pedersen HD. Epidemiology and inheritance of mitral valve prolapse in Dachshunds. J Vet Intern Med. 1999;13:448–56. doi: 10.1892/0891-6640(1999)013<0448:eaiomv>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 10.Hammer TA, Venta PJ, Eyster GE. The genetic basis of dilated cardiomyopathy in Doberman Pinschers. Animal Genetics. 1996;27:109. [Google Scholar]
- 11.Meurs KM. Insights into the hereditability of canine cardiomyopathy. Vet Clin North Am Small Anim Pract. 1998;28:1449–57. viii. doi: 10.1016/s0195-5616(98)50131-3. [DOI] [PubMed] [Google Scholar]
- 12.Dambach DM, Lannon A, Sleeper MM, Buchanan J. Familial dilated cardiomyopathy of young Portuguese water dogs. J Vet Intern Med. 1999;13:65–71. [PubMed] [Google Scholar]
- 13.Ostrander EA, Wayne RK. The canine genome. Genome Res. 2005;15:1706–16. doi: 10.1101/gr.3736605. [DOI] [PubMed] [Google Scholar]
- 14.Parker HG, Ostrander EA. Canine Genomics and Genetics: Running with the Pack. PLoS Genet. 2005;1:e58. doi: 10.1371/journal.pgen.0010058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parker HG, Kim LV, Sutter NB, et al. Genetic structure of the purebred domestic dog. Science. 2004;304:1160–4. doi: 10.1126/science.1097406. [DOI] [PubMed] [Google Scholar]
- 16.Priat C, Hitte C, Vignaux F, et al. A whole-genome radiation hybrid map of the dog genome. Genomics. 1998;54:361–78. doi: 10.1006/geno.1998.5602. [DOI] [PubMed] [Google Scholar]
- 17.Mellersh CS, Hitte C, Richman M, et al. An integrated linkage-radiation hybrid map of the canine genome. Mamm Genome. 2000;11:120–30. doi: 10.1007/s003350010024. [DOI] [PubMed] [Google Scholar]
- 18.Guyon R, Kirkness EF, Lorentzen TD, et al. Building comparative maps using 1. 5x sequence coverage: human chromosome 1p and the canine genome. Cold Spring Harb Symp Quant Biol. 2003;68:171–7. doi: 10.1101/sqb.2003.68.171. [DOI] [PubMed] [Google Scholar]
- 19.Guyon R, Lorentzen TD, Hitte C, et al. A 1-Mb resolution radiation hybrid map of the canine genome. Proc Natl Acad Sci U S A. 2003;100:5296–301. doi: 10.1073/pnas.0831002100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Breen M, Hitte C, Lorentzen TD, et al. An integrated 4249 marker FISH/RH map of the canine genome. BMC Genomics. 2004;5:1–11. doi: 10.1186/1471-2164-5-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hitte C, Madeoy J, Kirkness EF, et al. Facilitating genome navigation: survey sequencing and dense radiation-hybrid gene mapping. Nat Rev Genet. 2005;6:643–8. doi: 10.1038/nrg1658. [DOI] [PubMed] [Google Scholar]
- 22.van De Sluis B, Rothuizen J, Pearson PL, et al. Identification of a new copper metabolism gene by positional cloning in a purebred dog population. Hum Mol Genet. 2002;11:165–73. doi: 10.1093/hmg/11.2.165. [DOI] [PubMed] [Google Scholar]
- 23.Yuzbasiyan-Gurkan V, Blanton SH, Cao V, et al. Linkage of a microsatellite marker to the canine copper toxicosis locus in Bedlington terriers. Am J Vet Res. 1997;58:23–7. [PubMed] [Google Scholar]
- 24.Acland GM, Ray K, Mellersh CS, et al. Linkage analysis and comparative mapping of canine progressive rod-cone degeneration (prcd) establishes potential locus homology with retinitis pigmentosa (RP17) in humans. Proc Natl Acad Sci USA. 1998;96:3048–53. doi: 10.1073/pnas.95.6.3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Acland GM, Ray K, Mellersh CS, et al. A novel retinal degeneration locus identified by linkage and comparative mapping of canine early retinal degeneration. Genomics. 1999;59:134–42. doi: 10.1006/geno.1999.5842. [DOI] [PubMed] [Google Scholar]
- 26.Aguirre G, Lolley R, Farber D, et al. Rod-cone dysplasia in Irish Setter dogs: A defect in cyclic GMP metabolism in visual cells. Science. 1978;201:1133. doi: 10.1126/science.210508. [DOI] [PubMed] [Google Scholar]
- 27.Aguirre GD, Acland GM. Variation in retinal degeneration phenotype inherited at the prcd locus. Exp Eye Res. 1988;46:663–87. doi: 10.1016/s0014-4835(88)80055-1. [DOI] [PubMed] [Google Scholar]
- 28.Aguirre GD, Baldwin V, Pearce-Kelling S, et al. Congenital stationary night blindness in the dog: common mutation in the RPE65 gene indicates founder effect. Mol Vis. 1998;4:23. [PubMed] [Google Scholar]
- 29.Jonasdottir TJ, Mellersh CS, Moe L, et al. Genetic mapping of a naturally occurring hereditary renal cancer syndrome in dogs. Proc Natl Acad Sci U S A. 2000;97:4132–7. doi: 10.1073/pnas.070053397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lingaas F, Comstock KE, Kirkness EF, et al. A mutation in the canine BHD gene is associated with hereditary multifocal renal cystadenocarcinoma and nodular dermatofibrosis in the German Shepherd dog. Hum Mol Genet. 2003;12:3043–53. doi: 10.1093/hmg/ddg336. [DOI] [PubMed] [Google Scholar]
- 31.Lin L, Faraco J, Li R, et al. The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell. 1999;98:365–76. doi: 10.1016/s0092-8674(00)81965-0. [DOI] [PubMed] [Google Scholar]
- 32.Lingaas F, Aarskaug T, Sletten M, et al. Genetic markers linked to neuronal ceroid lipofuscinosis in English setter dogs. Anim Genet. 1998;29:371–6. doi: 10.1046/j.1365-2052.1998.295358.x. [DOI] [PubMed] [Google Scholar]
- 33.Chase K, Lawler DF, Adler FR, et al. Bilaterally asymmetric effects of quantitative trait loci (QTLs): QTLs that affect laxity in the right versus left coxofemoral (hip) joints of the dog (Canis familiaris) Am J Med Genet A. 2004;124:239–47. doi: 10.1002/ajmg.a.20363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lohi H, Young EJ, Fitzmaurice SN, et al. Expanded repeat in canine epilepsy. Science. 2005;307:81. doi: 10.1126/science.1102832. [DOI] [PubMed] [Google Scholar]
- 35.Chase K, Carrier DR, Adler FR, et al. Genetic basis for systems of skeletal quantitative traits: Principal component analysis of the canid skeleton. Proc Natl Acad Sci U S A. 2002;99:9930–5. doi: 10.1073/pnas.152333099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chase K, Carrier DR, Adler FR, et al. Interaction between the X chromosome and an autosome regulates size sexual dimorphism in Portuguese Water Dogs. Genome Res. 2005;15:1820–4. doi: 10.1101/gr.3712705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chase K, Adler FR, Miller-Stebbings K, Lark KG. Teaching a new dog old tricks: identifying quantitative trait loci using lessons from plants. J Hered. 1999;90:43–51. doi: 10.1093/jhered/90.1.43. [DOI] [PubMed] [Google Scholar]
- 38.Fondon JW, 3rd, Garner HR. Molecular origins of rapid and continuous morphological evolution. Proc Natl Acad Sci U S A. 2004;101:18058–63. doi: 10.1073/pnas.0408118101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kirkness EF. SINEs of canine genomic diversity. In: Ostrander EAUG, Lindblad-Toh K, editors. The Dog and Its Genome. 44. Cold Spring Harbor, NY: Cold Spring Harbor Press; 2005. pp. 209–19. [Google Scholar]
- 40.Patterson DF. Current Veterinary Therapy VII. Philadelphia: WB Saunders; 1980. A catalogue of genetic disorders of the dog. [Google Scholar]
- 41.Sargan D. IDID: inherited diseases in dogs: web-based information for canine inherited disease genetics. Mamm Genome. 2004;15:503–6. doi: 10.1007/s00335-004-3047-z. [DOI] [PubMed] [Google Scholar]
- 42.Meurs KM, Miller MW, Wright NA. Clinical features of dilated cardiomyopathy in Great Danes and results of a pedigree analysis: 17 cases (1990–2000) J Am Vet Med Assoc. 2001;218:729–32. doi: 10.2460/javma.2001.218.729. [DOI] [PubMed] [Google Scholar]
- 43.Werner P, Raducha MG, Prociuk U, et al. The keeshond defect in cardiac conotruncal development is oligogenic. Hum Genet. 2005;116:368–77. doi: 10.1007/s00439-004-1242-3. Epub 2005 Feb 12. [DOI] [PubMed] [Google Scholar]
- 44.Dokuparti MV, Pamuru PR, Thakkar B, et al. Etiopathogenesis of arrhythmogenic right ventricular cardiomyopathy. J Hum Genet. 2005;50:375–81. doi: 10.1007/s10038-005-0273-5. [DOI] [PubMed] [Google Scholar]
- 45.Pilichou K, Nava A, Basso C, et al. Mutations in desmoglein-2 gene are associated with arrhythmogenic right ventricular cardiomyopathy. Circulation. 2006;113:1171–9. doi: 10.1161/CIRCULATIONAHA.105.583674. [DOI] [PubMed] [Google Scholar]
- 46.Burkett EL, Hershberger RE. Clinical and genetic issues in familial dilated cardiomyopathy. J Am Coll Cardiol. 2005;45:969–81. doi: 10.1016/j.jacc.2004.11.066. [DOI] [PubMed] [Google Scholar]
- 47.Stabej P, Imholz S, Versteeg SA, et al. Characterization of the canine desmin (DES) gene and evaluation as a candidate gene for dilated cardiomyopathy in the Dobermann. Gene. 2004;340:241–9. doi: 10.1016/j.gene.2004.06.050. [DOI] [PubMed] [Google Scholar]
- 48.Stabej P, Leegwater PA, Imholz S, et al. The canine sarcoglycan delta gene: BAC clone contig assembly, chromosome assignment and interrogation as a candidate gene for dilated cardiomyopathy in Dobermann dogs. Cytogenet Genome Res. 2005;111:140–6. doi: 10.1159/000086383. [DOI] [PubMed] [Google Scholar]
- 49.Stabej P, Leegwater PA, Stokhof AA, et al. Evaluation of the phospholamban gene in purebred large-breed dogs with dilated cardiomyopathy. Am J Vet Res. 2005;66:432–6. doi: 10.2460/ajvr.2005.66.432. [DOI] [PubMed] [Google Scholar]
- 50.Meurs KM, Magnon AL, Spier AW, et al. Evaluation of the cardiac actin gene in Doberman Pinschers with dilated cardiomyopathy. Am J Vet Res. 2001;62:33–6. doi: 10.2460/ajvr.2001.62.33. [DOI] [PubMed] [Google Scholar]
- 51.Clark LA, Tsai KL, Steiner JM, et al. Chromosome-specific microsatellite multiplex sets for linkage studies in the domestic dog. Genomics. 2004;84:550–4. doi: 10.1016/j.ygeno.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 52.Boehnke M, Cox NJ. Accurate inference of relationships in sib-pair linkage studies. Am J Hum Genet. 1997;61:423–9. doi: 10.1086/514862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun L, Wilder K, McPeek MS. Enhanced pedigree error detection. Hum Hered. 2002;54:99–110. doi: 10.1159/000067666. [DOI] [PubMed] [Google Scholar]
- 54.O’Connell JR, Weeks DE. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet. 1998;63:259–66. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lathrop GM, Lalouel JM, Julier C, Ott J. Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci U S A. 1984;81:3443–6. doi: 10.1073/pnas.81.11.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ott J. How do you compute a lod score? Nature genetics. 1995;11:354–5. doi: 10.1038/ng1295-354. [DOI] [PubMed] [Google Scholar]
- 57.Cottingham RW, Jr, Idury RM, Schäffer AA. Faster sequential genetic linkage computations. Am J Hum Genet. 1993;53:252–63. [PMC free article] [PubMed] [Google Scholar]
- 58.Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES. Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet. 1996;58:1347–63. [PMC free article] [PubMed] [Google Scholar]
- 59.Markianos K, Daly MJ, Kruglyak L. Efficient multipoint linkage analysis through reduction of inheritance space. Am J Hum Genet. 2001;68:963–77. doi: 10.1086/319507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Andelfinger G, Wright KN, Lee HS, et al. Canine tricuspid valve malformation, a model of human Ebstein anomaly, maps to dog chromosome 9. J Med Genet. 2003;40:320–4. doi: 10.1136/jmg.40.5.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Patterson DF, Pexieder T, Schnarr WR, et al. A single major-gene defect underlying cardiac conotruncal malformations interferes with myocardial growth during embryonic development: studies in the CTD line of keeshond dogs. Am J Hum Genet. 1993;52:388–97. [PMC free article] [PubMed] [Google Scholar]
- 62.Vollmar AC. The prevalence of cardiomyopathy in the Irish wolfhound: a clinical study of 500 dogs. J Am Anim Hosp Assoc. 2000;36:125–32. doi: 10.5326/15473317-36-2-125. [DOI] [PubMed] [Google Scholar]
- 63.Mignot E, Wang C, Rattazzi C, et al. Genetic linkage of autosomal recessive canine narcolepsy with a mu immunoglobulin heavy-chain switch-like segment. Proc Natl Acad Sci USA. 1991;88:3475–8. doi: 10.1073/pnas.88.8.3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lindblad-Toh K, Wade CM, Mikkelsen TS, et al. Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature. 2005;438:803–19. doi: 10.1038/nature04338. [DOI] [PubMed] [Google Scholar]
- 65.Breen M, Jouquand S, Renier C, et al. Chromosome-specific single-locus FISH probes allow anchorage of an 1800-marker integrated radiation-hybrid/linkage map of the domestic dog genome to all chromosomes. Genome Res. 2001;11:1784–95. doi: 10.1101/gr.189401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Breen M, Bullerdiek J, Langford CF. The DAPI banded karyotype of the domestic dog (Canis familiaris) generated using chromosome-specific paint probes. Chromosome Res. 1999;7:401–6. doi: 10.1023/a:1009224232134. [DOI] [PubMed] [Google Scholar]
- 67.Yang F, O’Brien PC, Milne BS, et al. A complete comparative chromosome map for the dog, red fox, and human and its integration with canine genetic maps. Genomics. 1999;62:189–202. doi: 10.1006/geno.1999.5989. [DOI] [PubMed] [Google Scholar]
- 68.Kirkness EF, Bafna V, Halpern AL, et al. The dog genome: survey sequencing and comparative analysis. Science. 2003;301:1898–903. doi: 10.1126/science.1086432. [DOI] [PubMed] [Google Scholar]
- 69.Hyun C, Filippich LJ, Lea RA, et al. Prospects for whole genome linkage disequilibrium mapping in domestic dog breeds. Mamm Genome. 2003;14:640–9. doi: 10.1007/s00335-003-3006-0. [DOI] [PubMed] [Google Scholar]
- 70.Lou XY, Todhunter RJ, Lin M, et al. The extent and distribution of linkage disequilibrium in a multi-hierarchic outbred canine pedigree. Mamm Genome. 2003;14:555–64. doi: 10.1007/s00335-003-2272-1. [DOI] [PubMed] [Google Scholar]
- 71.Sutter NB, Eberle MA, Parker HG, et al. Extensive and breed-specific linkage disequilibrium in Canis familiaris. Genome Res. 2004;14:2388–96. doi: 10.1101/gr.3147604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kruglyak L. Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat Genet. 1999;22:139–44. doi: 10.1038/9642. [DOI] [PubMed] [Google Scholar]
- 73.Vila C, Savolainen P, Maldonado JE, et al. Multiple and ancient origins of the domestic dog [see comments] Science. 1997;276:1687–9. doi: 10.1126/science.276.5319.1687. [DOI] [PubMed] [Google Scholar]
- 74.Savolainen P, Zhang YP, Luo J, et al. Genetic evidence for an East Asian origin of domestic dogs. Science. 2002;298:1610–3. doi: 10.1126/science.1073906. [DOI] [PubMed] [Google Scholar]
- 75.Ostrander EA, Jong PM, Rine J, Duyk G. Construction of small-insert genomic DNA libraries highly enriched for microsatellite repeat sequences. Proc Natl Acad Sci U S A. 1992;89:3419–23. doi: 10.1073/pnas.89.8.3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Famula TR, Siemens LM, Davidson AP, Packard M. Evaluation of the genetic basis of tricuspid valve dysplasia in Labrador Retrievers. Am J Vet Res. 2002;63:816–20. doi: 10.2460/ajvr.2002.63.816. [DOI] [PubMed] [Google Scholar]
- 77.Meurs KM, Spier AW, Miller MW, et al. Familial ventricular arrhythmias in boxers. J Vet Intern Med. 1999;13:437–9. doi: 10.1892/0891-6640(1999)013<0437:fvaib>2.3.co;2. [DOI] [PubMed] [Google Scholar]
- 78.Crook A, Hill B, Dawson S. Canine Inherited Disorders Database. 1998. [PubMed] [Google Scholar]