

Abstract
We investigated whether antibodies against intracellular tumor-associated antigens support tumor-specific immunity when administered together with a treatment that destroys the tumor. We propose that released antigens form immune complexes with the antibodies, which are then efficiently taken up by dendritic cells. We cloned the first human monoclonal antibodies against the Cancer/Testis (CT) antigen, NY-ESO-1. We tested whether the monoclonal anti-NY-ESO-1 antibody (12D7) facilitates cross-presentation of a NY-ESO-1-derived epitope by dendritic cells to human CD8+ T cells, and whether this results in the maturation of dendritic cells in vitro. We investigated the efficacy of 12D7 in combination with chemotherapy using BALB/c mice bearing syngeneic CT26 tumors that express intracellular NY-ESO-1. Human dendritic cells that were incubated with NY-ESO-1:12D7 immune complexes efficiently stimulated NY-ESO-1157–165/HLA-A2-specific human CD8+ T cells to produce interferon-γ, whereas NY-ESO-1 alone did not. Furthermore, the incubation of dendritic cells with NY-ESO-1:12D7 immune complexes resulted in the maturation of dendritic cells. Treatment of BALB/c mice that bear CT26/NY-ESO-1 tumors with 5-fluorouracil (5-FU) plus 12D7 was significantly more effective than chemotherapy alone. We propose systemic injection of monoclonal antibodies (mAbs) against tumor-associated antigens plus a treatment that promotes the local release of those antigens resulting in immune complex formation as a novel therapeutic modality for cancer.
Keywords: NY-ESO-1, antibody, chemotherapy
Introduction
Cancer/Testis (CT) antigens form an extended family of proteins that are frequently expressed in a large variety of malignancies but are absent from healthy tissue, except for the testis and placenta. Cancer patients often develop spontaneous immune responses toward CT antigens, which illustrate their immunogenicity (1–3). Their apparent immunogenicity and unique expression pattern make CT antigens attractive targets for immunotherapy, and a number of clinical trials in which cancer patients were immunized with CT antigens in different forms have been completed, some of which show objective clinical responses (4–12).
Dendritic cell (DC) maturation is a key prerequisite for the activation of T cells, and moreover, antigen presentation by steady-state DCs results in peripheral tolerance induction, a process that is considered crucial for the protection against autoimmunity (13, 14). DC maturation usually is induced by infection or inflammation—or by adjuvants for that matter—and can be a local event. Insufficient maturation of tumor-associated DCs may be one of multiple reasons for the compromised response of tumor-infiltrating T cells compared to peripheral T cells (15, 16). Cross-presentation of sufficient amounts of tumor-derived antigens may be another limiting factor, especially because the number of tumor-associated DCs often is low and cross-presentation is inefficient (17, 18). Therefore, we developed a novel immunotherapeutic approach that combines enhanced cross-presentation of epitopes derived from intracellular proteins with concomitant DC maturation. We hypothesized that administration of monoclonal antibodies (mAbs) against CT antigens together with a therapy that releases these usually intracellular antigens may support the local formation of immune complexes, which are efficiently taken up by DCs (19, 20) resulting in increased presentation of CT antigen-derived epitopes to CD8+ T cells. Because there is evidence that the uptake of immune complexes by DCs through the activating receptor for IgG (FcγRIIA) results in DC maturation (21), the use of mAbs against CT antigens may serve both purposes: DC activation and enhanced cross-presentation.
The fact that NY-ESO-1 is one of the best-characterized and most immunogenic CT antigens known to date (22, 23) and is frequently expressed by tumors of different origin (6, 24) prompted us to clone human-derived mAbs against NY-ESO-1 from patients who had high serum levels of NY-ESO-1-specific IgG and, thus, presumably a high frequency of NY-ESO-1-specific B cells. The obvious advantage of cloning a therapeutic antibody from humans is that adverse side effects of such an antibody are very unlikely and that it therefore can relatively be quickly tested in clinical trials. We report here the generation of the first human-derived IgG1 mAbs against NY-ESO-1 and the selection of a lead development candidate (12D7). We show that 12D7 facilitates cross-presentation of a NY-ESO-1-derived epitope to CD8+ T cells, that 12D7:NY-ESO-1 immune complexes induce the maturation of human monocyte-derived DCs in vitro, and that 12D7 significantly enhances the therapeutic efficacy of chemotherapy using a preclinical syngeneic mouse model.
Results
Cloning of human-derived monoclonal antibodies from cancer patients
We cloned eight different NY-ESO-1-specific human-derived monoclonal antibodies (HD mAbs) from a melanoma patient, of which the following five were selected for further analysis based on their affinity to the target: 1D4, 12D7, 15B12, 30D6, and 31E4. All HD mAbs were of IgG1 isotype.
In vitro characterization of HD mAbs
To compare the binding properties of five different anti-NYESO-1 HD mAbs to recombinant NY-ESO-1 protein, we determined the half-maximal effective concentration (EC50) using a protein ELISA. All antibodies bound recombinant NY-ESO-1 produced in bacteria in the low pM range. Actual binding constants to recombinant NY-ESO-1 produced in bacteria and in eukaryotic cells were determined by surface plasmon resonance (Biacore Systems) (Table 1).
Table 1.
Binding of human monoclonal anti-NY-ESO-1 antibodies to NYESO-1. Comparison of EC50 and equilibrium affinity constants for the binding between NY-ESO-1 and different anti-NY-ESO-1 antibodies.
| Antibody | EC50 [pM] (prok. NY-ESO-1) | KD [M] (prok. NY-ESO-1) | KD [M] (euk. NY-ESO-1) |
|---|---|---|---|
| 12D7 | 1.14 | 2.08×10−10 | 1.56×10−10 |
| 1D4 | 2.23 | 1.62×10−9 | 2.24×10−10 |
| 30D6 | 1.09 | 4.35×10−9 | 2.65×10−9 |
| 31E4 | 9.52 | 1.9×10−8 | 2.23×10−8 |
| 15B12 | 72.6 | --- | --- |
| E978 control | 6.66 | 2.56×10−8 | 1.56×10−11 |
To determine the epitopes recognized by the different mAbs, we used a set of overlapping peptides spanning the complete NY-ESO-1 protein as coating antigen in ELISA. As shown in Figure 1A, 12D7 binds to a peptide representing the amino acids 11 to 30 from the NY-ESO-1 protein, but not to the two adjacent peptides that span amino acids 1–20 or 21–40. This suggests that the epitope recognized by 12D7 lies at the junction of these two peptides around amino acid 20 of NY-ESO-1. Figure 1B summarizes the epitope-specificity of all five anti-NY-ESO-1 antibodies. In addition, all antibodies were tested for binding to endogenous NY-ESO-1 from the human melanoma cell line SKMEL-37 by immunoprecipitation. All antibodies precipitate NYESO-1 from a cell lysate of an NY-ESO-1+ cell line (SK-MEL-37) (Figure 1C). Because 12D7 had the highest affinity for eukaryotic NY-ESO-1, we performed further experiments with this mAb.
Figure 1.

Epitope mapping of anti-NY-ESO-1 human monoclonal antibodies. (A) Representative peptide ELISA for antibody 12D7, where P1–P17 represent overlapping NYESO-1 peptides. (B) Overview of the specificities of different NY-ESO-1 specific human-derived mAbs. (C) Immunoprecipitation of NY-ESO-1 from a cell lysate of a NY-ESO-1+ cell line SK-MEL-37 or a NY-ESO-1-cell line A549 by human anti-NY-ESO-1 mAbs.
12D7 facilitates cross-presentation of NY-ESO-1 by DCs and induces concomitant DC maturation
To test whether 12D7 facilitates the cross-presentation of NYESO-1-derived epitopes in vitro, we generated monocyte-derived, HLA-A*0201+ DCs and fed them with 12D7:NY-ESO-1 immune complexes, NY-ESO-1, or 12D7. DCs were subsequently incubated with cloned NY-ESO-1157–165/HLAA*0201-specific CD8+ T cells, and the percentage of T cells that produced IFN-γ was used as readout for antigen recognition. Mature DCs fed with NY-ESO-1 protein induced IFN-γ production in a low but discernible percentage of T cells (Figure 2A, black bars), which did not occur when DCs were not matured (Figure 2A, white bars). DCs fed with 12D7:NY-ESO-1 immune complexes induced the production of IFN-γ in a much higher percentage of T cells and, importantly, also did so when DCs that were not deliberately matured were used (Figure 2A, compare black and white bars). None of the negative controls—DCs fed with 12D7, mock immune complexes, or medium—induced IFN-γ production (Figure 2A and data not shown). To exclude that our observations are a peculiarity of recombinant NY-ESO-1, we incubated 12D7 with a cell lysate of SK-MEL-37 cells, which naturally express NY-ESO-1, and subsequently fed this mixture to DCs. DCs fed with the 12D7:lysate or with 12D7:NY-ESO-1 presented NY-ESO-1-derived epitopes approximately equally well (Figure 2B).
Figure 2.

Human monoclonal anti-NY-ESO-1 antibody (12D7) facilitates cross-presentation of a NY-ESO-1-derived, HLA-A2-restriced epitope (NY-ESO-1157–165). (A) HLA-A2+, monocyte-derived DCs were incubated with 20 μg NY-ESO-1 protein, 200 μg human monoclonal anti-NY-ESO-1 antibody (12D7), with immune complexes (12D7:NY-ESO-1) or with media for 3 h, were washed and cultured for 36 h with (black bars) or without (white bars) 25 ng/mL TNF-α + 1 μg/mL sCD40L (maturation cocktail). 6 x 104 cloned, NY-ESO-1157–165 /HLA-A2-specific CD8+ T cells were added to 105 DCs in the presence of 10 μg/mL Brefeldin A, followed by a 5 h incubation and subsequent surface staining for CD8 and intracellular staining for IFN-γ. 10−6 M peptide was added to DCs as positive control. All cultures were performed in triplicate. (B) HLA-A2+, monocyte-derived DCs were incubated with 20 μg NY-ESO-1 protein, 200 μg human monoclonal anti-NY-ESO-1 antibody (12D7), with lysate of 107 NY-ESO-1+ SK-MEL-37 cells (lysate), with immune complexes (NY-ESO-1:12D7 or lysate:12D7), or with media (0) for 3 h, were washed and cultured for 36 h with 25 ng/mL TNF-α + 1 μg/mL sCD40L (maturation cocktail). 6 x 104 cloned, NY-ESO-1157–165 /HLA-A2-specific CD8+ T cells were added to 105 DCs in the presence of 10 μg/mL Brefeldin A, followed by a 5 h incubation and subsequent surface staining for CD8 and intracellular staining for IFN-γ. All cultures were performed at least in duplicate.
Because presentation of 12D7:NY-ESO-1 immune complexes seemed not to require deliberate DC maturation, we addressed whether the uptake of immune complexes, but not the uptake of uncomplexed protein, induced DC maturation in vitro. We therefore compared the expression of three surface molecules that are upregulated on mature DCs (CD83, CD86, and MHC class II) after incubation with media, 12D7, NY-ESO-1, or with 12D7:NY-ESO-1 immune complexes in the absence of maturation cocktail. We found that only immune complexes induced an upregulation of CD86, CD83, and MHC II (Figure 3A; left, middle, and right panels, respectively). We then compared the expression of CD83, CD86, and MHC II on DCs that were incubated with the maturation cocktail, with immune complexes, or with both, in order to determine the relative potency of immune complexes with respect to DC maturation. We found that immune complexes were almost as potent in inducing DC maturation as the classical maturation cocktail (sCD40L plus TNF-α) (Figure 3B). A combination of immune complexes plus maturation cocktail resulted in the most pronounced upregulation of CD86 and CD83 (Figure 3B; left and middle panels, respectively), whereas MHC II was not further upregulated compared to any of the two treatments alone (Figure 3B, right panels).
Figure 3.
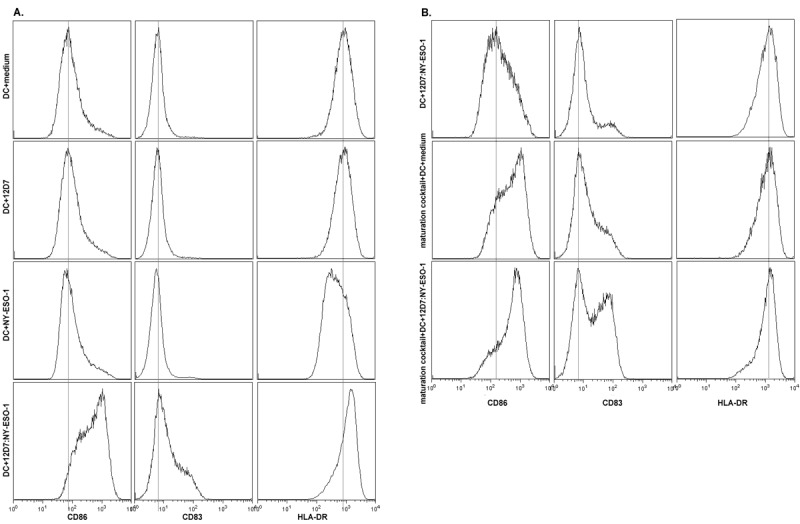
Immune complexes induce maturation of monocyte-derived DCs in vitro. (A) CD14-derived DCs were incubated with media, 200 μg NY-ESO-1, 20 μg 12D7 or pre-formed immune complexes of 20 μg 12D7 + 200 μg NY-ESO-1, and were analyzed 36 h later for surface expression of CD86, CD83, or MHC II. (B) CD14-derived DCs were incubated with preformed immune complexes of 20 μg 12D7 + 200 μg NY-ESO-1, maturation cocktail (sCD40L + TNF-α) or with preformed immune complexes plus maturation cocktail, and were analyzed 36 h later for surface expression of CD86, CD83, or MHC II.
12D7 increases the therapeutic efficacy of chemotherapy in mice with NY-ESO-1+ tumors
To test the therapeutic efficacy of 12D7 in vivo, we injected 106 syngeneic, NY-ESO-1-transfected CT26 tumor cells s.c. in BALB/c mice. To induce release of intracellular NY-ESO-1, mice were treated with 75 mg/kg 5-FU when tumors reached a size of approximately 25 mm2, which was typically around 2 weeks after injection of tumor cells. The treatment with 5-FU was repeated one week later and, in some groups, was combined with 100 μg 12D7 given systemically 2 d after each 5-FU injection. As can be seen from the growth curves, 5-FU has the expected therapeutic effect. Importantly, this was enhanced by 12D7 (Figure 4A). Treatment with 12D7 alone had no effect, presumably because the amount of spontaneously released antigen is not sufficient in this particular model. A compilation of end-point tumor sizes from 4 independent experiments shows a highly significant difference between mice treated with 5-FU plus 12D7, and mice treated with 5-FU alone (Figure 4B).
Figure 4.
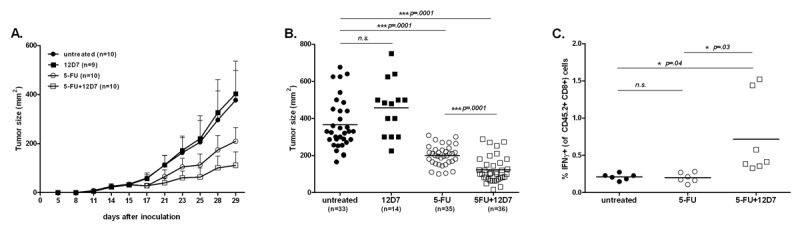
A human, monoclonal anti-NY-ESO-1 antibody (12D7) increases the therapeutic efficacy of 5-FU chemotherapy in mice bearing NY-ESO-1+ syngeneic tumors. Female BALB/c mice were injected s.c. with 106 CT26/NY-ESO-1+ cells and treatment was started when tumors reached a surface of approximately 25 mm2 (∼ d13–15). (A) Mice received 75 mg/kg 5-FU i.p. at days 15 and 22 and/or 100 μg 12D7 i.p. on days 17 and 24. The results are shown as mean±SD. A representative experiment of 4 experiments is shown. (B) Compilation of 4 independent experiments, each symbol represents the tumor surface of an individual mouse at the end of the experiment (d 29). (C) Mice were injected i.p. 1 week after the last injection with 12D7 (d 29) with 250 μg Brefeldin A and were euthanized 4 h later. Processing of tumors and staining with antibodies for CD45.2, CD8 (surface), and IFN-γ (intracellular) was performed in the presence of 10 μg/mL Brefeldin A. Each symbol represents values from individual mice at the end of the experiment.
To investigate whether treatment with 5-FU plus 12D7 supported tumor-specific immunity, we injected mice with Brefeldin A 4 h before euthanasia, followed by staining for CD45.2, CD8, and intracellular IFN-γ. This way of analysis shows which cells actually are making IFN-γ in vivo and not which cells potentially can do this upon in vitro restimulation with peptide. This method obviously does not allow discrimination between single peptide specificities, but it is of higher biological relevance (25) particularly because we envisaged that DC activation, which we have shown to occur upon cross-presentation (Figure 3), may also support the presentation of other epitopes besides those derived from NYESO-1. Treatment with 5-FU plus 12D7 supported CD8+ and effector function in the tumor (Figure 4C). Treatment with 5-FU (Figure 4C) or 12D7 (data not shown) did not have this effect.
Discussion
We hypothesized that antibodies against intracellular, tumor-associated antigens support tumor-specific immunity when used in combination with a therapy that induces cell death such as chemo- or radiotherapy. We envisaged that such antibodies form immune complexes with the released tumor antigens. These immune complexes are subsequently taken up with higher efficiency compared to protein (fragments) by DCs (26), which then cross-present relevant epitopes to local CD8+, tumor-specific T cells. This presumed sequence of events may be of particular interest as evidence is accumulating that both chemo- and radiotherapy support tumor-specific immunity (27), and we therefore reasoned that additional stimulation of tumor-specific immunity could further improve the efficacy of these standard therapies.
For this purpose, we have cloned the first fully human mAbs to NY-ESO-1 using Epstein-Barr virus (EBV)-transformed B cells from a melanoma patient and subjected those to preclinical experiments to obtain proof of principle. We found that 12D7, a fully human IgG1 mAb specific for the immunogenic CT antigen NY-ESO-1, supported cross-presentation of NY-ESO-1 in vitro resulting in an approximate 15-fold increase of the number of responding CD8+ T cells. Of the other four NY-ESO-1-specific mAbs we generated here, 1D4 and 30D6 improved cross-presentation of NY-ESO-1 (data not shown), whereas 15B12 and 31E4 seemed not effective (data not shown). This difference may be explained by the difference in affinity, as 15B12 did not show binding to NY-ESO-1 by Biacore—although it did bind weakly to NY-ESO-1 in ELISA—and 31E4 had at least a 1-log lower affinity than 12D7, 1D4, and 30D6. At present, we have no reason to think that the epitope recognized by the mAb impacts on its ability to support cross-presentation. Our observation that 12D7:NY-ESO-1 immune complexes are considerably less efficient than peptide-loaded DCs in stimulating IFN-γ production illustrates that cross-presentation is a rather inefficient process, but underscores the therapeutic potential of antibodies against tumor-associated antigens.
It is well accepted now that activation of T cells in vivo crucially depends on antigen presentation by mature or activated DCs (14, 28). Many cues, including inflammation and infection but also endogenous signals, can induce DC maturation (29), and the lack of such signals in the tumor environment may be one reason why tumor-infiltrating T cells often have compromised functions (16, 30). Because the uptake of immune complexes was shown to result in DC maturation (19), we specifically addressed this issue here. We found that the in vitro uptake of immune complexes resulted in DC maturation that was comparable to sCD40L plus TNF-α, which is a classical maturation cocktail. Therefore, the use of mAbs against CT antigens may serve both purposes: DC activation and enhanced cross-presentation at the relevant anatomic location. This is not trivial, as systemic activation of DCs may not be without risk as systemic side effects such as the release of cytokines or autoimmunity may ensue (31, 32).
We found that 12D7 improved the efficacy of chemotherapy in a preclinical mouse model of transplanted, syngeneic NY-ESO-1-expressing tumors, thus supporting our concept. Further support comes from the fact that more CD8+ T cells infiltrate the tumor and that those cells have increased effector function. By itself, however, 12D7 had no therapeutic effect, suggesting that the amount of released tumor antigen is limiting without deliberate destruction of the tumor. Our in vivo experiments require the binding of human IgG to mouse Fcγ receptors (FcγR), which was previously described (33, 34). Improved efficacy of chemotherapy by the use of tumor-associated antigen-specific antibodies will presumably work for chemotherapies especially, which are not immunosuppressive or—even more important—promote immunogenic cell death (35).
We propose the concept of antibody-facilitated T cell induction in cancer (AFTIC) as a novel type of immunotherapy. AFTIC is based on the application of mAbs against tumor-associated antigens, including CT antigens, plus a treatment that promotes the local release of those antigens, such as chemo- or radiotherapy. The locally released antigens and the mAb form immune complexes, which facilitate the uptake and subsequent presentation of antigen-derived peptides by tumor-associated DCs. As the uptake of immune complexes induces concomitant maturation of DCs, AFTIC supports boosting as well as de novo activation of tumor-specific CD8+ T cells. Furthermore, administration of antibodies against a particular tumor-associated antigen may promote the presentation of the same antigen when administrated as a cancer vaccine, thereby improving the efficacy of immunotherapy. Alternatively, better antigen presentation of immune complexes and concomitant DC maturation may support the activity of adoptively transferred T cells provided they have the same antigen specificity as the therapeutic antibodies.
Acknowledgments
We thank Alexandre Ruffieux and Victor Escalante (Institute of Laboratory Animal Science, University of Zürich) for expert animal care. This work was supported by Atlantic Philanthropies (New York, USA), the Cancer Research Institute, the Science Foundation for Oncology (Zürich), the Ellinger Foundation Zürich, the Hartmann Müller Foundation Zürich, and the Hanne Liebermann Foundation (Zürich).
Abbreviations
- CT
Cancer/Testis;
- DC
dendritic cell;
- mAb
monoclonal antibody
Materials and methods
Patient material
Serum and peripheral blood was collected from cancer patients. All patients were admitted at the University Hospital Zürich and provided written informed consent in accordance with the Declaration of Helsinki. The local ethics committee approved the study.
Memory B cell culture
PBMC were incubated with anti-CD22 coupled to magnetic beads (Miltenyi Biotec), PE-conjugated anti-IgD, and APC-conjugated antibodies to IgM, CD3, CD8, and CD56 (Becton Dickinson). B cells were isolated by positive selection of CD22+ cells using a midi-MACS device and LS columns (Miltenyi Biotec), followed by sorting PE-APC-cells using a MoFlo cell sorter (Beckman Coulter). CD22+ IgD- IgM- memory B cells were incubated with 10% EBV-containing supernatant from B95-8 cells (from European Collection of Cell Cultures, ECACC) in the presence of 2.5 μg/mL CpG 2006 at 37°C for 4 h. Cells were seeded in 96-well U-bottom plates at 10 cells per well plus 3 x 104 irradiated allogeneic PBMCs in RPMI 1640 medium supplemented with 10% human serum, antibiotics, 10% supernatant from B95-8 cells, and 2.5 μg/mL CpG 2006. Supernatants were tested for NY-ESO-1-specific antibodies after 2 weeks by ELISA.
Single cell-RT-PCR
B cell cultures were harvested and single cells were deposited into a 96-well PCR plate (Applied Biosystems) using a MoFlo XDP cell sorter (Beckman Coulter). RT-PCR was performed using random hexamer primers for cDNA synthesis and specific primers to amplify the immunoglobulin variable and constant regions. Immunoglobulin heavy and light chain variable regions were amplified using a nested PCR approach as described (36). Primer-encoded amino acid sequences and J-C regions of the antibodies were corrected to represent the authentic amino acid sequence as it occurred in the patient in a subsequent step prior to antibody production.
Antibody production and purification
293-T human embryonic kidney cells were transfected with 25 kDa branched polyethylenimine (PEI, Polysciences, Warrington, PA) plus DNA plasmids (heavy and light chain in equal ratios) in a 1.3:1 ratio and were incubated for 15 min at room temperature. Following transfection, the cells were cultured in serum free Opti-MEM I + GlutaMAX-I (Invitrogen) supplemented with 10 U penicillin-streptomycin (Lonza, Switzerland). After 72 h supernatants were collected and IgG was purified on a protein A column (GE Healthcare, Sweden) using FPLC (GE Healthcare, Sweden).
Biacore analysis
Antibody binding kinetics with NY-ESO-1 proteins derived from E. coli (LICR New York Branch) and HEK293 cells (OriGene Technologies, Inc.) were determined by Biacore technology (model Biacore 2000; Biacore AB) using CM5 sensor chips, EDC-NHS conjugation, and BIAevaluation software. Technical details have been described previously (37).
ELISA
• Protein or peptide ELISA
96-well half-area microtiter plates (Costar, USA) were coated with 30 μL/well of 1 μg/mL recombinant NY-ESO-1 protein, or 10 μg/mL 20-mer peptides spanning the entire NY-ESO-1 protein (Peptides & Elephants, Germany) diluted in PBS overnight at 4°C. After coating, plates were washed with PBS + 0.05% Tween-20 (PBS-T) and blocked for 1 h at room temperature with 2% BSA/PBS (Sigma). B cell-conditioned medium, patient serum, or recombinant antibody was incubated for 2 h at room temperature (RT) at indicated concentrations or dilutions in PBS. Plates were washed with PBS-T and incubated for 1 h at RT with HRPO-conjugated goat-anti-human Fcγ antibody (Jackson ImmunoResearch), diluted 1:4000 in 0.5% BSA/PBS, followed by measurement of the HRPO activity using a TMB substrate solution (Sigma, Buchs, Switzerland). The mouse IgG1 monoclonal anti-NY-ESO-1 antibody E978 (38) and HRPO-conjugated goat-anti-mouse Fcγ antibody (Jackson ImmunoResearch) at 1:4000 dilution in 0.5% BSA/PBS served as positive control.
• Cellular ELISA
4 x 104 SK-MEL-37 cells were seeded in a 96-well flat bottom plate and cultured under standard conditions overnight. Cells were fixed in ice-cold ethanol/acetone mix (1:1) for 15 min on ice. After two wash steps with PBS, cells were blocked and permeabilized with 100 μL of PBS + 0.5% BSA + 0.5% Triton X 100 for 2 h at 4°C. B cell-conditioned medium or recombinant antibody was incubated at indicated concentrations for 2 h at 4°C. Bound antibodies were detected after 1 h incubation at 4°C with HRPO-labeled goat anti-human Fc secondary antibody (Jackson ImmunoResearch).
Immunoprecipitation
SK-MEL-37 tumor cells were lysed with Triton X 100/Glycerol-based lysis buffer for 15 min at 4°C. Cell debris was separated by centrifugation at maximum speed in a table centrifuge and protein concentration of the supernatant analyzed by standard Bradford assay. 300 ng of antibody was used to precipitate NY-ESO-1 from 250 μg of SK-MEL-37 cell lysate in a 16 h incubation at 4°C. The immune complex was isolated by adding magnetic Protein G beads (New England Biolabs, Ipswich, MA) for 1 h at 4°C under constant agitation. Beads were washed, resuspended in NuPAGE LDS sample buffer (Invitrogen) and boiled prior to Gradient SDS Polyacrylamide Gel Electrophoresis (NuPAGE 4-12% Bis-Tris Gel, Invitrogen). NY-ESO-1 protein was detected by Western blot using murine antibody E978 (38).
In vitro cross-presentation assay
Human, HLA-A*0201/NY-ESO-1157–165 specific CD8+ T cells were cloned as previously described (39). To generate DCs, CD14+ cells were MACS-purified according to the manufacturer’s instructions (Miltenyi Biotech) from PBMC from HLA-A*0201+ healthy donors and cultured at 106 cells/mL in serum-free CellGro DC media (CellGenix), supplemented with 800 U/mL GM-CSF and 500 U/mL IL-4 (R&D Systems) to generate DCs. Medium was exchanged the following day and DCs were harvested on d 4 of culture and resuspended at 106/mL in Opti-MEM (Gibco). Immune complexes were generated by incubating 20 μg recombinant NY-ESO-1 with 200 μg 12D7 in a total volume of 500 μL Opti-MEM (Gibco) for 30 min at 37°C. Human IgG1 (Sigma Aldrich), 12D7 alone, or NY-ESO-1 alone were used as controls. Alternatively, 200 μg 12D7 was incubated with a lysate of an equivalent of 107 NY-ESO-1+ SKMEL-37 cells in 500 μL Opti-MEM. DCs (5x105 in 0.5 mL Opti-MEM) were added to the immune complexes and controls. The mixture was incubated at 37°C for 3 h. DCs were then centrifuged and resuspended in CellGro DC media at 106/mL. Hundred μL (105 DCs) were cultured in 96-well flatbottom plates at 37°C in the presence or absence of maturation cocktail (1 μg/mL soluble CD40L (sCD40L) trimer (PeproTech) plus TNF-α (25 ng/mL; R&D Systems)). After 36 h, approximately 6 x 105 HLA-A*0201/NY-ESO-1157–165 specific CD8+ T cells in 100 μL RPMI + 10% human serum + antibiotics + 20 μg/mL Brefeldin A (Sigma Chemicals) were added to the different DC-cultures. After 4 h, cultures were harvested in FACS buffer (PBS + 2% FCS + 2 mM EDTA + 0.05% NaN3) and surface stained with anti-CD8 followed by intracellular staining for IFN-γ as previously described (39). CD8+ T cells plus unloaded DCs served as negative control, and CD8+ T cells plus DCs with 10−6 M of NY-ESO-1157–165 (SLLMWITQC, Thermo Fisher Scientific) served as positive control. All cultures were performed at least in duplicates.
Mice and cell lines
BALB/c mice were originally obtained from Jackson Laboratories and were bred and kept under specific pathogen-free conditions in the Institute of Laboratory Animal Sciences (University of Zürich). Age- and sex-matched mice of 9–12 weeks old were used for all experiments. Mice were housed under specific pathogen-free conditions at University Hospital Zürich. All experiments were performed in agreement with the federal and cantonal laws on animal protection.
The colon carcinoma cell line CT26 was transfected to stably express intracellular NY-ESO-1 (40) and was cultured in RPMI + 10% FCS + antibiotics + 10 μg/mL puromycin under standard tissue culture conditions. CT26/NY-ESO-1 and the human melanoma cell line SK-MEL-37 were cultured in RPMI + 10% FCS + antibiotics under standard tissue culture conditions. 293-T cells were cultured in DMEM (Lonza, Switzerland) supplemented with 10% FCS (Linaris) and 10 U penicillin-streptomycin (Lonza, Switzerland) under standard tissue culture conditions.
Treatment of mice
Mice were injected s.c. into the right flank with 106 CT26/NYESO-1+ cells in 100 μL RPMI. The tumor surface was measured at least twice a week with a calliper. Treatment was started (d 0) when tumors reached a size of approximately 25 mm2. 5-Fluorouracil (5-FU, TEVA Pharma, Aesch, Switzerland) was diluted in saline and were given i.p. on d 0 and d 7 at 75 mg/kg, respectively. 12D7 (100 μg in 100 μL PBS) was given i.p. on d 2 and d 9. All animal experiments were performed in accordance with the Swiss federal and cantonal law on animal protection.
Flow cytometry
At the end of the experiment (1 week after the last injection of 12D7), mice were injected i.p. with 250 μg Brefeldin A and were euthanized 4 h later. Subsequent processing and staining was performed in the presence of 10 μg/mL Brefeldin A (25). Tumors were cut into small pieces and subsequently digested with 1.5 mg/mL collagenase + 100 μg/mL DNase for 1 h at 37°C followed by filtration through a 50 μm cell strainer. Single cell suspensions were surface stained in FACS buffer (FB, PBS + 2% FCS + 0.03% NaN3 + 20 mM EDTA) with anti-CD45.2 pacific blue and anti-CD8bPE. For intracellular staining to detect IFN-γ, cells were permeabilized with permeabilization buffer (PB, FB + 0.1% saponin) and stained intracellularly with anti-IFN-γ APC. All antibodies were obtained from BioLegend, San Diego, CA, USA. Samples were measured with a CyAn ADP9 (Beckman Coulter, Brea, CA, USA) and analyzed using FlowJo Analysis Software (Tree Star Inc., Ashland, OR, USA).
Statistical analysis
Statistics were done using an unpaired Student two-tailed t-test. Error bars represent SD. p values less than 0.05 were considered significant.
References
- 1.Jäger D, Jäger E, Knuth A. Immune responses to tumour antigens: implications for antigen specific immunotherapy of cancer. J Clin Pathol. 2001;54:669–674. doi: 10.1136/jcp.54.9.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Scanlan MJ, Güre AO, Jungbluth AA, Old LJ, Chen YT. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol Rev. 2002;188:22–32. doi: 10.1034/j.1600-065x.2002.18803.x. [DOI] [PubMed] [Google Scholar]
- 3.Scanlan MJ, Simpson AJ, Old LJ. The cancer/testis genes: review, standardization, and commentary. Cancer Immun. 2004;4:1. [PubMed] [Google Scholar]
- 4.Jäger E, Gnjatic S, Nagata Y, Stockert E, Jäger D, Karbach J, Neumann A, Rieckenberg J, Chen YT, Ritter G, Hoffman E, Arand E, Old LJ, Knuth A. Induction of primary NY-ESO-1 immunity: CD8+ T lymphocyte and antibody responses in peptide-vaccinated patients with NY-ESO-1+ cancers. Proc Natl Acad Sci U S A. 2000;97:12198–12203. doi: 10.1073/pnas.220413497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davis ID, Chen W, Jackson H, Parente P, Shackleton M, Hopkins W, Chen Q, Dimopoulos N, Luke T, Murphy R, Scott AM, Maraskovsky E, McArthur G, MacGregor D, Sturrock S, Tai TY, Green S, Cuthbertson A, Maher D, Miloradovic L, Mitchell SV, Ritter G, Jungbluth AA, Chen YT, Gnjatic S, Hoffman EW, Old LJ, Cebon JS. Recombinant NY-ESO-1 protein with ISCOMATRIX adjuvant induces broad integrated antibody and CD4(+) and CD8(+) T cell responses in humans. Proc Natl Acad Sci U S A. 2004;101:10697–10702. doi: 10.1073/pnas.0403572101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gnjatic S, Nishikawa H, Jungbluth AA, Güre AO, Ritter G, Jäger E, Knuth A, Chen YT, Old LJ. NY-ESO-1: review of an immunogenic tumor antigen. Adv Cancer Res. 2006;95:1–30. doi: 10.1016/S0065-230X(06)95001-5. [DOI] [PubMed] [Google Scholar]
- 7.Jäger E, Karbach J, Gnjatic S, Neumann A, Bender A, Valmori D, Ayyoub M, Ritter E, Ritter G, Jäger D, Panicali D, Hoffman E, Pan L, Oettgen H, Old LJ, Knuth A. Recombinant vaccinia/fowlpox NYESO-1 vaccines induce both humoral and cellular NY-ESO-1-specific immune responses in cancer patients. Proc Natl Acad Sci U S A. 2006;103:14453–14458. doi: 10.1073/pnas.0606512103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bender A, Karbach J, Neumann A, Jäger D, Al-Batran SE, Atmaca A, Weidmann E, Biskamp M, Gnjatic S, Pan L, Hoffman E, Old LJ, Knuth A, Jäger E. LUD 00-009: phase 1 study of intensive course immunization with NY-ESO-1 peptides in HLA-A2 positive patients with NY-ESO-1-expressing cancer. Cancer Immun. 2007;7:16. [PMC free article] [PubMed] [Google Scholar]
- 9.Nicholaou T, Ebert LM, Davis ID, McArthur GA, Jackson H, Dimopoulos N, Tan B, Maraskovsky E, Miloradovic L, Hopkins W, Pan L, Venhaus R, Hoffman EW, Chen W, Cebon J. Regulatory T-cell-mediated attenuation of T-cell responses to the NY-ESO-1 ISCO-MATRIX vaccine in patients with advanced malignant melanoma. Clin Cancer Res. 2009;15:2166–2173. doi: 10.1158/1078-0432.CCR-08-2484. [DOI] [PubMed] [Google Scholar]
- 10.Karbach J, Gnjatic S, Bender A, Neumann A, Weidmann E, Yuan J, Ferrara CA, Hoffmann E, Old LJ, Altorki NK, Jäger E. Tumor-reactive CD8+ T-cell responses after vaccination with NY-ESO-1 peptide, CpG 7909 and Montanide ISA-51: association with survival. Int J Cancer. 2010;126:909–918. doi: 10.1002/ijc.24850. [DOI] [PubMed] [Google Scholar]
- 11.Karbach J, Neumann A, Atmaca A, Wahle C, Brand K, von Boehmer L, Knuth A, Bender A, Ritter G, Old LJ, Jäger E. Efficient in vivo priming by vaccination with recombinant NY-ESO-1 protein and CpG in antigen naive prostate cancer patients. Clin Cancer Res. 2011;17:861–870. doi: 10.1158/1078-0432.CCR-10-1811. [DOI] [PubMed] [Google Scholar]
- 12.Valmori D, Souleimanian NE, Tosello V, Bhardwaj N, Adams S, O’Neill D, Pavlick A, Escalon JB, Cruz CM, Angiulli A, Angiulli F, Mears G, Vogel SM, Pan L, Jungbluth AA, Hoffmann EW, Venhaus R, Ritter G, Old LJ, Ayyoub M. Vaccination with NY-ESO-1 protein and CpG in Montanide induces integrated antibody/Th1 responses and CD8 T cells through cross-priming. Proc Natl Acad Sci U S A. 2007;104:8947–8952. doi: 10.1073/pnas.0703395104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hawiger D, Inaba K, Dorsett Y, Guo M, Mahnke K, Rivera M, Ravetch JV, Steinman RM, Nussenzweig MC. Dendritic cells induce peripheral T cell unresponsiveness under steady state conditions in vivo. J Exp Med. 2001;194:769–779. doi: 10.1084/jem.194.6.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Probst HC, McCoy K, Okazaki T, Honjo T, van den Broek M. Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat Immunol. 2005;6:280–286. doi: 10.1038/ni1165. [DOI] [PubMed] [Google Scholar]
- 15.Vicari AP, Chiodoni C, Vaure C, Ait-Yahia S, Dercamp C, Matsos F, Reynard O, Taverne C, Merle P, Colombo MP, O’Garra A, Trinchieri G, Caux C. Reversal of tumor-induced dendritic cell paralysis by CpG immunostimulatory oligonucleotide and anti-interleukin 10 receptor antibody. J Exp Med. 2002;196:541–549. doi: 10.1084/jem.20020732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Middel P, Brauneck S, Meyer W, Radzun HJ. Chemokine-mediated distribution of dendritic cell subsets in renal cell carcinoma. BMC Cancer. 2010;10:578. doi: 10.1186/1471-2407-10-578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Otahal P, Hutchinson SC, Mylin LM, Tevethia MJ, Tevethia SS, Schell TD. Inefficient cross-presentation limits the CD8+ T cell response to a subdominant tumor antigen epitope. J Immunol. 2005;175:700–712. doi: 10.4049/jimmunol.175.2.700. [DOI] [PubMed] [Google Scholar]
- 18.Petersen TR, Dickgreber N, Hermans IF. Tumor antigen presentation by dendritic cells. Crit Rev Immunol. 2010;30:345–386. doi: 10.1615/critrevimmunol.v30.i4.30. [DOI] [PubMed] [Google Scholar]
- 19.Regnault A, Lankar D, Lacabanne V, Rodriguez A, Théry C, Rescigno M, Saito T, Verbeek S, Bonnerot C, Ricciardi-Castagnoli P, Amigorena S. Fcgamma receptor-mediated induction of dendritic cell maturation and major histocompatibility complex class I-restricted antigen presentation after immune complex internalization. J Exp Med. 1999;189:371–380. doi: 10.1084/jem.189.2.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cioca DP, Deak E, Cioca F, Paunescu V. Monoclonal antibodies targeted against melanoma and ovarian tumors enhance dendritic cell-mediated cross-presentation of tumor-associated antigens and efficiently cross-prime CD8+ T cells. J Immunother. 2006;29:41–52. doi: 10.1097/01.cji.0000175496.51594.8b. [DOI] [PubMed] [Google Scholar]
- 21.Nimmerjahn F, Ravetch JV. Fc-receptors as regulators of immunity. Adv Immunol. 2007;96:179–204. doi: 10.1016/S0065-2776(07)96005-8. [DOI] [PubMed] [Google Scholar]
- 22.Jäger E, Chen YT, Drijfhout JW, Karbach J, Ringhoffer M, Jäger D, Arand M, Wada H, Noguchi Y, Stockert E, Old LJ, Knuth A. Simultaneous humoral and cellular immune response against cancer-testis antigen NY-ESO-1: definition of human histocompatibility leukocyte antigen (HLA)-A2-binding peptide epitopes. J Exp Med. 1998;187:265–270. doi: 10.1084/jem.187.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maio M, Coral S, Sigalotti L, Elisei R, Romei C, Rossi G, Cortini E, Colizzi F, Fenzi G, Altomonte M, Pinchera A, Vitale M. Analysis of cancer/testis antigens in sporadic medullary thyroid carcinoma: expression and humoral response to NY-ESO-1. J Clin Endocrinol Metab. 2003;88:748–754. doi: 10.1210/jc.2002-020830. [DOI] [PubMed] [Google Scholar]
- 24.Fratta E, Coral S, Covre A, Parisi G, Colizzi F, Danielli R, Marie Nicolay HJ, Sigalotti L, Maio M. The biology of cancer testis antigens: putative function, regulation and therapeutic potential. Mol Oncol. 2011;5:164–182. doi: 10.1016/j.molonc.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu F, Whitton JL. Cutting edge: re-evaluating the in vivo cytokine responses of CD8+ T cells during primary and secondary viral infections. J Immunol. 2005;174:5936–5940. doi: 10.4049/jimmunol.174.10.5936. [DOI] [PubMed] [Google Scholar]
- 26.Melief CJ. Cancer immunotherapy by dendritic cells. Immunity. 2008;29:372–383. doi: 10.1016/j.immuni.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 27.Lugade AA, Moran JP, Gerber SA, Rose RC, Frelinger JG, Lord EM. Local radiation therapy of B16 melanoma tumors increases the generation of tumor antigen-specific effector cells that traffic to the tumor. J Immunol. 2005;174:7516–7523. doi: 10.4049/jimmunol.174.12.7516. [DOI] [PubMed] [Google Scholar]
- 28.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 29.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 30.Zippelius A, Batard P, Rubio-Godoy V, Bioley G, Lienard D, Lejeune F, Rimoldi D, Guillaume P, Meidenbauer N, Mackensen A, Rufer N, Lubenow N, Speiser D, Cerottini JC, Romero P, Pittet MJ. Effector function of human tumor-specific CD8 T cells in melanoma lesions: a state of local functional tolerance. Cancer Res. 2004;64:2865–2873. doi: 10.1158/0008-5472.can-03-3066. [DOI] [PubMed] [Google Scholar]
- 31.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, Huhn RD, Song W, Li D, Sharp LL, Torigian DA, O’Dwyer PJ, Vonderheide RH. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331:1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rüter J, Antonia SJ, Burris HA, Huhn RD, Vonderheide RH. Immune modulation with weekly dosing of an agonist CD40 antibody in a phase I study of patients with advanced solid tumors. Cancer Biol Ther. 2010;10:983–993. doi: 10.4161/cbt.10.10.13251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Junttila TT, Parsons K, Olsson C, Lu Y, Xin Y, Theriault J, Crocker L, Pabonan O, Baginski T, Meng G, Totpal K, Kelley RF, Sliwkowski MX. Superior in vivo efficacy of afucosylated trastuzumab in the treatment of HER2-amplified breast cancer. Cancer Res. 2010;70:4481–4489. doi: 10.1158/0008-5472.CAN-09-3704. [DOI] [PubMed] [Google Scholar]
- 34.Loisel S, Ohresser M, Pallardy M, Daydé D, Berthou C, Cartron G, Watier H. Relevance, advantages and limitations of animal models used in the development of monoclonal antibodies for cancer treatment. Crit Rev Oncol Hematol. 2007;62:34–42. doi: 10.1016/j.critrevonc.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 35.Apetoh L, Ghiringhelli F, Tesniere A, Obeid M, Ortiz C, Criollo A, Mignot G, Maiuri MC, Ullrich E, Saulnier P, Yang H, Amigorena S, Ryffel B, Barrat FJ, Saftig P, Levi F, Lidereau R, Nogues C, Mira JP, Chompret A, Joulin V, Clavel-Chapelon F, Bourhis J, André F, Delaloge S, Tursz T, Kroemer G, Zitvogel L. Toll-like receptor 4-dependent contribution of the immune system to anticancer chemotherapy and radiotherapy. Nat Med. 2007;13:1050–1059. doi: 10.1038/nm1622. [DOI] [PubMed] [Google Scholar]
- 36.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Science. 2003;301:1374–1377. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 37.Rader C, Ritter G, Nathan S, Elia M, Gout I, Jungbluth AA, Cohen LS, Welt S, Old LJ, Barbas CF., 3rd The rabbit antibody repertoire as a novel source for the generation of therapeutic human antibodies. J Biol Chem. 2000;275:13668–13676. doi: 10.1074/jbc.275.18.13668. [DOI] [PubMed] [Google Scholar]
- 38.Stockert E, Jäger E, Chen YT, Scanlan MJ, Gout I, Karbach J, Arand M, Knuth A, Old LJ. A survey of the humoral immune response of cancer patients to a panel of human tumor antigens. J Exp Med. 1998;187:1349–1354. doi: 10.1084/jem.187.8.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nuber N, Curioni-Fontecedro A, Matter C, Soldini D, Tiercy JM, von Boehmer L, Moch H, Dummer R, Knuth A, van den Broek M. Fine analysis of spontaneous MAGE-C1/CT7-specific immunity in melanoma patients. Proc Natl Acad Sci U S A. 2010;107:15187–15192. doi: 10.1073/pnas.1002155107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mitsui J, Nishikawa H, Muraoka D, Wang L, Noguchi T, Sato E, Kondo S, Allison JP, Sakaguchi S, Old LJ, Kato T, Shiku H. Two distinct mechanisms of augmented antitumor activity by modulation of immunostimulatory/inhibitory signals. Clin Cancer Res. 2010;16:2781–2791. doi: 10.1158/1078-0432.CCR-09-3243. [DOI] [PubMed] [Google Scholar]