

Abstract
The use of zinc-finger nucleases (ZFNs) to permanently and precisely modify the human genome offers a potential alternative to cDNA-based gene therapy. The ΔF508 mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene is observed in ∼70% of patients with cystic fibrosis (CF) and is a candidate for ZFN-mediated repair. Here, we report the modular design and synthesis of a pair of ZFNs that can create a double-stranded break (DSB) 203 bp upstream of the ΔF508 lesion, resulting in a nonhomologous end-joining (NHEJ) frequency of 7.8%. In spite of this relatively long distance between the DSB and the ΔF508 mutation, homology-directed repair (HDR) could be detected when using a DNA donor containing part of the wild-type (WT) CFTR. The ZFN target half-sites in CFTR are separated by a 4-bp spacer, but efficient cleavage of synthetic targets with either a 4- or 6-bp spacer was observed in vitro. These ZFNs may be suitable for a genome-editing strategy using a partial cDNA sequence-containing exons 10–24 of CFTR to restore CFTR function to cells containing not only the ΔF508 mutation but also potentially any mutation in or downstream of exon 10.
Key words: AAV vectors, DNA repair, gene therapy, viral vectors, zinc-finger nuclease
Introduction
Cystic fibrosis (CF) is a chronic autosomal recessive disorder that affects the lungs and the digestive system of ∼70,000 people worldwide, and is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene.1,2 Nearly 2000 different disease-causing mutations have been identified, but the most common mutation (∼70% of patients3) is a three-base-pair deletion (CTT) in exon 10. This mutation causes an in-frame deletion of a phenylalanine residue (ΔF508) and results in the lack of a functional cAMP-gated chloride channel (CFTR) on the apical surface of secretory epithelial cells. The demonstration that the CFTR cDNA could correct the CF phenotype in cultured CF cells4 inspired a series of clinical trials to assess the feasibility of a therapeutic approach based on cDNA complementation of the underlying genetic defect.5 The cDNA-based gene therapy approach has not yet resulted in clinical benefit,6,7 however, a recent pre-clinical study concluded that a codon-optimized CpG-free CFTR cDNA construct, pGM169, delivered using the cationic liposome gene transfer agent GL67A, should be tested in both single- and multiple-dose clinical trials in CF patients.8 These trials are now in progress.
Two problems associated with using cDNA to complement a gene defect are duration of expression and correct regulation of expression due to the absence of the endogenous control elements. One option to overcome the short-term expression observed with cDNA delivery is to permanently correct mutations in the genome using zinc-finger nuclease (ZFN) homology-directed repair (HDR).9 By correcting mutations in the coding region of a gene whose control sequences are wild type (WT), this approach would have the added advantage of allowing the correct temporal and spatial expression of the repaired gene. ZFNs are synthetic restriction enzymes comprising three or more zinc-finger DNA-binding motifs linked to the nuclease domain of the FokI restriction enzyme.10 Each DNA-binding motif recognizes a 3-bp sequence of DNA. Targeting ZFNs to a unique genomic location is achieved by manipulation of a seven-amino-acid sequence in the α-helix of each DNA-binding motif.9,11–15 Modification of the nuclease domain can further increase specificity of ZFNs.16,17 ZFNs create a double-stranded break (DSB) that can be repaired by either non-homologous end joining (NHEJ), which typically results in the loss of genetic information,18 or homologous recombination using a sister chromatid or a donor DNA sequence to repair or precisely modify a target locus.9 ZFN-HDR has been used to correct mutations in many different genes both in vitro and in vivo.14,19–22 To date, ZFN-HDR of mutations in CFTR has not been reported; however, the use of the OPEN platform for ZFN design generated a pair of ZFNs that induced DSB formation in ∼1.2% of transfected cells.14 This relatively low level of cleavage of CFTR in cells (relative to other ZFN pairs generated in the same study against other target genes) led to the suggestion that some genes, such as CFTR, may be more difficult to target with ZFNs due to chromatin effects on DNA accessibility or target-site methylation.14
Here, we report the design and characterization of ZFNs targeted to an 18-bp recognition site in intron 9 of the human CFTR sequence, 203 bp upstream of the CTT deletion, which causes the ΔF508 mutation. ZFNs produced in an in vitro transcription/translation (IVTT) reaction can cleave both a synthetic target sequence and a polymerase chain reaction (PCR) product derived from cells corresponding to this region of the gene. When plasmids expressing the two ZFNs were transfected into a cell line derived from tracheal epithelial cells of a CF patient homozygous for the ΔF508 mutation, the NHEJ frequency was ∼8% of treated cells, nearly 7-fold higher than previously reported for ZFNs targeted against human CFTR.14 Repair of the CTT deletion in exon 10 of CFTR was observed using the ZFNs and a donor plasmid containing 4.3 kb of a wild-type (WT) sequence flanked by adeno-associated virus type 2-inverted terminal repeats (AAV-ITRs), but the overall efficiency was <1%.
Materials and Methods
Cell lines and transfections
Human bronchial epithelial (HBE) and cystic fibrosis tracheal epithelial (CFTE) cell lines were obtained from Prof. Dieter Gruenert (UCSF) and maintained in modified Eagle's medium (supplemented with 10% fetal calf serum, 1% l-glutamine, and 1% penicillin and streptomycin; Sigma) and incubated at 37°C and 5% CO2. 300,000 cells were transfected with a total of 4 μg plasmid DNA and 10 μL of lipofectamine 2000 (Invitrogen).
Selection of a pair of ZFN target sites close to the ΔF508 site in CFTR gene
An 800-bp region centered on the ΔF508 mutation within CFTR was initially searched using the sequence (NNC)3N4–6(GNN)3. The search was repeated at reduced stringency, allowing a single ANN, CNN, or TNN triplet to replace one of the GNN triplets.
Design and synthesis of ZFNs
For each ZFN, the sequence of the amino acids in the DNA-binding recognition helix of each of the three ZF domains was chosen from available databases of ZF domains that bind GNN12 or ANN11 triplets (Fig. 1A). These sequences were incorporated into the ZFP consensus sequence framework backbone.23,24 To facilitate subcloning, the sequence 5′-TCTAGAGCTAGC-3′ was included immediately upstream of the ATG codon (creating an NheI site), and the terminal C residue sequence of the ZFP backbone was replaced with the sequence 5′-ACTAGT-3′ (to create an SpeI site). The final-designed ZFP sequences were submitted to Geneart (Life Technologies) for codon optimization and synthesis as dsDNA molecules.
FIG. 1.

(A) The seven-amino-acid sequence of each ZF domain and its corresponding triplet. (B) Predicted sequence of the 277-bp region of CFTR in CFTE cells following repair showing location of ZFN target sites, the intron 9/exon 10 junction (in/EX), an A/G SNP (G in donor plasmids, A in the uncorrected CFTE cell line), a G residue (underlined) that creates an XhoI site, the CTT triplet, which is deleted in the ΔF508 mutation (ΔF508), and a C residue (underlined) that creates an XhoI site. The location of each ZFN target site is indicated by arrows above or below the sequence. The first base of each triplet is shown in bold. Intron sequences are in lowercase; exon sequences are in uppercase. Numbers within the sequence represent the size of sequence not shown in base pairs (Source: NCBI Reference Sequence: NC_000007.12). (C) Synthetic target sequences before cloning into the target plasmid. Nomenclature, for example, the target plasmid pL4R contains binding sites for the ZFNs CFi9L and CFi9R separated by a 4-bp spacer. ZFNs, zinc-finger nucleases; CFTE, cystic fibrosis trachael epithelial; CFTR, cystic fibrosis transmembrane conductance regulator; SNP, single-nucleotide polymorphism.
To create ZFN expression plasmids pCFi9-L and pCFi9-R, a three-way ligation was performed by inserting the appropriate 259-bp NheI–SpeI fragment from the respective ZFP plasmid and the 598-bp SpeI–BamHI fragment containing the FokI nuclease domain10 into the NheI–BamHI sites of pcDNA3.1+ vector (Invitrogen). Correct clones were identified by restriction enzyme analysis and verified by DNA sequencing (Eurofins MWG Operon). DNA sequencing identified a single amino acid mutation P202T in the FokI nuclease domain that was corrected by site-directed mutagenesis (SDM) (QuikChange, Stratagene) using the primers FokI-SDMFor and FokI-SDMRev.
The FokI domains of the ZFNs were modified by SDM (QuikChange, Stratagene) as previously described.16 The + variants (Q187E and I200L) were created using the oligonucleotides FokI+1For/FokI+1Rev and FokI+2For/FokI+2Rev. The − variants (E191K and I239K) were created using the oligonucleotides FokI−1For/FokI−1Rev and FokI−2For/FokI−2Rev.
Addition of nuclear localization signal to ZFNs
For ex vivo use, pZFN plasmids were modified to include the SV40 large T-antigen nuclear localization signal (NLS) sequence PKKKRKV in frame at the amino-terminus of the ZFN protein. The phosphorylated oligonucleotides NLS+ and NLS− were annealed to create a dsDNA molecule with NheI overhangs, and cloned into the unique NheI site of the pZFN plasmids. The correct orientation was determined by DNA sequencing (Eurofins MWG Operon).
Oligonucleotides
Oligonucleotides were from Eurofins MWG Operon (Table 1). Underlined sequences indicate restriction sites.
Table 1.
Oligonucleotides Used in This Study
| FokI-SDMFor | 5′-CACGAAACAAACATATCAACCCTAATGAATGGTGGAAAGTC-3′ |
| FokI-SDMRev | 5′-GACTTTCCACCATTCATTAGGGTTGATATGTTTGTTTCGTG-3′ |
| FokI+1For | 5′-GATGAAATGCAACGATATGTCAAAGAAAATCAAACACGAAAC-3′ |
| FokI+1Rev | 5′-GTTTCGTGTTTGATTTTCTTTGACATATCGTTGCA-3′ |
| FokI+2For | 5′-GCTTACACGATTAAATCATAAAACTAATTGTAATGGAGCTG-3′ |
| FokI+2Rev | 5′-CAGCTCCATTACAATTAGTTTTATGATTTAATCGTGTAAGC-3′ |
| FokI−1For | 5′-CCAAGCAGATGAAATGGAACGATATGTCGAAG-3′ |
| FokI−1Rev | 5′-CTTCGACATATCGTTCCATTTCATCTGCTTGG-3′ |
| FokI−2For | 5′-CAAACACGAAACAAACATCTCAACCCTAATGAATGGTG-3′ |
| FokI−2Rev | 5′-CACCATTCATTAGGGTTGAGATGTTTGTTTCGTGTTTG-3′ |
| CFTR-NHEJFor | 5′-ATCATGTGCCCCTTCTCTGT-3′ |
| CFTR-NHEJRev | 5′-TGCTTTGATGACGCTTCTGTAT-3′ |
| HBE-BstEIIFor | 5′-TGACTAGGTAACCGAGCCTTTTCTTTTTCATTG-3′ |
| HBE-MluIRev | 5′-CAACGCGTAGGCCCTAACTTTCATTCTTG-3′ |
| Donor-XhoIFor | 5′-GATTATGGGAGAACTCGAGCCTTCAGAGGG-3′ |
| Donor-XhoIRev | 5′-CCCTCTGAAGGCTCGAGTTCTCCCATAATC-3′ |
| Donor-ClaIFor | 5′-GTGTTTCCTATGATGAATATCGATACAGAAGCGTCATCAAAG-3′ |
| Donor-ClaIRev | 5′-CTTTGATGACGCTTCTGTATCGATATTCATCATAGGAAACAC-3′ |
| FP-e7 | 5′-AGAACTGAAACTGACTCGGAAGG-3′ |
| RP-e10WT | 5′-CTATATTCATCATAGGAAACACCAAAG-3′ |
| FP-i9.1 | 5′-AATTTTGTAAATTTGTTTCATC-3′ |
| RP-i10.1 | 5′-ACTTGCTTTGCCATTAACAGA-3′ |
| FP-e10.2 | 5′-6-FAM-CAGTTTTCCTGGATTATGCCTGGCA-3′ |
| RP-e10.2 | 5′-AGTTGGCATGCTTTGATGACGCT-3′ |
| NLS+ | 5′-CTAGCATGCCGAAGAAGAAGCGCAAGGTCA-3′ |
| NLS™ | 5′-CTAGTGACCTTGCGCTTCTTCTTCGGCATG-3′ |
Underlined sequences indicate restriction sites.
SDM, site-directed mutagenesis; CFTR, cystic fibrosis transmembrane conductance regulator; NHEJ, nonhomologous end joining; HBE, human bronchial epithelial; FP, forward primer; RP, reverse primer; NLS, nuclear localization signal.
Target plasmids (synthetic)
The 9-bp target sequences of the ZFNs, separated by either a 4- or 6-bp spacer sequence, were constructed as dsDNA oligos (Fig. 1C) and cloned into the unique Acc65I and XhoI sites of the pGL3 control (pCON) vector (Promega) to create pTarget plasmids. Clones were verified by DNA sequencing across the target region.
Amplification of target sequences from genomic DNA
Genomic DNA was extracted from HBE and CFTE cells using a DNeasy Blood and Tissue Kit (Qiagen). A 435-bp fragment spanning the target region was amplified by PCR using the primers CFTR-NHEJFor and CFTR-NHEJRev. The PCR product was purified using the StrataPrep DNA gel extraction kit (Stratagene) as per the manufacturer's protocol.
Donor plasmids
pITR-donor contained a 4330-bp BstEII–MluI fragment excised from a 4344-bp PCR product amplified using Platinum Taq HF (Invitrogen) and genomic DNA isolated from HBE cells using the primers HBE-BstEIIFor and HBE-MluIRev, and cloned into BstEII and MluI sites of pAAV-IRES-GFP (Stratagene).
pITR-donor-XC contained both a silent XhoI site 92 bp upstream of the CTT nucleotides and a silent ClaI site 19 bp downstream of the CTT nucleotides. The XhoI and ClaI sites were introduced by SDM (QuikChange II XL, Stratagene) using oligopairs Donor-XhoIFor and Donor-XhoIRev, and Donor-ClaIFor and Donor-ClaIRev, respectively.
In vitro transcription/translation
One microgram of pZFN expression plasmid was incubated in a 50-μL TnT T7 Quick-Coupled Transcription-Translation system reaction (Promega) for 2 h at 30°C. Protein yield was estimated using the Renilla luciferase reporter plasmid, pHRL, using the dual-luciferase reporter assay system (Promega) according to the manufacturer's guidelines (the Renilla cDNA is of a similar size to the ZFN cDNA and expressed from the same promoter).
In vitro cleavage assay
Approximately 10, 50, or 100 ng of protein was incubated with 1 μg of relevant pTarget plasmids or 100 ng of PCR product, and 10 U XbaI in restriction enzyme buffer #2 (New England Biolabs) supplemented with 10 μM ZnCl2, for 90 min at 37°C (1 μg plasmid:10 ng protein is roughly equivalent to a protein:DNA molar ratio of 1:1). DNA was purified using the StrataPrep DNA gel extraction kit (Stratagene) as per the manufacturer's protocol and then analyzed by 1% agarose gel electrophoresis.
PCR amplicons and CEL-I analysis
A 435-bp region of genomic DNA centered on the ZFN target site was amplified using Platinum Taq (Invitrogen) and primers CFTR-NHEJFor and CFTR-NHEJRev in a 50-μL reaction. Thirty microliters PCR product was incubated at 95°C for 5 min and then cooled slowly to room temperature to form heteroduplexes. Sixteen microliters of the heteroduplex reaction was incubated with 2 μL of Surveyor® nuclease (CEL-I; Transgenomic) and 2 μL of enhancer solution at 42°C for 20 min, and then inactivated with 2 μL of stop solution. The NHEJ frequency was calculated as described previously.23
Repair detection and quantification
Total RNA was isolated using TRIzol (Sigma), and the ImProm-II Reverse Transcription System (Promega) was used for synthesizing cDNA. cDNA was used as template for PCR (GoTaq; Promega) using forward primer (FP)-e7 and reverse primer (RP)-e10WT.
Genomic DNA was isolated and treated with DpnI (NEB) for 90 min at 37°C to restrict plasmid DNA. A 2.6-kb PCR product (PCR#1) was generated using Platinum Taq HF (Invitrogen) and primers FP-i9.1 and RP-i10.1. A fluorescent PCR was performed using a 1:10 dilution of PCR#1 as template and primers FP-e10.2 (FAM-labeled; Eurofins MWG Operon) and RP-e10.2.
If required, PCR products were TOPO-cloned (Invitrogen) before analysis by restriction enzyme (NEB) digestion. DNA was analyzed by 1% agarose gel electrophoresis, Lab Chip GX system with 5 K chip (Caliper Life Sciences), or 3130 Genetic Analyzer (Applied Biosystems). Eurofins MWG Operon was used for DNA sequencing.
Results
Selection of ZFN target sites, modular assembly, and in vitro DNA cleavage activity
A pair of three-finger ZFNs, CFi9-L and CFi9-R, was designed using the ZFP consensus sequence framework backbone.24,25 The sequence of amino acids in the DNA-binding recognition helix of each of the three ZF domains was chosen from available databases of ZF domains that bind GNN12 or ANN11 triplets (Fig. 1A).
To measure in vitro cleavage activity, CFi9L and CFi9R ZFNs were prepared by IVTT and incubated together with the target plasmid pL4R (see Fig. 1C) at three different protein: DNA ratios for 90 min. Cleavage of the target plasmid could be detected at a protein:DNA molar ratio of 1:1, and was essentially complete at ratios of 5:1 or higher (see Fig. 2A). No cleavage of the control vector (pCON), which lacks the ZFN-binding sites, was observed, even at a molar ratio of 10:1.
FIG. 2.
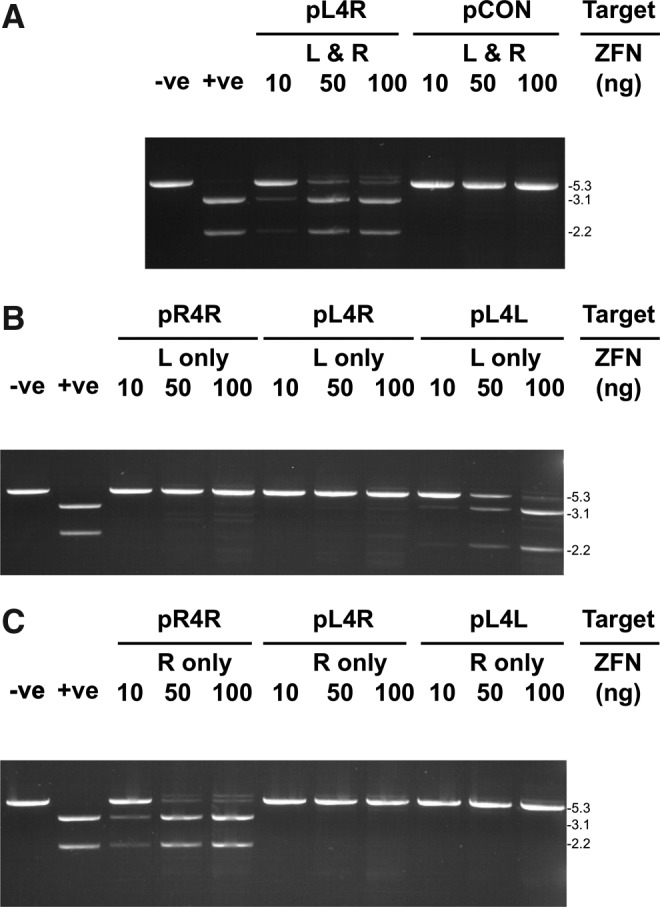
In vitro cleavage activity of ZFN-CFi9L and ZFN-CFi9R with synthetic target sequences. (A) One microgram target plasmids pL4R or pCON (pGL3-control) incubated with increasing amounts (10–100 ng) of ZFN CFi9L (L) and CFi9-R (R) for 90 min at 37°C and analyzed on a 1% agarose gel. (B) One microgram target plasmids (pR4R, pL4R, and pL4L) incubated with increasing amounts (10–100 ng) of ZFN CFi9L for 90 min at 37°C and analyzed on a 1% agarose gel. (C) One microgram target plasmids (pR4R, pL4R, and pL4L) incubated with increasing amounts (10–100 ng) of ZFN CFi9R for 90 min at 37°C and analyzed on a 1% agarose gel. On all gels, −ve is the pCON plasmid digested with XbaI only, and +ve is the pCON plasmid digested with XbaI and XhoI. XbaI was included in all digests to linearize plasmids, and thereby simplify detection of ZFN cleavage. Sizes (kb) of the linearized target plasmid or PCR product and cleavage products shown to the right of gels. The larger molecular weight band seen in some lanes at higher amounts of ZFNs is plasmid encoding ZFNs from IVTT reaction. CF, cystic fibrosis; PCR, polymerase chain reaction; IVTT, in vitro transcription/translation.
Both ZFNs were required to cleave the pL4R target; neither CFi9-L used alone (Fig. 2B) nor CFi9-R used alone (Fig. 2C) could cleave pL4R at any of the ratios tested. However, CFi9L could homodimerize to cut the pL4L target (which contains two CFi9L target sites arranged in an inverted orientation separated by a 4-bp spacer) in a dose-dependent manner, with an efficiency similar to that observed with the CFi9-L/CFi9-R pair and the pL4R target (Fig. 2B). Likewise, CFi9-R could homodimerize to completely cut pR4R (Fig. 2C). While these data confirm that CFi9-L and CFi9-R can heterodimerize to specifically cleave a synthetic sequence corresponding to the target site in intron 9 of CFTR, it also shows that ZFNs can homodimerize, which could lead to off-target cleavage events that are potentially toxic in cells.9,16,17 Of note, an alternate version of CFi9-R, in which the ZF1 domain designed to bind the AAT triplet had a recognition helix with the sequence VSSNLNV, did not form a functional heterodimer with CFi9-L (data not shown).
Modification of the FokI domain of ZFNs to prevent homodimerization
Amino acid changes in the FokI domain of ZFNs can prevent homodimerization and reduce their potential for off-target cleavage in cells, while still allowing them to function as heterodimers.16,17,26 The KK and EL mutations16 were introduced into the FokI domains of ZFNs CFi9L and CFi9R respectively, and their ability to cleave selected target plasmids was measured (ZFNs with a WT FokI domain are hereinafter suffixed WT to distinguish them from ZFNs containing a KK or EL variant FokI domain, suffixed KK or EL, respectively).
When used separately, neither CFi9LKK nor CFi9LEL could homodimerize to cleave the pL4L target (Fig. 3A, left panel). Based on the detection limit of ethidium bromide staining, this represents a ≥1000-fold reduction in ZFN homodimerization. Likewise, neither CFi9RKK nor CFi9REL could homodimerize to cleave the pR4R target (Fig. 3A, right panel). When combined as a heterodimeric pair, CFi9LEL and CFi9RKK could cleave the pL4R target (Fig. 3B), although with slightly reduced efficiency compared to CFi9LWT and CFi9RWT. However, the other heterodimeric pair, CFi9LKK and CFi9REL, was unable to cleave the pL4R target, even at a 10:1 molar ratio (Fig. 3B). When CFi9REL was used with CFi9RKK, full cleavage of pR4R was observed (data not shown), indicating that CFi9REL is functional. In contrast, when CFi9LEL was tested together with CFi9LKK, cleavage of pL4L was not detected (data not shown), indicating that CFi9LKK is nonfunctional.
FIG. 3.

Agarose gel analysis of target plasmids or PCR products incubated with CFi9L and CFi9R ZFNs (with WT, KK, or EL modifications of the FokI nuclease domain) produced by IVTT used alone or as pairs. All ZFNs tested at either three (A–C) or two (D) different DNA:protein ratios (amount of ZFN protein shown in ng). All plasmids (A–C) also digested with XbaI. (A) Cleavage of pL4L (left panel) or pR4R (right panel) using a single ZFN as shown. (B) Cleavage of pL4R using ZFN pairs as shown. (C) Cleavage of pL4R and pL6R using CFi9-LEL/CFi9-RKK together as shown. (D) Cleavage of a 435-bp PCR product centered on CFTR intron 9 derived from genomic DNA from CFTE cells using ZFN pairs as shown. Sizes (kb) of the linearized target plasmid and cleavage products shown to the right of gels. The larger molecular weight band seen in some lanes at higher amounts of ZFNs is plasmid encoding ZFNs from IVTT reaction. WT, wild type.
Overall, these data confirm that the CFi9-LEL/CFi9-RKK pair only functions efficiently as an obligate heterodimer. Although these EL/KK mutations prevent homodimerization, which should reduce off-target cleavage in ex vivo cell cultures, these modifications would not necessarily be expected to prevent the ZFNs binding to L–R target sites in the genome separated by larger spacer sequences. Indeed, the ZFN-LEL/ZFN-RKK pair can also cleave the pL6R target plasmid, which has a 6-bp spacer sequence (Fig. 3C).
Before testing for cleavage activity of genomic DNA in cells, and to confirm that CFi9-L/CFi9-R can cleave the bona fide target sequence in the human cell line (CFTE) derived from a CF patient homozygous for the ΔF508 mutation, the in vitro activity of the ZFNs was tested on a 435-bp PCR product derived from these cells. In addition, the CFi9-LEL/CFi9-RKK pair was modified to include a NLS (hereinafter referred to as CFi9-LEL-NLS/CFi9-RKK-NLS). The CFi9-LWT/CFi9-RWT pair efficiently cleaved this PCR product, with complete digestion at a ZFN:DNA ratio of 10:1 in 90 min (Fig. 3D). As with cleavage of synthetic targets, cleavage of the PCR product was slightly less efficient with both the CFi9-LEL/CFi9-RKK pair and the CFi9-LEL-NLS/CFi9-RKK-NLS pair compared to the CFi9-LWT/CFi9-RWT pair (Fig. 3D). However, there was no detectable difference between the in vitro cleavage activity between the CFi9-LEL/CFi9-RKK pair and the CFi9-LEL-NLS/CFi9-RKK-NLS pair. The CFi9-LEL-NLS/CFi9-RKK-NLS pair was used in subsequent ex vivo experiments.
ZFN cleavage activity in CF-patient-derived cells
ZFN-induced DSBs are normally repaired by NHEJ in the absence of a donor sequence. This DNA repair pathway results in the creation of small genomic insertions or deletions (indels) that can be detected and quantified using the CEL-I nuclease assay.27 As shown in Figure 4A, a dose-dependent increase in NHEJ frequency occurred in CFTE cells transfected with plasmids expressing either the CFi9-LWT/CFi9-RWT or the CFi9-LEL/CFi9-RKK pair; no indel formation was detected in mock transfected cells. A maximal level of NHEJ frequency of 7.8±0.9% occurred in cells transfected with 2 μg CFi9-LEL-NLS/CFi9-RKK-NLS plasmids (Fig. 4B, C); this ZFN pair was used for the subsequent repair experiments.
FIG. 4.

ZFN cleavage activity in CF-patient-derived cells. (A) Representative CEL-I assay performed on PCR products generated from genomic DNA isolated 72 h post-transfection with either pcDNA3.1+ (mock), increasing amounts of pCFi9-LWT/pCFi9-RWT (10–1000 ng), or increasing amounts of pCFi9-LEL/pCFi9-RKK (10–1000 ng). (B) Representative CEL-I assay performed on PCR products generated from genomic DNA isolated 72 h post-transfection with increasing amounts of pCFi9-LEL-NLS/pCFi9-RKK-NLS (200–2000 ng); cells incubated for 72 h at 37°C. (C) Bar graph showing the percentage NHEJ frequency. Data are expressed as mean±standard error of the mean (n=8–15). NLS, nuclear localization signal; NHEJ, nonhomologous end joining.
ZFN-induced repair of the ΔF508 mutation in CF-patient-derived cells
ZFN-induced DSBs can be repaired using a donor plasmid that contains the WT sequence flanked by relatively short homology arms delivered to cells in a plasmid.9 Use of the CFi9-LEL-NLS/CFi9-RKK-NLS ZFNs and a donor plasmid containing the WT exon 10 sequence (and flanking homology arms) from HBE cells should be able to correct the CTT deletion, and would also introduce the G variant of the well-characterized single-nucleotide polymorphism (SNP) (A/G rs213950; see Fig. 1B); both these changes occur within exon 10 and would therefore be incorporated into the CFTR mRNA. To detect correction of the CTT deletion, a reverse transcriptase–polymerase chain reaction assay was developed to amplify only corrected (i.e., CTT-containing) CFTR cDNA from a mixed population of WT and ΔF508 CFTR cDNAs. As shown in Figure 5, use of a forward primer that binds exon 7 (FP-e7), and a reverse primer that should bind only to the corrected CTT sequence in exon 10 (RP-e10WT), gave a 675-bp product from cDNA isolated from HBE cells (which contains only WT CFTR cDNA), but no product from cDNA isolated from CFTE cells (which contains only ΔF508 CFTR cDNA); glyceraldehyde 3-phosphate dehydrogenase could be amplified equally from cDNA from both cell types. The smaller PCR product generated from HBE cDNA most likely corresponds to a well-characterized exon 9-skipping splice variant,28 which should generate a product of 492 bp using these primers.
FIG. 5.

RT-PCR analysis of ZFN/donor-treated CFTE cells. RT-PCR was performed on total RNA extracted from HBE or CFTE cells using primers FP-e7 and RP-e10WT. All cells grown at 37°C for 72 h before RNA extraction unless indicated; the 37°C/30°C indicates that cells were grown for 24 h at 37°C and then 48 h at 30°C before RNA extraction. “200” or “2000” represents the total amount (ng) of ZFN plasmids per transfection. Two thousand nanograms donor plasmid used in all transfections (except Mock). RT-PCR, reverse transcriptase–polymerase chain reaction; HBE, human bronchial epithelial; FP, forward primer; RP, reverse primer.
Having verified that the PCR assay could only produce the 675-bp product from cDNA containing the CTT sequence, the assay was used to analyze cDNA from cells transfected with ZFNs and/or a donor plasmid (pITR-donor) containing a 4.3-kb sequence centered on WT exon 10 and adjacent intron sequences cloned in a plasmid that flanks the genomic donor sequence with adeno-associated virus type 2 (AAV-2)-inverted terminal-repeat (ITR) sequences. The 675-bp band was observed in cells transfected with the pITR-donor and 2000 ng pCFi9-LEL-NLS/pCFi9-RKK-NLS. Correction was dependent on the amount of ZFNs used; the 675-bp band was not detected in cells transfected with the pITR-donor and 200 ng pCFi9-LEL-NLS/pCFi9-RKK-NLS. If, however, cells were subjected to the transient 30°C cold-shift protocol,28 the 675-bp product could be detected when cells were transfected with as little as 200 ng pCFi9-LEL-NLS/pCFi9-RKK-NLS and pITR-donor. Of interest, the smaller band (492 bp) seen in HBEs was not seen in ZFN-HDR-corrected CFTEs, suggesting that CFTE cells have the (TG)10T9 sequence in intron 8, which prevents exon 9 skipping, and which is strongly linked to the ΔF508 mutation.29
Estimation of ZFN-HDR efficiency
To determine the efficiency of a gene repair, the experiment was repeated using the same ZFNs, but a modified donor (pITR-donor-XC) that also contains an XhoI site 92 bp upstream of the CTT insertion site and a ClaI site 19 bp downstream (see Fig. 1B). A two-step fluorescence-based PCR strategy was developed such that the amplicons from corrected DNA could be differentiated from noncorrected DNA by size (Fig. 6A). Using this assay, the CTT-containing sequence could be detected in a mixed population of genomic DNA extracted from untransfected HBE and CFTE cells, even when only 1% of template DNA contained the CTT sequence (see Fig. 6B). When the assay was performed on CFTE cells transfected with pITR-donor-XC only, no corrected sequence was observed. Similarly, when genomic DNA from CFTE cells transfected with both pITR-donor-XC and ZFNs was analyzed, no corrected PCR amplicons were detected, suggesting that the repair efficiency is <1%, below the limit of detection of this assay. Restriction analysis of cloned PCR#1 products from CFTE cells transfected with pITR-donorXC and ZFNs was unable to detect neither the XhoI site (0/100 clones) nor the ClaI site (0/44 clones), further suggesting that the repair level is <1% (data not shown).
FIG. 6.

Two-step fluorescence-based assay to detect HDR. (A) PCR#1 should amplify a 2.6-kb region of genomic DNA without creating a product from pITR-donorXC. Using the 2.6-kb product as a template, the second PCR (PCR#2) should generate a 101-bp uncorrected amplicon (A ΔCTT) in the absence of ZFN-HDR, or a 104-bp amplicon if the ΔCTT has been repaired; this corrected amplicon should also contain XhoI and ClaI restriction sites (G X+CTT C). (B) Relative levels of uncorrected and corrected PCR#2 amplicons generated from genomic DNA from HBE and CFTE cells mixed at percentages shown. (C) Relative levels of uncorrected and corrected PCR#2 amplicons generated from genomic DNA from CFTE cells transfected as shown. HDR, homology-directed repair.
Discussion
Here, we have described the modular design and synthesis of a pair of ZFNs that can bind and cleave intron 9 of human CFTR inducing an NHEJ frequency of 7.8%. This level of activity is similar to that reported for ZFNs targeting other mammalian genes,14,22,30 and >6-fold higher than previously reported for ZFNs targeted against human CFTR.14 Based on the proposal by Maeder et al.14 that the chromatin configuration and/or transcriptional status of a region may influence ZFN access to a target site, the higher activity of the ZFN pair reported here may be because the cell line used herein actively expresses CFTR mRNA,31 whereas the ZFNs described previously14 were evaluated in HEK cells that do not express CFTR.32 In addition, the overall architecture of these ZFNs may also contribute to the increased activity.
A recent study of zinc-finger specificity has shown that the interdomain linker is a major determinant of target-site selectivity33 and demonstrated that, in general, as the length of the spacer (bp) that separates the ZFN-binding sites increases, the length of the linker that joins the ZF domains to the nuclease domain must be increased to maintain efficient cleavage. Of interest, they found that only one 4-amino-acid-linker sequence (HxxH-LRGS-QLV) could cleave a 4-bp spacer, but with a very low efficiency. Of note, the ZFNs reported here efficiently cleave a 4-bp genomic site, but the linker used here (HxxH-TG-LV) was not tested in the study by Händel et al.33 This linker is shorter than the four-amino-acid linker shown above, which can recognize the 4-bp spacer, but longer than two other linkers that could not cleave a 4-bp spacer (δF: HXXH-TG-V and 0: HXXH-QLV), suggesting that linkers of two or three amino acids could further enhance cleavage of target sites with 4-bp spacers. It should also be noted that the HxxH-TG-LV linker used here allowed complete digestion of a 6-bp target site in vitro.
Repair of the CTT deletion in exon 10 of the CFTR gene (which causes the ΔF508 mutation) was observed using our ZFNs and a donor plasmid containing 4.3 kb of WT sequence flanked by AAV-ITRs, but the overall efficiency was <1%. To date, efficient ZFN-HDR has only been reported at or within a few bp of the ZFN-induced DSB.9,14 The low level of repair reported here is most likely a consequence of the distance (203 bp) of the CTT deletion from the ZFN target site. This distance between the repair site and the ZFN-induced DSB is within a range shown to be efficiently repaired using a plasmid-based model.34 However, in a study of gene targeting in a genomic context, it was shown that 80% of gene-targeting events extended no further than 45 bp from the DSB,35 although gene repair tracts did extend as far as 228 bp in 8% of corrected clones. In the same study, even a low level of heterology (1.2%) between donor and target was shown to decrease the level of gene repair by approximately sixfold; thus, the various differences between the pITR-donor-XC sequence and the target region (i.e., a SNP, two base changes to create the XhoI and ClaI sites, and the 3 bp CTT insertion) may also contribute to the low level of HDR. Finally, it has been reported that HDR efficiency can be reduced by approximately twofold using plasmids with AAV ITRs,36 although no detectable gene correction was observed by the two-step PCR assay when our ZFNs and the same 4.3-kb donor sequence was delivered using a plasmid that lacks ITRs (pUC-donor-XC; data not shown).
Thus, it would appear that efficient repair of the ΔF508 mutation in CFTR will require the use of ZFNs that can cleave closer to the CTT deletion site. Alternatively, the existing ZFNs could be used to drive the recombination or insertion of a minigene construct comprising exons 10–24 from the CFTR cDNA (with appropriate splice acceptor and polyA sites) into intron 9 to allow the production of a full-length-corrected CFTR mRNA. This approach has been successfully used in cells to correct the IL2RG locus19 and in vivo to correct human F9 in a murine model of hemophilia that subsequently restored hemostasis.21 The use of such a CFTR cDNA minigene approach should allow the correction of 83% of the known CF-causing mutations, including the six most common ones.37
Extensive in vitro analysis confirmed that CFi9-L and CFi9-R were able to heterodimerize and cleave the desired target sequence, but also showed that these ZFNs could homodimerize to cleave alternate target sequences. Using established modifications of the FokI nuclease domain,16 homodimerization of the CFi9-LEL and CFi9-RKK could be reduced by >99.9%, while still allowing the ZFNs to function as a pair to cleave the target DNA. However, these mutations would not be expected to prevent the ZFNs binding to L–R target sites in the genome separated by larger spacer sequences. Indeed, the ZFN-LEL/ZFN-RKK pair could cleave a target sequence with a 6-bp spacer sequence in vitro. Nonetheless, there was no detectable toxicity of any of the ZFNs in this report when tested in CFTE cells 72 h post-transfection in an established assay that uses green fluorescent protein expression as an indicator of cell viability (Supplementary Fig. S1). In contrast, the only other-reported ZFNs that can cleave CFTR in cells14 reduce cell viability by ∼44% using essentially the same assay,38 although in different cell lines (HEK293 and Huh7). A direct comparison of the ZFNs in the same cell line using more sensitive toxicity assays based on AAV's ability to integrate at DSBs39 could be informative.
In summary, we describe efficient ZFN-mediated cleavage of the human CFTR in cells, and repair of the ΔF508-causing mutation. While the repair efficiency is currently below that hypothesized to restore normal chloride efflux40 and/or normal airway surface liquid levels,41 it suggests that these ZFNs could be used to effect a genome-editing strategy using a cDNA minigene delivered by a suitable virus vector.19,21
Supplementary Material
Acknowledgments
The authors wish to thank Professor Srinivasan Chandrasegaran (Department of Environmental Health Sciences, Johns Hopkins University, Baltimore, MD) for the provision of the plasmid pET-15bZif-FN, Professor Dieter Gruenert (Department of Otolaryngology–Head and Neck Surgery, UCSF, San Francisco, CA) for CFTE and HBE cell lines, Dr. Collette Hand and Michael Skilling (Department of Pathology, University College Cork) for assistance with fluorescent PCR analysis, Dr. Sharon McKenna and Dr. Tracey O'Donovan (Cork Cancer Research Centre, Cork, Ireland) for assistance with FACS analysis, Miriam O'Sullivan for technical assistance, and Professor Edward J. Johns for long-term support of this project. This work was supported by Programme for Research in Third-Level Institutions Cycle 3/Higher Education Authority, Ireland, the Department of Physiology and College of Medicine and Health, University College Cork, and Cystinosis Foundation Ireland.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Riordan JR. Rommens JM. Kerem B, et al. Identification of the cystic fibrosis gene: cloning and characterisation of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- 2.Rommens JM. Iannuzzi MC. Kerem B, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science. 1989;245:1059–1065. doi: 10.1126/science.2772657. [DOI] [PubMed] [Google Scholar]
- 3.Kerem BS. Buchanan JA. Durie P, et al. DNA marker haplotype association with pancreatic sufficiency in cystic fibrosis. Am J Hum Genet. 1989;44:827–834. [PMC free article] [PubMed] [Google Scholar]
- 4.Rich DP. Anderson MP. Gregory RJ, et al. Expression of cystic fibrosis transmembrane conductance regulator corrects defective chloride channel regulation in cystic fibrosis airway epithelial cells. Nature. 1990;347:358–363. doi: 10.1038/347358a0. [DOI] [PubMed] [Google Scholar]
- 5.Zabner J. Couture LA. Gregory RJ, et al. Adenovirus-mediated gene transfer transiently corrects the chloride transport defect in nasal epithelia of patients with cystic fibrosis. Cell. 1993;75:207–216. doi: 10.1016/0092-8674(93)80063-k. [DOI] [PubMed] [Google Scholar]
- 6.Davies JC. Alton EW. Gene therapy for cystic fibrosis. Proc Am Thorac Soc. 2010;7:408–414. doi: 10.1513/pats.201004-029AW. [DOI] [PubMed] [Google Scholar]
- 7.Griesenbach U. Alton EW. Current status and future directions of gene and cell therapy for cystic fibrosis. BioDrugs. 2011;25:77–88. doi: 10.2165/11586960-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 8.McLachlan G. Davidson H. Holder E, et al. Pre-clinical evaluation of three non-viral gene transfer agents for cystic fibrosis after aerosol delivery to the ovine lung. Gene Ther. 2011;18:996–1005. doi: 10.1038/gt.2011.55. [DOI] [PubMed] [Google Scholar]
- 9.Urnov FD. Miller JC. Lee YL, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–651. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- 10.Kim YG. Cha J. Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to FokI cleavage domain. Proc Natl Acad Sci USA. 1996;93:1156–1160. doi: 10.1073/pnas.93.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dreier B. Beerli RR. Segal DJ, et al. Development of zinc finger domains for recognition of the 5′-ANN-3′ family of DNA sequences and their use in the construction of artificial transcription factors. J Biol Chem. 2001;276:29466–29478. doi: 10.1074/jbc.M102604200. [DOI] [PubMed] [Google Scholar]
- 12.Liu Q. Xia Z. Zhong X, et al. Validated zinc finger protein designs for all 16 GNN DNA triplet targets. J Biol Chem. 2002;277:3850–3856. doi: 10.1074/jbc.M110669200. [DOI] [PubMed] [Google Scholar]
- 13.Sera T. Uranga C. Rational design of artificial zinc-finger proteins using a nondegenerate recognition code table. Biochemistry. 2002;41:7074–7081. doi: 10.1021/bi020095c. [DOI] [PubMed] [Google Scholar]
- 14.Maeder ML. Thibodeau-Beganny S. Sander JD, et al. Oligomerised pool engineering (OPEN): an ‘open source’ protocol for making customised zinc-finger arrays. Nat Protoc. 2008;4:1471–1501. doi: 10.1038/nprot.2009.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sander JD. Maeder ML. Joung JK. Engineering designer nucleases with customized cleavage specificities. Curr Protoc Mol Biol. 2011;96:12.13.1–12.13.16. doi: 10.1002/0471142727.mb1213s96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miller JC. Holmes MC. Wang J, et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol. 2007;25:778–785. doi: 10.1038/nbt1319. [DOI] [PubMed] [Google Scholar]
- 17.Szczepek M. Brondani V. Büchel J, et al. Structure-based redesign of the dimerisation interface reduces the toxicity of zinc-finger nucleases. Nat Biotechnol. 2007;25:786–793. doi: 10.1038/nbt1317. [DOI] [PubMed] [Google Scholar]
- 18.Liu PQ. Chan EM. Cost GJ, et al. Generation of a triple-gene knockout mammalian cell line using engineered zinc-finger nucleases. Biotechnol Bioeng. 2010;106:97–105. doi: 10.1002/bit.22654. [DOI] [PubMed] [Google Scholar]
- 19.Lombardo A. Genovese P. Beausejour CM, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25:1298–1306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- 20.Zou J. Maeder ML. Mali P, et al. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell. 2009;5:97–110. doi: 10.1016/j.stem.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H. Haurigot V. Doyon Y, et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature. 2011;475:217–221. doi: 10.1038/nature10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sebastiano V. Maeder ML. Angstman JF, et al. In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells. 2011;29:1717–1726. doi: 10.1002/stem.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guschin DY. Waite AJ. Katibah GE, et al. A rapid and general assay for monitoring endogenous gene modification. Methods Mol Biol. 2010;649:247–256. doi: 10.1007/978-1-60761-753-2_15. [DOI] [PubMed] [Google Scholar]
- 24.Desjarlais JR. Berg JM. Use of a zinc-finger consensus sequence framework and specificity rules to design specific DNA binding proteins. Proc Natl Acad Sci USA. 1993;90:2256–2260. doi: 10.1073/pnas.90.6.2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mani M. Kandavelou K. Dy FJ, et al. Design, engineering, and characterization of zinc finger nucleases. Biochem Biophys Res Commun. 2005;335:447–457. doi: 10.1016/j.bbrc.2005.07.089. [DOI] [PubMed] [Google Scholar]
- 26.Guo J. Gaj T. Barbas CF., III Directed evolution of an enhanced and highly efficient FokI cleavage domain for zinc finger nucleases. J Mol Biol. 2010;400:96–107. doi: 10.1016/j.jmb.2010.04.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Perez EE. Wang J. Miller JC, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;26:808–816. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doyon Y. Choi VM. Xia DF, et al. Transient cold shock enhances zinc-finger nuclease-mediated gene disruption. Nat Methods. 2010;7:459–460. doi: 10.1038/nmeth.1456. [DOI] [PubMed] [Google Scholar]
- 29.Chu CS. Trapnell BC. Curristin S, et al. Genetic basis of variable exon 9 skipping in cystic fibrosis transmembrane conductance regulator mRNA. Nat Genet. 1993;3:151–156. doi: 10.1038/ng0293-151. [DOI] [PubMed] [Google Scholar]
- 30.Santiago Y. Chan E. Liu PQ, et al. Targeted gene knockout in mammalian cells by using engineered zinc-finger nucleases. Proc Natl Acad Sci USA. 2008;105:5809–5814. doi: 10.1073/pnas.0800940105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kunzelmann K. Schwiebert EM. Zeitlin PL, et al. An immortalized cystic fibrosis tracheal epithelial cell line homozygous for the delta F508 CFTR mutation. Am J Respir Cell Mol Bio. 1993;8:522–529. doi: 10.1165/ajrcmb/8.5.522. [DOI] [PubMed] [Google Scholar]
- 32.Bertrand CA. Zhang R. Pilewski JM, et al. SLC26A9 is a constitutively active, CFTR-regulated anion conductance in human bronchial epithelia. J Gen Physiol. 2009;133:421–438. doi: 10.1085/jgp.200810097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Händel EM. Alwin S. Cathomen T. Expanding or restricting the target site repertoire of zinc-finger nucleases: the inter-domain linker as a major determinant of target site selectivity. Mol Ther. 2009;17:104–111. doi: 10.1038/mt.2008.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Porteus MH. Mammalian gene targeting with designed zinc finger nucleases. Mol Ther. 2006;13:438–446. doi: 10.1016/j.ymthe.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 35.Elliott B. Richardson C. Winderbaum J, et al. Gene conversion tracts from double-strand break repair in mammalian cells. Mol Cell Biol. 1998;18:93–101. doi: 10.1128/mcb.18.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hirsch ML. Green L. Porteus MH, et al. Self-complementary AAV mediates gene targeting and enhances endonuclease delivery for double-strand break repair. Gene Ther. 2010;17:1175–1180. doi: 10.1038/gt.2010.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bobadilla JL. Macek M., Jr. Fine JP, et al. Cystic fibrosis: a worldwide analysis of CFTR mutations-correlation with incidence data and application to screening. Hum Mutat. 2002;19:575–606. doi: 10.1002/humu.10041. [DOI] [PubMed] [Google Scholar]
- 38.Cradick TJ. Keck K. Bradshaw S, et al. Zinc-finger nucleases as a novel therapeutic strategy for targeting hepatitis B virus DNAs. Mol Ther. 2010;18:947–954. doi: 10.1038/mt.2010.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Petek LM. Russell DW. Miller DG. Frequent endonuclease cleavage at off-target locations in vivo. Mol Ther. 2010;18:983–986. doi: 10.1038/mt.2010.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson LG. Olsen JC. Sarkadi B, et al. Efficiency of gene transfer for restoration of normal airway epithelial function in cystic fibrosis. Nat Genet. 1992;2:21–25. doi: 10.1038/ng0992-21. [DOI] [PubMed] [Google Scholar]
- 41.Zhang L. Button B. Gabriel SE, et al. CFTR delivery to 25% of surface epithelial cells restores normal rates of mucus transport to human cystic fibrosis airway epithelium. PLoS Biol. 2009;7:e1000155. doi: 10.1371/journal.pbio.1000155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.