

Abstract
Reovirus is a naturally occurring oncolytic virus that has shown preclinical efficacy in the treatment of a wide range of tumor types and has now reached phase III testing in clinical trials. The anti-cancer activity of reovirus has been attributed to both its direct oncolytic activity and the enhancement of anti-tumor immune responses. In this study, we have investigated the direct effect of reovirus on acute myeloid leukemia (AML) cells and its potential to enhance innate immune responses against AML, including the testing of primary samples from patients. Reovirus was found to replicate in and kill AML cell lines, and to reduce cell viability in primary AML samples. The pro-inflammatory cytokine interferon alpha (IFNα) and the chemokine (C-C motif) ligand 5 (known as RANTES [regulated upon activation, normal T-cell expressed, and secreted]) were also secreted from AML cells in response to virus treatment. In addition, reovirus-mediated activation of natural killer (NK) cells, within the context of peripheral blood mononuclear cells, stimulated their anti-leukemia response, with increased NK degranulation and IFNγ production and enhanced killing of AML targets. These data suggest that reovirus has the potential as both a direct cytotoxic and an immunotherapeutic agent for the treatment of AML.
Key words: acute myeloid leukemia, immunotherapy, NK cells, oncolytic virus, reovirus
Introduction
Reovirus is a naturally occurring oncolytic virus that has shown preclinical efficacy in the treatment of a wide range of human cancers, including melanoma, glioma, ovarian, breast, and colon.1–4 This nonenveloped, double-stranded RNA virus can cause infection of the respiratory and gastrointestinal tract, although this is usually asymptomatic. Reovirus has been reported to replicate preferentially in cells with an activated ras pathway, a mutation common to many different tumor types, although there is a current consensus that additional factors may be important for viral entry and tumor cytotoxicity.5,6 A number of phase I and II clinical trials have recently been completed using reovirus7–10, and a phase III trial for head and neck cancer is currently underway. The anti-cancer activity of reovirus has been attributed to both its direct cytotoxic effect on cancer cells and the induction of potent anti-tumor immune responses, which may be pivotal to the therapeutic effect of the virus. 2,11–13 With particular regard to the role of the innate immune response in reovirus therapy, we have previously shown that (1) VEGF-conditioned reovirus treatment is dependent on innate natural killer (NK) cells14; (2) a combination of cyclophosphamide/interleukin-2 enhances reovirus therapy via the hyperactivation of NK cells15; and (3) reovirus-infected human tumor cells stimulate dendritic cells to activate NK cells.12
In addition to the cytotoxicity toward cells derived from solid tumors, reovirus has also shown activity against hematological malignancies, inducing death in a range of lymphoid cell lines and in purging cancer cell lines of monocytic and myeloma origin in autografts.16–18 The activity of reovirus against these cells, along with its reported safety on systemic delivery and potential to enhance anti-tumor immunity, led us to investigate the use of reovirus in the treatment of acute myeloid leukemia (AML). This aggressive hematological malignancy is characterized by the proliferation and accumulation of abnormal myeloid precursor cells and is the most common form of acute leukemia in adults. Current chemotherapy regimens achieve remission in a substantial proportion of patients with AML. However, the problem of residual disease in AML patients after intensive chemotherapy indicates that remission duration is often short and overall survival remains poor, particularly in older patients, supporting the need for novel treatments that augment existing chemotherapy regimens. Moreover, there has been increased interest in the development of immune-mediated therapies that target AML, as the graft-versus-leukemia (GVL) effect has highlighted the importance of NK and T-cell effectors in the eradication of leukemic cells.19,20
In this study, we have demonstrated that reovirus decreases the viability of AML cell lines and primary blasts. In addition, reovirus stimulates the secretion of interferon alpha (IFNα) and chemokine (C-C motif) ligand 5 (known as RANTES [regulated upon activation, normal T-cell expressed, and secreted]) from these cells. We have also shown that reovirus activation of NK cells, within the context of peripheral blood mononuclear cells (PBMC), significantly increases the activity of NK cells against AML targets, as evidenced by higher levels of CD107 degranulation and IFNγ production on target recognition. This activation translates into increased AML cell lysis, with reovirus-activated NK cells displaying an enhanced capacity to kill AML cell lines. Both the stimulation of NK cells by reovirus and NK degranulation against AML blasts were evident when samples from patients were tested. This suggests that, in addition to its potential use as a direct cytotoxic agent for AML, reovirus may also be used in a novel approach to enhance anti-tumor innate immune responses in the treatment of AML.
Materials and Methods
Cell lines
The AML cell lines Kasumi-1 (myeloblast), THP-1 (monoblast), KG-1 (myeloblast), and ML-1 (myeloblast) were obtained from the Cancer Research UK cell bank (London, United Kingdom) and cultured in Roswell Park Memorial Institute (RPMI)-1640 (Sigma-Aldrich, Dorset, United Kingdom) supplemented with 10% (v/v) FCS (Biosera, Ringmer, United Kingdom) and 1% (v/v) L-glutamine (Sigma). Mouse fibroblast cells (L929) were also obtained from the Cancer Research UK cell bank and cultured in Dulbecco's modified Eagle's medium (DMEM; Sigma), supplemented with 10% fetal calf serum (FCS) and 1% L-glutamine. All cells were maintained at 37°C in a humidified atmosphere containing 5% CO2. All the cells were routinely tested for, and found to be free from, mycoplasma infection.
Reovirus
Reovirus type 3 Dearing strain (Reolysin®) was kindly provided by Oncolytics Biotech, Inc. (Calgary, Canada) in phosphate buffered saline (PBS) and stored neat at −80°C (for long-term storage) or 4°C (for short-term storage of approximately 2 weeks). Virus titer was determined by standard plaque assay using L929 cells (as described in a later section).
Patient samples
Leukemia cells were obtained from the peripheral blood of AML patients using Ficoll–Hypaque density-gradient centrifugation and cultured in Iscove's medium (Sigma) supplemented with 10% FCS and 1% L-glutamine. Blast phenotype was confirmed by flow cytometry using 10 μL of fluorescein isothiocyanate (FITC)-conjugated CD33 and 3 μL of PE-conjugated CD34 antibodies (BD Biosciences, Oxford, United Kingdom); blast purity in all the cases was more than 80% (data not shown). Written, informed consent was obtained from all patients in accordance with local institutional ethics review and approval.
Isolation of PBMC
Healthy donor blood was collected by the National Health Service Blood and Transplant (NHSBT) and processed in component donation leukocyte cones according to institutional guidelines. PBMC were then isolated from donated blood using Ficoll–Hypaque density-gradient centrifugation and maintained in RPMI supplemented with 10% FCS and 1% L-glutamine.
Flow cytometry
Cells were analyzed using an FACSCalibur flow cytometer, and data analysis was performed using the CellQuest©Pro software (v4.01) (both Becton Dickinson, Oxford, United Kingdom). To determine Jam-1 expression, cells were labeled with 3 μL of PE-conjugated Jam-1 antibody (Santa Cruz Biotechnologies, Wembley, United Kingdom) for 30 min at 4°C. The cells were then washed and fixed using 1% paraformaldehyde (PFA; Sigma).
Cell-surface reovirus binding
AML cell lines and primary samples were resuspended in ice-cold PBS prior to the addition of 10 plaque-forming units (pfu) of reovirus per cell and incubated on ice for 1 h. The cells were then washed and incubated with 1 μL of mouse anti-reovirus-σ3 antibody (Developmental Studies Hybridoma Bank, University of Iowa, Iowa, USA) for 30 min on ice. After an additional wash step, the cells were incubated for a further 30 min on ice with 2 μL of FITC-labeled goat anti-mouse antibody (BD Pharmingen, Oxford, United Kingdom). The cells were then fixed using 1% PFA and reovirus binding assessed by flow cytometry.
Cell viability using propidium iodide staining
AML cells were infected with a range of reovirus concentrations (0, 0.01, 0.1, 1 or 10 pfu/cell). At the indicated time points postinfection, the cells were harvested, washed, and stained with 5 μL of 0.05 mg/mL propidium iodide (PI; Sigma) for 15 min. The cells were acquired immediately by flow cytometry, and cell death was indicated by the percentage of cells staining positive for PI.
Chromium release assay
Cell-mediated cytotoxicity was measured using a standard 4 h chromium release assay, as previously described.21 Briefly, tumor cell targets (Kasumi-1, THP-1, KG-1 and ML-1) were labeled with 100 μCi 51Cr (PerkinElmer, Cambridge, United Kingdom) for 1 h and washed thrice in Hank's Balanced Salt Solution (HBSS; Sigma). The labeled cells were then co-cultured with PBMC at different effector:target (E:T) cell ratios. After 4 h, the cells were pelleted, and 50 μL of the supernatant was transferred to Luma scintillation plates (PerkinElmer) and left to dry overnight. Levels of 51Cr were determined using a Wallac Microbeta jet 1450 scintillation counter (Wallac EG & G Ltd, Milton Keynes, United Kingdom). Where indicated, the NK cells were depleted from PBMC using CD56 microbeads (Miltenyi-Biotec, Woking, United Kingdom), according to the manufacturer's instructions. Depletion effectively removed >90% of NK cells, and the resulting NK-depleted cell population was used in chromium release assays. In addition, cytotoxicity assays were performed in the presence or absence of the chelating agent, ethylene glycol tetraacetic acid (EGTA; 2 mM), which binds Ca2+, making it unavailable for granule exocytosis.22 In all cases, percentage target cell lysis was calculated using the following formula:
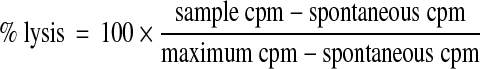 |
CD107 degranulation assay
Cell-surface CD107 expression was used as a marker of lymphocyte degranulation, as previously described.23 Briefly, PBMC from healthy donors (with or without previous overnight incubation with 0.1 pfu/cell reovirus) were cultured at a 10:1 ratio with AML cell line targets or primary blasts for 1 h at 37°C. Brefeldin A (5 μg/mL; BioLegend, Cambridge, United Kingdom) and 5 μL each of FITC-conjugated CD107a and CD107b (BD Pharmingen) antibodies were added to the cell co-culture and incubated for a further 4 h at 37°C. Cells were then washed and stained with 3 μL of CD3-PerCP (BD Pharmingen) and 3 μL of CD56-PE antibodies (Serotec, Kidlington, United Kingdom) for 30 min to allow identification of the NK cell population. The cells were fixed with 1% PFA, and CD107 expression was determined using flow cytometry. It is worth noting that since NK cells constitute ∼10% of the PBMC population, the NK:target cell ratio used in this assay was effectively 1:1.
Intracellular IFNγ staining
For intracellular IFNγ staining, PBMC were co-cultured with AML targets and treated as just described for the CD107 degranulation assay, without the addition of CD107 antibodies. After fixation, the cells were then permeabilized using 0.3% saponin (Sigma) for 15 min at room temperature (RT). FITC-conjugated IFNγ antibody (BD Pharmingen) was then added to the cells at a 1:50 dilution in 0.1% saponin for 30 min at RT, before acquisition using flow cytometry.
ELISA
Supernatants from reovirus-infected AML cells were collected at various time points postinfection and assessed for IFNα (MabTech AB, Buro, Germany) and RANTES (R&D Systems, Abingdon, United Kingdom) using matched paired antibodies, according to the manufacturer's instructions.
Plaque assay
Reovirus replication was determined by standard plaque assay using L929 cells. Infected AML cells and supernatant were subjected to three freeze-thaw cycles and diluted in serum-free media before being added to confluent L929 cells. The cells were incubated at 37°C for 2.5 h before overlay media, containing 1.6% (w/v) carboxymethylcellulose (Sigma) and DMEM supplemented with 10% FCS, was added to each well. After 72 h, the cells were washed and fixed using 0.1% (v/v) gluteraldehyde for 10 min. The plaques were then visualized using 0.5% crystal violet. Fold increase in virus titer was determined by comparison with the levels of input virus.
Statistical analysis
p-values were calculated using paired student's t-test or two-way ANOVA. Statistical significance is denoted by *p<0.05.
Results
Response of AML cell lines to reovirus
Reovirus has been reported as having efficient oncolytic activity against human cancer cell lines and primary ex-vivo tumor cells obtained from a range of different hematological malignancies.16–18 However, the susceptibility of AML cells to reovirus infection and oncolysis has not yet been determined. To begin to address the potential utility of reovirus in the treatment of AML, cell lines derived from either the peripheral blood or the bone marrow of patients with AML were assessed for the expression of Jam-1, the cell-surface receptor for reovirus.24 Jam-1 was expressed at the surface of all four AML cell lines studied: Kasumi-1, THP-1, KG-1, and ML-1 cells (Fig. 1a). Furthermore, reovirus attachment to AML cells was confirmed by FACS analysis for surface-bound virus (Fig. 1b), as previously described.25 Plaque assays were then used to determine virus replication. Despite reovirus binding to all the AML cell lines, only two of the cell lines, Kasumi-1 and THP-1, supported efficient reovirus replication (Fig. 1c). Replication was most efficient in Kasumi-1 cells, with a 1608-fold increase in replication at 4 days postinfection (note log scale). At the same time point, a fold-increase of 210 was observed in THP-1 cells, but only increases of 3- and 18-fold were found in KG-1 and ML-1 cells, respectively. This heterogeneity of productive infection, despite binding to all cell lines, is likely due to the recognized complexity of the mechanisms that control reovirus sensitivity. With regard to the ras status of these cell lines (a major determinant of reovirus sensitivity), mutations have been reported in Kasumi-1 and THP-1 cells (although only in THP-1 is the mutation activating), but not in KG-1 or ML-1.26 Therefore, the sensitivity of the cell lines to reovirus broadly correlates with their ras status.
FIG. 1.

Jam expression, cell-surface reovirus binding, and reovirus replication in AML cell lines. (a) PE-conjugated Jam-1 antibody was used to determine the cell-surface expression of Jam-1 in AML cell lines using flow cytometry. Jam-1 expression is shown by black lines, and gray lines indicate isotype-matched control staining. Data are representative of three independent experiments. (b) Reovirus binding at the surface of AML cell lines was detected using mouse anti-reovirus-σ3 and FITC-labeled goat anti-mouse antibodies. Representative binding from two independent experiments is shown by black lines, while gray lines indicate antibody binding in the absence of virus. (c) AML cell lines were treated with 0.1 pfu/cell reovirus for 2, 3, or 4 days. Reovirus replication was determined by plaque assay using L929 cells. Data show the mean+s.e.m. of three independent experiments. AML, acute myeloid leukemia; pfu, plaque-forming units; FITC, fluorescein isothiocyanate; s.e.m., standard error of the mean.
After the incubation of AML cell lines with reovirus, cell viability was measured using PI staining and flow cytometry. Both Kasumi-1 and THP-1 cells were highly susceptible to reovirus-induced cell death (Fig. 2a), correlating with their support of reovirus replication. At 3 days postinfection, a 37% decrease in the viability of Kasumi-1 cells was observed with 1 pfu/cell reovirus, and a 58% decrease was observed with 10 pfu/cell virus (p<0.0001 for both doses versus untreated controls). Further enhancement in cell death was seen at 4 days postinfection, with a decrease in cell viability of more than 70% after incubation with 10 pfu/cell reovirus. For THP-1 cells, a decrease in cell viability of 51% was observed after incubation of cells with 1 pfu/cell of reovirus for 3 days (p<0.0001). This was increased to 68% in the presence of 10 pfu/cell reovirus, but further death was not observed beyond this time point. In contrast, KG-1 and ML-1 cells remained relatively resistant to reovirus cytotoxicity, with viability affected only at later time points in response to large amounts of the virus (Fig. 2a). At 4 days postinfection, using 10 pfu/cell reovirus, the viability of KG-1 and ML-1 cells was only reduced by 19% and 15%, respectively (p<0.01). Similar changes in viability were observed using the WST-1 cell viability assay (data not shown). Hence, although levels of Jam-1 expression and reovirus binding did not correlate with replication in AML cells, productive infection was broadly linked to the amount of direct cell killing.
FIG. 2.

Reovirus-induced cell death and induction of cytokine release in AML cell lines. (a) Cells were infected with increasing amounts of virus ranging from 0 to 10 pfu/cell for 1–4 days. Cell viability was then determined using PI staining and flow cytometry. Results show mean±s.e.m. of three independent experiments. (b) AML cells were incubated with 0, 0.1, or 1 pfu/cell reovirus for 3 days before supernatant collection. The concentration of IFNα or RANTES was then determined by ELISA. Results indicate mean+s.e.m. of three independent experiments. IFNα, interferon alpha; PI, propidium iodide; RANTES, chemokine (C-C motif) ligand 5 (known as RANTES [regulated upon activation, normal T-cell expressed, and secreted]).
A range of pro-inflammatory cytokines and chemokines can be released from cells in response to virus infection (including reovirus), which may assist in the regulation of innate and adaptive immune responses.2,27 Therefore, the release of cytokines (IL-2, IL-4, IL-6, IL-8, IL-10, IL-12p40/70, IL-28, IFNα/β, GM-CSF, and TNFα) and the chemokine RANTES from reovirus-infected AML cells was assessed using ELISA. The type I interferon, IFNα, was secreted from THP-1 and KG-1 cells in response to virus infection, with negligible levels detected from Kasumi-1 cells (Fig. 2b). IFNα was not detected in supernatants from ML-1 cells, even after incubation with approximately 10 pfu/cell reovirus (data not shown). Therefore, high sensitivity to reovirus (Kasumi-1 and THP-1) was not linked to a low IFNα response (Kasumi-1 and ML-1), suggesting that reovirus is relatively resistant to type 1 IFNs, consistent with previous data in melanoma27 (no IFNβ was secreted by any AML cell line—data not shown). The chemokine RANTES was also detected in supernatants from all the AML cell lines, with higher levels detected in Kasumi-1, THP-1, and KG-1 cells in response to the virus (Fig. 2b). In addition, IL-8 was released from all cell lines in response to reovirus, with IL-6 secreted from only THP-1 cells (data not shown). There was no release of the pro-inflammatory cytokines IL-2, IL-4, IL-12p40/70, IL-28, GM-CSF, TNFα, or the anti-inflammatory cytokine IL-10 (data not shown).
The direct effect of reovirus on primary AML blasts
To increase the clinical relevance of this study, we next examined the effect of reovirus on primary AML cells. AML blasts were collected from the peripheral blood of patients and analyzed for Jam-1 expression, reovirus attachment, reovirus replication, cell viability, and cytokine secretion. The clinical characteristics of the patients are summarized in Table 1. Jam-1 expression was confirmed on the surface of all AML patient samples (n=10) assessed, with reovirus binding found on the surface of three out of the three samples tested (representative Jam-1 expression and reovirus binding are shown in Fig. 3a). Viral replication was analyzed in four AML samples, and significant reovirus replication (10-fold increase) was found in only one of the samples (data not shown). The direct effect of reovirus on the viability of primary AML blasts was determined using PI and flow cytometry. As shown in Figure 3b, reovirus led to a reduction in cell viability in 8 out of the 10 samples, despite variations in sample viability in the absence of a virus. We observed a maximum 44% decrease in viability (this sample is shown as a representative histogram of PI staining in Fig. 3b), with a statistically significant reduction in reovirus treatment across all samples (p=0.0046). In agreement with the cell line data, IFNα and RANTES were also found in supernatants from reovirus-infected primary AML cells, in a dose-dependent manner (Fig. 3c). Hence, consistent with the cell line data, primary AML cells undergo an inflammatory death on exposure to reovirus, albeit with less killing than seen in cell lines, and variable sensitivity among different patients.
Table 1.
Patient Clinical Data
| AML sample | Disease stage | FAB classification | WHO classification | Cytogenetics |
|---|---|---|---|---|
| AML 1 | Diagnosis | M4 | NOS with myelomonocytic differentiation | -Y |
| AML 2 | 1st relapse | M2 | AML with NPM1 mutation | -12p |
| AML 3 | 1st relapse | M2 | AML with NPM1 mutation | Normal |
| AML 4 | Diagnosis | M1 | NOS without maturation | +9, +21 |
| AML 5 | 1st relapse | M2 | NOS with maturation | Normal |
| AML 6 | Diagnosis | M2 | Biphenotypic—mixed B-myeloid. | 45∼48,XX,del(6)(q?),del(7) |
| Predominant clone (92%)=AML inv(16)(p13;q22) | (q?32),+8,+13,inv(16)(p13q22),+22 | |||
| AML 7 | Diagnosis | M1 | AML arising from transformation of MDS | Tetraploidy with 5q- |
| AML 8 | Diagnosis | M2 | AML arising from transformation of MDS | Normal |
| AML 9 | Refractory | M2 | AML NOS | Complex with complete/partial loss of ch5 |
| AML 10 | 1st relapse | M1 | AML arising from transformation of MDS | Complex with complete/partial loss of ch5 |
AML, acute myeloid leukemia; FAB, French–American–British; NOS, not otherwise specified; NPM, nucleophosmin; MDS, myelodysplastic syndrome.
FIG. 3.

The direct effect of reovirus on primary AML cells. (a) PE-conjugated Jam-1 antibody was used to determine the cell-surface expression of Jam-1 in primary AML samples using flow cytometry. Jam-1 expression is shown by black lines, and gray lines indicate isotype-matched control staining. Data are representative of 10 patient samples. Reovirus binding to the surface of primary AML cells was also confirmed using mouse anti-reovirus-σ3 and FITC-labeled goat anti-mouse antibodies. Representative binding from three independent experiments is shown by black lines, while gray lines indicate antibody binding in the absence of the virus. (b) Primary AML cells were incubated with either 0 or 10 pfu/cell reovirus for 7 days. Cell viability was assessed using PI staining and flow cytometry. Histograms show PI staining in the most susceptible AML sample in response to reovirus. Percentage viability is summarized in the line graph, where each line represents an individual patient (p=0.0046 across all samples). Disease stage, cytogenetics, and AML classification relating to these samples is as summarized in Table 1. (c) Primary AML cells were infected with 0, 1, or 10 pfu/cell reovirus. At 3 days postinfection, supernatants were collected and assessed for the presence of IFNα and RANTES by ELISA. The IFNα graph is representative of four out of four patient samples tested, whereas RANTES was found in one out of two samples.
Reovirus enhancement of NK cell activity against AML cell lines
It has become increasingly evident that the efficacy of oncolytic viruses is not only the result of direct virus cytotoxicity, but also due to the induction of anti-tumor immunity.2,11–13,27 The potential benefit of the activation of anti-tumor immunity in AML is already apparent from the recognized therapeutic effect of the GVL response. Therefore, the effect of reovirus on NK cell responses against AML was investigated, to determine the extent to which reovirus may be used to support innate anti-AML immunity.
We have previously shown that reovirus does not directly activate NK cells, but that dendritic cells can be stimulated by the virus, which, in turn, initiates NK cell effector function.11 In this study, we chose to test the response of PBMC to reovirus, as PBMC represent a physiologically relevant population of immune cells, including dendritic and NK cells, available from patients as well as normal donors. Furthermore, using this mixed population, rather than isolated and/or cultured cells, more closely reflects the cross-talk between different innate effectors, which occurs in vivo in the systemic administration of the virus. Within the mixed PBMC population, NK and other potential innate effector cells were identified via surface marker expression, to study their response to reovirus. The incubation of PBMC from healthy donors overnight with reovirus led to an ∼50% increase in the number of NK (CD56+ CD3−) cells expressing the early activation marker CD69 (Fig. 4a), indicating an increased state of activation in these cells. An increase in CD69 expression was not observed in either NK-T (CD56+ CD3+) or CD56− CD3+ T-cell populations within the PBMC (data not shown). To address whether these reovirus-activated NK cells could mount an effective cytotoxic response against AML cell targets, PBMC (with or without previous overnight activation with 0.1 pfu/cell reovirus) were incubated with each of the AML cell line targets for 4 h at a 10:1 ratio. A CD107 degranulation assay was then performed and used to identify the NK cells that were actively secreting cytotoxic granules.23 As shown in Figure 4b, when untreated PBMC were exposed to AML cell line targets, small increases (<10%) in NK cell degranulation were observed. Reovirus alone was not sufficient to enhance degranulation from the NK cells in the absence of tumor targets. Importantly, however, there was a marked increase in CD107 expression in NK cells in response to all four AML cell lines after the activation of PBMC with reovirus. In response to THP-1 and KG-1 cells, increases of 2.5- and 2.6-fold, respectively, were found in the percentage of degranulating NK cells, using reovirus-activated PBMC compared with nonactivated PBMC. An average of 9% of nonactivated NK cells degranulated in response to Kasumi-1 cells, compared with 34% of reovirus-activated PBMC. Significant enhancement in degranulation was also observed for ML-1 cells, with 25% of the NK cell population degranulating in response to these targets after reovirus activation.
FIG. 4.

Reovirus activates NK cells and enhances their activity against AML. Whole PBMC were isolated from blood of healthy donors, cultured overnight with 0 or 0.1 pfu/cell reovirus, and then assessed as follows: (a) Cell-surface expression of CD69 was assessed within the CD56+CD3− NK cell population by flow cytometry. Results indicate mean+s.e.m. of five independent experiments. *p<0.05. (b) PBMC were incubated with AML cell line targets at a ratio of 10:1, for a total of 5 h at 37°C. Cell-surface expression of CD107a/b was then assessed within the NK cell population. Dot plots are representative of NK cell degranulation in response to Kasumi-1 cells, while the histogram shows mean degranulation in response to all cell lines+s.e.m. of six independent experiments. *p<0.05. (c) PBMC were incubated with AML cell line targets at a ratio of 10:1, for a total of 5 h at 37°C. Intracellular IFNγ levels were then assessed within the NK cell population. Dot plots are representative of IFNγ production in response to Kasumi-1 cells, while the histogram shows mean production in response to all cell lines+s.e.m. of four independent experiments. *p<0.05. Ctl, no target control; NK, natural killer; PBMC, peripheral blood mononuclear cells.
In addition to degranulation, NK cells also secrete IFNγ after activation. Similar to the CD107 data, we found that the proportion of NK cells staining positive for intracellular IFNγ in response to AML targets was greater in PBMC which had been activated with reovirus (Fig. 4c), with fold increases of 3.5, 2.6, 2.8, and 3.3 for Kasumi-1, THP-1, KG-1, and ML-1 cells, respectively. Together, these data strongly suggest that the presence of reovirus enhances the potency of effector NK cells toward AML.
Reovirus enhancement of NK-mediated cytotoxicity against AML cell targets
To establish whether this increased NK cell activity translates into leukemic cell killing, chromium release assays were used to determine the susceptibility of AML cells to NK-mediated cell lysis. All cell lines were largely resistant to cell-mediated cytotoxicity after incubation with resting PBMC. However, the activation of PBMC with reovirus led to an increase in the lysis of Kasumi-1, THP-1, and KG-1 cells, but not ML-1 cells (Fig. 5a). For Kasumi-1 and KG-1 cells, a 2- to 3-fold increase in cell lysis was obtained with reovirus-activated PBMC at E:T cell ratios of 100:1 and 50:1, compared with lysis found with nonactivated PBMC. The monocytic THP-1 cells were particularly sensitive to reovirus-activated NK cells with a 9-fold increase in cell death at all E:T cell ratios of 25:1 and above. These results demonstrate that reovirus can improve innate immune-mediated killing of AML targets. The absence of ML-1 cell lysis is likely due to their resistance to perforin-mediated killing, as has been previously reported for ML-2 cells,28 which were originally derived from the same AML patient as ML-1.29
FIG. 5.

Reovirus enhances NK-mediated cytotoxicity against AML cells. Whole PBMC were isolated from blood of healthy donors, cultured overnight with 0 or 0.1 pfu/cell reovirus, and then assessed as follows: (a) PBMC were incubated with 51Cr-labeled AML cell lines for 4 h at different effector:target ratios. Percentage target cell lysis was then determined. Results show mean±s.e.m. of four independent experiments. (b) Whole PBMC, CD56-depleted PBMC, or PBMC treated with 2 mM EGTA were incubated with 51Cr-labeled Kasumi-1 or THP-1 cell lines for 4 h, at different effector:target ratios. Percentage target cell lysis was then determined. Results are representative of four independent experiments. EGTA, ethylene glycol tetraacetic acid.
Since whole PBMC were used in these killing assays, which contain multiple types of potential effector cells, the specific role of NK cells in AML cell lysis was further addressed by NK cell depletion. CD56 microbeads were used to remove NK cells from reovirus-activated PBMC, before their use in chromium release assays against Kasumi-1 and THP-1 targets. As shown in Figure 5b, depletion of the CD56-positive NK cell population substantially reduced AML cell killing by reovirus-activated PBMC, demonstrating that the NK cell population plays a major role in the observed cytotoxicity. To further characterize the mechanism of AML lysis, chromium release assays were also performed in the presence of 2 mM EGTA. The presence of this calcium chelating agent completely abolished cell lysis (Fig. 5b), indicating that the cell killing detected within this experiment was perforin mediated,22 as expected for NK cell cytotoxicity. This result is also consistent with previous data on the killing of melanoma tumor targets by isolated reovirus-treated dendritic cell-activated NK cells.11
Reovirus enhancement of NK cell responses toward AML in patient samples
To translate these findings further toward the clinical setting, we next wished to address whether NK cells from patients with AML could be activated by reovirus and whether reovirus-activated NK cells could target primary AML blasts. The addition of reovirus to a patient PBMC preparation (which also contained AML blasts) led to an increase in NK degranulation and IFNγ production in response to THP-1 targets (Fig. 6a), suggesting the potential functionality of reovirus-stimulated innate effectors in patients. This is particularly encouraging, as the immunotherapeutic potential of reovirus in enhancing anti-tumor activity against AML is likely to be even more effective in remission. This is because, when tumor burden is high on initial presentation or at relapse, immune effector cells may be suppressed,30 while NK cells from AML patients in remission function in a similar manner to those from healthy donors.31 In this context, and also to test patient AML blasts for their susceptibility as innate targets, we further investigated whether reovirus could enhance the activity of healthy donor NK cells against primary AML samples. As shown in Figure 6, reovirus increased NK cell activity against three different primary AML samples, with an increase in the percentage of NK cells degranulating after reovirus treatment (Fig. 6b; p<0.05 for targets 1 and 2; p=0.069 for target 3) and a rise in the percentage of NK cells positive for intracellular IFNγ (Fig. 6c; p=0.058 for target 1; p<0.05 for target 2; p=0.184 for target 3). Although we were not able to directly test the killing of primary AML blasts (as they could not be labeled with chromium), both NK cell degranulation and IFNγ production are likely to reflect functional cytotoxicity, as we have previously found a consistent association between these two assays and chromium release in other solid tumor models,11,12 as well as in AML cell lines, as demonstrated in Figures 4b/c and 5. Collectively, these findings support the use of reovirus that enhances innate immune responses against primary AML cell targets in patients.
FIG. 6.

Reovirus enhances the activity of NK cells toward AML in patient samples. (a) PBMC from an AML patient were incubated overnight in the presence of absence of 0.1 pfu/cell reovirus. Cells were then incubated with THP-1 cells (at a 10:1 ratio) for 5 h. Cell-surface CD107a/b expression within the NK cell population was determined using flow cytometry. (b) Healthy donor PBMC were incubated overnight in the presence or absence of 0.1 pfu/cell reovirus. PBMC were then incubated with primary AML blasts (at a 10:1 ratio) for 5 h. Cell-surface CD107a/b expression within the NK cell population was determined using flow cytometry. Each donor is represented by a separate line. p<0.05 for targets 1 and 2; p=0.069 for target 3. (c) Healthy donor PBMC were incubated overnight in the presence or absence of 0.1 pfu/cell reovirus. PBMC were then incubated with primary AML blasts (at a 10:1 ratio) for 5 h. Intracellular IFNγ production within the NK cell population was determined using flow cytometry. Graph shows mean+s.e.m. of four individual donors. p=0.058 for target 1; p<0.05 for target 2; p=0.184 for target 3).
Discussion
While standard chemotherapy regimens for AML achieve high rates of remission, this is often short lived, as the persistence of residual and refractory disease commonly leads to disease relapse. As a result, AML has one of the poorest overall survival rates of all types of leukemia, highlighting the need for novel drugs that could be used to augment current chemotherapy treatments. Progress in the treatment efficacy for elderly patients has been particularly limited, owing to complexities in both the biology of AML and the fitness of the patient for aggressive interventions.32 The use of oncolytic viruses that target hematological malignancies has been gaining interest, with efficient oncolytic activity observed in a range of lymphoma and leukemia cells.33–37 While to date the majority of these studies have focused on the direct cytolytic activity of these viruses, here, using reovirus as an example, we provide novel evidence which suggests that oncolytic viruses may also be used to support anti-leukemia immunity.
In this study, we initially investigated the direct effect of reovirus on AML cells, showing that reovirus can replicate and induce death in cell lines derived from AML of both myeloblastic and monocytic origin (Figs. 1c and 2a), particularly those with mutated ras. We also found that reovirus decreased cell viability in primary samples, albeit with varying efficiency (Fig. 3b). Although replication was found in only one of the four primary samples tested by plaque assay (data not shown), decreases in cell viability were found even in the absence of detectable replication. This lower sensitivity of primary patient AML samples to reovirus killing/replication in comparison to some immortalized cell lines is consistent with our previous data on melanoma and colorectal cancer, and highlights the importance of testing relevant clinical material wherever feasible (unpublished data).2 The KG-1 and ML-1 cell lines also remained fairly resistant to direct reovirus cytotoxicity (Fig. 2a), which, in conjunction with the primary AML cell data, suggests a degree of heterogeneity in the direct sensitivity of AML cells to reovirus. Since AML is itself a highly heterogenous disease, it would be interesting to correlate such sensitivity to the molecular profile of the cells,38 an approach that will require a much larger cohort of patients. A number of studies have previously investigated the oncolytic potential of reovirus in tumor cells of hematological origin, and a similar heterogeneity has been reported in the sensitivity of lymphoid cell lines to reovirus.16,17
In our study, we also found that reovirus induced secretion of the pro-inflammatory cytokine IFNα and the chemokine RANTES from infected AML cell lines and primary blasts (Figs. 2b and 3c). In addition to playing a role in the anti-viral response, IFNα has also been shown to activate dendritic cells and other immune effector cells that may support the development of anti-leukemic immune responses39–42; there is also evidence that IFNα may have direct anti-leukemia effects against AML.43 Furthermore, it is noteworthy that, despite the production of IFNα, significant viral replication was still detected in some cell lines, as previously observed for IFNβ and melanoma.27 This suggests that an effective type 1 IFN response from reovirus-infected malignant cells, potentially capable of supporting anti-tumor immunity,12 does not necessarily abrogate virus replication and direct oncolysis. This is the first time that IFNα from tumor cells in response to reovirus has been detected; however, IFNα from dendritic cells directly infected with reovirus had been previously observed, suggesting that this response may be common to cells of myeloid origin.11
An early observation of the GVL effect in patients after allogeneic bone marrow transplantation,20 and the prolonged remission of patients with relapsed chronic myeloid leukemia after donor lymphocyte transfusion,44 revealed the potential of the immune system to eradicate leukemia cells. Therefore, given current interest in the use of immunotherapy to target leukemia,45–47 and our previous data showing that reovirus can enhance anti-tumor immunity,11–13,27 we investigated the effect of reovirus on NK cell responses against AML.
During high leukemic cell burden, the function of patient NK cells may be impaired, as NK cells from AML patients have been shown to express lower levels of activating receptors, compared with normal NK cells.48,49 Nonetheless, we found that AML patient NK cells can be activated by reovirus to increase their degranulation against the myeloblastic cell line THP-1 (Fig. 6a). However, in the clinical setting, reovirus would not replace chemotherapy, which is generally effective in reducing the bulk of tumor cells. Rather, we anticipate that reovirus would be administered in combination with, or post-, chemotherapy, with the aim of targeting minimal residual disease and preventing disease relapse. Since previous studies have demonstrated that NK cells expanded from AML and acute lymphoblastic leukemia patients in complete remission are functionally equivalent to those from normal donors,31,50 it is likely that, following immune recovery after chemotherapy, patients' NK cells would function normally. Hence, the experiments presented in this study, describing normal donor NK cell interactions with AML targets, remain highly relevant to the clinical scenario of patients in partial or complete remission after initial treatment for AML.
Here, we have shown for the first time that reovirus can be used to activate NK cells which display enhanced activity against AML cell lines, as well as toward primary blasts (Figs. 4 and 6). In a novel approach, NK cells were activated by reovirus within the context of a whole PBMC population, which is a clinically more relevant scenario than adding reovirus to isolated NK cells and also allows cross-talk between potential virus-responsive and anti-tumor effector immune cell subsets. We hypothesize that this activation of NK cells is the result of reovirus activation of dendritic cells and subsequent cross-talk with NK cells, as isolated NK cells are not activated by reovirus, whereas dendritic cells pulsed with reovirus can stimulate NK cell function.11 The idea that dendritic cells are responsible for reovirus detection within PBMC in our study is further supported by the broader data implicating dendritic cells as sensors of viral infection that are able to then activate NK cell innate immune responses.51
The increased levels of CD107 degranulation (Figs. 4b and 6a) and IFNγ production (Figs. 4c and 6b) found in the NK cell population in response to AML targets after reovirus-mediated activation suggest that reovirus increases the functional potential of NK cells against AML cells. Importantly, we have shown that this activation of NK cells by reovirus translates into more efficient killing of AML targets compared with nonactivated NK cells (Fig. 5a). Depletion of the CD56-positive NK cell population led to a significant reduction in target cell lysis, confirming that the reovirus enhancement of AML cell killing was largely mediated by NK cells (Fig. 5b). Although all these experiments involved an allogeneic mismatch between reovirus-activated NK cells and their AML targets, these findings are encouraging for the incorporation of reovirus into current allogeneic NK cell/bone marrow transplant-based treatment strategies. We also demonstrated that all the killing within these assays was perforin mediated, as the inclusion of 2 mM EGTA within the chromium release assay led to a complete abolition of cell-mediated killing (Fig. 5b). The absence of cell-mediated cytotoxicity toward ML-1 cells (Fig. 5a) suggests that these cells possess a mechanism, such as resistance to perforin-mediated killing,52 which protects them from NK-mediated lysis, despite efficient NK degranulation and IFNγ production in response to ML-1 targets (Fig. 4b, c). Indeed, as just mentioned, it has been reported that perforin is unable to attach to the surface of ML-2 cells, which renders them resistant to NK-mediated killing28 and offers a likely explanation for the resistance of ML-1 cells, as ML-1 and ML-2 cells were derived from the same patient. Nevertheless, the enhanced killing of three out of the four AML targets by reovirus-activated NK cells provides encouraging evidence that reovirus may boost anti-leukemia immune responses. Furthermore, the killing of KG-1 cells by reovirus-activated NK cells (Fig. 5) highlights the potential for immune-mediated reovirus therapy even when direct oncolysis is limited.
Our ongoing studies are addressing the feasibility of reovirus delivery to AML cells in humans in vivo. Since the majority of the population has been infected with reovirus at an early age, the presence of neutralizing anti-reovirus antibodies may be expected to hinder the systemic administration of the virus. However, our recently completed clinical trial (REO13) has confirmed that after intravenous delivery to colorectal cancer patients, reovirus can be found in association with both mononuclear and granulocyte immune cells in the blood, as well as in metastatic disease in the liver (data submitted). Furthermore, a recent study has demonstrated the efficient delivery of vaccinia virus to residual lymphoma cells using cytokine-induced killer cells as a carrier vehicle.53 In conjunction with other studies, these findings suggest that there are strategies which may be used to circumvent the potential limitations of systemic delivery imposed by pre-existing immunity to reovirus.25
There is increasing evidence that the efficacy of oncolytic viruses may be optimized by a combination with other anti-cancer agents54–56 and this has been translated to clinical trials.7,9 Recently, the use of certain virus combinations has also led to a synergistic increase in cancer cell death in some models.57,58 Since a number of other oncolytic viruses, including adenovirus, myxoma virus, and vesicular stomatitis virus, have also been shown to induce death in leukemic cells of myeloid origin, future work should also include testing reovirus in combination with other viruses for the treatment of leukemia.33,36,37,59,60
This study, using patient material as well as cell lines, provides evidence that reovirus can be directly cytotoxic against AML cells. We have also demonstrated that reovirus can activate NK cells within a whole PBMC population and that this reovirus-mediated activation of NK cells can enhance their cytolytic activity toward AML targets. These findings support the potential utility of reovirus in the treatment of residual and/or relapsed AML, both as a useful addition to chemotherapy for direct cytolysis and as an immunotherapeutic agent stimulating anti-leukemic innate immunity. In the context of its established clinical safety record, these results support the incorporation of reovirus into the design of future clinical trials for the treatment of AML.
Acknowledgments
This study was supported by Yorkshire Cancer Research. The authors would also like to thank Oncolytics for the provision of Reolysin.
Author Disclosure Statement
Oncolytics Biotech, Inc.: K.J.H./R.V./H.P./A.M., commercial research grants; M.C., employee.
References
- 1.Alloussi SH. Alkassar M. Urbschat S, et al. All reovirus subtypes show oncolytic potential in primary cells of human high-grade glioma. Oncol Rep. 2011;26:645–649. doi: 10.3892/or.2011.1331. [DOI] [PubMed] [Google Scholar]
- 2.Errington F. White CL. Twigger KR, et al. Inflammatory tumour cell killing by oncolytic reovirus for the treatment of melanoma. Gene Ther. 2008;15:1257–1270. doi: 10.1038/gt.2008.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hata Y. Etoh T. Inomata M, et al. Efficacy of oncolytic reovirus against human breast cancer cells. Oncol Rep. 2008;19:1395–1398. [PubMed] [Google Scholar]
- 4.Hirasawa K. Nishikawa SG. Norman KL, et al. Oncolytic reovirus against ovarian and colon cancer. Cancer Res. 2002;62:1696–1701. [PubMed] [Google Scholar]
- 5.Alain T. Kim TS. Lun X, et al. Proteolytic disassembly is a critical determinant for reovirus oncolysis. Mol Ther. 2007;15:1512–1521. doi: 10.1038/sj.mt.6300207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marcato P. Shmulevitz M. Pan D, et al. Ras transformation mediates reovirus oncolysis by enhancing virus uncoating, particle infectivity, and apoptosis-dependent release. Mol Ther. 2007;15:1522–1530. doi: 10.1038/sj.mt.6300179. [DOI] [PubMed] [Google Scholar]
- 7.Comins C. Spicer J. Protheroe A, et al. REO-10: A phase I study of intravenous reovirus and docetaxel in patients with advanced cancer. Clin Cancer Res. 2010;16:5564–5572. doi: 10.1158/1078-0432.CCR-10-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harrington KJ. Karapanagiotou EM. Roulstone V, et al. Two-stage phase I dose-escalation study of intratumoral reovirus type 3 dearing and palliative radiotherapy in patients with advanced cancers. Clin Cancer Res. 2010;16:3067–3077. doi: 10.1158/1078-0432.CCR-10-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lolkema MP. Arkenau HT. Harrington K, et al. A phase I study of the combination of intravenous reovirus type 3 Dearing and gemcitabine in patients with advanced cancer. Clin Cancer Res. 2011;17:581–588. doi: 10.1158/1078-0432.CCR-10-2159. [DOI] [PubMed] [Google Scholar]
- 10.Vidal L. Pandha HS. Yap TA, et al. A phase I study of intravenous oncolytic reovirus type 3 Dearing in patients with advanced cancer. Clin Cancer Res. 2008;14:7127–7137. doi: 10.1158/1078-0432.CCR-08-0524. [DOI] [PubMed] [Google Scholar]
- 11.Errington F. Steele L. Prestwich R, et al. Reovirus activates human dendritic cells to promote innate antitumor immunity. J Immunol. 2008;180:6018–6026. doi: 10.4049/jimmunol.180.9.6018. [DOI] [PubMed] [Google Scholar]
- 12.Prestwich RJ. Errington F. Steele LP, et al. Reciprocal human dendritic cell-natural killer cell interactions induce antitumor activity following tumor cell infection by oncolytic reovirus. J Immunol. 2009;183:4312–4321. doi: 10.4049/jimmunol.0901074. [DOI] [PubMed] [Google Scholar]
- 13.Prestwich RJ. Ilett EJ. Errington F, et al. Immune-mediated antitumor activity of reovirus is required for therapy and is independent of direct viral oncolysis and replication. Clin Cancer Res. 2009;15:4374–4381. doi: 10.1158/1078-0432.CCR-09-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kottke T. Hall G. Pulido J, et al. Antiangiogenic cancer therapy combined with oncolytic virotherapy leads to regression of established tumors in mice. J Clin Invest. 2010;120:1551–1560. doi: 10.1172/JCI41431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kottke T. Thompson J. Diaz RM, et al. Improved systemic delivery of oncolytic reovirus to established tumors using preconditioning with cyclophosphamide-mediated Treg modulation and interleukin-2. Clin Cancer Res. 2009;15:561–569. doi: 10.1158/1078-0432.CCR-08-1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alain T. Hirasawa K. Pon KJ, et al. Reovirus therapy of lymphoid malignancies. Blood. 2002;100:4146–4153. doi: 10.1182/blood-2002-02-0503. [DOI] [PubMed] [Google Scholar]
- 17.Alain T. Muzik H. Otsuka S, et al. Susceptibility of mantle cell lymphomas to reovirus oncolysis. Leuk Res. 2010;34:100–108. doi: 10.1016/j.leukres.2009.05.020. [DOI] [PubMed] [Google Scholar]
- 18.Thirukkumaran CM. Luider JM. Stewart DA, et al. Reovirus oncolysis as a novel purging strategy for autologous stem cell transplantation. Blood. 2003;102:377–387. doi: 10.1182/blood-2002-08-2508. [DOI] [PubMed] [Google Scholar]
- 19.Barrett AJ. Understanding and harnessing the graft-versus-leukaemia effect. Br J Haematol. 2008;142:877–888. doi: 10.1111/j.1365-2141.2008.07260.x. [DOI] [PubMed] [Google Scholar]
- 20.Weiden PL. Flournoy N. Thomas ED, et al. Antileukemic effect of graft-versus-host disease in human recipients of allogeneic-marrow grafts. N Engl J Med. 1979;300:1068–1073. doi: 10.1056/NEJM197905103001902. [DOI] [PubMed] [Google Scholar]
- 21.Errington F. Jones J. Merrick A, et al. Fusogenic membrane glycoprotein-mediated tumour cell fusion activates human dendritic cells for enhanced IL-12 production and T-cell priming. Gene Ther. 2006;13:138–149. doi: 10.1038/sj.gt.3302609. [DOI] [PubMed] [Google Scholar]
- 22.Warren HS. Smyth MJ. NK cells and apoptosis. Immunol Cell Biol. 1999;77:64–75. doi: 10.1046/j.1440-1711.1999.00790.x. [DOI] [PubMed] [Google Scholar]
- 23.Betts MR. Brenchley JM. Price DA, et al. Sensitive and viable identification of antigen-specific CD8+ T cells by a flow cytometric assay for degranulation. J Immunol Methods. 2003;281:65–78. doi: 10.1016/s0022-1759(03)00265-5. [DOI] [PubMed] [Google Scholar]
- 24.Barton ES. Forrest JC. Connolly JL, et al. Junction adhesion molecule is a receptor for reovirus. Cell. 2001;104:441–451. doi: 10.1016/s0092-8674(01)00231-8. [DOI] [PubMed] [Google Scholar]
- 25.Ilett EJ. Barcena M. Errington-Mais F, et al. Internalization of oncolytic reovirus by human dendritic cell carriers protects the virus from neutralization. Clin Cancer Res. 2011;17:2767–2776. doi: 10.1158/1078-0432.CCR-10-3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Morgan MA. Dolp O. Reuter CW. Cell-cycle-dependent activation of mitogen-activated protein kinase kinase (MEK-1/2) in myeloid leukemia cell lines and induction of growth inhibition and apoptosis by inhibitors of RAS signaling. Blood. 2001;97:1823–1834. doi: 10.1182/blood.v97.6.1823. [DOI] [PubMed] [Google Scholar]
- 27.Steele L. Errington F. Prestwich R, et al. Pro-inflammatory cytokine/chemokine production by reovirus treated melanoma cells is PKR/NF-kappaB mediated and supports innate and adaptive anti-tumour immune priming. Mol Cancer. 2011;10:20. doi: 10.1186/1476-4598-10-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lehmann C. Zeis M. Schmitz N. Uharek L. Impaired binding of perforin on the surface of tumor cells is a cause of target cell resistance against cytotoxic effector cells. Blood. 2000;96:594–600. [PubMed] [Google Scholar]
- 29.Ohyashiki K. Ohyashiki JH. Sandberg AA. Cytogenetic characterization of putative human myeloblastic leukemia cell lines (ML-1, -2, and -3): Origin of the cells. Cancer Res. 1986;46:3642–3647. [PubMed] [Google Scholar]
- 30.Orleans-Lindsay JK. Barber LD. Prentice HG. Lowdell MW. Acute myeloid leukaemia cells secrete a soluble factor that inhibits T and NK cell proliferation but not cytolytic function—implications for the adoptive immunotherapy of leukaemia. Clin Exp Immunol. 2001;126:403–411. doi: 10.1046/j.1365-2249.2001.01692.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Torelli GF. Guarini A. Palmieri G, et al. Expansion of cytotoxic effectors with lytic activity against autologous blasts from acute myeloid leukaemia patients in complete haematological remission. Br J Haematol. 2002;116:299–307. [PubMed] [Google Scholar]
- 32.Erba HP. Has there been progress in the treatment of older patients with acute myeloid leukemia? Best practice & research. Clin Haematol. 2010;23:495–501. doi: 10.1016/j.beha.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 33.Kim M. Madlambayan GJ. Rahman MM, et al. Myxoma virus targets primary human leukemic stem and progenitor cells while sparing normal hematopoietic stem and progenitor cells. Leukemia. 2009;23:2313–2317. doi: 10.1038/leu.2009.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parrula C. Fernandez SA. Zimmerman B, et al. Measles virotherapy in a mouse model of adult T-cell leukaemia/lymphoma. J Gen Virol. 2011;92:1458–1466. doi: 10.1099/vir.0.028910-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patel B. Dey A. Ghorani E, et al. Differential cytopathology and kinetics of measles oncolysis in two primary B-cell malignancies provides mechanistic insights. Mol Ther. 2011;19:1034–1040. doi: 10.1038/mt.2011.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang G. Li G. Liu H, et al. E1B 55-kDa deleted, Ad5/F35 fiber chimeric adenovirus, a potential oncolytic agent for B-lymphocytic malignancies. J Gene Med. 2009;11:477–485. doi: 10.1002/jgm.1326. [DOI] [PubMed] [Google Scholar]
- 37.Yang CM. Liu H. Yang XD, et al. [Inhibitive effects of chimeric oncolytic adenovirus SG235 on leukemia cells in vitro] Zhejiang Da Xue Xue Bao Yi Xue Ban [J Zhejiang Univ Med Sci] 2010;39:226–230. doi: 10.3785/j.issn.1008-9292.2010.03.002. (In Chinese.) [DOI] [PubMed] [Google Scholar]
- 38.Marcucci G. Haferlach T. Dohner H. Molecular genetics of adult acute myeloid leukemia: prognostic and therapeutic implications. J Clin Oncol. 2011;29:475–486. doi: 10.1200/JCO.2010.30.2554. [DOI] [PubMed] [Google Scholar]
- 39.Arico E. Castiello L. Urbani F, et al. Concomitant detection of IFNalpha signature and activated monocyte/dendritic cell precursors in the peripheral blood of IFNalpha-treated subjects at early times after repeated local cytokine treatments. J Transl Med. 2011;9:67. doi: 10.1186/1479-5876-9-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brassard DL. Grace MJ. Bordens RW. Interferon-alpha as an immunotherapeutic protein. J Leukoc Biol. 2002;71:565–581. [PubMed] [Google Scholar]
- 41.Hansen ML. Woetmann A. Krejsgaard T, et al. IFN-alpha primes T- and NK-cells for IL-15-mediated signaling and cytotoxicity. Mol Immunol. 2011;48:2087–2093. doi: 10.1016/j.molimm.2011.07.008. [DOI] [PubMed] [Google Scholar]
- 42.Takaoka A. Yanai H. Interferon signalling network in innate defence. Cell Microbiol. 2006;8:907–922. doi: 10.1111/j.1462-5822.2006.00716.x. [DOI] [PubMed] [Google Scholar]
- 43.Anguille S. Lion E. Willemen Y, et al. Interferon-alpha in acute myeloid leukemia: An old drug revisited. Leukemia. 2011;25:739–748. doi: 10.1038/leu.2010.324. [DOI] [PubMed] [Google Scholar]
- 44.Kolb HJ. Mittermuller J. Clemm C, et al. Donor leukocyte transfusions for treatment of recurrent chronic myelogenous leukemia in marrow transplant patients. Blood. 1990;76:2462–2465. [PubMed] [Google Scholar]
- 45.Barrett AJ. Le Blanc K. Immunotherapy prospects for acute myeloid leukaemia. Clin Exp Immunol. 2010;161:223–232. doi: 10.1111/j.1365-2249.2010.04197.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smits EL. Berneman ZN. Van Tendeloo VF. Immunotherapy of acute myeloid leukemia: current approaches. Oncologist. 2009;14:240–252. doi: 10.1634/theoncologist.2008-0165. [DOI] [PubMed] [Google Scholar]
- 47.Smits EL. Lee C. Hardwick N, et al. Clinical evaluation of cellular immunotherapy in acute myeloid leukaemia. Cancer Immunol Immunother. 2011;60:757–769. doi: 10.1007/s00262-011-1022-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sanchez-Correa B. Gayoso I. Bergua JM, et al. Decreased expression of DNAM-1 on NK cells from acute myeloid leukemia patients. Immunol Cell Biol. 2012;90:109–115. doi: 10.1038/icb.2011.15. [DOI] [PubMed] [Google Scholar]
- 49.Sanchez-Correa B. Morgado S. Gayoso I, et al. Human NK cells in acute myeloid leukaemia patients: Analysis of NK cell-activating receptors and their ligands. Cancer Immunol Immunother. 2011;60:1195–1205. doi: 10.1007/s00262-011-1050-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Torelli GF. Guarini A. Maggio R, et al. Expansion of natural killer cells with lytic activity against autologous blasts from adult and pediatric acute lymphoid leukemia patients in complete hematologic remission. Haematologica. 2005;90:785–792. [PubMed] [Google Scholar]
- 51.Andrews DM. Andoniou CE. Scalzo AA, et al. Cross-talk between dendritic cells and natural killer cells in viral infection. Mol Immunol. 2005;42:547–555. doi: 10.1016/j.molimm.2004.07.040. [DOI] [PubMed] [Google Scholar]
- 52.Otten HG. Van Ginkel WG. Hagenbeek A, et al. Prevalence and clinical significance of resistance to perforin- and FAS-mediated cell death in leukemia. Leukemia. 2004;18:1401–1405. doi: 10.1038/sj.leu.2403414. [DOI] [PubMed] [Google Scholar]
- 53.Contag CH. Sikorski R. Negrin RS, et al. Definition of an enhanced immune cell therapy in mice that can target stem-like lymphoma cells. Cancer Res. 2010;70:9837–9845. doi: 10.1158/0008-5472.CAN-10-2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Heinemann L. Simpson GR. Boxall A, et al. Synergistic effects of oncolytic reovirus and docetaxel chemotherapy in prostate cancer. BMC Cancer. 2011;11:221. doi: 10.1186/1471-2407-11-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang B. Sikorski R. Kirn DH. Thorne SH. Synergistic anti-tumor effects between oncolytic vaccinia virus and paclitaxel are mediated by the IFN response and HMGB1. Gene Ther. 2011;18:164–172. doi: 10.1038/gt.2010.121. [DOI] [PubMed] [Google Scholar]
- 56.Pandha HS. Heinemann L. Simpson GR, et al. Synergistic effects of oncolytic reovirus and cisplatin chemotherapy in murine malignant melanoma. Clin Cancer Res. 2009;15:6158–6166. doi: 10.1158/1078-0432.CCR-09-0796. [DOI] [PubMed] [Google Scholar]
- 57.Alkassar M. Gartner B. Roemer K, et al. The combined effects of oncolytic reovirus plus Newcastle disease virus and reovirus plus parvovirus on U87 and U373 cells in vitro and in vivo. J Neurooncol. 2011;104:715–727. doi: 10.1007/s11060-011-0606-5. [DOI] [PubMed] [Google Scholar]
- 58.Le Boeuf F. Diallo JS. Mccart JA, et al. Synergistic interaction between oncolytic viruses augments tumor killing. Mol Ther. 2010;18:888–895. doi: 10.1038/mt.2010.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lichty BD. Stojdl DF. Taylor RA, et al. Vesicular stomatitis virus: A potential therapeutic virus for the treatment of hematologic malignancy. Hum Gene Ther. 2004;15:821–831. doi: 10.1089/hum.2004.15.821. [DOI] [PubMed] [Google Scholar]
- 60.Meng HT. Li L. Liu H, et al. Homoharringtonine acts synergistically with SG235-TRAIL, a conditionally replicating adenovirus, in human leukemia cell lines. Acta Pharmacologica Sinica. 2009;30:1529–1536. doi: 10.1038/aps.2009.147. [DOI] [PMC free article] [PubMed] [Google Scholar]