

Abstract
Low levels of the adipocyte-secreted protein adiponectin correlate with albuminuria in both mice and humans, but whether adiponectin has a causative role in modulating renal disease is unknown. Here, we first generated a mouse model that allows induction of caspase-8–mediated apoptosis specifically in podocytes upon injection of a construct-specific agent. These POD-ATTAC mice exhibited significant kidney damage, mimicking aspects of human renal disease, such as foot process effacement, mesangial expansion, and glomerulosclerosis. After the initial induction, both podocytes and filtration function recovered. Next, we crossed POD-ATTAC mice with mice lacking or overexpressing adiponectin. POD-ATTAC mice lacking adiponectin developed irreversible albuminuria and renal failure; conversely, POD-ATTAC mice overexpressing adiponectin recovered more rapidly and exhibited less interstitial fibrosis. In conclusion, these results suggest that adiponectin is a renoprotective protein after podocyte injury. Furthermore, the POD-ATTAC mouse provides a platform for further studies, allowing precise timing of podocyte injury and regeneration.
CKD, either as a primary renal abnormality or resulting from systemic conditions, such as diabetes or hypertension, afflicts an increasing number of Americans each year.1 Reduced GFR, which is a hallmark of CKD, may be present in upwards of 20 million patients in the United States.1,2 Within the glomerulus, podocytes form intricate foot processes, whose specialized contacts wrap glomerular capillaries forming the slit diaphragms that restrict large proteins from entering the urine. Selective injury to podocytes can result in primary diseases, such as FSGS with podocyte loss.3,4 Podocyte loss also occurs in systemic diseases, such as early diabetic nephropathy,5 obesity, and the metabolic syndrome,6 resulting in a sclerosing lesion and potential progression to CKD.7 Although the subsequent progression of these nephropathies of diverse causes may follow different paths, the loss of or injury to podocytes has become increasingly appreciated as a common starting point.
Albuminuria and diminished GFR are hallmarks of failing glomerular function leading to progressive sclerosis and eventual renal failure. Several rodent models have been developed to address specific aspects of this progression. Rodent remnant models, such as uninephrectomies and 5/6 nephrectomies, and various chemical injury models have multiple effects on different nephron structures.8,9 Genetic manipulations of podocytes in mice and rats have demonstrated the importance of specific surface and structural proteins on podocyte function or rendered the models’ podocytes more susceptible to injury or cytotoxic ablation.4,10–12 Correlating many of these models to initial human podocyte loss, however, has not always been possible because of design limitations or non-physiologic pathologic progression. Here we present a novel model of podocyte apoptosis that relies on the endogenous caspase-8 apoptotic pathway to permit inducible, selective ablation of podocytes from the glomerulus.
We generated a mouse using the human podocin promoter (Nphs2) to drive expression of a mutant FKBP fusion protein that, once dimerized, induces physiologic, caspase-8–mediated apoptosis in podocytes. Dimerization and caspase-8 activation occurs upon injection of a low-molecular-weight, construct-specific agent (AP20187) that uniquely binds only to the FKBP mutant protein.13 This ATTAC (apoptosis through targeted activation of caspase-8) gene construct expression strategy has been successfully used in adipocytes,13 pancreatic β cells,14 and cardiac myocytes,15 with each model demonstrating dose-dependent apoptosis with restored tissue function over time. The endogenous nature of ATTAC-induced ablation is also not inflammatory.16 Our new podocyte-specific POD-ATTAC mouse thus allows us to mimic the first step in many forms of human glomerulosclerosis—loss of podocytes—and study the resultant nephropathic progression, potential recovery, and long-term sequelae.
Adiponectin is an adipocyte-secreted protein that has positive effects on insulin sensitivity and cardiovascular disease.17,18 Unlike other adipokines that increase with weight gain, serum adiponectin is inversely correlated with obesity. Low adiponectin has been correlated with albuminuria in both mice and humans, and obesity (and, therefore, low adiponectin) may be causative in some patients with FSGS.6,7,19,20 Adiponectin knockout (KO) mouse models demonstrate increased podocyte injury and albuminuria, with adiponectin therapies potentially restoring some renal function.21,22 Interestingly, the relationship is inversed in human CKD: Adiponectin levels are greatly elevated in both children and adults with CKD, with serum levels directly correlated to proteinuria.20,23 Whether adiponectin merely correlates with the disease states associated with CKD or plays a direct role in renal function remains to be clarified. We have recently demonstrated that transgenic mice overexpressing adiponectin (we call them “adipo Tg” mice) are protected from caspase-8–mediated apoptosis and loss of organ function.15 These mice and our adiponectin KO mice (all crossed with our POD-ATTAC mice) furnish us with a valuable model to examine the role of adiponectin levels in the context of specific, inducible podocyte loss.
The POD-ATTAC (POD) mouse exhibits inducible and titratable podocyte reduction and dose-dependent changes in GFR and albuminuria. Minimal nephropathic changes occur at low induction, but glomerular histologic changes approximating human FSGS are seen with higher dimerizer dose. At intermediate dimerizer doses, POD mice recover podocyte density, reverse foot process effacement, and restore filtration function, but at higher doses they develop interstitial fibrosis and fail to recover. Adiponectin KO POD mice are in every aspect worse off than wild-type adiponectin POD mice after equivalent podocyte ablation. Adipo Tg-POD mice lose numbers of podocytes similar to those lost by wild-type mice but are protected from fibrosis and are better able to restore function. With the ability to present a range of nephropathic features dependent on the degree of podocyte loss, the POD-ATTAC mouse provides a tightly controlled novel murine platform for studying the progression of CKD with the potential of recovery.
Results
POD-ATTAC Mice Demonstrate a Dose-Dependent Inducible Reduction in Podocytes with Potential Recovery
Dimerization of the mutant FKBP–caspase-8 (Supplemental Figure 1) in POD-ATTAC mice resulted in podocyte apoptosis as early as 2–4 hours after treatment, with a loss of podoplanin protein and the presence of cleaved caspase-3+ podocytes seen through 12 hours (Figure 1A and Supplemental Figure 2). Albuminuria in spot urine was first measurable at 12 hours after treatment (Supplemental Figure 1). Apoptotic podocytes were observed at 48 hours by transmission electron microscopy (TEM) (Supplemental Figure 2). Prior uses of the ATTAC transgene expression have demonstrated dose-dependent induction of apoptosis.13–15 Injections of 0.1, 0.2, and 0.5 μg of the dimerizer AP20187 per g body weight, or daily 0.2 µg/g injections for 5 days, resulted in a dose-dependent reduction in podocytes, as judged by Wilms tumor gene 1–positive (WT1+) nuclei per glomerular area (see Concise Methods), from each glomerulus at 48 hours by ANOVA (F[4,20]=22.36 P<0.01; Figure 1B and Supplemental Figure 2). Dimerization of 0.2 µg/g resulted in a reduction of WT1+ nuclei by 41.4%, a 50.3% loss at 0.5 µg/g, and, despite only two doses into the five-dose regimen, a 72.9% reduction with repeated daily doses from POD glomeruli, with each reduction significant by Tukey multiple comparison test. In addition to the reduction in podocyte density, expression of the podocyte membrane protein podoplanin was also markedly reduced (Figure 1C and Supplemental Figure 2), an early sign of podocyte dysfunction or potentially dedifferentiation that may occur upon injury. Loss of susceptible podocytes occurred rapidly after dimerization, disrupting glomerular histologic features and leading to mild, although not statistically significant, glomerular hypertrophy over time at high doses (Supplemental Figure 3). The lowest number of podocytes per glomerular area was reached at 7 days for each of the different injection protocols (Figure 1D and Supplemental Figure 2), resulting in percentage podocyte reductions of 33.1%, 61.8%, 60.0%, and 78.5% for the four groups. With rapid apoptosis, the further decline in numbers could indicate dedifferentiation or loss of podocytes beyond the initial apoptotic induction. TEM confirmed glomerular basement membranes lacking podocytes at day 7 (Supplemental Figure 2). This dimerizer dose-dependent effect on podocyte numbers was demonstrated through 60 days after injection (Figure 1D).
Figure 1.

The POD-ATTAC mouse demonstrates inducible, podocyte-specific apoptosis in a dimerizer dose-dependent manner. (A) Cleaved caspase-3 labeling (red, arrows), downstream of ATTAC dimerization, can be seen in podocytes (green, podoplanin) as soon as 4 hours after a high-dose AP20187 injection in POD-ATTAC glomeruli; apoptosis is still present at 12 hours. Bar = 50 µm. (B) The number of WT1+ podocyte nuclei per glomerular area (µm2) remaining counted in over 50 glomeruli in mice treated with different concentrations of AP20187 (in µg/g body weight) are reduced in a dose-dependent manner 2 days after injection. Each dot represents a unique mouse. (C) Podocyte numbers, identified by WT1+ nuclei (red), are reduced at day 2 (podoplanin, green). Positive “red” red blood cells within the vascular space were not counted. Bar = 50 µm. (D) Podocyte counting of WT1+ nuclei 2, 7, 28, and 60 days after dimerization indicate a potential restoration of podocyte numbers per glomerular area over time, particularly at lower doses. Each dot represents a unique mouse. (E) Podocyte densities (WT1 nuclei, red) may increase in mice treated at low dimerizer concentrations, although restoration is limited at higher doses. Podoplanin expression may be restored in some glomeruli, green. Bar = 50 µm. *0.05>P>0.01, **0.01>P>0.001, ***P<0.001 from day 0; #0.05>P>0.01, ###P<0.001 from the lowest count (day 7).
POD-ATTAC mice receiving lower-dose (0.1 µg/g) dimerizer demonstrated significant initial loss of podocytes and restoration in WT1+ podocyte density by days 28 and 60, (F[4,16]=14.75, P<0.001; Figure 1D). Mice receiving 0.2 µg/g recovered some cells beyond day 28 (F[4,18]=9.81, P<0.001; Figure 1D). POD-ATTAC mice receiving 0.5 µg/g dimerizer, or the more aggressive multiple daily injections, exhibited highly significant reductions in WT1+ podocyte density but did not exhibit a change or increase in podocytes after day 7 (F[4,20]=31.16, P<0.0001; F[4,16]=35.12, P<0.0001, respectively). A concurrent increase in or restoration of podoplanin expression in intact glomeruli of low-dose treated mice was observed by day 28 (Figure 1E); levels remained low at higher doses.
Induction of Caspase-8 in Podocytes Leads to Dose-Dependent Changes in Renal Function and Structure, Followed by Recovery
Loss of podocytes in the POD-ATTAC mouse resulted in varying degrees of reduced glomerular filtration. With 0.1 and 0.2 μg/g dimerizer, creatinine clearance (ClCr) did not change (Figure 2A). With 0.5 µg/g, there was a decreasing trend in ClCr from 7 to 28 days, with some recovery at 60 days, although this was not statistically significant by ANOVA (F[6,37]=2.288, P=0.06). Repeat dimerizer treatments led to a sustained decline in ClCr (Figure 2A) and elevation of serum creatinine concentrations (Supplemental Figure 4). Urinary protein excretion was significantly increased at all doses (Supplemental Figure 4), and although urinary albumin was elevated, albumin excretion rates were not consistently high (Supplemental Figure 4). Urinary albumin-to-creatinine ratios, however (Figure 2B), were increased with podocyte loss at higher doses. The measured values at the terminal points significantly varied for each mouse, with potential recovery at 0.2 µg/g and 0.5 µg/g, possibly dependent on each animal’s initial degree of podocyte loss (F[5,28]=2.456, P=0.06 and F[5,28]=4.815, P=0.003, respectively). POD-ATTAC mice receiving repeat dimerizer doses (0.2×5) failed to recover at all (F[5,21]=4.040, P=0.01) and showed a continued decrease in function, with several mice dying before the termination of their study (Supplemental Figure 5).
Figure 2.

Renal filtration function is restored over time after induction of podocyte apoptosis in POD-ATTAC mice. (A) Creatinine clearance (ClCr, µl/min) is minimally disturbed in POD-ATTAC mice at low dimerizer doses, in line with their reduced podocyte loss. Although podocyte numbers did not generally return at 0.5 µg/g, GFR is restored over time in some mice. GFR continues to decline with repeat treatments. (B) Urinary albumin-to-creatine ratio (mg/mg) is greatly increased at 7 days after injection in all mice, with the magnitude dependent on dimerizer dose. Mice appear to recover with only one injection. Each dot represents a unique mouse. (C) Electron microscopy of POD-ATTAC mice treated with 0.5 µg/g highlights the podocyte foot processes (arrows) that completely disappear with foot process effacement at day 7. Podocyte foot processes return by day 28, along with a restoration of filtration. Bars = 2 µm. *0.05>P>0.01, **0.01>P>0.001 from day 0.
Depending on the dose of dimerizer and number of podocytes affected, renal tissues exhibited histologic changes reminiscent of human glomerular abnormalities with mesangial expansion and glomerulosclerosis (Supplemental Figure 3). TEM revealed foot process effacement at 2 and 7 days in remaining podocytes, mirroring the loss of filtration barrier (Figure 2C and Supplemental Figures 2 and 3) and increased albumin-to-creatinine ratios. Although not indicated by podocyte cell numbers or clear functional recovery, TEM of mice receiving even 0.5 µg/g dimerizer did show evidence of restoration of podocyte foot processes at the filtration barrier (Figure 2C and Supplemental Figure 3). In combination, these observations establish the POD-ATTAC model as a potent model of kidney injury with functional podocyte recovery potential at lower insult levels.
Adiponectin KO and Adipo Tg Mice Have Normal Renal Function
To study the role of adiponectin levels on podocyte loss and renal function upon induction of podocyte apoptosis, adiponectin KO24 and adiponectin-overexpressing adipo Tg mice25 were crossed with the POD-ATTAC (POD) model, generating adiponectin KO POD-ATTAC (POD-KO) and adipo Tg POD-ATTAC (POD-Tg) mice. Baseline serum levels of adiponectin in POD-Tg mice of 22.3 µg/ml were nearly three-fold higher than wild-type adiponectin levels (P<0.0001; Figure 3A). Unchallenged, these mouse models present the same baseline serum glucose levels (Supplemental Figure 5).24–26 WT1+ podocytes per glomerulus in these mouse lines were equivalent to those in wild-type mice before dimerizer treatment (Figure 3B), but normalizing for the larger glomerular area of the POD-Tg mice (Figure 3B and Supplemental Figure 5) made the POD-Tg podocytes significantly less dense (P<0.01). Although decreased renal function has been reported in one adiponectin KO mouse derivation,22 our adiponectin KO mouse exhibited normal ClCr, albumin excretion rate, and urinary albumin-to-creatine ratio, mirroring other reports in another distinct KO mouse description (Figure 3D).21 Functional measurements in POD-Tg mice revealed no renal impairment or enhancement under normal conditions (Figure 3D). Hence, our three POD-ATTAC lines had similar renal function at baseline before ATTAC induction.
Figure 3.
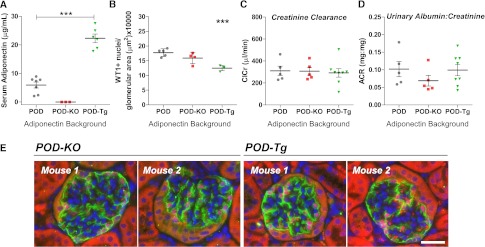
Adiponectin null POD-KO and adiponectin-overexpressing POD-Tg mice show no alteration in baseline renal function. (A) Adiponectin levels in POD-Tg mice are two to three times greater than in wild-type POD serum (P<0.01). Adiponectin is undetectable in POD-ATTAC adiponectin KO mice. (B) POD-KO have similar numbers of WT1+ podocyte densities, whereas POD-Tg mice have fewer WT1+ podocytes per glomerular area. (C) Functionally, POD-KO and POD-Tg mice vary in their baseline ClCr and (D) urinary albumin-to-creatinine ratio (ACR) but are statistically equivalent to wild-type mice. (E) Glomeruli of both POD-KO and POD-Tg mice appear normal at baseline (podoplanin, green; WT1, red). Bar = 50 µm. ***P<0.001 from POD d0 baseline versus adiponectin mouse models.
Adiponectin Improves Podocyte Recovery and Reconstitution of Kidney Function
POD-KO and POD-Tg mice were treated with 0.5 µg/g body weight dimerizer. This high dose in wild-type adiponectin POD mice induced significant loss but not total failure and death. This allows us to determine whether low adiponectin worsens the POD pathology or limits recovery and, conversely, whether heightened adiponectin prevents podocyte apoptosis or promotes recovery of renal function.
POD-KO mice exhibited a rapid loss in podocytes (F[4,13]=11.56, P<0.001), but the lack of adiponectin did not increase the number of podocytes ablated after dimerization compared with POD mice at day 2 or 7 (Figure 4A). Unlike in some POD mice, however, POD-KO podocytes failed to recover, and the glomeruli ranged from normal in appearance to FSGS and disruption of podoplanin labeling (Figure 4B and Supplemental Figure 5). Ablation of podocytes did not significantly alter POD-KO serum glucose levels (Supplemental Figure 5). Tubule epithelial cells were also positive for WT1 in late-stage POD-KO sections (Supplemental Figure 5), suggesting cell differentiation27 and further deviation from normalcy. Adiponectin null POD-KO mice demonstrated reduced ClCr (F[4,20]=2.076, P=0.1220; Figure 4C). Despite a similar number of podocytes lost compared with POD, POD-KO mice exhibited severe albuminuria (Figure 4D) with urinary albumin-to-creatinine ratio still extremely high at 28 days in some POD-KO mice (F[4,22]=5.096, P=0.005). Surviving POD-KO mice—approximately 30% died acutely, with additional losses over time (Supplemental Figure 5)—eventually exhibited reduced albuminuria despite their early massive peak response, suggesting progression to ESRD.
Figure 4.
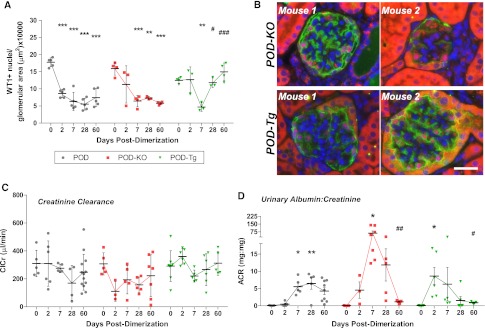
Adiponectin aids in restoring podocytes and renal function, but not in the initial protection of podocytes from apoptosis. (A) At 0.5 µg/g dimerization, adiponectin null POD-KO mice progressively lose WT1+ podocytes after injection. POD-Tg mice lose an equivalent number of podocytes by day 7 but demonstrate increased WT1+ nuclei per glomerular area by day 28. (B) Return of WT1+ nuclei, red, and filtration membrane protein podoplanin, green, in POD-Tg mice at day 60, but not in all glomeruli of POD-KO mice. Note the increased WT1 labeling in tubule epithelium. Bar = 50µm. (C) ClCr (µl/min) is protected in POD-Tg mice to an extent similar to that seen in POD mice, whereas POD-KO mice fair worse. (D) Urinary albumin-to-creatinine ratios (ACRs) are initially far worse in both POD-KO and POD-Tg mice than in wild-type adiponectin POD mice. By day 28, however, POD-Tg mice demonstrate reduced albuminuria compared with POD mice. Albumin-to-creatinine ratios in POD-KO mice were surprisingly reduced, an effect attributable to ESRD. *0.05>P>0.01, **0.01>P>0.001, ***P<0.001 from the baseline (day 0 or POD d0 baseline versus adiponectin mouse models); #0.05>P>0.01, ##0.01>P>0.001, ###P<0.001 from the peak or bottom of time course data.
In contrast to adiponectin overexpression conferring strong cytoprotective actions in past ATTAC mouse models,15 POD-Tg mice demonstrated a loss of podocyte density at 7 days (F[4,12]=10.28, P<0.001). By day 28, however, a marked recovery of WT1+ podocytes per glomerular area was observed in POD-Tg mice that continued through day 60 (P<0.01). Concurrently, podoplanin expression was also restored by day 60, suggesting restoration of the filtration barrier (Figure 4B) with glomerular recovery. In contrast to the POD-KO mice, POD-Tg mice maintained normal ClCr (Figure 4C). Despite a similar percentage of podocytes lost at day 7, POD-Tg mice exhibited heightened albuminuria (Figure 4D) compared with POD mice (F[4,27]=3.473, P=0.02). Adiponectin-overexpressing POD-Tg mice did, however, quickly recover, with restoration of WT1 and podoplanin expression, and presented less albumin excretion by 28 days, despite their early heightened peak levels (Figure 4D and Supplemental Figure 1). POD-Tg mice saw significant improvements that were greater than in POD mice; most POD-Tg mice exhibited functional ratios of <1.0 by day 60 and exhibited glomerular hypotrophy or contraction rather than hypertrophy (F[4,11]=12.29, P<0.001) over time (Supplemental Figure 5).
Adiponectin Overexpression Limits Renal Fibrosis after Podocyte Loss
In addition to glomerular injury, a prominent and common pathway of progression to ESRD is widespread renal interstitial fibrosis that is beyond glomerulosclerosis. After caspase-8 activation in podocytes, POD mice develop renal fibrosis. A low dimerizer dose of 0.2 µg/g resulted in few podocytes lost, and fibrosis was limited 28 days after injection (Figure 5A) as judged by trichrome staining. With more podocytes affected at higher doses, by 2 weeks mice began to develop fibrosis that progressed through day 28 (Supplemental Figure 6). At 28 days, fibrosis was seen not only in and around each glomerulus but throughout the renal cortex (Figure 5A). Large fibrotic areas may be the result of entire nephrons lost.
Figure 5.

POD-Tg mice are protected from interstitial fibrosis resulting from dose-dependent podocyte loss. (A) At 28 days, mice receiving the 0.2 µg/g dose exhibit little fibrosis, whereas the 0.5 µg/g dose results in significant matrix deposition (blue) as visualized by trichrome staining. POD-KO mice exhibit levels similar to those in POD mice. POD-Tg mice, however, are greatly protected at the 0.5 µg/g dose. Minimal fibrosis is evident in the glomeruli of low-dose dimerized or POD-Tg mice. Bar, 10× = 200 µm; 40× = 50 µm. (B) At 60 days. mice may exhibit some recovery from fibrosis, but POD-Tg mice are best off. Bar, 10× = 200 µm; 40× = 50 µm. (C) Interstitial fibrosis quantification revealed a dose-dependent increase in fibrotic area. (D) Both POD and POD-Tg mice demonstrate a significant reduction interstitial fibrosis from day 28 to day 60; POD-KO mice do not significantly recover. *0.05>P>0.01, **0.01>P<0.001 from day 0 or POD d0 baseline versus adiponectin mouse models; #0.05>P>0.01 from the peak of time course data.
Blinded quantification of interstitial fibrosis scores (Figure 5C) demonstrated significant fibrosis at 28 days at higher doses (for 0.5 µg/g, F[2,13]=6.478, P=0.0112; for 0.2 µg/g × 5, F[2,11]=8.214, P=0.01). At day 60, 0.5 µg/g-treated mice exhibited significant recovery (P<0.05), but in repeat-dose mice, fibrosis remained. Quantified fibrosis in both POD-KO and POD-Tg mouse kidney sections was significantly increased, as in POD mice, at 28 days (Figure 5D). POD-Tg mice, however, exhibited a significant reduction in interstitial fibrosis at day 60 (F[2,11]=7.758, P=0.08), whereas POD-KO did not (F[2,10]=5.332, P=0.03). This limited propensity for chronic fibrosis may facilitate the greater restoration of renal function measured in POD-Tg mice.
Peroxisome Proliferator-Activated Receptor-γ Agonist Treatment Raises Adiponectin Levels but Cannot Rescue Renal Function
An effect triggered by peroxisome proliferator-activated receptor-γ (PPARγ) agonists is increased serum adiponectin levels.28 We thus sought to determine whether PPARγ agonist treatment, through its effects on adiponectin, could preserve or rescue renal function in POD-ATTAC mice in a manner similar to the effects seen with specific genetic elevation of adiponectin (as in the POD-Tg mouse). To observe the potential effects of PPARγ agonists beyond the induction of adiponectin, POD-KO mice were also treated. PPARγ agonist [2-(2-(4-phenoxy-2-propylphenoxy)ethyl)indole-5-acetic acid, or COOH] treatment for 1 week before dimerizer injection significantly increased serum adiponectin levels to 11.08 µg/ml in POD mice (Figure 6A; P<0.01), although levels were still significantly lower than those seen in POD-Tg mice (P<0.01). Upon induction of podocyte apoptosis with a single 0.5 µg/g dimerizer dose, COOH-treated POD and POD-KO mice lost 63.3% and 55.0% of WT1+ cells per area, respectively (F[4,11]=20.23, P<0.0001 and F[4,10]=6.677, P=0.007, respectively; Figure 6B). Higher adiponectin levels with COOH treatment resulted in a small increase in podocyte density by day 60, although not as pronounced as in POD-Tg mice, with changing glomerular areas (Supplemental Figure 5). Functionally, there was no difference between POD-KO-COOH and POD-COOH mice before dimerization, but induction of podocyte apoptosis resulted in lowered ClCr for both groups, similar to absence of PPARγ agonism (Figure 6C) (F[4,32]=3.733, P=0.01; F[4,37]=4.860, P=0.003, respectively). Urinary albumin-to-creatinine ratios were dramatically increased 2 days after dimerization in COOH-treated mice (F[4,37]=22.07, P<0.0001)—with POD-KO-COOH mice displaying the highest levels measured (F[4,32]=18.03, P<0.0001; Figure 6D). Although these levels declined by 28 days, they were similar to those in mice not exposed to the PPARγ agonist; some mice continued to exhibit elevated levels. The absence of adiponectin in the KO animals probably unmasked yet-unidentified renal changes with PPARγ agonist use.29 Trichrome staining revealed marked fibrosis at 28 days in both POD and POD-KO mice treated with PPARγ agonists (Figure 6F). Fibrosis in COOH-treated POD-KO mice was worsened, as expected, at days 28 and 60 (F[2,10]=8.226, P=0.008). Unlike the improvement in fibrosis measured in the adiponectin-overexpressing POD-Tg mouse model (Figure 5), PPARγ agonist–treated mice did not exhibit a reduced interstitial fibrosis score (F[2,11]=5.029, P=0.03). In total, this highlights the potent effects that adiponectin exerts on functional reconstitution of the system and perhaps uncovers some negative effects that PPARγ agonists can exert on the kidney that cannot be offset by the elevated adiponectin levels.
Figure 6.

PPARγ agonist treatment increases serum adiponectin levels but cannot protect renal function. (A) Serum adiponectin is increased with PPARγ agonist treatment (COOH), but not to the levels of POD-Tg mice. (B) Podocyte numbers were not significantly restored at 0.5 µg/g in POD-KO mice with COOH, but recovery of WT1+ podocytes per glomerular area was increased by PPARγ agonist treatment in POD mice. (C) PPARγ agonist–treated POD-KO and POD mice demonstrate loss of ClCr (µl/min) similar to that of untreated mice at 0.5 µg/g. (D) Albuminuria is increased 10 times over mice not treated with PPARγ agonist at day 2 and, although it is reduced by day 28, is not improved by PPARγ agonist treatment. (E) PPARγ agonist treatment does not protect POD-ATTAC mice from interstitial fibrosis or aid in recovery through day 60. Bar, 10× = 200 µm; 40× = 50 µm. (F) Quantified interstitial fibrosis of PPARγ agonist–treated mice demonstrated equivalent, or worse, fibrosis compared with untreated POD mice at days 28 and 60. *0.05>P>0.01, **0.01>P>0.001, ***P<0.001 from the baseline (day 0 or POD d0 baseline versus adiponectin mouse models or COOH treatment); #0.05>P>0.01, ###P<0.001 from the peak of time course data
Discussion
Our POD-ATTAC mouse permits dose-dependent induction of podocyte loss, taking advantage of a physiologically relevant, endogenous cell death mechanism involving caspase-8–mediated apoptosis. Targeting the podocyte directly allows the introduction of a highly targeted lesion and makes the POD-ATTAC mouse more relevant to human glomerulopathies initiated by podocyte loss.3,4 Uninephrectomies and 5/6 nephrectomies induce glomerulosclerosis in the residual kidney but are clearly not glomerulus specific.9 Likewise, chemical injury models cause direct simultaneous damage to multiple cell types, such as renal tubules.8 Several diabetic models exhibit some forms of nephropathy but are metabolically more complicated than podocyte-specific injuries. Genetic mouse models deficient in key slit diaphragm proteins, such as nephrin or podocin, for example, develop early severe nephritis with multiple secondary effects and die shortly after birth.4 The most specific models developed to date that permit selective deletion of podocytes are podocin promoter expression of diphtheria toxin in rats12 and nephrin-driven expression of human CD25 in mice.11 Alternatively, podocin promoter–driven expression of nitroreductase in mice10 and zebrafish30 have induced podocyte loss. These pathways induce necrosis rather than cell-specific apoptosis. With the ability to present models simulating human nephropathies, such as FSGS, in an acute dose-dependent manner, the POD-ATTAC mouse provides an ideal murine platform for the study of podocyte loss and mechanisms driving CKD progression. Most important, the POD-ATTAC mouse demonstrates recovery of podocyte density and restores filtration function to varying degrees with dimerizer dose (and initial podocyte loss), permitting therapeutic examination of the different pathways to recovery. Specifically targeting podocytes also makes the POD-ATTAC model ideally suited to determine from where new podocytes may arise in the adult.31,32 The combination of precisely timed induction of podocyte apoptosis and the potential for recovery in the POD-ATTAC mouse present great promise that this model will be useful for future renal studies.
Regulation of glomerular filtration and urinary protein excretion requires processes and cell types beyond the filtration slit barrier provided by podocytes. Initial podocyte loss leads to foot process effacement and dedifferentiation or loss of additional podocytes, along with changes in function and numbers of mesangial, parietal epithelial, glomerular endothelial, and tubule epithelial cells.3,33,34 These downstream events lead to basement membrane thickening and interstitial inflammation and fibrosis. The POD-ATTAC mouse, although initiated by a discrete podocyte lesion, eventually elicits a more complex whole organ functional response. Our previously published ATTAC mouse models have demonstrated reversibility and physiologic effects associated with the specific loss of the targeted cell types, such as pancreatic β cells,14 and show that “physiologic” apoptosis may limit inflammatory processes that are generally associated with more brute-force cell ablation approaches that trigger a necrotic response.16 At low doses, restoration of WT1+ podocyte density and a reduction in albuminuria were demonstrated in the POD-ATTAC mouse. The increased podocyte reduction at high dimerizer doses, however, induces changes throughout the kidney that limit the reversibility and restoration of function previously documented in other ATTAC models.13,14 Protection beyond the podocyte itself is therefore necessary to limit the progressive injury and development of interstitial fibrosis at high dimerizer doses.
Direct roles for adipokines, such as adiponectin, leptin, and vaspin, have been difficult to discern because of metabolic changes often coincident with their altered levels in conditions with renal abnormalities.20,35,36 Adiponectin levels are lower in obese patients; obese patients often demonstrate proteinuria, and some develop frank FSGS lesions.20 Adiponectin levels and adiponectin complex distribution are altered in patients with type 2 diabetes.28,37 Whether lack of adiponectin correlates with or contributes to impaired renal function is not clear. On the other hand, adiponectin levels are increased in CKD20,23 and in type 1 diabetes,38 an effect not due to failed urinary clearance.39 One of the adiponectin KO mouse models demonstrated baseline renal impairment that was corrected with adiponectin treatment.22 Another adiponectin KO mouse exhibited normal baseline function but a worsened response to 5/6 nephrectomy, including increased fibrosis that was corrected by adenoviral adiponectin.21 Our adiponectin KO mouse was functionally normal at baseline but worse than wild-type POD-ATTAC mice after podocyte ablation. Adiponectin null POD-KO mice were unable to recover WT1+ cell densities or function in the absence of near-ESRD. POD-KO mice also exhibited tubule epithelial cell WT1 labeling after dimerization, similar to a previous report linking differentiation with altered renal function.27 Reduced adiponectin levels in obesity would probably limit the potential for patient recovery in FSGS. Weight loss increases adiponectin levels and has demonstrated the ability to reduce obesity-related FSGS; our findings further reinforce the utility of this prescribed lifestyle change for obese patients.40,41
The adiponectin-overexpressing POD-Tg mice lost an equivalent number of podocytes after dimerizer treatment, unlike past ATTAC models in which the adiponectin transgene reduced apoptosis in the target cells.15 Oddly, POD-Tg had increased albuminuria (compared with POD mice) after podocyte loss and glomerular contraction. The recovery of both podocyte cell numbers and filtration function, however, was greatly increased in our POD-Tg mice that also exhibited reduced interstitial fibrosis. Overexpression of adiponectin did not, therefore, protect podocytes directly from injury but may have reduced the downstream effect of podocyte loss on other renal cells because of its antiapoptotic15 and antiinflammatory roles.15,42 Direct pharmacologic AMP-activated protein kinase activation, another proposed mechanism of adiponectin action, has demonstrated the ability to limit renal dysfunction induced by a high-fat diet.43 It is therefore clear that adiponectin is, overall, a potently renoprotective protein. The increased levels observed in CKD may be the result of metabolic changes in adipose tissue44,45 and probably represent a compensatory upregulation over the course of CKD, without which CKD could take a more rapid negative outcome.
The aforementioned conclusions are based on our past in vivo work with the ATTAC cassette in which cell-specific apoptosis was unambiguously demonstrated in wild-type, adiponectin KO, and adiponectin Tg backgrounds.13–15 Although TEM indeed reveals apoptotic podocytes and podocyte loss, this large decline in WT1+ nuclei numbers should also be detectable in histologic sections, such as in the form of TUNEL-positive podocytes. The difficulty of demonstrating this phenomenon with this second method is due to the very rapid onset of apoptosis and the loss of cells through urinary secretion of apoptotic podocytes.34 This is in contrast to pancreatic β cells or adipocytes that are tissue-bound and hence more easily detectable. However, we cannot rule out the possibility that podocytes dedifferentiate upon induction of caspase-8 activation. Direct podocyte dedifferentiation, injury, or a mesenchymal transition after caspase-8 dimerization may thus offer an alternate explanation for the loss of WT1 labeling and reduced surface podoplanin expression in glomerular immunohistochemistry.10,46 Our reduced podocyte counts may also result from changes in the remaining podocytes not directly ablated by dimerization after loss of their neighbors. Multiple models, in vivo and in vitro, describe the loss of traditional podocyte markers and function upon injury.10,46,47,48 Circulating metabolites, glucose levels, or, potentially, the lack of or heightened levels of adiponectin may affect this transition or injury.49 Functional recovery or recovery of WT1+ nuclear numbers at low dimerizer doses or in the POD-Tg mouse may therefore be a return to podocyte quiescence and health. Future studies with more detailed analysis of this process through lineage tracing studies will have to address these questions in greater detail.
Adiponectin secretion from adipose tissue and circulating adiponectin levels are increased with PPARγ agonist compounds.28,29,50 These drugs have been prescribed to treat type 2 diabetes with great success, although the adverse effect of fluid retention or edema may be linked to some cardiovascular complications with the drugs.51 Changes in fluid retention have been linked to PPARγ-mediated changes in salt absorption and aquaporin protein expression in renal tubule epithelial cells.29,52,53 When used in our POD-ATTAC model, adiponectin serum levels were significantly elevated. After podocyte loss—just as in untreated mice—PPARγ agonist–treated mice exhibited massive albuminuria. Although these levels were reduced over time, the overall recovery by PPARγ agonist treatment was no better than that measured in untreated POD mice. The small elevation in adiponectin levels was unable to compensate for the systemic fluid balance challenges and other potential ill effects presented by the PPARγ agonist, but the fact that adiponectin null POD-KO mice fared significantly worse than wild-type mice on the drug again highlights the beneficence of adiponectin.
The novel POD-ATTAC mouse provides a murine platform for inducible, titratable podocyte-specific apoptosis. Depending on the number of podocytes ablated, this model presents human disease–like abnormalities and potential recovery. Use of a murine platform permits rapid introduction of other experimental molecular factors through crossing with transgenic or KO mouse lines. Crossing with adiponectin KO and adiponectin-overexpressing Tg mice has further reinforced the view that adiponectin is important as a protective adipokine after podocyte injury. With the ability to titrate podocyte loss and disease severity and the ability to recover podocyte numbers and renal function, the POD-ATTAC mouse is an ideal mouse model for continued study of renal pathology progression and efficacy of treatment.
Concise Methods
Generation of POD-ATTAC Mice
The 2.5-kb human podocin promoter (Nphs2) was amplified from a vector containing the promoter region with a Kpn I site at the 5′ end and a Not I site at the 3′ end, respectively.54 The promoter fragment was then inserted into a pCR4TA vector (Invitrogen, Carlsbad, CA) and verified by sequencing (Genewiz, South Plainfield, NJ). The FAT-ATTAC construct13 was partially digested by Kpn I and Not I and the 6-kb fragment backbone containing the FKBP-caspase 8 fusion protein and rabbit β globin 3′ untranslated region was extracted. The 2.5-kb promoter region was released from the pCR4TA vector by Kpn I and Not I enzymatic digestion and inserted into the linearized 6-kb backbone vector. The fragment containing the podocin promoter, FKBP-caspase 8 fusion protein, and 3′ untranslated region was released by KpnI and Sac I digestion purified and subjected for pro-nuclear injection into FVB embryos (Supplemental Figure 1). The components and dimerizer were purchased from Invitrogen.
A total of 32 founders were obtained, and 6 of them were ATTAC transgene positive. The founders were then crossed to the parental FVB strain. Four of them showed germline transmission of the transgene (Supplemental Figure 1). To compare the transgene expression levels across the four lines, kidneys were harvested and total RNA isolated (RNEasy, Qiagen, Germantown, MD). cDNA was produced by reverse transcription (Invitrogen), and RT-PCR was performed for the level of the caspase-8 fusion protein as previously described;13,14 mouse podocin gene expression was used as a reference. The line with highest expression was identified and used for all experiments (Supplemental Figure 1). mRNA expression levels in various tissues was determined by quantitative PCR to demonstrate kidney-specific expression (Supplemental Figure 1).
All POD-ATTAC mice were bred as heterozygotes and genotyped by PCR as above. FVB homozygote adiponectin null (KO) and hemizygote adiponectin-overexpressing mice (adipo Tg), both previously characterized,24,25 were crossed with POD-ATTAC (POD) mice to generate KO POD-ATTAC and adipo Tg POD-ATTAC mice on pure FVB backgrounds. All animals were maintained with ad libitum access to water and a normal chow diet (Harlan2916) throughout unless they were treated with PPARγ agonists (see the section on experimental treatments). All animal protocols were approved by the Institutional Animal Care and Use Committee at the University of Texas Southwestern Medical Center.
Experimental Treatments
Mice used in PPARγ agonist studies were provided powdered chow diet containing 0.5 mg/g COOH, a nonthiazolidine agonist (a kind gift from Merck & Co., Inc.55) beginning 1 week before dimerizer administration. The diet was maintained for the duration of the experimental period, with fresh preparations provided twice weekly.
Dimerization Treatment and Fluid Collections
The dimerizer AP20187 was prepared according to the manufacturer’s recommendation (Ariad Pharmaceuticals, Boston, MA; Invitrogen) and injected intraperitoneally according to the treatment group. Groups 1, 2, and 3 received a single dose of 0.1 µg/g, 0.2 µg/g, and 0.5 µg/g body weight, respectively. Group 4 received 0.2 µg/g daily for 5 consecutive days. Concentrations were prepared such that dose volumes averaged 120 µl. Vehicle injections of the same volumes (minus AP20187) were used as controls. Fluids and tissue were collected on days 2, 7, 14, 28, and 60 after initial injections: Mice were placed into individual urine collection metabolic chambers (Harvard Apparatus, Holliston, MA) and provided ad libitum powdered chow and water for 48 hours before experimental collection for acclimatization. Urine was collected for 24 hours from 9 a.m. on the noted sampling date after injection through 9 a.m. the following day, when the mice were euthanized by isoflurane overdose and sera were collected. Twenty-four-hour urine volumes were measured and up to 1000 µl was saved and briefly spun (3 minutes at 300 g) to remove debris before analyses. Serum glucose and adiponectin concentrations were analyzed by plate assays from Sigma and Millipore, respectively.
Albumin and Creatinine Measurements
Serum and urine creatinine concentrations were measured using a P/ACE MDQ Capillary Electrophoresis System and photodiode detector (Beckman-Coulter, Fullerton, CA) at 214 nm. Samples were individually diluted with HPLC-grade H2O so as to fall within the calibration curve (0.4–6 mg/dl creatinine; Sigma-Aldrich, St. Louis, MO). Urine total protein concentrations were first determined using a BCA assay (Fisher, Waltham, MA); dilutions of 1:40–1:400 were necessary with standard prepared in PBS. Specific urine albumin concentrations were determined using the Exocell mouse albumin ELISA kit (Exocell, Philadelphia, PA) according to manufacturer’s instructions; urine dilutions of 1:20 up to 1:10,000 in assay buffer were necessary. Measured 24-hour urine volumes were applied to calculate creatinine, protein, and albumin clearance rates. Functional data are presented as average ± SEM for clarity in the respective time courses.
Tissue Preparation and Histology
Kidneys were dissected immediately after euthanasia and submersion fixed in 10% buffered formalin (Fisher Scientific, Fair Lawn, NJ) for 24 hours or frozen-embedded in optimal cutting temperature medium and stored at −80°C. Fixed kidneys were rinsed in PBS, then stored in 70% ethanol for processing. Whole kidneys were then paraffin embedded and cut into 3-µm-thick sagittal sections. Frozen kidneys were cryosectioned into 12-µm-thick sections. For pathologic examination, sections were stained with protocols for periodic acid-Schiff (PAS) staining to demarcate glomerular filtration membranes. PAS sections were also used to calculate average glomerular cross-sectional areas. Trichrome staining was used to demonstrate tissue fibrosis. Stained sections were imaged using a Nikon Coolscope VS microscopy system.
Immunohistochemistry and Cell Labeling
After rehydration of embedded tissues, podocyte filtration membranes were specifically labeled on renal sections with an anti-podoplanin primary antibody (goat polyclonal, 1:100; R&D Systems, Minneapolis, MN) and detected by an Alexafluor488-conjugated donkey anti-goat secondary antibody (1:300; Invitrogen). After citrate buffer antigen retrieval, podocyte nuclei used for counting were labeled with an anti-WT1 primary antibody (rabbit polyclonal, 1:50; Santa Cruz Biotechnology, Santa Cruz, CA) followed by an Alexa Fluor 594–conjugated donkey anti-rabbit secondary antibody (1:300; Invitrogen). Cleaved caspase-3 (rabbit monoclonal,1:50; Cell Signaling Technology, Inc., Danvers, MA) was used to detect apoptotic podocytes. DAPI was used to label nuclei. Frozen kidney sections, after 2 minutes in −20°C methanol, were labeled for WT1, podoplanin, and cleaved caspase-3, as above, without antigen retrieval. Immunofluorescence was imaged using a Zeiss AxioObserver fluorescence microscopy system and Zeiss Axiocam MRc camera with exposure times for WT1, casp-3, and podoplanin fixed across all samples. Brightness and contrast were adjusted in Adobe Photoshop equivalently across the entire image panel. The red channel was strongly enhanced on paraffin sections to better visualize the signals.
Electron Microscopy
A small piece (approximately 40 mm3) of renal cortex was fixed in a solution of 2.5% glutaraldehyde in 10% buffered formalin. Tissues were transferred to 4% paraformaldehyde at 4°C for 4 hours before preparation. After staining with uranyl acetate, sections were visualized with a JEOL 1200 EX transmission electron microscope.
Podocyte Counting
The average number of podocytes per glomerulus was determined by counting WT1+ nuclei surrounded by podoplanin labeling for ≥50 random glomeruli per section. Counts were all performed on sections cut through the sagittal center of the kidney (such that the medulla was substantial, not just cortical sections), starting at the renal hilum and recording the number of WT1+ cells in each glomerulus up around the cortex until 50 glomeruli had been counted. Because no bias was given to the area of glomerulus present on the section, WT1+ cell numbers per glomerulus ranged from 2 to 15 for normal kidneys. The number counted for each section was averaged across the 50 glomeruli. In disrupted kidneys, when podoplanin expression was greatly reduced, WT1+ nuclei in correct anatomic positions were deemed sufficient to define podocytes. A section from at least three animals for each group and time point were counted to compute the average using GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA). WT1+ cell numbers were divided by the average glomerular cross-sectional area as quantified by blinded area measures performed in Image J software (National Institutes of Health, Bethesda, MD) on three 5× images of PAS-stained sections from three mice from each time point and treatment. The initial pixel areas were converted to µm2, and the final podocyte counts are reported as WT1+ cells/glomerular area (µm2) × 10,000 for each terminal mouse sample ± SD.
Interstitial Fibrosis Quantification
Fibrosis density—defined as interstitial fibrosis score—was quantified as the area of collagen deposition divided by the area of renal tubules and glomeruli as blue/red percentage averaged across the five images as previously described, with minor modification.56 After blinded capture of five cortex micrographs (approximately the same locations on each blinded section per slide) at 20× of trichrome-stained kidney sections, all proteinaceous casts were carefully erased using the “magic wand” tool in Adobe Photoshop. Each image was then evaluated by a technician blinded to the experimental conditions: For each independent sample, collagen deposition (blue pixels) and renal tubules and glomeruli (red pixels) were first defined for their respective blue or red intensity values using the first of the five images. The total “blue” pixel and “red” areas across all five images was then determined using Image J software. Presented scores are the average score ± SD of three or more animals. Images are presented with the proteinaceous casts in place (i.e., lacking the deletions necessary for quantification).
Statistical Analyses
All data are graphed as dot plots; each dot represents the terminal collection of a single mouse. Although using only one time point from each animal scatters the data, the individual points reveal the range of effects and overall trends. One-way ANOVA with Tukey post-test was performed for all time-course and dose-dependent data in GraphPad Prism software. P and F values of the ANOVA are reported significance is indicated on the graphs as follows: P>0.05 not significant, one asterisk 0.05>P>0.01, two asterisks 0.01>P>0.001, and three asterisks P<0.001 from the baseline (day 0 or POD d0 baseline versus adiponectin mouse models or COOH treatment). Similar to the asterisk convention, a pound sign indicates significant changes from the peak or bottom of time course data by the same Tukey test comparisons. Differences in serum adiponectin levels were compared by paired two-tailed t tests.
Disclosures
None.
Supplementary Material
Acknowledgments
We thank the O’Brien Kidney Research Core Center at the University of Texas Southwestern Medical Center at Dallas for all creatinine measurements. J.M.R. is supported by a National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases postdoctoral fellowship F32DK085935-01A1. These studies were also supported by the National Institutes of Health (grants R01-DK55758, R01-CA112023, RC1-DK086629, and P01-DK088761-01 to P.E.S.).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
This article contains supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2012040414/-/DCSupplemental.
References
- 1.Tonna SJ: Invited commentary: Defining incident chronic kidney disease in epidemiologic study settings. Am J Epidemiol 170: 425–427, 2009 [DOI] [PubMed] [Google Scholar]
- 2.Coresh J, Selvin E, Stevens LA, Manzi J, Kusek JW, Eggers P, Van Lente F, Levey AS: Prevalence of chronic kidney disease in the United States. JAMA 298: 2038–2047, 2007 [DOI] [PubMed] [Google Scholar]
- 3.D’Agati VD: Podocyte injury in focal segmental glomerulosclerosis: Lessons from animal models (a play in five acts). Kidney Int 73: 399–406, 2008 [DOI] [PubMed] [Google Scholar]
- 4.Kwoh C, Shannon MB, Miner JH, Shaw A: Pathogenesis of nonimmune glomerulopathies. Annu Rev Pathol 1: 349–374, 2006 [DOI] [PubMed] [Google Scholar]
- 5.Susztak K, Raff AC, Schiffer M, Böttinger EP: Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 55: 225–233, 2006 [PubMed] [Google Scholar]
- 6.Darouich S, Goucha R, Jaafoura MH, Zekri S, Ben Maiz H, Kheder A: Clinicopathological characteristics of obesity-associated focal segmental glomerulosclerosis. Ultrastruct Pathol 35: 176–182, 2011 [DOI] [PubMed] [Google Scholar]
- 7.Ritz E, Koleganova N, Piecha G: Is there an obesity-metabolic syndrome related glomerulopathy? Curr Opin Nephrol Hypertens 20: 44–49, 2011 [DOI] [PubMed] [Google Scholar]
- 8.Kirchhoff F, Krebs C, Abdulhag UN, Meyer-Schwesinger C, Maas R, Helmchen U, Hilgers KF, Wolf G, Stahl RA, Wenzel U: Rapid development of severe end-organ damage in C57BL/6 mice by combining DOCA salt and angiotensin II. Kidney Int 73: 643–650, 2008 [DOI] [PubMed] [Google Scholar]
- 9.Susztak K, Bitzer M, Meyer TW, Hostetter TH: Animal models of renal disease. Kidney Int 73: 526–528, 2008 [DOI] [PubMed] [Google Scholar]
- 10.Macary G, Rossert J, Bruneval P, Mandet C, Bélair MF, Houillier P, Duong Van Huyen JP: Transgenic mice expressing nitroreductase gene under the control of the podocin promoter: A new murine model of inductible glomerular injury. Virchows Arch 456: 325–337, 2010 [DOI] [PubMed] [Google Scholar]
- 11.Matsusaka T, Xin J, Niwa S, Kobayashi K, Akatsuka A, Hashizume H, Wang QC, Pastan I, Fogo AB, Ichikawa I: Genetic engineering of glomerular sclerosis in the mouse via control of onset and severity of podocyte-specific injury. J Am Soc Nephrol 16: 1013–1023, 2005 [DOI] [PubMed] [Google Scholar]
- 12.Wharram BL, Goyal M, Wiggins JE, Sanden SK, Hussain S, Filipiak WE, Saunders TL, Dysko RC, Kohno K, Holzman LB, Wiggins RC: Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol 16: 2941–2952, 2005 [DOI] [PubMed] [Google Scholar]
- 13.Pajvani UB, Trujillo ME, Combs TP, Iyengar P, Jelicks L, Roth KA, Kitsis RN, Scherer PE: Fat apoptosis through targeted activation of caspase 8: A new mouse model of inducible and reversible lipoatrophy. Nat Med 11: 797–803, 2005 [DOI] [PubMed] [Google Scholar]
- 14.Wang ZV, Mu J, Schraw TD, Gautron L, Elmquist JK, Zhang BB, Brownlee M, Scherer PE: PANIC-ATTAC: A mouse model for inducible and reversible beta-cell ablation. Diabetes 57: 2137–2148, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, Davis KE, Bikman BT, Halberg N, Rutkowski JM, Wade MR, Tenorio VM, Kuo MS, Brozinick JT, Zhang BB, Birnbaum MJ, Summers SA, Scherer PE: Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med 17: 55–63, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischer-Posovszky P, Wang QA, Asterholm IW, Rutkowski JM, Scherer PE: Targeted deletion of adipocytes by apoptosis leads to adipose tissue recruitment of alternatively activated M2 macrophages. Endocrinology 152: 3074–3081, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shetty S, Kusminski CM, Scherer PE: Adiponectin in health and disease: Evaluation of adiponectin-targeted drug development strategies. Trends Pharmacol Sci 30: 234–239, 2009 [DOI] [PubMed] [Google Scholar]
- 18.Wang ZV, Scherer PE: Adiponectin, cardiovascular function, and hypertension. Hypertension 51: 8–14, 2008 [DOI] [PubMed] [Google Scholar]
- 19.Sharma K: The link between obesity and albuminuria: Adiponectin and podocyte dysfunction. Kidney Int 76: 145–148, 2009 [DOI] [PubMed] [Google Scholar]
- 20.Zoccali C, Mallamaci F: Adiponectin and leptin in chronic kidney disease: Causal factors or mere risk markers? J Ren Nutr 21: 87–91, 2011 [DOI] [PubMed] [Google Scholar]
- 21.Ohashi K, Iwatani H, Kihara S, Nakagawa Y, Komura N, Fujita K, Maeda N, Nishida M, Katsube F, Shimomura I, Ito T, Funahashi T: Exacerbation of albuminuria and renal fibrosis in subtotal renal ablation model of adiponectin-knockout mice. Arterioscler Thromb Vasc Biol 27: 1910–1917, 2007 [DOI] [PubMed] [Google Scholar]
- 22.Sharma K, Ramachandrarao S, Qiu G, Usui HK, Zhu Y, Dunn SR, Ouedraogo R, Hough K, McCue P, Chan L, Falkner B, Goldstein BJ: Adiponectin regulates albuminuria and podocyte function in mice. J Clin Invest 118: 1645–1656, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lo MM, Salisbury S, Scherer PE, Furth SL, Warady BA, Mitsnefes MM: Serum adiponectin complexes and cardiovascular risk in children with chronic kidney disease. Pediatr Nephrol 26: 2009–2017, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nawrocki AR, Rajala MW, Tomas E, Pajvani UB, Saha AK, Trumbauer ME, Pang Z, Chen AS, Ruderman NB, Chen H, Rossetti L, Scherer PE: Mice lacking adiponectin show decreased hepatic insulin sensitivity and reduced responsiveness to peroxisome proliferator-activated receptor gamma agonists. J Biol Chem 281: 2654–2660, 2006 [DOI] [PubMed] [Google Scholar]
- 25.Combs TP, Pajvani UB, Berg AH, Lin Y, Jelicks LA, Laplante M, Nawrocki AR, Rajala MW, Parlow AF, Cheeseboro L, Ding YY, Russell RG, Lindemann D, Hartley A, Baker GR, Obici S, Deshaies Y, Ludgate M, Rossetti L, Scherer PE: A transgenic mouse with a deletion in the collagenous domain of adiponectin displays elevated circulating adiponectin and improved insulin sensitivity. Endocrinology 145: 367–383, 2004 [DOI] [PubMed] [Google Scholar]
- 26.Asterholm IW, Scherer PE: Enhanced metabolic flexibility associated with elevated adiponectin levels. Am J Pathol 176: 1364–1376, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller-Hodges E, Hohenstein P: WT1 in disease: Shifting the epithelial-mesenchymal balance. J Pathol 226: 229–240, 2012 [DOI] [PubMed] [Google Scholar]
- 28.Pajvani UB, Hawkins M, Combs TP, Rajala MW, Doebber T, Berger JP, Wagner JA, Wu M, Knopps A, Xiang AH, Utzschneider KM, Kahn SE, Olefsky JM, Buchanan TA, Scherer PE: Complex distribution, not absolute amount of adiponectin, correlates with thiazolidinedione-mediated improvement in insulin sensitivity. J Biol Chem 279: 12152–12162, 2004 [DOI] [PubMed] [Google Scholar]
- 29.Tiwari S, Blasi ER, Heyen JR, McHarg AD, Ecelbarger CM: Time course of AQP-2 and ENaC regulation in the kidney in response to PPAR agonists associated with marked edema in rats. Pharmacol Res 57: 383–392, 2008 [DOI] [PubMed] [Google Scholar]
- 30.Zhou W, Hildebrandt F: Inducible podocyte injury and proteinuria in transgenic zebrafish. J Am Soc Nephrol 23: 1039–1047, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Appel D, Kershaw DB, Smeets B, Yuan G, Fuss A, Frye B, Elger M, Kriz W, Floege J, Moeller MJ: Recruitment of podocytes from glomerular parietal epithelial cells. J Am Soc Nephrol 20: 333–343, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ronconi E, Sagrinati C, Angelotti ML, Lazzeri E, Mazzinghi B, Ballerini L, Parente E, Becherucci F, Gacci M, Carini M, Maggi E, Serio M, Vannelli GB, Lasagni L, Romagnani S, Romagnani P: Regeneration of glomerular podocytes by human renal progenitors. J Am Soc Nephrol 20: 322–332, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brosius FC, 3rd, Alpers CE, Bottinger EP, Breyer MD, Coffman TM, Gurley SB, Harris RC, Kakoki M, Kretzler M, Leiter EH, Levi M, McIndoe RA, Sharma K, Smithies O, Susztak K, Takahashi N, Takahashi T, Animal Models of Diabetic Complications Consortium : Mouse models of diabetic nephropathy. J Am Soc Nephrol 20: 2503–2512, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun D, Zhao X, Meng L: Relationship between urinary podocytes and kidney diseases. Ren Fail 34: 403–407, 2012 [DOI] [PubMed] [Google Scholar]
- 35.Hida K, Wada J, Eguchi J, Zhang H, Baba M, Seida A, Hashimoto I, Okada T, Yasuhara A, Nakatsuka A, Shikata K, Hourai S, Futami J, Watanabe E, Matsuki Y, Hiramatsu R, Akagi S, Makino H, Kanwar YS: Visceral adipose tissue-derived serine protease inhibitor: A unique insulin-sensitizing adipocytokine in obesity. Proc Natl Acad Sci U S A 102: 10610–10615, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mathew AV, Okada S, Sharma K: Obesity related kidney disease. Curr Diabetes Rev 7: 41–49, 2011 [DOI] [PubMed] [Google Scholar]
- 37.Hamilton MP, Gore MO, Ayers CR, Xinyu Wu, McGuire DK, Scherer PE: Adiponectin and cardiovascular risk profile in patients with type 2 diabetes mellitus: Parameters associated with adiponectin complex distribution. Diab Vasc Dis Res 8: 190–194, 2011 [DOI] [PubMed] [Google Scholar]
- 38.Perseghin G, Lattuada G, Danna M, Sereni LP, Maffi P, De Cobelli F, Battezzati A, Secchi A, Del Maschio A, Luzi L: Insulin resistance, intramyocellular lipid content, and plasma adiponectin in patients with type 1 diabetes. Am J Physiol Endocrinol Metab 285: E1174–E1181, 2003 [DOI] [PubMed] [Google Scholar]
- 39.von Eynatten M, Liu D, Hock C, Oikonomou D, Baumann M, Allolio B, Korosoglou G, Morcos M, Campean V, Amann K, Lutz J, Heemann U, Nawroth PP, Bierhaus A, Humpert PM: Urinary adiponectin excretion: a novel marker for vascular damage in type 2 diabetes. Diabetes 58: 2093–2099, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fowler SM, Kon V, Ma L, Richards WO, Fogo AB, Hunley TE: Obesity-related focal and segmental glomerulosclerosis: Normalization of proteinuria in an adolescent after bariatric surgery. Pediatr Nephrol 24: 851–855, 2009 [DOI] [PubMed] [Google Scholar]
- 41.Praga M, Hernández E, Andrés A, León M, Ruilope LM, Rodicio JL: Effects of body-weight loss and captopril treatment on proteinuria associated with obesity. Nephron 70: 35–41, 1995 [DOI] [PubMed] [Google Scholar]
- 42.Berg AH, Scherer PE: Adipose tissue, inflammation, and cardiovascular disease. Circ Res 96: 939–949, 2005 [DOI] [PubMed] [Google Scholar]
- 43.Declèves AE, Mathew AV, Cunard R, Sharma K: AMPK mediates the initiation of kidney disease induced by a high-fat diet. J Am Soc Nephrol 22: 1846–1855, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baldwin W, McRae S, Marek G, Wymer D, Pannu V, Baylis C, Johnson RJ, Sautin YY: Hyperuricemia as a mediator of the proinflammatory endocrine imbalance in the adipose tissue in a murine model of the metabolic syndrome. Diabetes 60: 1258–1269, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.D’Apolito M, Du X, Zong H, Catucci A, Maiuri L, Trivisano T, Pettoello-Mantovani M, Campanozzi A, Raia V, Pessin JE, Brownlee M, Giardino I: Urea-induced ROS generation causes insulin resistance in mice with chronic renal failure. J Clin Invest 120: 203–213, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boini KM, Xia M, Xiong J, Li C, Payne LP, Li PL: Implication of CD38 gene in podocyte epithelial-to-mesenchymal transition and glomerular sclerosis. J Cell Mol Med 16: 1674–1685, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harvey SJ, Jarad G, Cunningham J, Goldberg S, Schermer B, Harfe BD, McManus MT, Benzing T, Miner JH: Podocyte-specific deletion of dicer alters cytoskeletal dynamics and causes glomerular disease. J Am Soc Nephrol 19: 2150–2158, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Herman-Edelstein M, Thomas MC, Thallas-Bonke V, Saleem M, Cooper ME, Kantharidis P: Dedifferentiation of immortalized human podocytes in response to transforming growth factor-β: A model for diabetic podocytopathy. Diabetes 60: 1779–1788, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stieger N, Worthmann K, Schiffer M: The role of metabolic and haemodynamic factors in podocyte injury in diabetes. Diabetes Metab Res Rev 27: 207–215, 2011 [DOI] [PubMed] [Google Scholar]
- 50.Gealekman O, Burkart A, Chouinard M, Nicoloro SM, Straubhaar J, Corvera S: Enhanced angiogenesis in obesity and in response to PPARgamma activators through adipocyte VEGF and ANGPTL4 production. Am J Physiol Endocrinol Metab 295: E1056–E1064, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hurren KM, Taylor TN, Jaber LA: Antidiabetic prescribing trends and predictors of thiazolidinedione discontinuation following the 2007 rosiglitazone safety alert. Diabetes Res Clin Pract 93: 49–55, 2011 [DOI] [PubMed] [Google Scholar]
- 52.Endo Y, Suzuki M, Yamada H, Horita S, Kunimi M, Yamazaki O, Shirai A, Nakamura M, Iso-O N, Li Y, Hara M, Tsukamoto K, Moriyama N, Kudo A, Kawakami H, Yamauchi T, Kubota N, Kadowaki T, Kume H, Enomoto Y, Homma Y, Seki G, Fujita T: Thiazolidinediones enhance sodium-coupled bicarbonate absorption from renal proximal tubules via PPARγ-dependent nongenomic signaling. Cell Metab 13: 550–561, 2011 [DOI] [PubMed] [Google Scholar]
- 53.Panchapakesan U, Pollock C, Saad S: Review article: Importance of the kidney proximal tubular cells in thiazolidinedione-mediated sodium and water uptake. Nephrology (Carlton) 14: 298–301, 2009 [DOI] [PubMed] [Google Scholar]
- 54.Moeller MJ, Sanden SK, Soofi A, Wiggins RC, Holzman LB: Two gene fragments that direct podocyte-specific expression in transgenic mice. J Am Soc Nephrol 13: 1561–1567, 2002 [DOI] [PubMed] [Google Scholar]
- 55.Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, Schraw T, Durand JL, Li H, Li G, Jelicks LA, Mehler MF, Hui DY, Deshaies Y, Shulman GI, Schwartz GJ, Scherer PE: Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest 117: 2621–2637, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hu MC, Shi M, Zhang J, Quiñones H, Griffith C, Kuro-o M, Moe OW: Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol 22: 124–136, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.