

Abstract
Glycerol trinitrate reductase (NerA) from Agrobacterium radiobacter, a member of the old yellow enzyme (OYE) family of oxidoreductases, was expressed in and purified from Escherichia coli. Denaturation of pure enzyme liberated flavin mononucleotide (FMN), and spectra of NerA during reduction and reoxidation confirmed its catalytic involvement. Binding of FMN to apoenzyme to form the holoenzyme occurred with a dissociation constant of ca. 10−7 M and with restoration of activity. The NerA-dependent reduction of glycerol trinitrate (GTN; nitroglycerin) by NADH followed ping-pong kinetics. A structural model of NerA based on the known coordinates of OYE showed that His-178, Asn-181, and Tyr-183 were close to FMN in the active site. The NerA mutation H178A produced mutant protein with bound FMN but no activity toward GTN. The N181A mutation produced protein that did not bind FMN and was isolated in partly degraded form. The mutation Y183F produced active protein with the same kcat as that of wild-type enzyme but with altered Km values for GTN and NADH, indicating a role for this residue in substrate binding. Correlation of the ratio of KmGTN to KmNAD(P)H, with sequence differences for NerA and several other members of the OYE family of oxidoreductases that reduce GTN, indicated that Asn-181 and a second Asn-238 that lies close to Tyr-183 in the NerA model structure may influence substrate specificity.
Nitroglycerin (glycerol trinitrate [GTN]) has been widely used for more than a century as an explosive (22) and as a vasodilator in the treatment of angina (17). During its production, large quantities of washing wastewaters, saturated with GTN, are commonly transferred to lagoons or soakaways, resulting in actual or potential contamination of soils. Nitrate esters, such as GTN, are known to persist in the environment for significant periods of time (28), and their biodegradation presents a truly xenobiotic challenge to microorganisms due to the rarity of naturally occurring analogues.
Several bacterial strains that can biodegrade GTN have been isolated previously (2, 4, 16, 24-26). These bacteria generally utilize GTN as a sole source of nitrogen by removing either one or two nitro groups from GTN to form isomers of glycerol dinitrate and glycerol mononitrate. Most exhibit no biodegradation of glycerol mononitrate and are therefore incapable of completely mineralizing GTN. To date, only one axenic bacterial strain, a Rhodococcus species, has been shown to achieve complete denitration of GTN (14), although complete denitration has also been demonstrated previously in mixed bacterial populations (1, 23) and in cultures of Penicillium corylophilum Dierckx (29).
The enzymes involved in the removal of the first nitro group from GTN have been isolated and characterized for some bacterial strains. These include NerA from Agrobacterium radiobacter (20), pentaerythritol tetranitrate reductase (Onr) from Enterobacter cloacae (2, 9), and xenobiotic reductases XenA and XenB from Pseudomonas putida and Pseudomonas fluorescens, respectively (3, 4). All four enzymes exhibit similar properties in that they are oxidoreductases that convert GTN to glycerol dinitrate and nitrite by using either NADH or NADPH as a reducing agent, and they are members of the old yellow enzyme (OYE) family of oxidoreductases (27). Crystallization of OYE in the presence of an inhibitor, p-hydroxybenzaldehyde, and subsequent mutagenesis studies have shown that His-191, Asn-194, and Tyr-196 are involved in OYE's activity toward its substrates, including substituted phenols, cyclohexanone, and GTN (6, 8, 11, 15). The aim of the work presented here was to establish unequivocally the involvement of flavin mononucleotide (FMN) as a prosthetic group in the action of NerA on GTN, the kinetic form of the reaction, and the effect on the kinetics of mutations at the corresponding His, Asn, and Tyr residues.
MATERIALS AND METHODS
Materials.
A stock solution of GTN (97% pure by high-performance liquid chromatography analysis [26]; 5% [vol/vol] in ethanol) was kindly provided by EXCHEM, Derbyshire, United Kingdom. Restriction enzymes, T4 DNA ligase, deoxynucleotide triphosphates (dNTPs), and Vent DNA polymerase were obtained from New England Biolabs, Hitchin, United Kingdom. TaqDNA polymerase was obtained from Promega, Southampton, United Kingdom. PCR primers were synthesized by Life Technologies, Ltd., Paisley, United Kingdom. Bradford reagent was obtained from Bio-Rad Laboratories Ltd., Hemel Hempstead, United Kingdom. Blue Sepharose, Q Sepharose, phenyl-Sepharose, and PD10 desalting columns were obtained from Amersham Pharmacia Biotech, Little Chalfont, United Kingdom. Antibiotics were obtained from Melford Laboratories, Ltd., Ipswich, United Kingdom. All other chemicals were obtained from either Fisher Scientific, Loughborough, United Kingdom, or from Sigma-Aldrich Company, Ltd., Poole, United Kingdom.
Bacterial growth media.
Luria-Bertani (LB) medium and LB agar were obtained in capsule form from Anachem, Luton, United Kingdom. M9 salts (10×) contained Na2HPO4, 60 g · liter−1; KH2PO4, 30 g · liter−1; NaCl, 5 g · liter−1; and NH4Cl, 10 g · liter−1 at pH 7.4. RM medium contained 1× M9 salts, 2% (wt/vol) Casamino Acids, 1% (vol/vol) glycerol, 1 mM MgCl2, and 100 μg of ampicillin per ml. RMG-agar plates consisted of 1× M9 salts, 2% (wt/vol) Casamino Acids, 0.5% (wt/vol) glucose, 1 mM MgCl2, 250 μg of carbenicillin per ml, and 1.5% (wt/vol) agar. Induction medium consisted of 1× M9 salts, 0.2% (wt/vol) Casamino Acids, 0.5% (wt/vol) glucose, 1 mM MgCl2, and 100 μg of ampicillin per ml.
DNA manipulations.
Genomic and plasmid DNAs were extracted by using QiaTip 100 columns and miniprep kits, respectively (both from Qiagen, Crawley, United Kingdom). Restriction digest mixtures typically consisted of approximately 1 to 1.5 μg of DNA, 5 to 10 U of restriction enzyme, and a 1× concentration of the appropriate buffer in a final volume of 20 μl and were incubated for 2 to 3 h at the temperature recommended by the manufacturer (New England Biolabs). Ligation reactions were carried out using T4 DNA ligase according to the manufacturer's instructions. DNA sequences were determined by using an ABI PRISM dye terminator cycle-ready reaction kit (Applied Biosystems, Warrington, United Kingdom).
PCR screening reaction mixtures (10 μl) consisted of 100 ng of each primer, 1× PCR buffer, 1 mM MgCl2, 2.5 μg of bovine serum albumin per μl, 0.2 mM of each dNTP (dATP, dCTP, dGTP, and dTTP), and 0.5 U of TaqDNA polymerase. A portion of the bacterial colony was mixed with the reaction mixture and then sealed in 10-μl glass capillary tubes. The tubes were heated in a thermal cycle (Rapidcycler PCR machine; Idaho Technology, Salt Lake City, Utah) which consisted of denaturing at 96°C for 5 min to lyse the cells, followed by 30 cycles of 96°C for 20 s, 56°C for 20 s, and 72°C for 90 s. PCR products were visualized on 1% (wt/vol) agarose gels.
Introduction of nerA into the PL expression system.
The PL expression system allows the tightly regulated, tryptophan-inducible expression of heterologous genes from the pLEX vector (Invitrogen, Ltd., Paisley, United Kingdom). The coding region of nerA was amplified by PCR from A. radiobacter genomic DNA. The reaction mixture (25 μl) contained approximately 100 ng of genomic DNA, 100 ng each of primer NerA3 (5′-CAA CCG ACC AGA AAG TGA AGT CAT ATG ACC AGT CTT-3′) and NerA2 Stop (5′-CGT CTT TTT CAT TGC TAT TGG GCG AGG GCC GGA TAG-3′), 1× PCR buffer, 0.2 mM of each dNTP, and 0.5 U of Vent DNA polymerase. The thermal cycler (MWG-Biotech) program consisted of initial denaturation at 94°C for 2 min, followed by 30 cycles of 94°C for 1 min, 58°C for 1 min, and 72°C for 2 min, with a final extension step of 10 min at 72°C. The single product visualized on a 1% (wt/vol) agarose gel was ligated into pCR-Blunt and transformed into Escherichia coli One Shot Top10 competent cells according to the manufacturer's instructions (Invitrogen, Ltd.). Recombinant clones were identified by PCR screening using M13F (5′-GTT TTC CCA GTC ACG AC-3′) and M13R (5′-CAG GAA ACA GCT ATG AC-3′) primers, which anneal to the vector at regions on either side of the multiple cloning site. The orientation of the gene insert was then determined by digestion with NdeI and either BamHI or XbaI.
The nerA gene was excised from pCR-BluntnerA with NdeI and BamHI and ligated into NdeI/BamHI-digested pLEX. The ligation mixture was transformed into competent E. coli GI724 cells according to the manufacturer's instructions (Invitrogen, Ltd.), and recombinant clones were identified by plasmid extraction and restriction digestion with NdeI and BamHI. The fidelity of the PCR amplification of nerA was confirmed by sequencing.
Production of NerA.
The pLEXnerA construct was transformed into E. coli GI724 cells and grown overnight at 30°C on RMG-agar plates. RM medium was inoculated with a single colony and grown overnight at 30°C and with shaking at 100 rpm. The overnight culture was used to inoculate (2% [vol/vol]) induction medium in which the bacteria were grown at 30°C and with shaking at 100 rpm until the culture attenuance at 550 nm reached 0.5. Protein expression was induced by the addition of tryptophan (final concentration, 100 μg/ml) and further incubation for 2.5 h at 37°C. The bacteria were harvested by centrifugation (6,000 × g, 30 min), and the cell pellets were stored at −20°C until required.
Purification of NerA. (i) Preparation of cell extracts.
Pellets of tryptophan-induced cells from 5 liters of cell culture were resuspended in 1/100 of their original culture volume in 50 mM potassium phosphate buffer (pH 6.5) supplemented with 100 μg of FMN per ml and then incubated on ice for 30 min. Chilled cells were ruptured by passage three times through a French pressure cell (American Instruments Co., Bethesda, Md.) at 126 MPa. Residual whole cells and cell debris were removed from the lysate by centrifugation (10,000 × g, 1 h) and filtration through 0.2-μm-pore-size filters (Sartorius, Ltd., Epsom, United Kingdom).
(ii) Affinity chromatography.
Freshly prepared cell extracts were desalted by size exclusion chromatography by using PD10 columns and were then applied to a 250-ml column containing Blue Sepharose that had been preequilibrated with 50 mM potassium phosphate buffer (pH 6.5). The loaded column was washed with 3 column volumes of phosphate buffer to remove unbound proteins. NerA was eluted with 2 column volumes of 1 mM NADH in phosphate buffer. Residual bound proteins were eluted from the column with 3 column volumes of 2 M NaCl in phosphate buffer. Fractions were collected throughout and analyzed for enzyme activity and protein concentration.
(iii) Ion-exchange chromatography.
Fractions of highest activity from step (ii) were pooled and diluted five times with distilled water to reduce the phosphate buffer concentration to 10 mM. The diluted sample was applied to a Q Sepharose column (50 ml) that had been preequilibrated with 10 mM phosphate buffer (pH 6.5). Unbound proteins were washed from the column with 3 column volumes of 10 mM phosphate buffer. Bound proteins were then eluted with a linear gradient of 0 to 0.3 M NaCl in 10 mM phosphate buffer over 3 column volumes, followed by 3 column volumes of 1 M NaCl in phosphate buffer. Fractions were collected throughout and analyzed for enzyme activity and protein concentration.
(iv) Hydrophobic-interaction chromatography.
Fractions of highest activity from step iii were pooled and diluted 1:1 with 3 M (NH4)2SO4 in 10 mM phosphate buffer (pH 6.5). The sample was then applied to a 50-ml phenyl-Sepharose column that had been preequilibrated with 10 mM phosphate buffer containing 1.5 M (NH4)2SO4. The column was washed with 3 column volumes of buffered 1.5 M (NH4)2SO4. Bound proteins were eluted with a decreasing linear salt gradient (1.5 to 0 M) over 4 column volumes, followed by 3 column volumes of 10 mM phosphate buffer (pH 6.5). Fractions were collected throughout and analyzed for enzyme activity and protein concentration.
Standard protein assay.
Protein concentrations were determined by using the method of Bradford (5) and bovine serum albumin standards.
Standard enzyme assays.
Unless otherwise stated, enzyme activity was determined by measuring the concentration of nitrite produced from GTN in the presence of NADH under standard conditions. Samples (20 μl) were mixed with 60 μl of phosphate buffer (pH 6.5), 100 μl of 1 mM GTN, and 20 μl of 1 mM NADH, both of which were prepared in the same buffer. Assay mixtures were incubated at 30°C for 15 min and were then acidified by the addition of 200 μl of 60% (vol/vol) acetic acid and centrifuged (20,000 × g, 10 min) to remove any precipitated protein. The concentration of nitrite in the supernatant was then determined by using a modification (26) of the method of Litchfield (13). One unit of activity was defined as the amount of enzyme required to release 1 μmol of nitrite per min from GTN under the standard assay conditions.
PAGE.
Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed by the method of Laemmli (12). Native PAGE was performed by the same method except that SDS and β-mercaptoethanol were omitted. Gels were stained with Coomassie blue R-350 or with a silver staining kit (Amersham Pharmacia Biotech). For two-dimensional gel electrophoresis, isoelectric focusing in the first dimension was achieved on 18-cm nonlinear (pH 3 to 10) immobilized pH gradient (IPG) strips on an IPGphor system (Amersham Pharmacia Biotech). The strip was rehydrated by incubating at 20°C for 12 h in rehydration buffer [8 M urea, 2% (wt/vol) 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), 18 mM dithiothreitol, 2% IPG buffer, and trace amounts of bromophenol blue] containing the test sample (430 μg). Isoelectric focusing was performed in three stages: (i) 500 V for 1 h, (ii) 1,000 V for 1 h, and finally, (iii) 8,000 V until 45 to 60 kV · h had been achieved. After focusing, the strip was incubated for 30 min in equilibration buffer (50 mM Tris-HCl [pH 6.8], 6 M urea, 30% [vol/vol] glycerol, 2% [wt/vol] SDS, and trace amounts of bromophenol blue) containing 43 mM dithiothreitol, followed by a additional 30 min in equilibration buffer containing 90 mM iodoacetamide. The equilibrated strip was transferred to a 12 to 14% ExcelGel XL (Amersham Pharmacia Biotech), and SDS-PAGE was performed by using a Multiphor II system at 15°C in three steps: (i) 45 min with upper limits set at 20 mA, 1,000 V, and 40 W; (ii) 5 min at 40 mA, 1,000 V, and 40 W; and (iii) 2 h 40 min at 40 mA, 1,000 V, and 40 W. The IPG strip was removed from the surface of the gel after step i, and the cathode buffer strip was moved forward to cover the area of the removed IPG strip after step ii.
Protein spots were visualized by rinsing the gel twice in fixing solution (ethanol-acetic acid-water, 4:1:5 [vol/vol/vol]) for 1 h and staining overnight with prefiltered (0.2 μm) Coomassie blue R-350 (0.1% [wt/vol]) in destaining solution (ethanol-acetic acid-water, 25:8:67 [vol/vol/vol]). The gel was washed twice in deionized water and transferred to destaining solution. The destained gel was kept in 10% (vol/vol) methanol during spot analysis. Spots were excised and identified by mass fingerprinting and fragmentation analysis (Protein and Nucleic Acid Chemistry Facility, Cambridge University, Cambridge, United Kingdom).
Amino acid analysis.
Protein supplemented with 0.5 ml of 6 N HCl and 15 nmol of norleucine (internal standard) was sparged with nitrogen and then sealed under vacuum and hydrolyzed at 110°C for 18 h. Hydrolyzed samples were dried under vacuum over NaOH and analyzed on a Biochrom 20 amino acid analyzer (Amersham Pharmacia Biotech) according to the manufacturer's instructions.
Analysis for flavin prosthetic groups.
Purified NerA was boiled in 50 mM phosphate buffer (pH 6.5) for 5 min, and the denatured protein was removed by centrifugation in a microcentrifuge (20,000 × g, 10 min). Supernatant samples (10, 20, and 40 μl) were loaded onto two 20-by-20-cm K6F fluorescent silica gel (60 Å) plates with a layer thickness of 250 μm (Whatman, Inc.) alongside standards of flavin adenine dinucleotide (FAD) and FMN. The plates were eluted in either solvent A (2% [wt/vol] Na2HPO4 in water) or solvent B (n-butanol, water, acetic acid, and methanol [14:14:1:16 by volume]). Air-dried plates were examined under UV light for areas of quenched fluorescence, and the corresponding Rf values were calculated.
Spectral properties of NerA.
UV-visible spectra of NerA were obtained by using a Hewlett-Packard 8452 diode array spectrophotometer and sealable 3.5-ml quartz cuvettes (Spectrocell, Oreland, Pa.). Enzyme solutions were prepared in 50 mM phosphate buffer (pH 6.5) and made anaerobic by degassing with argon for 2 h. For reduction experiments, the NerA was reduced with either dithionite or NADH, both of which were dissolved in the same buffer as was the enzyme. The reduced enzyme was then reoxidized either by allowing air into the cuvette or by adding GTN dissolved in a small volume of ethanol.
Determination of kinetic constants.
Kinetic parameters were determined by measuring rates of disappearance of NADH in the presence of enzyme and various concentrations of substrates. Reaction mixtures were prepared in triplicate in multiwell plates. Each well contained 50 mM phosphate buffer (pH 6.5), NADH (4 to 10 μM), GTN (0 to 1,000 μM), and enzyme (0.5 to 1.0 μg/ml) in a total volume of 0.2 ml. Plates were agitated to ensure good mixing and incubated at 30°C in a Thermomax microtiter plate reader (Molecular Devices, Wokingham, United Kingdom). Disappearance of NADH was monitored by loss of absorbance at 340 nm over a period of 5 min, with measurements recorded automatically every 10 s, and initial rates were calculated over the initial linear part of each curve.
Site-directed mutation.
Specific codon changes, namely H178A, N181A, and Y183F, were introduced into the nerA sequence by using the QuikChange site-directed mutagenesis kit from Stratagene (Cambridge, United Kingdom) according to the manufacturer's instructions. PCR mixtures consisted of 16 to 160 ng of pCR-BluntnerA plasmid DNA, 2.5 U of Pfu Turbo DNA polymerase, 1× Pfu Turbo DNA polymerase buffer, 125 ng of each of the relevant primers, and a 0.4 mM concentration of each dNTP. The PCR thermal cycle was 95°C for 5 min, followed by 30 cycles of 95°C for 1 min, 55°C for 1 min, and 68°C for 10 min. The presence of a product of the correct size was checked by agarose gel electrophoresis. Template DNA was removed by incubating 20 μl of the PCR mixture with 8 U of DpnI at 37°C for 1 h, which was followed by a 20-min step at 80°C to inactivate the enzyme. The digested material was then transformed into E. coli One Shot Top10 competent cells according to the manufacturer's instructions (Invitrogen) and grown overnight at 37°C on LB agar plates containing 250 μg of carbenicillin per ml. The colonies obtained were inoculated into 10 ml of LB medium containing 50 μg of ampicillin per ml and grown overnight at 37°C and with shaking at 200 rpm. Plasmid DNA was extracted, and the presence of the desired mutation was confirmed by sequencing. The mutant nerA sequences were excised from pCR-Blunt and transferred into the pLEX vector as described for wild-type NerA. The entire coding region of nerA was then sequenced to ensure that only the desired mutations had been introduced. Protein was produced and purified from mutant NerA genes as described for the wild type.
RESULTS
Purification of recombinant NerA.
Typical progress of the purification is shown in Table 1. A single peak of NerA activity was obtained from both the affinity and ion-exchange chromatography steps, and a 3.4-fold purification was achieved by their combined use. However, the protein was not entirely pure at this stage (Fig. 1), and an additional hydrophobic-interaction chromatography stage was introduced. This yielded two major protein peaks, the first of which (peak 1) was yellow and exhibited enzyme activity toward GTN, while the other (peak 2) demonstrated only weak GTN reductase activity and was colorless. SDS-PAGE (Fig. 1) showed that both peaks contained a single band of the same molecular mass. Hence, although the protein present in peak 2 from the phenyl-Sepharose column was found to have very little activity toward GTN, it had the same molecular mass as that of the active protein in peak 1 and was abundant. This finding indicated that peak 2 might be the NerA apoenzyme lacking a flavin cofactor (see below).
TABLE 1.
Summary of the purification of recombinant NerA
| Purification stepa | Total vol (ml) | Protein concn (mg/ml) | Total protein (mg) | Enzyme activity (U/ml) | Total enzyme activity (U) | Sp act (U/mg) | Yield (%) | Purification (n-fold) |
|---|---|---|---|---|---|---|---|---|
| Cell extract | 65 | 7.24 | 471 | 6.35 | 413 | 0.88 | 100 | 1 |
| Affinity chromatography | 37 | 3.70 | 137 | 7.07 | 262 | 1.91 | 63 | 2.2 |
| Ion-exchange chromatography | 8.5 | 7.14 | 60.7 | 21.4 | 182 | 2.99 | 44 | 3.4 |
| HIC peak 1 | 27.5 | 0.82 | 22.6 | 2.52 | 69.3 | 3.07 | 17 | 3.5 |
| HIC peak 2 | 20 | 0.85 | 17.0 | 0.001 | 0.02 | 0.001 | —b | —b |
HIC, hydrophobic-interaction chromatography.
This fraction is the protein lacking FMN and therefore lacking enzyme activity (see text).
FIG. 1.

Silver-stained SDS-PAGE gel of the purification steps of NerA showing cell extract (lane a), pooled samples obtained after affinity chromatography (lane b), and ion-exchange chromatography (lane c). Hydrophobic-interaction chromatography yielded peak 1 (lane d) and peak 2 (lane e). M indicates the molecular mass marker.
Amino acid analysis.
Amino acid analysis showed that the relative molar amounts of Lys, Ser, and Gly were in the ratio of 1.0:1.0:2.2 and 1.0:1.1:2.4 for peaks 1 and 2, respectively, indicating that both peaks consisted of the same protein. The theoretical ratio for NerA is 1.0:1.0:2.7 (20). Assuming that peaks 1 and 2 are NerA apo- and holoenzymes, each amino acid content was used to estimate the protein concentration present. Peaks 1 and 2 were found to contain protein concentrations of 7.2 ± 0.7 μM (mean plus standard deviation; n = 3) and 40 ± 2 μM (mean plus standard deviation; n = 3), respectively.
Identification of flavin prosthetic group.
The identity of the prosthetic group of NerA, released by boiling the native enzyme, was determined by comparison of the Rf values obtained from thin-layer chromatography of the dissociated yellow ligand with those of FAD and FMN standards. In solvent system A, the Rf values of FAD, FMN, and the NerA ligand were 0.68, 0.49, and 0.43, and in solvent B, the Rf values were 0.72, 0.53, and 0.51, respectively. Thus, the NerA ligand was quite distinct from FAD but was very similar to FMN in terms of chromatographic behavior.
Reflavination of apoenzyme.
FMN was added to the putative apoenzyme in peak 2 obtained from the phenyl-Sepharose purification step in an attempt to restore activity toward GTN. Enzyme solution (10.6 μM in 50 mM phosphate buffer [pH 6.5]) was incubated with 0, 5, 10, 20, or 100 μM FMN (final concentrations), respectively, for 2.5 h at 4°C. Unincorporated FMN was removed by passing each sample (500 μl) through a 5-ml HiTrap desalting column preequilibrated with 50 mM phosphate buffer (pH 6.5). Fractions containing protein were pooled and assayed for enzyme activity and protein concentration. Specific activity increased with increasing concentrations of FMN up to 20 μM (Fig. 2).
FIG. 2.

Activation of NerA apoenzyme by titration with FMN. Aliquots from peak 2 were supplemented with the indicated concentrations of FMN and assayed for activity. The solid line is the theoretical curve based on a simple association of the apoenzyme with FMN, with an apparent dissociation constant of 0.1 ± 0.1 μM and maximum specific activity of the fully flavinated holoenzyme of 2.6 ± 0.1 U/mg. The dashed line represents conditions in which no dissociation occurs, giving a maximum specific activity of 2.6 ± 0.1 U/mg.
Assuming that the binding of FMN to enzyme is a simple equilibrium described by the dissociation constant (K = [E][F]/[EF]), then
 |
(1) |
where [E], [F] and [EF] are concentrations of the apoenzyme, FMN, and holoenzyme, respectively, and [E]0 = [E] + [EF] and [F]0 = [F] + [EF]. Assuming that only EF is active, the measured specific activity, SA, during titration of apoenzyme with FMN is related to [EF] by:
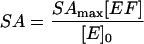 |
(2) |
where SAmax is the maximum observed specific activity. Thus, combining the two equations:
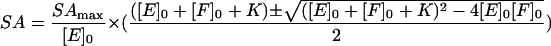 |
(3) |
The data in Fig. 2 were fitted to equation 3 by using Sigma Plot (Jandel Scientific), with [SA] and [F]0 (concentrations described as micromolar) as dependent and independent variables, respectively, [E]0 equal to 10.6 μM (based on the protein concentration used), and SAmax and K as parameters. The values of SAmax and K giving the best fit were 2.6 ± 0.1 U/mg and 0.1 ± 0.1 μM, respectively.
The maximum specific activity achieved (2.6 U/mg) on reflavination of the protein in peak 2 was comparable to the specific activity of the flavinated NerA protein obtained from peak 1 of the phenyl-Sepharose purification step (3.1 U/mg).
Figure 3 shows that native PAGE resolved peak 1 protein (active, yellow, higher electrophoretic mobility) from peak 2 (inactive, colorless, lower mobility). When peak 2 was titrated with increasing amounts of FMN, the intensity of the slower band decreased and the intensity of the faster band increased, a result consistent with binding of FMN to the apoenzyme (peak 2) to form the holoenzyme (peak 1). At 5 μM FMN, a concentration corresponding to approximately half of the protein concentration (10.6 μM), two bands of approximately equal density were observed, and the specific activity of the enzyme (Fig. 2) was approximately half that of the maximum specific activity obtained. At 10 μM FMN, most of the protein was present as holoenzyme, with only small amounts present in the apoenzyme form. These results are consistent with tight binding of one FMN molecule per molecule of enzyme. Incubation with 20 or 100 μM FMN resulted in essentially complete flavination of the enzyme as shown by saturation in the specific activity curve and near-absence of the apoenzyme on the gel. The faster-moving band produced by the addition of FMN to the colorless inactive protein in peak 2 corresponded exactly to the protein obtained from peak 1, which contained active, yellow protein. The data in Fig. 2 and 3 thus collectively show that the production and purification procedure for NerA resulted in a mixture of apo- and holoenzymes and that the apoenzyme form was converted to the holoenzyme (with high recovery of activity) by titration with FMN, which became tightly bound.
FIG. 3.

Conversion of NerA apoenzyme to holoenzyme by titration with FMN. Samples of NerA apoenzyme were treated with increasing concentrations of FMN and subjected to nondenaturing gel electrophoresis. Lanes 1 to 5 correspond to the same five FMN-treated samples that were assayed in Fig. 2. Lanes 6 and 7 are untreated samples of peaks 1 and 2 from the hydrophobic-interaction chromatography that contained fully active, yellow protein and inactive, colorless protein, respectively.
Spectral properties of NerA.
The visible spectrum (Fig. 4A) of the active NerA protein (peak 1) exhibited absorbance peaks at 360 and 460 nm, with shoulders at 430 and 490 nm, readings which are typical of flavinated proteins. In contrast, the inactive, colorless protein in peak 2 did not exhibit any absorbance peaks in the 300-to-800-nm range (data not shown).
FIG. 4.

Spectral properties of the active, yellow holoenzyme. (A) Spectra measured before (a) and after (b) reduction with 60 mM sodium dithionite and after reoxidation of the enzyme by exposure to air for 30 min (c). (B) Spectra of NerA during reduction by titration with NADH. After degassing, NerA was treated with NADH at final concentrations (millimolar) of 0 (a), 0.08 (b), 0.24 (c), 0.45 (d), and 0.54 (e). In all cases, protein concentration was 16 μM in 50 mM potassium phosphate buffer (pH 6.5).
Figure 4A shows that addition of excess dithionite to NerA in the absence of air caused complete reduction of the bound FMN to the dihydro state and that reduced NerA could be reoxidized by the allowing air into the sample. The FMN present in NerA could also be reduced in the absence of air by the addition of NADH (Fig. 4B), increasing concentrations of which caused progressive diminution in the 460-nm absorbance spectrum. Complete reoxidation of the NADH-treated enzyme was achieved by the addition of GTN dissolved in ethanol (data not shown) in the absence of air. The addition of ethanol alone to the cuvette did not cause reoxidation of the reduced enzyme.
Steady-state kinetics for NerA.
Two-substrate reactions in which substrate Ax transfers group x to substrate B can occur by either (i) a compulsory-order mechanism, in which the two substrates bind to the enzyme in a specific order, (ii) a random-order mechanism, in which there is no specific order of binding, or (iii) a double-displacement or ping-pong mechanism, in which Ax binds to and transfers group x to the enzyme and A is released prior to binding of the second substrate B (21). All three mechanisms are described by a single Michaelis-Menten-type equation (equation 4) that contains four constants: Vmax, a Michaelis constant for each substrate (KmAx and KmB), and KsAx, which is the apparent dissociation constant for the release of the substrate Ax from the enzyme-substrate complex before binding of the second substrate.
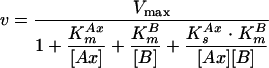 |
(4) |
When the double-displacement or ping-pong mechanism is operative, the two substrates never meet on the surface of the enzyme, and there is therefore an obligatory step, namely the transfer of x from Ax to the enzyme and the release of A, which must occur before B can bind. The step in which A is released from the enzyme is essentially irreversible during initial rate measurements because the concentration of A in the system at time zero is zero. This makes the apparent dissociation constant KsAx also zero (21), and equation 4 can thus be simplified to:
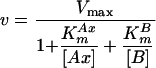 |
(5) |
To determine the kinetic constants and the mechanism of catalysis, we measured initial reaction rates at various concentrations of NADH and GTN in the presence of 12.5 nM NerA. For each set of reactions at a fixed NADH concentration, a plot of velocity versus GTN concentration was constructed (Fig. 5), and the complete data set, taking all such plots together, was fitted to equations 4 and 5. A good fit was achieved only with equation 5, in which KsAx = 0, suggesting that a ping-pong mechanism was operative with values of KmNADH, KmGTN, and kcat (kcat equals Vmax/[E0]) as shown in Table 2. Operation of a ping-pong mechanism was confirmed by classical double-reciprocal plots of 1/v versus 1/[GTN] at each NADH concentration, which gave the characteristic parallel lines (7, 21).
FIG. 5.

Kinetic analysis of NerA. Different symbols represent data obtained on three separate occasions. Data were fitted to equations 4 and 5, but the latter (derived from the former by setting the value of KsAx to 0) gave the better fit (solid lines), implying that a ping-pong mechanism was operative. Data are means, and the error bars indicate standard deviations.
TABLE 2.
Correlation of kinetic parameters with structural features for NerA and its homologs
| Enzyme | Reference | kcat (s−1)a | KmGTN (μM) | KmNAD(P)H (μM) | KmGTN/ KmNAD(P)H | Amino acid residue at siteb
|
|||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | ||||||
| NerA | This study | 8.8 ± 0.8 | 705 ± 74 | 8 ± 1 | 88 | Tyr-160 | Ile-177 | Asn-181 | Asn-238 |
| XenB | 4 | —c | 110 ± 10 | 5 ± 1 | 22 | Tyr-155 | Ile-172 | Asn-176 | His-233 |
| OYE | 15 | 5.5 | 380 | 7.5 | 51 | Tyr-173 | Ile-190 | Asn-194 | Asn-251 |
| Onr | 9 | 16.6 ± 1.0 | 39.3 ± 3.5 | 107 ± 10 | 0.37 | Phe-164 | Leu-181 | His-185 | Phe-241 |
| XenA | 4 | —c | 52 ± 4 | 28 ± 2 | 1.86 | Phe-160 | Leu-177 | His-181 | Phe-232 |
Where indicated, errors arise from regression to kinetic equations.
Numbering includes the N terminus as Met-1, except for OYE (for which the original numbering omitting Met is traditionally used).
Blehert et al. quoted Vmax without specifying the enzyme concentration.
NerA mutants.
In OYE, His-191 and Asn-194 form hydrogen bonds to phenolic ligands that bind in the active site, stabilizing the phenolate form involved in charge transfer interactions with FMN, and they are also predicted to interact with the nicotinamide ring of NADPH in the active site (6). Tyr-196 in OYE also contributes H-bonding interactions with substrates during reoxidation of FMN (11). All three residues are conserved at corresponding positions (His-178, Asn-181, and Tyr-183) in NerA (20), and we therefore examined the effects of mutating each of these residues in NerA.
NerA mutant H178A behaved in a manner similar to that of the wild-type protein in terms of its elution from both the affinity and ion-exchange chromatography columns. The purified protein appeared yellow and therefore presumably contained bound FMN. However, the change of His-178 to Ala completely abolished its activity.
In contrast with the wild-type NerA, the mutated enzyme N181A could not be eluted from the affinity chromatography column with 1 mM NADH alone, and inclusion of a salt gradient (0 to 2 M NaCl) with 1 mM NADH was necessary. The eluted protein was colorless and exhibited no activity toward GTN. During ion-exchange chromatography, a higher salt concentration (∼0.6 M) was necessary to elute the mutant protein than that needed for the wild-type protein (∼0.3 M). The final stage (hydrophobic-interaction chromatography) produced a single colorless, inactive protein peak, as was expected for a protein lacking FMN. SDS-PAGE revealed the presence of three major components corresponding to 40, 30, and 15 kDa in the samples eluted from each of the chromatography steps (Fig. 6). Size exclusion chromatography failed to separate any components, suggesting that they were associated in some way. Two-dimensional gel electrophoresis revealed the presence of four major peptide spots that were identified by mass fingerprinting as fragments of the NerA protein. The fragments corresponded with proteolytic cleavage of NerA in the regions Ser-126 to Lys-127, Lys-321 to Lys-329, and Thr-354. Examination of a model structure of NerA (produced using OYE coordinates [8], the published amino acid sequence alignment with NerA [20], and ExPASy Swiss model [10, 18, 19]) indicated that these three sites are all at exposed sites on the protein surface, suggesting that the mutant protein was subject to limited proteolysis but that the fragments produced remained associated. Attempts to produce activity toward GTN by reintroducing FMN into the purified mutant protein (as described for the wild-type protein) were unsuccessful.
FIG. 6.

SDS-PAGE gel of the purification steps of NerA N181A showing whole lysate of cells prior to protein induction (lane a) whole lysate of cells 4 h after protein induction (lane b), and cell extract of induced cells (lane c). The dominant peak was collected during elution from affinity chromatography (lane d), ion-exchange chromatography (lane e), hydrophobic-interaction chromatography (lane f), and size exclusion chromatography (lane g). M indicates the molecular mass marker.
The Y183F mutant of NerA exhibited similar properties to those of the wild-type enzyme in terms of its protein purification profiles, with the exception that a single protein peak was eluted from the hydrophobic-interaction chromatography column, and this protein was colorless and inactive. However, this apoprotein was easily reflavinated by incubation with FMN overnight at 4°C. Kinetic data obtained in a similar manner to that described for the wild-type enzyme could not be fitted to equation 5, and double-reciprocal plots of 1/v versus 1/[GTN] were not parallel, showing that a ping-pong mechanism was no longer operative. In fact, the data generated primary plots (1/v versus either 1/[GTN] or 1/[NADH]) and secondary plots (intercepts and slopes from primary plots versus either 1/[NADH] or 1/[GTN]) that were similar to those predicted for uncompetitive substrate inhibition in an ordered sequential mechanism (7). From these secondary plots, KmNADH, KmGTN, KsNADH, and kcat were estimated to be 15 μM, 368 μM, 29 μM, and 9.4 s−1, respectively.
DISCUSSION
Hitherto, the conclusion of the involvement of FMN in the reduction of GTN by NerA to release nitrite has been based on similarity of the predicted protein sequence with those of other enzymes known to include FMN as a prosthetic group (20). In the present work, we demonstrated by thin-layer chromatography in different solvent systems that denaturation of the native active enzyme liberated a yellow ligand that was chromatographically distinct from FAD but very similar to FMN. In addition, the NerA holoenzyme exhibited a UV-visible absorbance spectrum that is characteristic of flavinated proteins, whereas the NerA apoenzyme exhibited no such absorbances. In contrast with previous studies of related enzymes, including Onr from E. cloacae PB2 (2), XenA from P. putida (4), and XenB from P. fluorescens (4), the fortuitous production in our expression system of a mixture of NerA apo- and holoenzymes separable by hydrophobic-interaction chromatography or nondenaturing gel electrophoresis (Fig. 2, peaks 1 and 2) allowed us to demonstrate unequivocally for the first time that the addition of FMN converted the inactive apoenzyme to the fully active holoenzyme (Fig. 2 and 3), with a dissociation constant on the order of 10−7 M. The FMN-treated apoenzyme had activity, electrophoretic, and spectral properties identical to those of the native holoenzyme. In addition, the spectral changes associated with the addition of the reducing agents dithionite or NADH, and with the reoxidation by admission of oxygen or GTN, were all entirely consistent with the involvement of FMN in the redox cycling in this enzyme.
Kinetic studies indicated that NerA operates via a ping-pong mechanism, as do Onr (9) and OYE (15). Table 2 shows kinetic constants for NerA and several of its homologs. Values of kcat for enzymes (NerA and Onr) from strains isolated for their ability to use nitrate esters as sole sources of nitrogen were a little higher than that of the archetypal OYE, but whether this is sufficient to indicate a possible adaptation is not clear.
The enzymes may be divided on a kinetic basis into two subgroups (Table 2); members of the first subgroup I (NerA, XenB, and OYE) have relatively high values of the ratio KmGTN/KmNAD(P)H (>20), whereas the second subgroup II (Onr and XenA) have relatively low values (<2). Scrutiny of the sequence alignments made by Blehert et al. (3) for positions at which the amino acid is common within a subgroup but different between subgroups shows four such positions (Table 2). Of these, site 1 has Tyr and Phe in subgroups I and II, respectively, but this site is on the exterior surface and is some distance from the active site in OYE (and so presumably in the others) and is therefore unlikely to be the cause of the intersubgroup differences in Km. At site 2, amino acids in subgroups I and II (Ile and Leu, respectively) are functionally conserved and are oriented away from the active site; therefore, they are also unlikely to account for the kinetic differences.
Site 3 contains Asn and His in subgroups I and II, respectively, and it is immediately adjacent to the substrate-binding site in OYE (8) and the corresponding putative site in the model for NerA. This finding, together with the correlation of the high Km ratios with Asn and low ratios with His (Table 2), suggests that these residues contribute to the observed differences in ratios of Km values among the five enzymes. The importance of this Asn-181 in NerA was confirmed in the present work by the N181A mutation, which resulted in loss of both FMN binding and activity toward GTN. Two-dimensional gel electrophoresis and peptide mass fingerprinting showed that the mutant protein had succumbed to partial proteolysis, although the fragments appeared to remain associated because size exclusion chromatography failed to separate the peptides formed despite large differences in their size. Brown et al. (6) also mutated the corresponding Asn-194 in OYE to a histidine but were unable to produce the mutant protein in amounts sufficient for purification. A susceptibility to proteolysis similar to that observed in our work with the NerA mutant may now account for this. However, Brown et al. were able to purify a double-OYE mutant protein (H191N/N194H), the structure of which suggested that, as in NerA, the N194 was important for FMN binding.
In OYE, Asn-194 acts alongside His-191 in substrate-binding and FMN-binding roles (6, 11). The conversion of the corresponding NerA His-178 to an alanine residue completely abolished NerA activity toward GTN. This abolition was not due to the loss of the FMN prosthetic group since the mutant protein still appeared yellow. In OYE, the corresponding H191N mutation similarly caused a very marked decrease in the rate of reoxidation of reduced enzyme by GTN. However, there was sufficient activity to show that the mutation decreased the binding affinity of OYE for substituted phenols but increased its binding affinity for GTN (6, 15). This increase in GTN-binding affinity was attributed to the creation of more room in the active site, making it possible for other amino acid residues to interact with GTN but also altering the position of GTN within the active site to one that was less favorable for the reaction to occur, leading to a decrease in the reaction rate (15). The OYE His-191, which is highly conserved in this family of proteins, thus appears to be essential for orienting GTN (15) and other substrates (6) in the active site so that hydride transfer from FMN can take place. A similar role in NerA would account for its indispensability.
Site 4 contains Asn or His in subgroup I enzymes but Phe in subgroup II (Table 2). In OYE, this Asn-251 lies some distance above the si face of FMN but lies close to Tyr-196, which is itself situated above the plane of a bound substrate molecule (8, 27) and is involved in GTN binding (by hydrogen bonding to a nitrate group) rather than in the catalytic process (15). This role in substrate binding rather than oxidation-reduction is supported by our kinetic data for the NerA Y183F mutation, which produced changes in Km values, but not kcat. Kohli and Massey (11) noted that in the oxidation of OYE by 2-cyclohexenone, the amidic side chain of Asn-251 may promote proton transfer from Tyr-196 in OYE by increasing the acidity of the tyrosine phenolic OH group. The subgroup II enzymes have Phe in place of Asn or His at this position (Table 2), which is unable to promote, and might actually suppress, proton transfer from the neighboring tyrosine. Thus, the presence of Phe in subgroup II in place of Asn or His in subgroup I may change the way that Tyr interacts with one or other or both substrates, and this may be a contributing factor in lowering the KmGTN/KmNAD(P)H ratio (Table 2). The fact that the Y183F mutation in NerA also reduced the KmGTN/KmNAD(P)H ratio (from 88 to about 25) supports this argument because this mutation completely prevents proton release by removing the phenolic OH altogether. A further effect of this Y183F mutation in NerA was to change the form of the steady-state kinetics from ping pong to one which fitted equations for a compulsory-order mechanism exhibiting substrate inhibition. Given this important but unexplained change and the highly conserved nature of the Tyr residue further investigation of the role of adjacent amino acids in moderating the action of Tyr and in determining substrate specificity of this group of enzymes would now seem appropriate.
Acknowledgments
We gratefully acknowledge financial support from the Biotechnology and Biological Sciences Research Council (United Kingdom) and the Erasmus exchange program.
We thank Len Packman, Cambridge University, for conducting the peptide mass fingerprinting.
REFERENCES
- 1.Accashian, J. V., R. T. Vinopal, B. J. Kim, and B. F. Smets. 1998. Aerobic growth on nitroglycerin as the sole carbon, nitrogen, and energy source by a mixed bacterial culture. Appl. Environ. Microbiol. 64:3300-3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Binks, P. R., C. E. French, S. Nicklin, and N. C. Bruce. 1996. Degradation of pentaerythritol tetranitrate by Enterobacter cloacae PB2. Appl. Environ. Microbiol. 62:1214-1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blehert, D. S., B. G. Fox, and G. H. Chambliss. 1999. Cloning and sequence analysis of two Pseudomonas flavoprotein xenobiotic reductases. J. Bacteriol. 181:6254-6263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blehert, D. S., K. L. Knoke, B. G. Fox, and G. H. Chambliss. 1997. Regioselectivity of nitroglycerin denitration by flavoprotein nitroester reductases purified from two Pseudomonas species. J. Bacteriol. 179:6912-6920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 6.Brown, B. J., Z. Deng, P. A. Karplus, and V. Massey. 1998. On the active site of old yellow enzyme. Role of histidine 191 and asparagine 194. J. Biol. Chem. 273:32753-32762. [DOI] [PubMed] [Google Scholar]
- 7.Cleland, W. W. 1970. Steady state kinetics, p. 1-63. In P. D. Boyer (ed.), The enzymes—kinetics and mechanism, 3rd ed., vol. II. Academic Press, New York, N.Y. [Google Scholar]
- 8.Fox, K. M., and P. A. Karplus. 1994. Old yellow enzyme at 2Å resolution—overall structure, ligand-binding, and comparison with related flavoproteins. Structure 2:1089-1105. [PubMed] [Google Scholar]
- 9.French, C. E., S. Nicklin, and N. C. Bruce. 1996. Sequence and properties of pentaerythritol tetranitrate reductase from Enterobacter cloacae PB2. J. Bacteriol. 178:6623-6627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guex, N., and M. C. Peitsch. 1997. SWISS-MODEL and the Swiss-pdbViewer: an environment for comparative protein modelling. Electrophoresis 18:2714-2723. [DOI] [PubMed] [Google Scholar]
- 11.Kohli, R. M., and V. Massey. 1998. The oxidative half-reaction of old yellow enzyme. The role of tyrosine 196. J. Biol. Chem. 273:32763-32770. [DOI] [PubMed] [Google Scholar]
- 12.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 13.Litchfield, M. H. 1971. Aspects of nitrate ester metabolism. J. Pharm. Sci. 60:1599-1607. [DOI] [PubMed] [Google Scholar]
- 14.Marshall, S. J., and G. F. White. 2001. Complete denitration of nitroglycerin by bacteria isolated from a washwater soakaway. Appl. Environ. Microbiol. 67:2622-2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meah, Y., B. J. Brown, S. Chakraborty, and V. Massey. 2001. Old yellow enzyme: reduction of nitrate esters, glycerin trinitrate, and propylene 1,2-dinitrate. Proc. Natl. Acad. Sci. USA 98:8560-8565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Meng, M., W. Q. Sun, L. A. Geelhaar, G. Kumar, A. R. Patel, G. F. Payne, M. K. Speedie, and J. R. Stacy. 1995. Denitration of glycerol trinitrate by resting cells and cell extracts of Bacillus thuringiensis/cereus and Enterobacter agglomerans. Appl. Environ. Microbiol. 61:2548-2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Murrell, W. 1879. Nitroglycerine as a remedy for angina pectoris. Lancet i:80-81, 151-152, 225-227. [Google Scholar]
- 18.Peitsch, M. C. 1995. Protein modeling by e-mail. Bio/Technology 13:658-660. [Google Scholar]
- 19.Schwede, T., J. Kopp, N. Guex, and M. C. Peitsch. 2003. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31:3381-3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Snape, J. R., N. A. Walkley, A. P. Morby, S. Nicklin, and G. F. White. 1997. Purification, properties, and sequence of glycerol trinitrate reductase from Agrobacterium radiobacter. J. Bacteriol. 179:7796-7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tipton, K. F. 1974. Enzyme kinetics, p. 227-251. In A. T. Bull, J. R. Lagnado, J. O. Thomas, and K. F. Tipton (ed.), Companion to biochemistry—selected topics for further study. Longman, London, United Kingdom.
- 22.Urbanski, T. 1965. Chemistry and technology of explosives. PWN—Polish Scientific Publishers, Warsaw, Poland.
- 23.Wendt, T. M., J. H. Cornell, and A. M. Kaplan. 1978. Microbial degradation of glycerol nitrates. Appl. Environ. Microbiol. 36:693-699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.White, G. F., and J. R. Snape. 1996. Bacterial biodegradation of nitrate esters., p. 145-156. In A. V. Kaffka (ed.), Sea-dumped chemical weapons: aspects, problems and solutions. Kluwer Academic Press, Dordrecht, The Netherlands.
- 25.White, G. F., J. R. Snape, and S. Nicklin. 1996. Bacterial biodegradation of glycerol trinitrate. Int. Biodeterior. Biodegrad. 38:77-82. [Google Scholar]
- 26.White, G. F., J. S. Snape, and S. Nicklin. 1996. Biodegradation of glycerol trinitrate and pentaerythritol tetranitrate by Agrobacterium radiobacter. Appl. Environ. Microbiol. 62:637-642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Williams, R. E., and N. C. Bruce. 2002. “New uses for an old enzyme”—the old yellow enzyme family of flavoenzymes. Microbiology 148:1607-1614. [DOI] [PubMed] [Google Scholar]
- 28.Williams, R. E., and N. C. Bruce. 2000. The role of nitrate ester reductase enzymes in the biodegradation of explosives, p. 161-184. In J. C. Spain, J. B. Hughes, and H.-J. Knackmuss (ed.), Biodegradation of nitroaromatic compounds and explosives. CRC Press LLC, Boca Raton, Fla.
- 29.Zhang, Y. Z., S. T. Sundaram, A. Sharma, and B. W. Brodman. 1997. Biodegradation of glyceryl trinitrate by Penicillium corylophilum Dierckx. Appl. Environ. Microbiol. 63:1712-1714. [DOI] [PMC free article] [PubMed] [Google Scholar]