

Abstract
The chemolithoautotroph NT-26 oxidizes arsenite to arsenate by using a periplasmic arsenite oxidase. Purification and preliminary characterization of the enzyme revealed that it (i) contains two heterologous subunits, AroA (98 kDa) and AroB (14 kDa); (ii) has a native molecular mass of 219 kDa, suggesting an α2β2 configuration; and (iii) contains two molybdenum and 9 or 10 iron atoms per α2β2 unit. The genes that encode the enzyme have been cloned and sequenced. Sequence analyses revealed similarities to the arsenite oxidase of Alcaligenes faecalis, the putative arsenite oxidase of the beta-proteobacterium ULPAs1, and putative proteins of Aeropyrum pernix, Sulfolobus tokodaii, and Chloroflexus aurantiacus. Interestingly, the AroA subunit was found to be similar to the molybdenum-containing subunits of enzymes in the dimethyl sulfoxide reductase family, whereas the AroB subunit was found to be similar to the Rieske iron-sulfur proteins of cytochrome bc1 and b6f complexes. The NT-26 arsenite oxidase is probably exported to the periplasm via the Tat secretory pathway, with the AroB leader sequence used for export. Confirmation that NT-26 obtains energy from the oxidation of arsenite was obtained, as an aroA mutant was unable to grow chemolithoautotrophically with arsenite. This mutant could grow heterotrophically in the presence of arsenite; however, the arsenite was not oxidized to arsenate.
Arsenic is ubiquitous in the environment and is most commonly found in an insoluble form associated with rocks and minerals (11). In soluble forms, arsenic occurs as trivalent arsenite [As(III)] and pentavalent arsenate [As(V)]. Arsenate, a phosphate analogue, can enter cells via the phosphate transport system and is toxic because it can interfere with normal phosphorylation processes by replacing phosphate. It was recently demonstrated that arsenite enters cells at a neutral pH by aqua-glyceroporins (glycerol transport proteins) in bacteria, yeasts, and mammals (23, 39), and its toxicity lies in its ability to bind sulfhydryl groups of cysteine residues in proteins, thereby inactivating them.
Arsenite is considered to be more toxic than arsenate and can be oxidized to arsenate chemically or microbially (12, 16). The arsenite-oxidizing bacteria so far isolated either can gain energy from arsenite oxidation (25, 32, 33) or have been proposed to do so as part of a detoxification process (14, 15, 27, 30, 36). Chemolithoautotrophic arsenite oxidation, for which oxygen is used as the terminal electron acceptor, arsenite is the electron donor, and carbon dioxide is the carbon source, has to date only been reported for organisms isolated from gold mines (32, 33). Aerobic growth with arsenite as the electron donor is exergonic, generating a substantial amount of free energy (32).
Arsenite oxidation by the chemolithoautotrophic arsenite oxidizer NT-26, a member of the α-Proteobacteria, has been studied in detail (32). Preliminary biochemical studies showed the NT-26 arsenite oxidase (Aro) to be periplasmic and its synthesis to be induced by arsenite (32). The heterotrophic arsenite oxidizer Alcaligenes faecalis, a member of the β-Proteobacteria (33), contains a membrane-bound arsenite oxidase that has been purified and characterized (3, 9, 21) and was previously shown to be a member of the dimethyl sulfoxide (DMSO) reductase family of molybdoenzymes (17, 20). The enzyme consists of two heterologous subunits (α1β1) and has a native molecular mass of 100 kDa (3, 13). The large catalytic subunit (825 residues) contains a molybdenum site, consisting of a molybdenum atom coordinated to two pterin cofactors, and a [3Fe-4S] cluster (13). The small subunit (133 residues) contains a Rieske-type [2Fe-2S] cluster (13).
Recently, the arsenite oxidase genes of the heterotrophic arsenite oxidizer ULPAs1, a member of the β-Proteobacteria, were cloned and sequenced (24, 36). Two genes, designated aoxA and aoxB, involved in arsenite oxidation were identified by transposon mutagenesis. The putative products encoded by aoxA (AoxA; 173 residues) and aoxB (AoxB; 826 residues) showed a high degree of sequence identity to the β (65%) and α (72%) subunits, respectively, of the A. faecalis arsenite oxidase. The ULPAs1 enzyme has not been studied except to say that it was found to be associated with spheroplasts (24); presumably, this means that it is located in the inner membrane.
This report describes the study of the NT-26 arsenite oxidase. We describe the purification and preliminary characterization of the enzyme together with the cloning, sequencing, and molecular analysis of the genes. We also present data supporting the role for this enzyme in energy generation and some preliminary data on the mechanism used by NT-26 to cope with the toxicity of arsenate.
MATERIALS AND METHODS
Growth conditions.
NT-26 was grown aerobically at 28°C in a minimal salts medium (MSM) containing arsenite (5 mM) and yeast extract (0.04%), as described previously (32). For purification of the arsenite oxidase (Aro), NT-26 was grown in 5-liter batch cultures. Cultures were harvested during late exponential phase (after 14 h of growth) at a final optical density (A600) of 0.14 to 0.15. Typical cell yields were 2 g of cells (wet weight) per 5 liters.
For DNA isolation experiments, NT-26 was grown in MSM containing yeast extract (0.08%) until stationary phase, at a final optical density (A600) of 0.22.
For testing of its resistance to arsenate and arsenite, NT-26 was grown in MSM containing yeast extract (0.08%) with and without various concentrations of arsenate (5, 10, 20, 50, and 100 mM) and arsenite (5, 10, and 20 mM). Growth was observed by measuring the optical densities (A600) after 48 h of incubation.
Purification of the arsenite oxidase.
Enzyme purification was performed at room temperature unless otherwise specified. Cells were chilled on ice (0°C), harvested by centrifugation for 20 min at 21,000 × g (4°C), and resuspended in 10 mM Tris-HCl (pH 8). The periplasmic fraction was prepared as described previously (32). The proteins in the periplasm (30.6 mg of protein with 2.9 U of total activity) were precipitated with ammonium sulfate (50 to 80% saturation). The precipitated material, which contained NT-26 Aro, was collected by centrifugation for 30 min at 38,700 × g (4°C) and was resuspended in 2 ml of 50 mM morpholineethanesulfonic acid (MES)-1 M NH4SO4 (pH 5.5) (7.4 mg of protein with 1.2 U of total activity). The sample was centrifuged at 38,700 × g to remove insoluble material, filtered through a 0.22-μm-pore-size filter, and loaded onto a phenyl Sepharose high-performance hydrophobic interaction column (HiLoad 16/10; Amersham Pharmacia Biotech) that had been equilibrated with 50 mM MES-1 M NH4SO4 (pH 5.5). During the chromatographic steps, the protein concentration was determined by measuring the absorbance at 280 nm. After washing of the column with 2 volumes of 50 mM MES-1 M NH4SO4 (pH 5.5), Aro was eluted from the column with an NH4SO4 gradient starting at 1 M and decreasing to 0 M. Aro eluted at an NH4SO4 concentration of 0.10 to 0.12 M. The fractions containing Aro were pooled and concentrated by centrifugation in a Centriprep-30 concentrator (Amicon) (0.41 mg of protein with 0.7 U of total activity). The sample was then loaded onto a Superdex 200 gel filtration column (Amersham Pharmacia Biotech) which had been equilibrated with 50 mM MES (pH 5.5). Aro was eluted with 50 mM MES (pH 5.5), and the fractions were pooled and concentrated as described above (0.17 mg of protein with 0.4 U of total activity). Freshly purified Aro was used for all analyses.
Total cell extracts were prepared as described previously (32) (59.4 mg of protein with 2.3 U of total activity).
Bradford reagent (7) was used to determined protein concentrations. Bovine serum albumin served as the standard.
Enzyme assay.
Aro activity was measured, as described previously, in the determined optimum buffer, 50 mM MES (pH 5.5) (32). The concentration of arsenite included in the assay was 2.5 mM (except for experiments to determine the Km and Vmax, in which the concentration of arsenite ranged from 0.02 to 2.5 mM).
Determination of the apparent molecular mass.
The native molecular mass of Aro was determined by gel filtration chromatography through a Superdex 200 (Amersham Pharmacia Biotech) column as described previously (18).
Protein gel electrophoresis, protein transfer, and cofactor analyses.
Protein gel electrophoresis and the transfer of the AroA and AroB subunits to polyvinylidene difluoride membranes were done as described previously (10, 18). For determination of the cofactor composition, freshly purified Aro (0.068 to 0.069 mg) was hydrolyzed (with nitric acid), and metal (i.e., molybdenum, iron, tungsten, selenium, cobalt, copper, and zinc) detection was performed by the VIEPS School of Geosciences, Monash University (Victoria, Australia). Cofactor determination was performed with fresh Aro on three separate occasions.
Cloning and sequencing of the aro genes.
A degenerate oligonucleotide was designed from the AroA N-terminal sequence. The primer was called AroA1 and had the sequence 5′-GGITATCATGCITATACITGGCCIAT-3′. The [γ-32P]ATP-labeled AroA1 oligonucleotide was used to screen two NT-26 genomic libraries, which were constructed by partially digesting the chromosomal DNA with BamHI and HindIII (1). Hybridization (with 5× SSPE [1× SSPE is 0.18 M NaCl, 10 mM NaH2PO4, and 1 mM EDTA, pH 7.7], 5× Denhardt's solution, 0.5% sodium dodecyl sulfate [SDS] [pH 7.2], and salmon sperm DNA) was performed at 37°C, and the membranes were washed twice for 15 min with each of the following: (i) 2× SSPE-0.1% SDS (room temperature), (ii) 1× SSPE-0.1% SDS (37°C), and (iii) 0.1× SSPE-0.1% SDS (37°C).
The large aro clones were subcloned to facilitate the sequencing of both strands. Primer walking was also used to complete the sequencing of the aro genes. Sequencing was performed at the Australian Genome Research Facility (Brisbane, Queensland, Australia) with an ABI Prism 210 capillary DNA sequencer and genetic analyzer. DNA sequences were assembled by use of DNASIS (V2-5) for Windows (Hitachi Software Genetic Systems). Database searches were performed by use of BLASTP (2) at the Australian National Genomic Information Service. Multiple sequence alignments were performed by use of DNASIS (V2-5) for Windows (Hitachi Software Genetic Systems).
Mutagenesis.
A 659-bp portion of the aroA gene (BamHI-SacI) was cloned into the suicide plasmid pJP5603 (Kmr) (26) for the purpose of disrupting it by insertional mutagenesis. This plasmid, pAroAΔ1, was transformed into Escherichia coli strain S17-1 λpir and transferred into a spontaneous rifampin-resistant NT-26 mutant by conjugation, and the mixture was plated onto minimal salts agar (1.5%) plates containing 0.08% yeast extract, rifampin (100 μg/ml), and kanamycin (5 μg/ml). Transconjugants were purified and tested for the ability to oxidize 5 mM arsenite. One mutant was chosen for further study. Insertion of the plasmid into aroA was confirmed by Southern hybridization at 55°C with wash solutions that were identical to those described for the cloning of the aro genes (see above).
The mutant was tested for the ability to oxidize arsenite chemolithoautotrophically and heterotrophically in MSM containing 5 mM arsenite with and without 0.04% yeast extract. For testing of the specific activity of Aro, the mutant was grown in 2 liters of batch culture in MSM containing 5 mM arsenite and 0.04% yeast extract. Total cell extracts were prepared as described previously (32).
Detection of R773 ars genes in NT-26.
Plasmid pWSU1 (31) containing the R773 arsenic resistance genes (arsRDABC) was used as a template for PCR amplification of the arsA, arsB, and arsC genes as described previously (19). Southern hybridization against NT-26 chromosomal and plasmid DNA was carried out by using [α-32P]dCTP-labeled probes at temperatures ranging from 40 to 60°C; digested pWSU1 served as the positive control. The wash conditions used were the same as those described for the cloning of the aro genes (see above).
Nucleotide sequence accession number.
The DNA sequences described in this report have been submitted to the GenBank database under accession number AY345225.
RESULTS
Purification and preliminary characterization of NT-26 arsenite oxidase.
NT-26 Aro was purified from the periplasmic fraction, followed by ammonium sulfate precipitation, hydrophobic interaction, and gel filtration chromatography, which led to a 60-fold enrichment of the enzyme. After the final purification step, Aro was found to be >99% pure (Fig. 1) and to consist of two heterologous subunits, of 98 kDa (AroA) and 14 kDa (AroB). The native molecular mass of the enzyme, based on gel filtration chromatography, was found to be 219 kDa (data not shown), suggesting that the subunits are present in a 2:2 stoichiometry and therefore an α2β2 configuration.
FIG. 1.
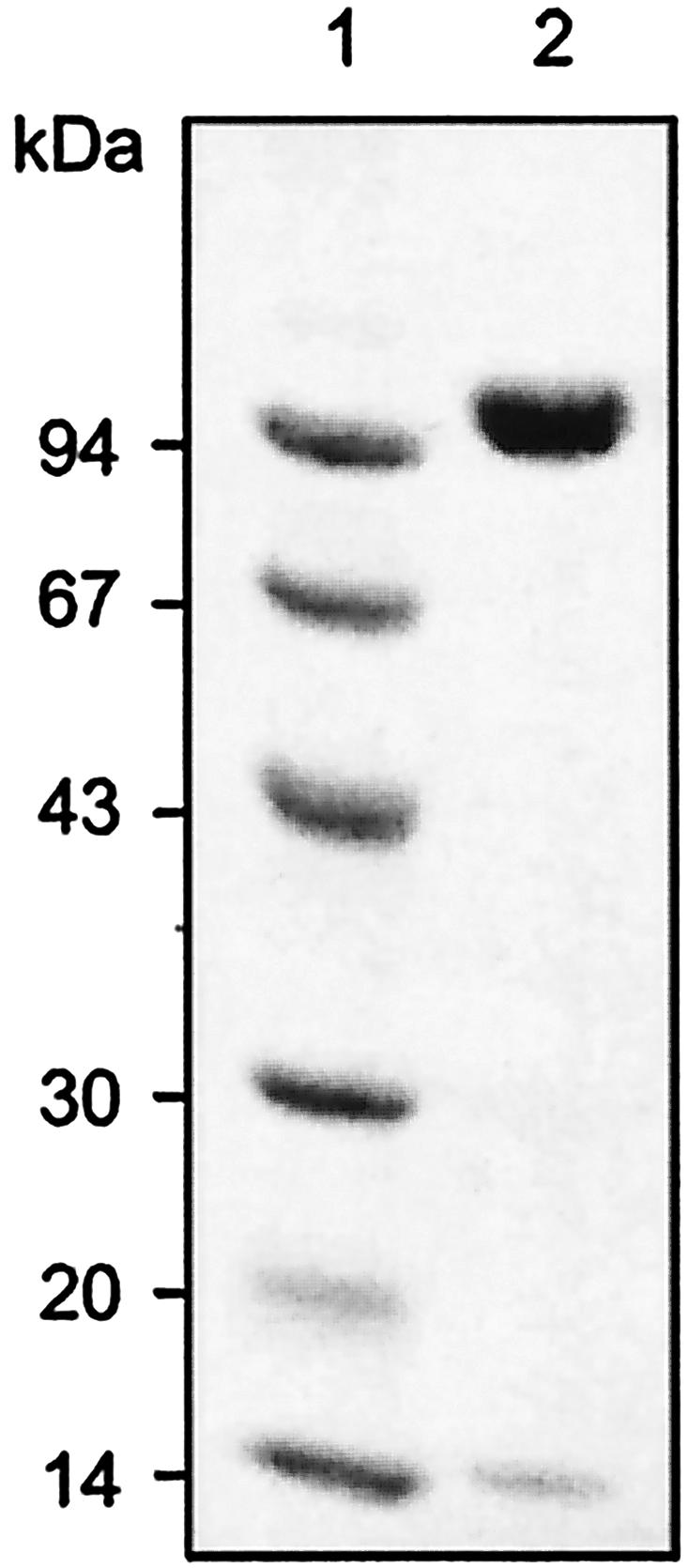
SDS-polyacrylamide (12.5%) gel of purified NT-26 arsenite oxidase. Proteins were stained with Coomassie blue R350. Lane 1, molecular mass standards phosphorylase b (94 kDa), albumin (67 kDa), ovalbumin (43 kDa), carbonic anhydrase (30 kDa), trypsin inhibitor (20 kDa), and α-lactalbumin (14 kDa); lane 2, purified arsenite oxidase.
With DCIP (2,4-dichlorophenolindophenol) as the artificial electron acceptor, NT-26 Aro exhibited a Vmax of 2.4 μmol of arsenite oxidized · min−1 · mg of protein−1, which corresponds to an enzyme turnover of 8.6 s−1 (based on the native molecular mass of 219 kDa). The Km for arsenite was found to be 61 μM.
The purified Aro protein was analyzed for the presence of cofactors. Based on the estimated molecular mass of 219 kDa, analysis by inductively coupled plasma mass spectroscopy revealed the presence of 2.02 ± 0.08 mol of Mo and 9.2 ± 0.6 mol of Fe per mol of enzyme (α2β2). These results suggest that Aro contains molybdenum as a cofactor and iron-sulfur clusters as prosthetic groups.
The N-terminal sequences of both Aro subunits, AroA (40 residues) and AroB (50 residues), were determined: the AroA sequence is AFKRHIDRLPIIPADAKKHNVTCHGCIVGCGYHAYTWPIN and the AroB sequence is ATAAAGVEYPANRLANISELTLNEPLDVAYPDEDAAGVLLKLGTRVEGGV. The N-terminal sequence of AroA contains the conserved cysteine cluster Cys-X2-Cys-X3-Cys that is found in the molybdenum-containing subunits of molybdoenzymes (17).
Cloning and characterization of the aro genes.
The locations of the aro genes within the NT-26 clones were mapped, and the genes were subcloned and sequenced. Two open reading frames, of 528 and 2,538 bp (Fig. 2), were identified as being aroB (175 amino acids, 18,354 Da) and aroA (855 amino acids, 93,267 Da), respectively, as the deduced amino acid sequences contained regions of identity at the N terminus to the N-terminal sequences of the individual subunits (see above). The genes are transcribed in the same direction, and an intergenic region of 12 bp occurs between them.
FIG. 2.

Diagrammatic representation of aro operon, consisting of aroB and aroA. The direction of transcription is represented by arrows, and the 12-bp intergenic region between aroB and aroA is shaded. Restriction sites are also shown.
The N-terminal sequence of AroA corresponded to the deduced sequence for amino acid positions 2 to 40. Only the N-terminal methionine was absent from the mature protein, presumably due to posttranslational cleavage. The N-terminal sequence of AroB, however, corresponded to the deduced amino acid sequence for positions 41 to 91. This suggests that AroB was probably synthesized in a precursor form with a 40-amino-acid N-terminal signal sequence (Fig. 3). This signal displays certain characteristics associated with the Tat (twin-arginine translocation) signal sequences (4, 5). The Tat pathway functions to transport folded proteins, predominantly proteins containing redox cofactors involved in respiratory and photosynthetic electron transport chains across the cytoplasmic membrane. These sequences can be up to 58 amino acids long and contain the following three conserved regions: (i) an N-terminal positively charged region (n-region) that harbors the conserved RRXFL motif (the AroB motif is RRQFL), (ii) a hydrophobic α-helical region (a more frequent occurrence of glycine residues and significantly lower abundance of lysine residues are present in Tat signal sequences), and (iii) a C-domain, usually an alanine, which is the cleavage site of the signal peptidase (Fig. 3). The presence of a Tat signal sequence in AroB and the absence of one in AroA suggest that both proteins are exported to the periplasm as a folded complex, using the AroB signal sequence for export. The only other examples of a small subunit directing the export of itself and a large subunit are the NiFe and Fe hydrogenases (38).
FIG. 3.

Sequence alignment of the NT-26 AroB N terminus with those of the ULPAs1 putative protein AoxA (accession no. AAN05580), the A. faecalis (A.f) arsenite oxidase β-subunit (accession no. nrp 1G8K_B), and putative proteins of C. aurantiacus (C.a), Aeropyrum pernix (A.p), and S. tokodaii (S.t); accession numbers for the last three sequences are indicated in the figure. The gaps upstream of the A. faecalis β-subunit are due to the absence of the leader sequence; the sequence is based on the processed protein (13). The conserved twin-arginine motifs are indicated by asterisks, and the cleavage site for AroB is indicated by an arrow.
The AroA sequence shows the highest identity with the molybdenum-containing α-subunit of the A. faecalis arsenite oxidase (49.2%) and with AoxB of ULPAs1 (48.4%), involved in arsenite oxidation. Interestingly, putative proteins of unknown function in Chloroflexus aurantiacus and two members of the Archaea, Aeropyrum pernix and Sulfolobus tokodaii, also displayed high sequence identities to AroA (49.2, 33.8, and 27.3%, respectively). The conserved motif, Cys-X2-Cys-X3-Cys-X70-Ser, that binds the [3Fe-4S] cluster in the α-subunit of the A. faecalis arsenite oxidase (13) is present in AroA (X23-Cys-X2-Cys-X3-Cys-X69-Ser) and the other sequences. Moreover, the four amino acids (His, Glu, Arg, and His) that are predicted to be involved in binding arsenite also appear to be conserved in AroA (His199, Glu207, Arg447, and His451) and the other sequences, except in the case of S. tokodaii, in which two amino acids vary (i.e., Glu and Arg). Apart from these proteins, AroA showed similarities to the molybdenum-containing subunits of the formate dehydrogenases and assimilatory nitrate reductases.
AroB showed the highest sequence identities to a putative protein of Aeropyrum pernix (55.9%), to the respective arsenite oxidase subunits of ULPAs1 (AoxA; 52.4%) and A. faecalis (β; 44.8%) (note that the percent identity calculated for the A. faecalis β-subunit lacks the leader sequence, as it is based on the mature protein sequence obtained from the crystal structure of the enzyme), and to putative proteins from C. aurantiacus (48%) and S. tokodaii (42%). Other similarities were with Rieske-type iron-sulfur proteins that are part of cytochrome bc1 or b6f complexes. These proteins have a conserved cysteine motif, Cys-X-His-X15-Cys-X2-His, in common that binds the [2Fe-2S] cluster.
aroA mutant is unable to oxidize arsenite.
For a further demonstration that NT-26 Aro is involved in energy generation, the aroA gene was inactivated by a single crossover with the construct pAroAΔ1. The mutant failed to grow chemolithoautotrophically with arsenite as the electron donor but did grow in MSM containing 5 mM arsenite and yeast extract, although in the latter case arsenite was not oxidized to arsenate (data not shown). Aro activity in whole-cell extracts was not detectable (<0.0002 μmol of arsenite oxidized · min−1 · mg of protein−1), consistent with the disruption of the aroA gene. The fact that the NT-26 mutant still grows in the presence of arsenite further illustrates the fact that the oxidation of arsenite by this organism is a metabolic process.
NT-26 contains an R773 plasmid-borne arsC homologue.
Because NT-26 oxidizes arsenite to arsenate, one possible mechanism by which it copes with the toxic effects of arsenate was explored. Although the arsenite is oxidized to arsenate in the periplasm, it may be possible that small amounts of arsenate enter the cell. Arsenic resistance has been studied in detail and the genes that encode these arsenic resistance systems (ars) are widespread in both gram-negative and gram-positive bacteria (23, 34). The ars operon of the R factor R773 comprises five genes, arsRDABC (8). The arsC gene encodes a small (16 kDa), soluble arsenate reductase, ArsC, which reduces arsenate to arsenite in the cytoplasm. The products of the arsA and arsB genes act as an arsenite efflux pump, and the arsR and arsD gene products are involved in the regulation of the ars operon. Initial experiments demonstrated that the growth of NT-26 was not affected by arsenate concentrations up to 100 mM; this was also the case for the aroA mutant (A600 values of the wild type and the mutant with 100 mM arsenate were 0.110 and 0.115, respectively, and without arsenate were 0.100 and 0.105, respectively). No significant difference in the growth of NT-26 and the aroA mutant was observed with increasing concentrations of arsenite (i.e., growth of both was inhibited with arsenite concentrations of >10 mM; data not shown). We determined whether NT-26 contained homologues of the R773 arsA, arsB, and arsC genes. An arsC homologue was detected on the NT-26 21-kb plasmid by Southern hybridization (Fig. 4). No arsA and arsB homologues were detected on either the chromosome or plasmid of NT-26 (data not shown).
FIG. 4.

Southern hybridization of NT-26 plasmid DNA with an α-32P-labeled arsC probe. DNA markers of λ (HindIII) are indicated to the left. Lane 1, pWSU1 (contains arsRDABC genes of R773) digested with EcoRI; lanes 2 and 3, NT-26 plasmid DNA digested with EcoRI and BamHI, respectively.
DISCUSSION
This study is the first to describe arsenite oxidation by a chemolithoautotrophic arsenite oxidizer. The arsenite oxidase of the heterotrophic bacterium A. faecalis has been studied in depth (3, 9, 13, 21); however, little is known at the molecular genetic level. It remains unclear whether this organism gains energy from the oxidation of arsenite to arsenate. The electron transport chain is thought to consist of the arsenite oxidase (the molybdenum and Rieske Fe-S subunits) and a periplasmic electron acceptor (either azurin or cytochrome c) (3). Given this, it is difficult to understand that this reaction is not an exergonic one. Perhaps this will become clearer with the isolation of arsenite oxidase-deficient mutants. The arsenite oxidase genes of another heterotrophic arsenite-oxidizing bacterium, ULPAs1, have been cloned and sequenced, but the enzyme has not been studied (24). The arsenite oxidase genes of this organism were identified by transposon mutagenesis (24). This organism, like A. faecalis, has been suggested to oxidize arsenite as part of a detoxification process. Interestingly, insertion mutations in both arsenite oxidase genes did not prevent the organism from growing in the presence of arsenite, therefore suggesting that this putative arsenite oxidase is not solely involved in detoxification.
NT-26 Aro is another example of a molybdenum-containing enzyme of the DMSO reductase family. Both Aro and the arsenite oxidase of A. faecalis share similarities, but they also have some distinct differences (3). The most interesting observation is the different subunit configuration of the enzymes; NT-26 Aro has an α2β2 configuration and A. faecalis arsenite oxidase has an α1β1 configuration. Interestingly, another arsenite oxidase, from the heterotrophic bacterium NT-14 (like A. faecalis, it is a member of the β-Proteobacteria [33]), also contains two heterologous subunits, but they are in an α3β3 configuration (R. N. vanden Hoven and J. M. Santini, unpublished data). These enzymes all catalyze the same reaction yet differ in their subunit configurations. There is no obvious explanation for this phenomenon, but perhaps the characterization of more arsenite oxidases will provide us with some insight.
Based on the crystal structure of the A. faecalis arsenite oxidase, this enzyme is the first example of a new subgroup of the DMSO reductase family (13). In the other three subgroups of this family, the molybdenum atom is coordinated by the side chain of a serine, cysteine, or selenocysteine residue. In the A. faecalis arsenite oxidase, the corresponding residue is Ala199 and there is no covalent bond between the molybdenum atom and the protein (13). NT-26 Aro may represent the second member of this new subgroup of the DMSO reductase family, as it also contains the corresponding Ala in the AroA sequence (Ala203).
Purified Aro has been shown to contain 2.02 ± 0.08 mol of Mo and 9.2 ± 0.6 mol of Fe per mol of enzyme (α2β2). The iron analyses correspond well with the amino acid sequences, which suggest a [3Fe-4S] cluster in AroA and a [2Fe-2S] cluster in AroB. The deduced amino acid sequence of AroA suggests that this polypeptide binds the molybdopterin cofactor. AroA shares similarities with the large subunits of the arsenite oxidases and a number of putative proteins from two members of the Crenarchaeota, Aeropyrum pernix and S. tokodaii, and a photosynthetic bacterium, C. aurantiacus. The deduced AroB sequence predicts this polypeptide to bind the Rieske-like [2Fe-2S] cluster. This Rieske subunit, like that of A. faecalis, is unique among molybdoenzymes. The AroB sequence shares similarities with the small subunits of the arsenite oxidases and putative proteins from Aeropyrum pernix, S. tokodaii, and C. aurantiacus. Surprisingly, the highest sequence identity was to the putative protein of Aeropyrum pernix (55.9%) rather than to the arsenite oxidase subunits. AroB also shares similarities to the Rieske proteins of the cytochrome bc1 and b6f complexes. Rieske proteins all share a conserved cysteine cluster whose side chains are responsible for coordinating the Fe atoms of the Fe-S cluster. To date, all Rieske proteins have been demonstrated to be membrane bound (6). NT-26 Aro is periplasmic, and AroB may be the first example of a periplasmic Rieske protein. Like Rieske proteins, AroB contains a conserved twin-arginine motif at its N terminus (Fig. 3). The Rieske protein of the cytochrome b6f complex in chloroplasts is targeted in a Tat-dependent manner, with the uncleaved Tat signal peptide doubling as a signal anchor sequence (22). Both the A. faecalis and ULPAs1 arsenite oxidases have been found associated with spheroplasts, suggesting that the Rieske subunit is either attached to the membrane or perhaps strongly associated to the membrane via another protein. The latter explanation is probably the most plausible, as (i) the predicted amino acid sequences of both subunits do not contain putative transmembrane domains (data not shown) (the only hydrophobic region in AoxA occurs at the N terminus before the putative cleavage sequence), (ii) the signal sequence of the β-subunit of the A. faecalis arsenite oxidase is cleaved (13), and (iii) the A. faecalis arsenite oxidase is soluble (3).
The possibility that the homologues of AroA and AroB are involved in arsenite oxidation is supported by the fact that their genes, like those of NT-26, are directly adjacent to one another in the genomes and show a conserved order, with the homologue for aroB upstream of the homologue for aroA. It is plausible that these organisms can oxidize arsenite, given that they were isolated from hot springs, in the cases of S. tokodaii (35) and C. aurantiacus (28), or a coastal solfataric vent, in the case of Aeropyrum pernix (29), which can contain elevated concentrations of arsenic (37).
NT-26 gains energy from the oxidation of arsenite to arsenate as it grows chemolithoautotrophically (32). NT-26 is not alone in this ability, as five other members of the α-Proteobacteria (33) and one member of the γ-Proteobacteria (25) also oxidize arsenite chemolithoautotrophically; the latter organism, MLHE-1, uses nitrate as the terminal electron acceptor instead of oxygen. An NT-26 aroA mutant was unable to grow chemolithoautotrophically with arsenite as the electron donor, but it did grow heterotrophically in the presence of arsenite. Disruption of the aroA gene was confirmed, and no Aro activity was detected in total cell extracts. These results, together with the observation that NT-26 oxidizes arsenite chemolithoautotrophically, provide further evidence that this organism gains energy from arsenite oxidation.
Arsenite oxidation by NT-26 occurs in the periplasm. During this process, arsenate may enter the cell. We therefore decided to examine one possible mode by which NT-26 copes with arsenate. NT-26 was found to be resistant to high levels of arsenate. The growth of NT-26 and the aroA mutant was not affected by arsenate concentrations up to 100 mM. These concentrations are well above those reported for other organisms (34). NT-26 was also found to contain a homologue of the R773 arsenate reductase gene arsC on its 21-kb plasmid (Fig. 4). No arsA or arsB homologues were detected. The absence of arsA and arsB homologues, the proteins of which act as an arsenite efflux pump, is quite surprising. It is possible that arsA and arsB homologues were not detected in NT-26 because they are significantly different from those of R773 or that arsenite exits the cell by diffusion or through the glycerol transport system.
In summary, this paper describes the first arsenite oxidase from a chemolithoautotrophic bacterium, NT-26. This enzyme is involved in metabolism, not detoxification, as has been proposed for the arsenite oxidases from heterotrophs. Perhaps the study of the electron transport chains involved in arsenite oxidation will ultimately clarify whether the heterotrophic arsenite-oxidizing bacteria gain energy from arsenite oxidation.
Acknowledgments
This work was supported by an Australian Research Council (ARC) grant (DP0209802) to J.M.S. J.M.S. is a recipient of an ARC Australian Postdoctoral fellowship. R.N.V. is a recipient of an Australian Postgraduate Award.
We thank B. Rosen for plasmid pWSU1, J. Pemberton for plasmid pJP5603, D. Flood for technical assistance, J. F. Stolz for calculating percent identities, and I. Streimann. We also extend our thanks to two of the reviewers of this manuscript for their constructive comments.
REFERENCES
- 1.Allen, L. N., and R. S. Hanson. 1985. Construction of broad-host-range cosmid cloning vectors: identification of genes necessary for growth of Methylobacterium organophilum on methanol. J. Bacteriol. 161:955-962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403-410. [DOI] [PubMed] [Google Scholar]
- 3.Anderson, G. L., J. Williams, and R. Hille. 1992. The purification and characterisation of arsenite oxidase from Alcaligenes faecalis, a molybdenum-containing hydroxylase. J. Biol. Chem. 267:23674-23682. [PubMed] [Google Scholar]
- 4.Berks, B. C. 1996. A common export pathway for proteins binding complex redox cofactors? Mol. Microbiol. 22:393-404. [DOI] [PubMed] [Google Scholar]
- 5.Berks, B. C., F. Sargent, and T. Palmer. 2000. The Tat protein export pathway. Mol. Microbiol. 35:260-274. [DOI] [PubMed] [Google Scholar]
- 6.Berry, E. A., M. Guergova-Kuras, L. Huang, and A. R. Crofts. 2000. Structure and function of cytochrome bc complexes. Annu. Rev. Biochem. 69:1005-1075. [DOI] [PubMed] [Google Scholar]
- 7.Bradford, M. M. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilising the principle of protein-dye binding. Anal. Biochem. 72:248-254. [DOI] [PubMed] [Google Scholar]
- 8.Chen, C. M., T. K. Misra, S. Silver, and B. P. Rosen. 1986. Nucleotide sequence of the structural genes for an anion pump. The plasmid encoded arsenical resistance operon. J. Biol. Chem. 261:15030-15038. [PubMed] [Google Scholar]
- 9.Conrads, T., C. Hemann, G. N. George, I. J. Pickering, R. C. Prince, and R. Hille. 2002. The active site of arsenite oxidase from Alcaligenes faecalis. J. Am. Chem. Soc. 124:11276-11277. [DOI] [PubMed] [Google Scholar]
- 10.Edman, P., and G. Begg. 1967. A protein sequenator. Eur. J. Biochem. 1:80-91. [DOI] [PubMed] [Google Scholar]
- 11.Ehrlich, H. L (ed.). 1996. Geomicrobiology. Marcel Dekker Inc., New York, N.Y.
- 12.Ehrlich, H. L. 2002. Bacterial oxidation of As(III) compounds, p. 313-327. In W. T. Frankenberger (ed.), Environmental chemistry of arsenic. Marcel Dekker Inc., New York, N.Y.
- 13.Ellis, P. J., T. Conrads, R. Hille, and P. Kuhn. 2001. Crystal structure of the 100 kDa arsenite oxidase from Alcaligenes faecalis in two crystal forms at 1.64 Å and 2.03 Å. Structure 9:125-132. [DOI] [PubMed] [Google Scholar]
- 14.Gihring, T. M., and J. F. Banfield. 2001. Arsenite oxidation and arsenate respiration by a new Thermus isolate. FEMS Microbiol. Lett. 204:335-340. [DOI] [PubMed] [Google Scholar]
- 15.Gihring, T. M., G. K. Druschel, R. J. Mccleskey, R. J. Hamers, and J. F. Banfield. 2001. Rapid arsenite oxidation by Thermus aquaticus and Thermus thermophilus: field and laboratory investigations. Environ. Sci. Technol. 35:3857-3862. [DOI] [PubMed] [Google Scholar]
- 16.Inskeep, W. P., T. R. McDermott, and S. Fendorf. 2002. Arsenic(V)/(III) cycling in soils and natural waters: chemical and microbiological processes, p. 183-215. In W. T. Frankenberger (ed.), Environmental chemistry of arsenic. Marcel Dekker Inc., New York, N.Y.
- 17.Kisker, C., H. Schindelin, D. Baas, J. Rétey, R. U. Mechenstock, and P. M. H. Kroneck. 1999. A structural comparison of molybdenum-cofactor containing enzymes. FEMS Microbiol. Rev. 22:503-521. [DOI] [PubMed] [Google Scholar]
- 18.Krafft, K., and J. M. Macy. 1998. Purification and characterisation of the respiratory arsenate reductase of Chrysiogenes arsenatis. Eur. J. Biochem. 255:647-653. [DOI] [PubMed] [Google Scholar]
- 19.Macy, J. M., J. M. Santini, B. V. Pauling, A. H. O'Neill, and L. I. Sly. 2000. Two new arsenate/sulfate-reducing bacteria: mechanisms of arsenate reduction. Arch. Microbiol. 173:49-57. [DOI] [PubMed] [Google Scholar]
- 20.McEwan, A. G., J. P. Ridge, and C. A. McDevitt. 2002. The DMSO reductase family of microbial molybdenum enzymes: molecular properties and role in the dissimilatory reduction of toxic elements. Geomicrobiol. J. 9:3-21. [Google Scholar]
- 21.McNellis, L., and G. L. Anderson. 1998. Redox-state dependent chemical inactivation of arsenite oxidase. J. Inorg. Biochem. 69:253-257. [Google Scholar]
- 22.Molik, S., I. Karnauchov, C. Weidlich, R. G. Herrmann, and R. B. Klösgen. 2001. The Rieske Fe/S protein of the cytochrome b6/f complex in chloroplasts. J. Biol. Chem. 276:42761-42766. [DOI] [PubMed] [Google Scholar]
- 23.Mukhopadhyay, R., B. P. Rosen, L. Phung, and S. Silver. 2002. Microbial arsenic: from geocycles to genes and enzymes. FEMS Microbiol. Rev. 26:311-325. [DOI] [PubMed] [Google Scholar]
- 24.Muller, D., D. Lièvremont, D. Dancheva Simeonova, J.-C. Hubert, and M.-C. Lett. 2003. Arsenite oxidase aox genes from a metal-resistant β-proteobacterium. J. Bacteriol. 185:135-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oremland, R. S., S. E. Hoeft, J. M. Santini, N. Bano, R. A. Hollibaugh, and J. T. Hollibaugh. 2002. Anaerobic oxidation of arsenite in Mono Lake water and by a facultative, arsenite-oxidising chemoautotroph, strain MLHE-1. Appl. Environ. Microbiol. 68:4795-4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Penfold, R. J., and J. M. Pemberton. 1992. An improved suicide vector for construction of chromosomal insertion mutations in bacteria. Gene 118:145-156. [DOI] [PubMed] [Google Scholar]
- 27.Phillips, S. E., and M. L. Taylor. 1976. Oxidation of arsenite to arsenate by Alcaligenes faecalis. Appl. Environ. Microbiol. 32:392-399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pierson, B. K., and R. W. Castenholz. 1971. Bacteriochlorophylls in gliding filamentous prokaryotes from hot springs. Nature 233:25-27. [DOI] [PubMed] [Google Scholar]
- 29.Sako, Y., N. Nomura, A. Uchida, Y. Ishida, H. Morii, Y. Koga, T. Hoaki, and T. Maruyama. 1996. Aeropyrum pernix gen. nov., sp. nov., a novel aerobic hyperthermophilic Archaeon growing at temperatures up to 100°C. Int. J. Syst. Bacteriol. 46:1070-1077. [DOI] [PubMed] [Google Scholar]
- 30.Salmassi, T. M., K. Venkateswaren, M. Satomi, K. H. Nealson, D. K. Newman, and J. G. Hering. 2002. Oxidation of arsenite by Agrobacterium albertimagni, AOL15, sp. nov., isolated from Hot Creek, California. Geomicrobiol. J. 19:53-66. [Google Scholar]
- 31.San Francisco, M. J. D., C. L. Hope, J. B. Owolabi, L. S. Tisa, and B. P. Rosen. 1990. Identification of the metalloregulatory element of the plasmid-encoded arsenical resistance operon. Nucleic Acids Res. 18:619-624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Santini, J. M., L. I. Sly, R. D. Schnagl, and J. M. Macy. 2000. A new chemolithoautotrophic arsenite-oxidizing bacterium isolated from a gold mine: phylogenetic, physiological and preliminary biochemical studies. Appl. Environ. Microbiol. 66:92-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Santini, J. M., L. I. Sly, A. Wen, D. Comrie, P. De Wulf-Durand, and J. M. Macy. 2002. New arsenite-oxidizing bacteria isolated from Australian gold mining environments—phylogenetic relationships. Geomicrobiol. J. 19:67-76. [Google Scholar]
- 34.Silver, S., L. T. Phung, and B. P. Rosen. 2002. Arsenic metabolism: resistance, reduction and oxidation, p. 247-272. In W. T. Frankenberger (ed.), Environmental chemistry of arsenic. Marcel Dekker Inc., New York, N.Y.
- 35.Suzuki, T., T. Iwasaki, T. Uzawa, K. Hara, N. Nemoto, T. Kon, T. Ueki, A. Yamagishi, and T. Oshima. 2002. Sulfolobus tokodaii sp nov (f. Sulfolobus sp. strain 7), a new member of the genus Sulfolobus isolated from Beppu Hot Springs, Japan. Extremophiles 6:39-44. [DOI] [PubMed] [Google Scholar]
- 36.Weeger, W., D. Lièvremont, M. Perret, F. Lagarde, J.-C. Hubert, M. Leroy, and M.-C. Lett. 1999. Oxidation of arsenite to arsenate by a bacterium isolated from an aquatic environment. BioMetals 12:141-149. [DOI] [PubMed] [Google Scholar]
- 37.Wilkie, J. A., and J. G. Hering. 1998. Rapid oxidation of geothermal arsenic(III) in streamwaters of the Eastern Sierra Nevada. Environ. Sci. Technol. 32:657-662. [Google Scholar]
- 38.Wu, L.-F., A. Chanal, and A. Rodrigue. 2000. Membrane targeting and translocation of bacterial hydrogenases. Arch. Microbiol. 173:319-324. [DOI] [PubMed] [Google Scholar]
- 39.Wysocki, R., C. C. Chéry, D. Wawrzycka, M. Van Hulle, R. Cornelis, J. M. Thevelein, and M. J. Tamás. 2001. The glycerol channel Fps1p mediates the uptake of arsenite and antimonite in Saccharomyces cerevisiae. Mol. Microbiol. 40:1391-1401. [DOI] [PubMed] [Google Scholar]