

Abstract
The interaction between hydrogen metabolism, respiration, and photosynthesis was studied in vivo in whole cells of Synechocystis sp. strain PCC 6803 by continuously monitoring the changes in gas concentrations (H2, CO2, and O2) with an online mass spectrometer. The in vivo activity of the bidirectional [NiFe]hydrogenase [H2:NAD(P) oxidoreductase], encoded by the hoxEFUYH genes, was also measured independently by the proton-deuterium (H-D) exchange reaction in the presence of D2. This technique allowed us to demonstrate that the hydrogenase was insensitive to light, was reversibly inactivated by O2, and could be quickly reactivated by NADH or NADPH (+H2). H2 was evolved by cells incubated anaerobically in the dark, after an adaptation period. This dark H2 evolution was enhanced by exogenously added glucose and resulted from the oxidation of NAD(P)H produced by fermentation reactions. Upon illumination, a short (less than 30-s) burst of H2 output was observed, followed by rapid H2 uptake and a concomitant decrease in CO2 concentration in the cyanobacterial cell suspension. Uptake of both H2 and CO2 was linked to photosynthetic electron transport in the thylakoids. In the ndhB mutant M55, which is defective in the type I NADPH-dehydrogenase complex (NDH-1) and produces only low amounts of O2 in the light, H2 uptake was negligible during dark-to-light transitions, allowing several minutes of continuous H2 production. A sustained rate of photoevolution of H2 corresponding to 6 μmol of H2 mg of chlorophyll−1 h−1 or 2 ml of H2 liter−1 h−1 was observed over a longer time period in the presence of glucose and was slightly enhanced by the addition of the O2 scavenger glucose oxidase. By the use of the inhibitors DCMU [3-(3,4-dichlorophenyl)-1,1-dimethylurea] and DBMIB (2,5-dibromo-3-methyl-6-isopropyl-p-benzoquinone), it was shown that two pathways of electron supply for H2 production operate in M55, namely photolysis of water at the level of photosystem II and carbohydrate-mediated reduction of the plastoquinone pool.
Cyanobacteria are oxygenic photosynthetic prokaryotes containing two photosystems, PS II and PS I, linked by a linear electron transport chain similar to that of higher plant or algal chloroplasts. Much attention has been focussed on H2-producing cyanobacteria in the hope that the photosynthetic reactions producing reductant from water can be coupled to the reduction of protons to generate molecular hydrogen at the expense of solar energy (3, 11, 24, 31, 32, 39). In cyanobacteria, two different enzymes capable of H2 production in vivo have been described, nitrogenase and reversible hydrogenase. The first enzyme, which occurs only in N2-fixing strains, reduces protons to H2 concomitantly with the reduction of N2 to NH4+. In these organisms, however, H2 production is limited due to an efficient recycling of H2 by an uptake [NiFe]hydrogenase, which is present in all known N2-fixing strains (31, 39). Accordingly, it has been demonstrated that mutations impairing the activity of uptake hydrogenase (26, 41) or inactivating the structural hupSL genes (13) can stimulate H2 photoproduction in N2 fixers. The other type of hydrogenase found in cyanobacteria is the reversible [NiFe]hydrogenase (42). The first molecular characterization of this enzyme in Anabaena and Anacystis species showed that it belonged to the class of NAD(P)-reducing hydrogenases homologous to the previously described HoxFUYH complex of Ralstonia eutropha or Nocardia opaca (33). The reversible hydrogenase is not ubiquitous enzyme in cyanobacteria, but it has been found both in N2 fixers and non-N2 fixers.
The complete genome sequence of Synechocystis sp. strain PCC 6803 (18, 27) and a survey based on a molecular approach (40) showed the presence of genes encoding the reversible hydrogenase and the absence of both nitrogenase and uptake hydrogenase. The Synechocystis reversible hydrogenase is a pentameric [NiFe] enzyme utilizing NAD(P) as a substrate (4, 34). The HoxY and HoxH subunits form the [NiFe]hydrogenase moiety, while three other subunits (HoxU, HoxF, and HoxE), homologous to subunits of complex I of respiratory chains, contain NAD(P), flavin mononucleotide, and FeS binding sites (3, 6, 18, 33, 34; also reviewed in reference 44). Since no hydrogenase activity has been found in a hydrogenase deletion mutant (ΔhoxH), it has been concluded that in Synechocystis sp. strain PCC 6803 only the bidirectional HoxEFUYH hydrogenase functions for H2 uptake or H2 production (2).
In Synechocystis, following anoxic incubation in the dark, H2 photoproduction has been described as limited to a low-amplitude transient outburst (1, 9). The authors hypothesized that it was driven by counteracting evolution and uptake reactions. As only the reversible hydrogenase is present in Synechocystis strain PCC 6803, variations in amplitude and direction of the H2 flux may rely on two main factors: the hydrogenase activity (related to enzyme amounts and to its activation status) and the redox status of the donor/acceptor NAD(P)H/NAD(P) pool.
The activity of hydrogenases is generally inhibited by O2. The reversible hydrogenases of cyanobacteria are considered to be more O2 sensitive than the uptake hydrogenases (14, 39). In the case of Synechocystis strain PCC 6803, H2 production has been observed only in anoxia (1, 9), but interestingly, the hydrogenase is constitutively expressed in the presence of O2 and its suppression was reported to induce a perturbation in photosynthetic activity (2), indicating that it might be active under such conditions.
In cyanobacterial cells, the redox status of the NAD(P)H/NAD(P) pools is essentially controlled by the activities of photosynthesis and respiration. Photosynthetic electron transport takes place in the thylakoid membrane, while respiratory electron transport occurs in both the thylakoid and the cytoplasmic membranes. In the thylakoids, the respiratory and photosynthetic electron transport chains have electron carriers in common (cytochrome b6f complex and the plastoquinone [PQ] pool; see Fig. 1). The photosynthetic electron transport chain reduces NADP to NADPH, which is the electron donor used for photosynthetic CO2 fixation and is also the preferential substrate of the respiratory complex I of cyanobacteria, also called the type I NADPH-dehydrogenase complex NDH-1 (8, 25), which is largely confined to the thylakoid membrane (30).
FIG. 1.

Proposed electron transfer pathways in the light involved in H2 production and uptake in Synechocystis sp. strain PCC 6803. Abbreviations: H2ase, hydrogenase; PS I, photosystem I; PS II, photosystem II; cyt, cytochrome; cyt b6f, cytochrome b6f complex; cyt ox, cytochrome aa3 oxidase; SDH, succinate dehydrogenase; NDH-1, type I NAD(P)H-dehydrogenase; PQ, plastoquinone; PC, plastocyanin; FNR, ferredoxin-NADP+ reductase; Fd, ferredoxin. Electron transfers are represented by solid arrows; DCMU and DBMIB inhibition sites are indicated by thick white arrows. Enzyme names are in italics. Grey flash arrows indicate pathways which are impaired in the ndhB mutant M55.
NDH-1 is encoded by the ndh genes, which number only 11 in the Synechocystis genome (18, 27), instead of the 14 found in other, related bacterial complexes. The three missing genes are those coding for the peripheral diaphorase subunits. It has been suggested that these subunits might be used simultaneously by the reversible hydrogenase and the respiratory complex I (3, 4, 6, 35), as is shown in Fig. 1. On the other hand, the bidirectional hydrogenase is absent in a significant number of cyanobacterial strains (40), and there is evidence that there is not a common use of the diaphorase subunits by the hydrogenase and NDH-1 (5). Different cyanobacterial mutants affected in the NDH-1 complex have been obtained. The large hydrophobic subunit, NdhB, is a core membrane component of the complex, and the ndhB gene is present in a single copy in the genome. Therefore, the ndhB-deficient mutant M55 is totally devoid of the NDH-1 complex (28), in contrast to other ndh mutants affected in multicopy genes, such as ndhD and ndhF (22, 29). M55 has a high CO2-requiring phenotype and is characterized by a low respiration rate and a reduced activity of cyclic electron flow around PS I (28). As a result, M55 shows a highly impaired photosynthetic activity under normal CO2 concentrations (Fig. 1), with the NADP pool remaining almost entirely in the reduced state (8).
The goal of this study was to understand the nature of the interactions between hydrogenase function, respiratory activity, and photosynthesis, a prerequisite to looking for appropriate strains allowing economical and efficient H2 production. To this end, we have used time-resolved mass spectrometry to measure H2, O2, and CO2 fluxes in wild-type (WT) Synechocystis and in the NDH-1-deficient mutant M55 and to monitor hydrogenase activity by the proton-deuterium (H-D) exchange reaction in the D2-H2O system. While a low and transitory rate of H2 production was observed in the WT during dark-to-light transition, the NDH-1-deficient mutant was able to sustain significant H2 production in the light. We discuss the mechanisms underlying the competition between NDH-1, CO2 fixation, and hydrogenase for reducing equivalents in Synechocystis (Fig. 1) and their potential use for improving the H2-producing capacities of this organism.
MATERIALS AND METHODS
Strains and growth conditions.
WT Synechocystis strain PCC 6803 and mutant strains were grown autotrophically in liquid aerated modified Allen's medium (16) at 34°C under continuous illumination, using two fluorescent tubular lamps, which provided an average light intensity of 70 μmol of photons m−2 s−1. The high CO2- requiring mutant M55 (ndhB::Kmr cartridge) (kindly provided by T. Ogawa) was grown in medium supplemented with 100 μg of kanamycin ml−1 and 10 mM NaHCO3 (28).
Cell fractionation.
Cells grown autotrophically were pelleted, resuspended in 50 mM morpholineethanesulfonic acid (MES)-KOH buffer, pH 6, supplemented with 2% (wt/vol) bovine serum albumin before two passages through a French press (16,000 psi). Cell debris were removed from the cell extract by centrifugation at 3,000 × g for 10 min.
Mass spectrometric measurements of gas exchange.
Cultures were harvested in the exponential or plateau phase of growth, centrifuged, washed in distilled water, and resuspended in 35 mM HEPES-NaOH buffer, pH 7.2, or in 50 mM MES-KOH buffer, pH 6.0. The cell suspension was placed in the measuring chamber (1.5 ml) of a mass spectrometer (model MM 8-80; VG Instruments, Cheshire, United Kingdom). The bottom of the chamber (Hansatech electrode type) was sealed by a Teflon membrane, allowing dissolved gases to be directly introduced through a vacuum line into the ion source of the mass spectrometer. The chamber was thermostated at 30°C, and the cell suspension was stirred continuously by a magnetic stirrer. Light was supplied to the suspension by a fiber optic illuminator (Schott, Mainz, Germany). Experiments were performed at 300 μmol of photons m−2 s−1 of incident light. Inhibitors (DCMU [3-(3,4-dichlorophenyl)-1,1-dimethylurea] and DBMIB (2,5-dibromo-3-methyl-6-isopropyl-p-benzoquinone) when used were introduced directly into the chamber with a Hamilton syringe about 10 min before experiments. Before hydrogenase activity was studied, cells were subjected to anaerobic dark adaptation in HEPES-NaOH buffer (pH 7.2); after transfer to the measuring chamber of the mass spectrometer, the cells were sparged with argon, the Ar was removed, the vessel was closed, and the cells were left to consume the residual O2 (or, when indicated, O2 was removed by O2 scavengers).
The principle of the kinetic measurements of H2 production, H2 uptake, H-D exchange, O2 uptake or production, and CO2 uptake has been described previously (17). The spectrometer sequentially scans the abundance of the different gases (H2, D2, HD, O2, and CO2) by automatically adjusting the magnet current to the corresponding mass peaks (m/e = 2, 4, 3, 32, and 44, respectively). Measuring one mass peak typically takes 0.5 s. Mass peaks are continuously recorded during the experiment, every 2 s for fast-kinetic measurements, e.g., for light-to-dark transitions, or every 10 s for measuring slower kinetics, e.g., during hydrogenase induction. The amperometric signal collected by the spectrometer is directly proportional to the gas concentration in the chamber, the proportionality coefficient varying from one mass to the other according to the ionization properties of the corresponding gas. In order to calculate the rates of gas exchange, the time derivation of gas concentrations has to be corrected for the slow but significant rate of gas consumption by the mass spectrometer, which is superimposed on production or uptake rates. The consumption of gases by the mass spectrometer was assayed in cell-free buffer. It showed first-order kinetics, with time constants around 0.09 min−1 for H2, 0.08 min−1 for D2, and 0.024 min−1 for O2; CO2 consumption by the apparatus was negligible under the test conditions. The setup also allowed us to determine the in vivo activity of hydrogenase by the use of hydrogen isotopes for the H-D exchange reaction, in which D2 disappears and is quantitatively replaced by HD and H2. Calculation of hydrogen gas exchange has been elaborated as described below.
Modeling of the H-D exchange reaction catalyzed by hydrogenase enzymes in the presence of various relative concentrations of D2 and of H2-consuming or H2-producing processes.
As a first approximation for modeling H-D exchange, we will assume that the cleavage is heterolytic and that the D+ concentration in water is negligible, so that each D2 exchange event leads to 1 HD molecule and each exchange of an HD molecule leads with a 0.5 probability to either 1 H2 or 1 HD molecule, the H2 interaction with the hydrogenase being neutral in this respect. When reducing or oxidizing equivalents are provided to the hydrogenase, H2 production or uptake can be superimposed on the H-D exchange activity. We will consider that production and uptake are mutually exclusive at a given time and are therefore equal to the net rate of change in hydrogen concentration (all isotopic species combined). We will also assume that the probability that one hydrogen species interacts with the hydrogenase (either for H-D exchange or for uptake) is equal to its proportion in the mix and that H2 production, apart from the H-D exchange, yields only H2 coming from water protons. In the following equations, the total concentration of hydrogen species (Σ) = [D2] + [HD] + [H2]; dΣ/dt is then the net production or uptake of hydrogen (<0 in case of uptake, >0 in case of production); Vexch is the turnover rate of hydrogen species at the hydrogenase active site leading to the H-D exchange reaction (i.e., the H-D exchange activity of the enzyme); and τ is the isotopic ratio of D in hydrogen: τ = ([D2] + 1/2[HD])/Σ. A simple model for H-D exchange combined with hydrogen production or uptake can then be formulated (Table 1) (in order to simplify the equations, the rates described are those corrected for consumption by the mass spectrometer).
TABLE 1.
Simple model for H-D exchange combined with H2 production or uptake
| Equation | Exchange | Production (dΣ/dt > 0) | Uptake (dΣ/dt < 0) |
|---|---|---|---|
| 1 | 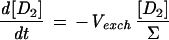 |
+0 | 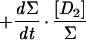 |
| 2 | 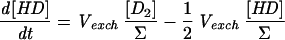 |
+0 | 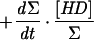 |
| 3 | 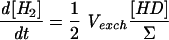 |
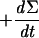 |
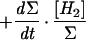 |
The hydrogenase activity can then be deduced from D2, HD, and H2 concentration changes, by simple rearrangement of the equations (Table 1). In the case of a simple H-D exchange (dΣ/dt = 0), adding equations 2 and 3 (×2) (Table 1, Exchange column) gives
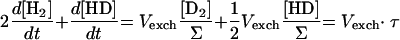 |
which gives
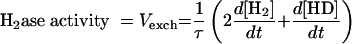 |
An example of calculations in the case of the H-D exchange reaction only (in the absence of electron donors or acceptors) is shown in Fig. 2. These results, obtained with a pure hydrogenase sample, validate the calculations and show that the method is suitable for assaying the H-D exchange rate for a wide range of D2 concentrations.
FIG. 2.

H2 and HD production in exchange with D2 uptake catalyzed by Desulfovibrio fructosovorans [NiFe]hydrogenase. (A) Real concentrations of the hydrogen species present in the vessel (D2 concentration reaches zero). (B) Gas concentration changes corrected for consumption by the apparatus (shown is the equivalence between D2 uptake and H2 plus HD production). (C) Hydrogenase activity, calcu-lated as 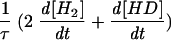
, is expressed in nanomoles per milliliter per minute (or micromolar per minute). The sum (Σ) of D2 plus H2 plus HD concentrations is calculated as described in Materials and Methods. Σ remains constant since only the exchange reaction between hydrogen isotopes and protons of the medium is taking place here. Vexch and hydrogenase activity are then confounded.
When H2 production occurs simultaneously with H-D exchange (dΣ/dt > 0), addition of equations 2 and 3 (×2) (Table 1, Exchange and Production columns) yields
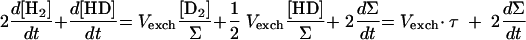 |
which gives
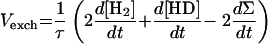 |
Then
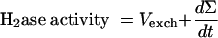 |
When uptake occurs simultaneously with H-D exchange (dΣ/dt < 0), addition of the terms of equations 2 and 3 (×2) (Table 1, Exchange and Uptake columns) finally yields
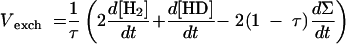 |
Then
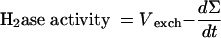 |
The mathematical treatment developed in this report extends the H-D exchange interpretation method, previously essentially applied to the initial rates of H2 and HD production, to the whole period during which isotopes are present (i.e., about 20 to 30 min in our setup). The treatment allows monitoring of changes in hydrogenase activity over time and enables us to evaluate whether the measured concentration changes of the hydrogen species are simply related to the H-D exchange reaction or are accompanied by H2 production or uptake.
Other measurements.
Pigments were extracted by methanol from pelleted cells, and the chlorophyll concentration in the centrifuged cell extract was measured at 665 nm (23).
Chemicals.
Chemicals were all supplied by Sigma-Aldrich (Saint Louis, Mo.).
RESULTS
Anaerobic hydrogenase activation and H2 production in darkness.
Synechocystis cells were placed in the dark in the online measuring chamber of the mass spectrometer. No H2 was detected in the medium when O2 was present. The cells were then made anaerobic by the addition of glucose and glucose oxidase in the presence of catalase. H2 gas production began around 1 to 2 min after O2 was completely depleted from the medium (Fig. 3). The H2 concentration in the medium increased progressively, reaching a stable value when H2 production by the cells was compensated for by H2 consumption by the mass spectrometer. The maximal H2 production rate (around 0.35 nmol of H2 ml−1 min−1) was sustained during 2 to 3 min at the beginning of the production period and progressively decreased to a lower steady-state rate (around 0.15 nmol of H2 ml−1 min−1). The activity of hydrogenase was measured by the H-D exchange reaction assay in separate experiments repeated under identical conditions. In the presence of O2, H-D exchange was not detectable (Fig. 3). Five minutes after anaerobiosis had been reached, the hydrogenase activity assayed by the H-D exchange reaction was 3.5 nmol ml−1 min−1 (or 350 nmol min−1 mg of chlorophyll (Chl)−1) and increased only slightly during the following 30 min of incubation. This fast activation indicated that the hydrogenase was present in cells taken from oxic conditions but required anaerobiosis to become active. Moreover, the rate of H2 production in the dark represented only a fraction of the potential hydrogenase activity, indicating that the limiting factor for H2 production was not hydrogenase but rather the NAD(P)H supply, which under these conditions is produced from fermentation (38).
FIG. 3.

Anaerobic H2 production in darkness. A cell suspension of Synechocystis sp. strain PCC 6803 (10 μg of Chl ml−1) in 35 mM HEPES buffer, pH 7.2, was placed in the measuring chamber of the online mass spectrometer, and catalase (500 U), glucose (5 mM), and glucose oxidase (30 U) were added to make the medium totally anaerobic. The concentrations of H2 (straight line, “recorded”) and O2 (dotted line) were recorded every 10 s at mass peaks 2 and 32, respectively. The curve labeled “corrected” shows H2 evolution in the medium corrected for the consumption by the apparatus and thus directly reflects H2 production by the cells. For the determination of hydrogenase activity by the H-D exchange reaction (•), the cells were preincubated in darkness under the same conditions as those described above for the indicated periods of time and then sparged with D2 and hydrogenase activity, based on the rates of H2 and HD formation, was calculated as described in Materials and Methods and in the legend to Fig. 2C.
O2 and H2 exchange during dark-light transitions in WT Synechocystis.
O2 and H2 exchange were then measured during dark-light transitions in WT Synechocystis cells adapted to anaerobic conditions. Anaerobiosis was obtained by flushing the sample with argon, closing the vessel, and letting the cells consume the residual O2 in the dark. When O2 was fully consumed, H2 production started. After stabilization of the H2 level (about 10 min), corresponding to a production rate of 0.1 μmol of H2 mg of Chl−1 h−1, the light was switched on. A sudden, short (less than 30-s) burst of H2 (initial rate, 1.2 μmol of H2 mg of Chl−1 h−1) was observed, followed by a rapid H2 uptake consuming all H2 present in the medium (Fig. 4A). This burst preceded photosynthetic O2 production, which began after 1 min of illumination. When the light was switched off, O2 was consumed by respiration and H2 production resumed once anaerobiosis was achieved. Figure 4B shows results of the same type of experiment carried out in the presence of DCMU, an inhibitor of PS II. In the light, the addition of DCMU prevented photosynthetic O2 production (Fig. 4B) while the H2 exchange pattern was not modified. H2 production started as soon as the light was switched off, probably due to the absence of O2.
FIG. 4.

Mass spectrometric measurements of H2, O2, and CO2 exchange during dark-light-dark transitions in WT Synechocystis cells adapted to dark anaerobic conditions. Shown are effects of the PS II inhibitor DCMU. (A) Transitory H2 output and H2 uptake at the onset of light. Cells of Synechocystis sp. strain PCC 6803 (10 μg of Chl ml−1) were preincubated anaerobically in darkness in the measuring chamber of the mass spectrometer, and H2 production was allowed to proceed in the dark. The cells were then illuminated briefly (2 min), and the concomitant changes in concentrations of dissolved gases H2, O2, and CO2 were measured every 4 s. (B) Same conditions as for panel A but in the presence of 75 μM DCMU. Dark periods are represented on the x axis by black bars, and the light period is represented by a white bar.
Oxygen inhibition of hydrogenase activity, measured by the H-D exchange reaction.
Hydrogenase activity was assessed during dark-light-dark transitions by injecting D2 and monitoring the H-D exchange (Fig. 5). Under these conditions, the O2 exchange pattern (Fig. 5 A and B) was similar to that observed in the previous experiment. In the light, hydrogenase activity (Fig. 5C) was inhibited as soon as O2 appeared in the medium and was restored in the dark once O2 had been consumed (Fig. 5A). In this experiment, the global hydrogen flux can be followed on the curve representing the sum [D2] + [H2] + [HD], corrected for instrument consumption (Σ). When hydrogenase was active, significant hydrogen consumption was observed in the light. When the hydrogenase activity was inhibited, the hydrogen consumption stopped, with the total hydrogen concentration (Σ) remaining constant (Fig. 5C). In the presence of DCMU, no O2 was produced (Fig. 5B) and the hydrogenase activity remained practically constant during the light period, resulting in a continuous hydrogen (Σ) uptake in the light (Fig. 5D).
FIG. 5.

Mass spectrometric measurements of O2 and CO2 exchange and H-D exchange during dark-light-dark transitions in WT Synechocystis cells adapted to dark anaerobic conditions in the presence of D2. Shown are effects of the PS II inhibitor DCMU. (A and B) Time course of O2 and CO2 concentration changes. (C and D) Time course of hydrogenase activity measured by the H-D exchange reaction and of the sum (Σ) of D2 plus H2 plus HD concentrations, corrected for the apparatus consumption, determined in the presence of 400 μM D2 without further addition (A and C) or in the presence of 75 μM DCMU (B and D). Dark periods are represented on the x axis by black bars, and the light period is represented by a white bar.
Involvement of hydrogenase in CO2 fixation.
CO2 exchange was also recorded in the gas exchange experiments (Fig. 4 and 5). In control experiments, the light-induced CO2 exchange pattern observed in the presence of D2 (Fig. 5A) qualitatively resembled that in the absence of D2 (Fig. 4A): a rapid drop in CO2 concentration at light onset due to the activation of CO2-concentrating mechanisms, followed by a secondary CO2 uptake process corresponding to CO2 fixation by the Calvin cycle (19, 28). In the presence of DCMU, only a transient CO2 uptake, corresponding to the concentration phase, was observed in the absence of D2 (Fig. 4B) while a sustained CO2 uptake, resulting in a net CO2 consumption (compare differences in steady-state CO2 levels in the dark before and after illumination), was recorded in the presence of D2 (Fig. 5B). These differences in CO2 uptake can be compared to the differences in hydrogen uptake patterns. In Fig. 4B, the small H2 amount produced in the dark was rapidly consumed. On the other hand, in Fig. 5D, the high D2 concentration added to measure H-D exchange allowed sustenance of continuous hydrogen (Σ) consumption during the light period. The amount of CO2 fixed in the light in the presence of DCMU (Fig. 5B) was about 3 μM, which corresponds to ca. 30 μM of dissolved carbon (CO2 + HCO3−) at pH 7.2, while the consumption of hydrogen (decrease of Σ during the light period) was about 70 μM. This indicates that under these conditions hydrogen is the main source of electrons for CO2 fixation (2 H2 molecules provide 4 reducing equivalents, which are needed for reduction of 1 CO2 molecule). Interestingly, this amount is of the same order of magnitude as the photosynthetic O2 production measured in the control experiment (Fig. 5A). Light-driven H2, O2, and CO2 exchange were inhibited by the cytochrome b6f inhibitor DBMIB (20 μM) (data not shown), indicating that electron transfer from the PQ pool to PS I is essential for all the light-dependent activities discussed here. (It was checked that neither DCMU nor DBMIB affected hydrogenase activity, as monitored by H-D exchange in the dark in intact cells and extracts.)
H2 production in the light in the Synechocystis mutant M55, deficient in the NDH-1 complex.
Since NADPH produced by photosynthetic electron transfer or by oxidation of H2 may enter the respiratory chain via NDH-1, we studied photobiological gas exchange in the NDH-1-deficient mutant M55 (Fig. 6). When the light was switched on after anaerobic adaptation, the M55 mutant showed H2 production in the light, with an initial rate (5 μmol of H2 mg Chl−1 h−1) higher than that observed in the WT and with a much longer duration (Fig. 6A). This H2 production was accompanied by a simultaneous O2 production, while CO2 uptake was negligible. The O2 production rate was 25-fold lower than in the WT. The absence of apparent H2 uptake in the NDH-1-deficient mutant during illumination indicates that the uptake observed in the WT was related to the activity of the NDH-1 complex. H2 production in M55 stopped however after 4 to 5 min of illumination. The light-dependent H2 production was completely inhibited by DCMU (Fig. 6B), indicating that essentially electrons originating from water photolysis at PS II were used for H2 production. Addition of glucose to DCMU-treated cells increased the light-dependent H2 production (Fig. 6C), showing that electrons originating from the fermentative metabolism of glucose can reach PS I by a PS II-independent electron transfer pathway. DBMIB inhibited light-induced H2 production occurring in the presence of glucose (Fig. 6D), showing that both PS II-dependent and PS II-independent pathways rely on PQ reoxidation by the cytochrome b6f complex (Fig. 1).
FIG. 6.

H2 and O2 concentration changes in darkness and during a 5-min illumination period in the Synechocystis ndhB mutant M55. A culture of M55 (optical density at 730 nm = 1.5) was adapted to dark anaerobic conditions (A), in the presence of DCMU (75 μM) (B), in the presence of glucose (10 mM) and DCMU (75 μM) (C), or in the presence of glucose and DBMIB (20 μM) (D) in a closed reaction vessel connected to a mass spectrometer. The concentrations of H2, O2, and CO2 in the medium were continuously recorded. Dark periods are represented on the x axis by black bars, and the light period is represented by a white bar.
Glucose addition also had significant effects on H2 exchange in the dark. In M55, hydrogenase induction under anaerobiosis in the dark (assayed by H-D exchange) was comparable to that observed in the WT (data not shown), but H2 production in darkness was barely detectable (Fig. 6A and B) unless exogenous substrate (succinate or glucose) was added (Fig. 6C and D). This is probably due to the poor CO2 fixation capacity of M55 cells, which do not accumulate high levels of endogenous substrates during the growth phase and therefore cannot support significant H2 production in the dark. However, dark H2 production in the absence of an exogenous supply was observed with M55 cells taken from the stationary phase, probably because, having stopped growing, they had accumulated endogenous substrates (data not shown).
Sustained H2 photoproduction in the light in the ndhB-defective Synechocystis M55 mutant.
As shown in Fig. 7A, light-induced H2 production in M55 stopped after a few minutes. This might have been due to the observed increase in O2 concentration, which could have inhibited hydrogenase. In the presence of glucose, the concentration of O2 remained low and H2 production was sustained in the light for at least 25 min (Fig. 7B). The addition of glucose oxidase, by further decreasing the O2 level, slightly stimulated the rate of H2 production (Fig. 7C). In this case a production rate of ca. 6 μmol of H2 mg Chl−1 h−1 (or 2 ml of H2 liter−1 h−1) was observed during the whole light period, a value in the upper range of hydrogenase-mediated H2 production by cyanobacteria (31). In the presence of glucose, DCMU had a limited effect (ca. 20% inhibition) on H2 photoproduction by M55 during the whole light period (data not shown), indicating that both PS II-dependent and PS II-independent pathways of PQ reduction may durably contribute to the supply of electrons for H2 production.
FIG. 7.

Mass spectrometric measurements of H2, O2, and CO2 exchange in the Synechocystis ndhB mutant M55 in darkness and during a 25-min illumination period. Cells were adapted for 1 h under anaerobiosis in the dark in a closed vessel connected to a mass spectrometer, and the concentrations of H2, O2, and CO2 in the medium were continuously monitored. (A) Control, (B) with 10 mM glucose, and (C) with 10 mM glucose plus glucose oxidase plus catalase. Conditions were the same as for Fig. 6 except for the duration of the illumination period. Dark periods are represented on the x axis by black bars, and the light period is represented by a white bar.
Both NADH and NADPH act as electron donors and activators for the bidirectional hydrogenase.
The ability of NADH or NADPH to provide electrons for H2 production was tested in cell extracts. As observed in intact cells, no H2 was evolved before O2 was totally removed from the medium. Both nucleotides could serve as electron donors for H2 production with comparable efficiencies. Final rates of H2 production were around 0.09 μmol of H2 mg Chl−1 h−1 with 1 mM of either NADPH or NADH (Fig. 8). The low rate found in cell extracts may be explained by a partial inactivation of hydrogenase by O2 during the preparation of the extract. A lag time of a few minutes was observed between NADPH addition and H2 production (Fig. 8), which might correspond to the time necessary to activate the enzyme.
FIG. 8.

H2 evolution catalyzed by a Synechocystis cell extract with NADH or NADPH as the electron donor. The extract (200 μg of Chl) was gassed with argon, the chamber was closed, and O2 (monitored at mass peak 32) was removed by the addition of catalase, glucose, and glucose oxidase. NADH (1 mM) (dashed line) or NADPH (1 mM) (solid line) was added at 2 min (arrow).
The ability of reduced nucleotides to activate the enzyme was tested by measuring the hydrogenase activity with the H-D exchange assay. In aerobic extracts saturated with D2, no activity was detected until O2 was removed. When the extract was placed under anaerobic conditions, hydrogenase activity was barely detectable. Upon NADH addition, the hydrogenase activity rose rapidly (Fig. 9, left panel), reaching its steady-state level within minutes. When NADH was added before anaerobiosis was reached, no activation of the hydrogenase was observed in the presence of O2 but hydrogenase activity started as soon as O2 was removed (data not shown). NADPH was also able to activate the hydrogenase to comparable levels, but after a lag phase of a few minutes (Fig. 9 right panel), accounting for the delay in H2 production observed in the experiment for which results are shown Fig. 8. On the other hand, NAD+ or NADP+ added at a high concentration (5 mM) did not interfere with the NADH or NADPH activation process (data not shown). At pH 6, the Km value for hydrogenase activation was 12 μM for NADH and 100 μM for NADPH. Since these experiments were conducted with cell extracts, it cannot be excluded that NADH can be formed from NADPH by a transhydrogenase reaction and that NADH is the true activator of the hydrogenase. The cellular localization of hydrogenase was assessed by the H-D exchange assay performed with cell extracts in membrane and in cytosolic fractions (after NADH activation). The membrane fraction was found to retain 10 to 25% of hydrogenase activity, and the supernatant fraction retained around 70% (in WT as well as in mutant cells).
FIG. 9.

Activation by NADH (left) or NADPH (right) of the hydrogenase activity in cell extracts of WT Synechocystis sp. strain PCC 6803 at pH 6.0. NADH or NADPH (0.2 mM) was added after the extract (73 μg of Chl) saturated with D2 was made anaerobic as described in the legend to Fig. 3. Two minutes after the O2 concentration had reached zero, the concentration changes in D2, HD, H2, and O2 were measured at mass peaks 4, 3, 2, and 32, respectively. Hydrogenase activity and the corrected total concentration of hydrogen species (Σ) were calculated as described in Materials and Methods and depicted in Fig. 2.
DISCUSSION
How does the NDH-1-deficient mutant of Synechocystis sustain photobiological hydrogen gas production?
During a dark-to-light transition in anoxia, a low and transitory H2 production was observed in WT Synechocystis cells. In contrast, the NDH-1-deficient mutant M55 was able to sustain significant H2 production in the light. Suppression of the NADPH-consuming enzyme NDH-1 therefore provoked a long-term accumulation of reducing equivalents in the light which was sufficient to maintain activity of the NADPH-dependent, reversible hydrogenase. Due to the difference in electrochemical potentials (ca. 320 mV for the NAD(P)/NAD(P)H couple and 420 mV for H+/H2), the reversible hydrogenase can evolve H2 only in the case of a very high (ca. 99.9%) reduction of the NAD(P) pool. It is likely therefore that H2 production depends on the ability of the cells to maintain such a high reduction state. In Synechocystis strain PCC 6803, the size and redox state of the pyridine nucleotide pools have been determined in aerobically grown WT and NDH-1 mutant cells (8). While the NAD reduction level was 70% in both strains, the reduction level of NADP shifted from 50% in the WT to 100% in the NDH-1 mutant (8), indicating that the mutant is specifically impaired in NADPH oxidation. In anoxia, it is likely that reduction of the NAD(P) pools is even higher, making H2 evolution possible. The total NADP content was found to be more than 10 times that of NAD, in both WT and NDH-1 mutant cells (8). As the enzyme catalyzes electron transfer at comparable rates using NADH or NADPH (34; this study), the predominance of the NADP pool suggests that H2 uptake or production observed in our study is essentially due to reactions of the hydrogenase with NADP or NADPH.
Apart from its interaction with hydrogenase, NADP reduction in Synechocystis is driven in the dark essentially by carbohydrate metabolism and in the light by photosynthesis. On the other hand, NADPH oxidation is primarily driven in the dark by respiration (through NDH-1) and in the light by the Calvin cycle. Recently, two functionally distinct classes of the NDH-1 complex have been identified. The first class, containing the NdhD1 or NdhD2 subunit, catalyzes NADPH-dependent PQ reduction and is involved in PS I cyclic electron transport and respiration, while the second class, containing the NdhD3 or NdhD4 subunit, does not directly oxidize NADPH but is involved in CO2 uptake (22, 29). In NdhB− mutants such as M55, both types of NDH-1 complex are absent. However, it would be of interest to determine how independent alteration of the NdhD1/NdhD2 or NdhD3/NdhD4 subunits, might interfere with H2 production.
Sources of reducing power for H2 production.
Hydrogenase-dependent H2 evolution in cyanobacteria can be supported in the dark by endogenous substrates or by exogenous electron donors such as glucose (14). This was also the case with Synechocystis in our experiments. In the light, the existence of simultaneous O2 and H2 outputs in the NDH-1-deficient mutant and their inhibition by DCMU (Fig. 6) show that the water-oxidizing photosynthetic electron transport chain can also serve as an electron donor for H2 production. In vivo, H2 evolution associated with water photolysis via a reversible hydrogenase was first observed by Laczkó (20) with Anabaena cylindrica cells grown at high light intensities.
In the M55 mutant strain, electron flow from PS II to hydrogenase stopped after a few minutes. Glucose addition allowed sustained H2 production, probably through two main mechanisms. First, the presence of glucose considerably lowers net O2 production in the light. This can be explained either by a stimulation of respiratory O2 consumption or by an inhibition of PS II due to PQ overreduction. We showed that addition of glucose to PS II-inhibited M55 cells restored H2 photoproduction by a pathway involving PQ. This indicates that glucose-stimulated PS II-independent reduction of PQ can also provide electrons to PS I for H2 production. The identity of this pathway remains to be elucidated. In any case, it must lead to an increase in the level of reduction of the NAD(P) pool, suggesting that the mechanism for PQ reduction is different from those classically described for cyclic electron transfer around PS I, which consume NADPH or reduced ferredoxin. A probable explanation for our observations is that nonphotochemical reduction of PQ by products of glucose metabolism such as succinate (7, 8) could sustain NADPH production by PS I and therefore play a significant role in H2 production.
Sinks of reducing power for H2 uptake.
In the WT, H2 is efficiently taken up in the light before inactivation of the hydrogenase by photosynthetic O2 (Fig. 4). H2 uptake did not occur at significant rates in the NDH-1-deficient mutant (Fig. 6 and 7), which is essentially affected in NADPH-mediated PQ reduction and in CO2 uptake. Therefore, the H2 oxidation observed in the WT proceeded essentially via oxidation of NADPH by the electron transport chains and by CO2 fixation (Fig. 1). In the presence of high amounts of hydrogen (D2), under microaerobic conditions, hydrogen oxidation could be sustained in the light and efficiently serve as an electron donor for CO2 fixation, hydrogen being almost as efficient as PS II in this case (Fig. 5). Therefore, although it has been stated that in cyanobacteria, CO2 fixation by the Calvin cycle proceeds at a much higher rate than H2 uptake or production by the bidirectional hydrogenase (35), it appears that under our experimental conditions, in anoxia (or microaerobiosis) and in the presence of hydrogen, hydrogen consumption is of the same magnitude as photosynthetic activity and could therefore significantly contribute to CO2 fixation.
Characteristics of Synechocystis hydrogenase and physiological implications.
In cell extracts, Synechocystis hydrogenase was inactivated by O2 in a reversible manner. It could be reactivated in anoxia in the presence of hydrogen, this activation being considerably stimulated (within minutes) by NADH or NADPH at catalytic concentrations. Most [NiFe]hydrogenases are inactive in the presence of O2. They need to be reductively activated, for example by a long incubation (several hours) with H2 in the absence of O2 and in the presence of a reductant such as dithionite (10, 21) or NADH (14, 15, 37). But the activation that we observed in Synechocystis, occurring at low concentrations of NAD(P)H and being unaltered by NAD(P)+ addition, appears to be a specific phenomenon.
The activation observed in vivo in anoxia in the dark is then probably due to a combination of O2 scavenging and of NAD(P) reduction within the cells. Inactivation of hydrogenase in the light in vivo can be attributed to a direct effect of PS II-produced O2. Other interpretations, such as an independent consequence of illumination or a deactivation through the decrease of the NAD(P)H/NAD(P)+ ratio, can be discarded. Indeed, in the light and in the presence of DCMU, the hydrogenase was not inactivated, although the Calvin cycle was functional and was able to reoxidize NADPH quickly. Therefore, although the bidirectional hydrogenase in Synechocystis is constitutively expressed in the presence of O2 (2), it probably plays a role mainly under anaerobic or microaerobic conditions and, as suggested by Appel et al. (2), at the onset of light before the enzyme is inactivated by photosynthetic O2.
We can compare the features of the Synechocystis hydrogenase to those of the homologous NAD+-dependent hydrogenase of R. eutropha (formerly Alcaligenes eutrophus), which has been extensively studied and is often referred to as a prototype for this kind of hydrogenase. The R. eutropha enzyme is highly insensitive to O2 (36), is clearly NAD preferring, and is activated by NADH, but activation is prevented by NAD+ (37). Thus, despite sequence homologies between the Synechocystis and R. eutropha enzymes, their characteristics are not identical. Another example of a [NiFe]hydrogenase insensitive to O2 is the H2 sensor HupUV of Rhodobacter capsulatus, which is able to catalyze H-D exchange in the presence of high levels of O2 (43). In both cases, O2 insensitivity has been assumed to result from steric hindrance preventing O2 access to Ni. In the case of the R. eutropha enzyme, additional CN− ligands bound to the NiFe metallocenter may protect the enzyme against O2 inactivation (12). In the case of the H2 sensor HupUV, a partial blocking of the gas channel close to the active site, due to the presence of bulkier amino acid residues, might limit access to Ni to very small molecules (e.g., H2) (45). These recent advances in unraveling the O2 resistance mechanism of [NiFe]hydrogenases could open a way to increase the O2 resistance of the Synechocystis hydrogenase.
Acknowledgments
Generous gifts of strain M55 by T. Ogawa and of the Desulfovibrio fructosovorans [NiFe]hydrogenase by C. Hatchikian and critical reading of the manuscript by J. C. Willison are gratefully acknowledged.
This work was supported by the CNRS ENERGIE program.
REFERENCES
- 1.Abdel-Basset, R., and K. P. Bader. 1998. Physiological analyses of the hydrogen gas exchange in cyanobacteria. J. Photochem. Photobiol. B Biol. 43:146-151. [Google Scholar]
- 2.Appel, J., S. Phunpruch, K. Steinmuller, and R. Schulz. 2000. The bidirectional hydrogenase of Synechocystis sp PCC 6803 works as an electron valve during photosynthesis. Arch. Microbiol. 173:333-338. [DOI] [PubMed] [Google Scholar]
- 3.Appel, J., and R. Schulz. 1998. Hydrogen metabolism in organisms with oxygenic photosynthesis: hydrogenases as important regulatory devices for a proper redox poising? J. Photochem. Photobiol. B Biol. 47:1-11. [Google Scholar]
- 4.Appel, J., and R. Schulz. 1996. Sequence analysis of an operon of a NAD(P)-reducing nickel hydrogenase from the cyanobacterium Synechocystis sp PCC 6803 gives additional evidence for direct coupling of the enzyme to NAD(P)H-dehydrogenase (complex I). Biochim. Biophys. Acta 1298:141-147. [DOI] [PubMed] [Google Scholar]
- 5.Boison, G., H. Bothe, A. Hansel, and P. Lindblad. 1999. Evidence against a common use of the diaphorase subunits by the bidirectional hydrogenase and by the respiratory complex I in cyanobacteria. FEMS Microbiol. Lett. 174:159-165. [Google Scholar]
- 6.Boison, G., O. Schmitz, B. Schmitz, and H. Bothe. 1998. Unusual gene arrangement of the bidirectional hydrogenase and functional analysis of its diaphorase subunit HoxU in respiration of the unicellular cyanobacterium Anacystis nidulans. Curr. Microbiol. 36:253-258. [DOI] [PubMed] [Google Scholar]
- 7.Cooley, J. W., C. A. Howitt, and W. F. Vermaas. 2000. Succinate:quinol oxidoreductases in the cyanobacterium Synechocystis sp. strain PCC 6803: presence and function in metabolism and electron transport. J. Bacteriol. 182:714-722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooley, J. W., and W. F. J. Vermaas. 2001. Succinate dehydrogenase and other respiratory pathways in thylakoid membranes of Synechocystis sp. strain PCC 6803: capacity comparisons and physiological function. J. Bacteriol. 183:4251-4258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cournac, L., F. Mus, L. Bernard, G. Guedeney, P. Vignais, and G. Peltier. 2002. Limiting steps of hydrogen production in Chlamydomonas reinhardtii and Synechocystis PCC 6803 as analysed by light-induced gas exchange transients. Int. J. Hydrog. Energy 27:1229-1237. [Google Scholar]
- 10.Fernandez, V. M., R. Aguirre, and E. C. Hatchikian. 1984. Reductive activation and redox properties of hydrogenase from Desulfovibrio gigas. Biochim. Biophys. Acta 790:1-7. [Google Scholar]
- 11.Hansel, A., and P. Lindblad. 1998. Towards optimization of cyanobacteria as biotechnologically relevant producers of molecular hydrogen, a clean and renewable energy source. Appl. Microbiol. Biotechnol. 50:153-160. [Google Scholar]
- 12.Happe, R. P., W. Roseboom, G. Egert, C. G. Friedrich, C. Massanz, B. Friedrich, and S. P. J. Albracht. 2000. Unusual FTIR and EPR properties of the H2-activating site of the cytoplasmic NAD-reducing hydrogenase from Ralstonia eutropha. FEBS Lett. 466:259-263. [DOI] [PubMed] [Google Scholar]
- 13.Happe, T., K. Schutz, and H. Bohme. 2000. Transcriptional and mutational analysis of the uptake hydrogenase of the filamentous cyanobacterium Anabaena variabilis ATCC 29413. J. Bacteriol. 182:1624-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Houchins, J. P. 1984. The physiology and biochemistry of hydrogen metabolism in cyanobacteria. Biochim. Biophys. Acta 768:227-255. [Google Scholar]
- 15.Hyman, M. R., and D. J. Arp. 1988. Reversible and irreversible effects of nitric oxide on the soluble hydrogenase from Alcaligenes eutrophus H16. Biochem. J. 254:469-475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Joset, F., T. Buchou, C.-C. Zhang, and R. Jeanjean. 1988. Physiological and genetic analysis of the glucose-fructose permeation system in two Synechocystis species. Arch. Microbiol. 149:417-421. [Google Scholar]
- 17.Jouanneau, Y., B. C. Kelley, Y. Berlier, P. A. Lespinat, and P. M. Vignais. 1980. Continuous monitoring, by mass spectrometry, of H2 production and recycling in Rhodopseudomonas capsulata. J. Bacteriol. 143:628-636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaneko, T., S. Sato, H. Kotani, A. Tanaka, E. Asamizu, Y. Nakamura, N. Miyajima, M. Hirosawa, M. Sugiura, S. Sasamoto, T. Kimura, T. Hosouchi, A. Matsuno, A. Muraki, N. Nakazaki, K. Naruo, S. Okumura, S. Shimpo, C. Takeuchi, T. Wada, A. Watanabe, M. Yamada, M. Yasuda, and S. Tabata. 1996. Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res. 3:109-136. [DOI] [PubMed] [Google Scholar]
- 19.Kaplan, A., and L. Reinhold. 1999. CO2 concentrating mechanisms in photosynthetic microorganisms. Annu. Rev. Plant Physiol. Plant Mol. Biol. 50:539-570. [DOI] [PubMed] [Google Scholar]
- 20.Laczkó, I. 1986. Appearance of a reversible hydrogenase activity in Anabaena cylindrica grown in high light. Physiol. Plant. 67:634-637. [Google Scholar]
- 21.Lissolo, T., S. Pulvin, and D. Thomas. 1984. Reactivation of the hydrogenase from Desulfovibrio gigas by hydrogen. Influence of redox potential. J. Biol. Chem. 259:11725-11729. [PubMed] [Google Scholar]
- 22.Maeda, S., M. R. Badger, and G. D. Price. 2002. Novel gene products associated with NdhD3/D4-containing NDH-1 complexes are involved in photosynthetic CO2 hydration in the cyanobacterium, Synechococcus sp. PCC7942. Mol. Microbiol. 43:425-435. [DOI] [PubMed] [Google Scholar]
- 23.Marker, A. E. H. 1972. The use of acetone and methanol in the estimation of chlorophyll in the presence of pheophytin. Freshwater Biol. 2:361-385. [Google Scholar]
- 24.Markov, S. A., M. J. Bazin, and D. O. Hall. 1995. The potential of using cyanobacteria in photobioreactors for hydrogen production. Adv. Biochem. Eng. Biotechnol. 52:59-86. [Google Scholar]
- 25.Mi, H., T. Endo, U. Schreiber, T. Ogawa, and K. Asada. 1995. Thylakoid membrane-bound, NADPH-specific pyridine nucleotide dehydrogenase complex mediates cyclic electron transport in the cyanobacterium Synechocystis PCC 6803. Plant Cell Physiol. 36:661-668. [Google Scholar]
- 26.Mikheeva, L. E., O. Schmitz, S. V. Shestakov, and H. Bothe. 1995. Mutants of the cyanobacterium Anabaena variabilis altered in hydrogenase activities. Z. Naturforsch. C 50:505-510. [Google Scholar]
- 27.Nakamura, Y., T. Kaneko, M. Hirosawa, N. Miyajima, and S. Tabata. 1998. CyanoBase, a WWW database containing the complete nucleotide sequence of the genome of Synechocystis sp. strain PCC6803. Nucleic Acids Res. 26:63-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogawa, T. 1991. A gene homologous to the subunit-2 gene of NADH dehydrogenase is essential to inorganic carbon transport of Synechocystis PCC6803. Proc. Natl. Acad. Sci. USA 88:4275-4279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohkawa, H., H. B. Pakrasi, and T. Ogawa. 2000. Two types of functionally distinct NAD(P)H dehydrogenases in Synechocystis sp strain PCC6803. J. Biol. Chem. 275:31630-31634. [DOI] [PubMed] [Google Scholar]
- 30.Ohkawa, H., M. Sonoda, M. Shibata, and T. Ogawa. 2001. Localization of NAD(P)H dehydrogenase in the cyanobacterium Synechocystis sp. strain PCC 6803. J. Bacteriol. 183:4938-4939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pinto, F. A. L., O. Troshina, and P. Lindblad. 2002. A brief look at three decades of research on cyanobacterial hydrogen evolution. Int. J. Hydrog. Energy 27:1209-1215. [Google Scholar]
- 32.Rao, K. K., and D. O. Hall. 1996. Hydrogen production by cyanobacteria: potential, problems and prospects. J. Mar. Biotechnol. 4:10-15. [Google Scholar]
- 33.Schmitz, O., G. Boison, R. Hilscher, B. Hundeshagen, W. Zimmer, F. Lottspeich, and H. Bothe. 1995. Molecular biological analysis of a bidirectional hydrogenase from cyanobacteria. Eur. J. Biochem. 233:266-276. [DOI] [PubMed] [Google Scholar]
- 34.Schmitz, O., G. Boison, H. Salzmann, H. Bothe, K. Schutz, S. H. Wang, and T. Happe. 2002. HoxE—a subunit specific for the pentameric bidirectional hydrogenase complex (HoxEFUYH) of cyanobacteria. Biochim. Biophys. Acta 1554:66-74. [DOI] [PubMed] [Google Scholar]
- 35.Schmitz, O., and H. Bothe. 1996. NAD(P)+-dependent hydrogenase activity in extracts from the cyanobacterium Anacystis nidulans. FEMS Microbiol. Lett. 135:97-101. [Google Scholar]
- 36.Schneider, K., R. Cammack, H. G. Schlegel, and D. O. Hall. 1979. The iron-sulphur centres of soluble hydrogenase from Alcaligenes eutrophus. Biochim. Biophys. Acta 578:445-461. [DOI] [PubMed] [Google Scholar]
- 37.Schneider, K., and H. G. Schlegel. 1976. Purification and properties of soluble hydrogenase from Alcaligenes eutrophus H 16. Biochim. Biophys. Acta 452:66-80. [DOI] [PubMed] [Google Scholar]
- 38.Stal, L. J., and R. Moezelaar. 1997. Fermentation in cyanobacteria. FEMS Microbiol. Rev. 21:179-211. [Google Scholar]
- 39.Tamagnini, P., R. Axelsson, P. Lindberg, F. Oxelfelt, R. Wunschiers, and P. Lindblad. 2002. Hydrogenases and hydrogen metabolism of cyanobacteria. Microbiol. Mol. Biol. Rev. 66:1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tamagnini, P., J. L. Costa, L. Almeida, M. J. Oliveira, R. Salema, and P. Lindblad. 2000. Diversity of cyanobacterial hydrogenases, a molecular approach. Curr. Microbiol. 40:356-361. [DOI] [PubMed] [Google Scholar]
- 41.Tsygankov, A. A., A. S. Fedorov, S. N. Kosourov, and K. K. Rao. 2002. Hydrogen production by cyanobacteria in an automated outdoor photobioreactor under aerobic conditions. Biotechnol. Bioeng. 80:777-783. [DOI] [PubMed] [Google Scholar]
- 42.Vignais, P. M., B. Billoud, and J. Meyer. 2001. Classification and phylogeny of hydrogenases. FEMS Microbiol. Rev. 25:455-501. [DOI] [PubMed] [Google Scholar]
- 43.Vignais, P. M., L. Cournac, E. C. Hatchikian, S. Elsen, L. Serebryakova, N. Zorin, and B. Dimon. 2002. Continuous monitoring of the activation and activity of [NiFe]-hydrogenases by membrane-inlet mass spectrometry. Int. J. Hydrog. Energy 27:1441-1448. [Google Scholar]
- 44.Vignais, P. M., J. C. Willison, and A. Colbeau. 2004. H2 respiration. In D. Zannoni (ed.), Respiration in Archaea and Bacteria, vol 2. Diversity of prokaryotic respiratory systems, in press. Kluwer Academic Publishers, Dordrecht, The Netherlands.
- 45.Volbeda, A., Y. Montet, X. Vernede, E. C. Hatchikian, and J. C. Fontecilla-Camps. 2002. High-resolution crystallographic analysis of Desulfovibrio fructosovorans [NiFe]hydrogenase. Int. J. Hydrog. Energy 27:1449-1461. [Google Scholar]