

Abstract
Bacterial surface carbohydrates are important pathogenic factors in gram-negative pneumonia infections. Among these factors, O antigen has been reported to protect pathogens against complement-mediated killing. To examine further the role of O antigen, we insertionally inactivated the gene encoding a galactosyltransferase necessary for serotype O1 O-antigen synthesis (wbbO) from Klebsiella pneumoniae 43816. Analysis of the mutant lipopolysaccharide by sodium dodecyl sulfate-polyacrylamide gel electrophoresis confirmed the absence of O antigen. In vitro, there were no detectable differences between wild-type K. pneumoniae and the O-antigen-deficient mutant in regard to avid binding by murine complement C3 or resistance to serum- or whole-blood-mediated killing. Nevertheless, the 72-h 50% lethal dose of the wild-type strain was 30-fold greater than that of the mutant (2 × 103 versus 6 × 104 CFU) after intratracheal injection in ICR strain mice. Despite being less lethal, the mutant organism exhibited comparable intrapulmonary proliferation at 24 h compared to the level of the wild type. Whole-lung chemokine expression (CCL3 and CXCL2) and bronchoalveolar inflammatory cell content were also similar between the two infections. However, whereas the wild-type organism produced bacteremia within 24 h of infection in every instance, bacteremia was not seen in mutant-infected mice. These results suggest that during murine pneumonia caused by K. pneumoniae, O antigen contributes to lethality by increasing the propensity for bacteremia and not by significantly changing the early course of intrapulmonary infection.
Gram-negative infection is an important contributor to morbidity and mortality in hospitalized, immunosuppressed, and otherwise chronically ill patients. Under these circumstances, Klebsiella pneumoniae is among the most commonly encountered species; in a recent analysis of over 35,000 bacterial isolates from intensive care units in the United States, K. pneumoniae was the third most commonly isolated organism overall and the species most commonly identified in blood cultures, comprising 20% of all cases (17). A similar pattern has recently been noted in European hospitals (10).
Previous work has shown that two surface carbohydrate structures of K. pneumoniae, its capsular polysaccharide (CPS) and the O-antigen portion of its lipopolysaccharide (LPS), are important pathogenic determinants (1, 6). As the outermost components of the bacterial surface, these structures are among the first to be encountered by the innate immune system and are believed to defend against opsonization (by complement and other innate opsonins) and phagocytosis (18). Recent work has demonstrated that, of the two, CPS rather than O antigen is the more critical contributor to virulence in Klebsiella (4, 6). Genetic disruption of the gene cluster responsible for CPS production renders this organism, at the least the clinically prevalent K2 serotype, virtually nonpathogenic. While CPS is of undisputed importance, 80% of all clinical Klebsiella isolates, and 90% of all strains isolated from the blood, are CPS serotypeable, suggesting that in infected hosts the presence of CPS is largely a precondition of infection and that other virulence factors, such as O antigen, may still account for some of the variability in the severity of illness encountered in patients (7, 8).
The O antigens of Klebsiella sp. represent a family of molecules comprised of repeating carbohydrate subunits linked to the core antigen of LPS. In the case of serotype O1, one of the most frequently clinically isolated serotypes, O antigen is made of two disaccharides, d-galactan I [→3)-β-Galf-(1→3)-α-Galp-(1→] and d-galactan II [→3)-β-Galp-(1→3)-α-Galp-(1→] (12, 13, 24). d-galactan II forms the distal end of the O chains. The subunits are assembled into chains within the bacterial cytosol and subsequently transported to the outer membrane for linkage via an α-GlcN group to the lipid A and core antigen components of LPS (22). The synthetic enzymes and transport mechanism used in this process are encoded by the gene cluster rfb, and the process is initiated by two galactosyltransferases encoded by the genes wbbM and wbbO (5, 9).
In the present work, we further investigated the contribution of O antigen to the development of gram-negative pneumonia in a mouse model. Using an allelic replacement strategy to insertionally inactivate the wbbO gene from a virulent strain of K. pneumoniae O1:K2, we examined host response to wild-type and O-antigen-deficient bacteria in terms of complement binding, susceptibility to serum and whole-blood bactericidal activity, host lethality, and pulmonary inflammatory response. Our results demonstrate an important role for O antigen in this model system and suggest that the role of O antigen in the pathogenesis of lethal pneumonia is due in part to promoting bacteremia.
MATERIALS AND METHODS
Bacterial strains and plasmids used in this study are shown in Table 1. Six- to 10-week-old female ICR mice (Jackson Laboratories, Bar Harbor, Maine) maintained under specific pathogen-free conditions were used in most experiments. A C3 knock-out strain on a C57Bl/6J background (B6.129S4-C3tm1Crr) and C3-sufficient C57Bl/6J mice were obtained from Jackson Laboratories (23). Bacteria were grown in tryptic soy broth or on tryptic soy blood agar (Difco, Detroit, Mich.) at 37°C. In all experiments, bacteria at mid-log growth were washed and then resuspended in appropriate buffer and antibiotics. A previously determined relationship between the turbidity of the suspended organism at A600 and viable counts was used to quantify bacteria. Antisera and antibodies included goat anti-mouse C3 antiserum (ICN/Cappel, Aurora, Ohio), rabbit anti-K2 capsular antigen (Denka Seiken, Tokyo, Japan), rabbit anti-d-galactan I and II polyclonal antisera (generated in C. Whitfield's laboratory), and appropriate horseradish peroxidase- or alkaline phosphatase-conjugated secondary antibodies (Amersham Biosciences, Piscataway, N.J., and Jackson Laboratories). Cytokine enzyme immunoassay kits were purchased from R & D Systems, Minneapolis, Minn. All other reagents were obtained from Sigma Chemical Co., St. Louis, Mo., unless otherwise noted. The local animal use committee approved all in vivo experimental protocols.
TABLE 1.
Bacterial strains, plasmids, and primers used in this study
| Strain, plasmid, or primer | Description | Reference or source |
|---|---|---|
| K. pneumoniae | ||
| 43816 | Serotype O1:K2 | ATCC |
| 43816-01 | 43816 with a plasmid cointegrate in wbbO | This study |
| CWK2 | An O1:K− strain used as a positive control for LPS electrophoresis analysis | 24 |
| E. coli | ||
| BW20677 | E. coli for conjugation of suicide plasmid pLD55, pir+ | 14 |
| DH5α | F−φ80lacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rK−,mK+) phoA supE44 thi-1 gyrA96 relA1 λ− | Invitrogen |
| Plasmids | ||
| pWW84 | pUC containing kanamycin resistance cartridge | 16 |
| pBC KS+ | pBluescript with chloramphenicol resistance cassette | Stratagene |
| pLD55 | pBluescript II (SK+) F1(+)ori lacZαTetsoriRR6Kλ | 14 |
| Primers | ||
| 5′ wbbO (internal fragment) | CGGGATCCGGTGCAGGCATTAATCCAGAGA (BamH1 site underlined) | This study |
| 3′ wbbO (internal fragment) | ACGCGTCGACACCCATTTCAACACGTGCTTTAGG (Sal1 site underlined) | This study |
Construction of an O-antigen-deficient K. pneumoniae mutant.
The O1 and K2 serotypes of K. pneumoniae 43816 were confirmed by PCR of O1-specific rfb sequences and by anti-K2 antisera, respectively (data not shown). A wbbO mutant of K. pneumoniae 43816 was constructed by using a conditionally replicative allelic replacement vector (14). wbbO is the last gene of the operon involved in serotype O1 O-antigen biosynthesis, and its product is the first dedicated enzyme in the assembly pathway for the O1 antigen (5, 9). A 512-bp fragment spanning nucleotides 529 to 1041 of the O1 serotype wbbO gene (National Center for Biotechnology Information accession no. L31762) was cloned via PCR by using BamHI and SalI primer extensions (Table 1). The resulting fragment was inserted into the multiple cloning site of plasmid pBC and transformed into E. coli DH5α. An EcoR1 fragment containing 165 bp was removed, the ends were filled, and the fragment was replaced by a kanamycin resistance cassette (16). The fragment containing the insertionally inactivated wbbO gene was then cloned into the pir-dependent vector pLD55 and transformed into E. coli BW20677 (Π+). The construct was transferred from E. coli BW20677 to K. pneumoniae 43816 by conjugation to allow high-efficiency transfer. Recombinants were screened on kanamycin (50 μg/ml) tryptic soy blood agar. For reasons that have not been determined, it was not possible to generate a second crossover event to facilitate true allelic exchange. Instead, a mutant was selected with a plasmid cointegrate that insertionally inactivated wbbO. As this is the last gene in the operon, polarity effects are not an issue. The mutant containing the inactivated wbbO gene was named 43816-01.
Characterization of LPS.
LPS-containing cell lysates were purified from mid-log growth phase bacteria by using the sodium dodecyl sulfate (SDS)-proteinase K whole-cell method, as previously described (11). Samples were separated by SDS-polyacrylamide gel electrophoresis (PAGE). For analysis of LPS, gels were fixed in 40% ethanol-5% acetic acid with 300 mM periodic acid and then silver stained by using a modification of a previously published method (21). To detect d-galactan I and II content, LPS was transferred from PAGE gels to polyvinylidene difluoride membranes (Schleicher and Schuell Bioscience, Inc., Keene, N.H.). Immunoblots were probed with polyclonal rabbit serum against d-galactan I or II.
C3 opsonization assay.
C3 binding was assessed by using microtiter wells coated with ethanol-killed bacterial as previously described (25). Pooled fresh mouse serum was diluted from 1:2 to 1:32 in triethanolamine buffer (2.8% triethanolamine in 130 mM NaCl, 150 μM CaCl2, 500 μM MgCl2 [pH 7.35]) and then applied to bacteria-coated wells. The wells were incubated for 30 min at 37°C. C3 binding was assessed by using goat anti-mouse C3 antibody paired with an appropriate horseradish peroxidase-conjugated secondary antibody. The specificity of C3 binding was confirmed with parallel experiments performed in the presence of 10 mM EDTA.
Serum killing assay.
Bacterial susceptibility to complement-mediated killing was determined by incubating bacterial suspensions with normal mouse serum (NMS; initial dose, ∼103 CFU/100 μl) as recently described (25). Samples were incubated at 37°C, and aliquots were taken at baseline and at 15, 30, and 60 min and cultured quantitatively by serial dilution on tryptic soy blood agar.
Whole-blood killing assay.
Fresh whole blood was collected from the abdominal vena cava of normal mice into a lepirudin-filled syringe (final concentration, 50 μg/ml). Lepirudin (Hoechst Marion Roussel, Research Triangle Park, N.C.) is a direct thrombin inhibitor which does not inhibit complement activity in whole-blood assays (15). Blood was then mixed at a ratio of 1:1 with RPMI 1640 and distributed into microtiter wells. Cells of the wild type and wbbO mutant were added in quantities of 100 to 10,000 CFU per 100 μl of blood. Wells were incubated at 37°C in 5% CO2 for 2 h and then quantitatively cultured for bacterial content. Results were displayed as bacterial content at 2 h versus content of the initial inoculum. As an additional control, E. coli DH5α (a rough, nonvirulent strain) was studied in parallel experiments.
Murine pneumonia model.
Mice were anesthetized with 2% inhaled isofluorane (Abbott Laboratories, North Chicago, Ill.) and suspended vertically by their maxillary incisors. In each mouse, the trachea was exposed through a midline neck incision, and 30 μl of bacterial suspension was delivered intratracheally by a pipetter. The incision was closed with a surgical staple.
Survival analysis and determination of the 50% lethal dose (LD50).
For survival studies, mice received either wild-type or O-antigen-deficient bacteria in intratracheal doses ranging from 102 to 105 CFU. Following inoculation, animals were returned to standard housing and observed for 7 days. A census of survivors was taken daily. Based on these data, the probabilities of survival on postinoculation days 2 to 7 were modeled for each dose and each bacterial strain by using a logistical regression method provided in the software SAS 8.2 (SAS/STAT User's Guide, 4th ed., SAS Institute, Cary, N.C.) as previously described (20). The logistical model of probability of survival was constructed as follows:
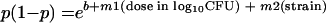 |
where p is the probability of survival on any experimental day and b and m1−2 are the y intercept and dose- and strain-related coefficients (in this case, the strain coefficient is −1 for mutant organisms and 1 for wild-type organisms), respectively. The statistical model returns values for b, m1, and m2 for each day of the experiment as well as levels of statistical significance between strains.
From these results, the 3-day LD50s of the wild-type and mutant strains were determined (2 × 103 CFU and 6 × 104 CFU, respectively). In subsequent in vivo experiments, two doses of the mutant were compared to doses of the wild type: 2 × 103 CFU (hereafter referred to as the isometric dose) and 6 × 104 CFU (hereafter referred to as the isolethal dose). In this manner, the bacterial strains could be compared not just in terms of the pattern of illness caused by an equal burden of bacteria but also in terms of the pattern of illness caused by an equally lethal exposure.
Lung bacterial clearance and development of bacteremia.
At 24 h postinoculation, animals were reanesthetized, and blood was collected in a sterile fashion. Lungs were flushed with sterile saline via the pulmonary artery and homogenized in sterile saline. Bacterial content was determined by plating serial 10-fold dilutions on tryptic soy blood agar and counting colonies following 24 h of incubation at 37°C.
Whole-lung cytokine measurement.
At 24 h after inoculation, animals were euthanized, and their pulmonary vasculature was flushed with sterile saline. Harvested lungs were homogenized in a 2× protease inhibitor cocktail (Complete; Roche Diagnostics GmbH, Mannheim, Germany), sonicated, and then passed through a 0.22-μm-pore-size filter before analysis. CCL3, CXCL2, interleukin-10 (IL-10), IL-12p40, and IL-18 were assayed by using commercially available murine enzyme immunoassay kits (R & D Systems) to measure CC and CXC chemokine levels and cytokine levels related to Th1-Th2 adaptive immune responses. Levels were standardized to total soluble lung protein.
Lung inflammatory cell content.
Airspace inflammatory cells were collected by bronchoalveolar lavage (BAL) 24 h after inoculation. Lungs of euthanized animals were lavaged three times with phosphate buffered saline. Recovered cells were counted with an electronic particle counter (Z-1; Beckman Coulter, Fullerton, Calif.) and reported as cells per 100 μl of recovered BAL fluid (BALF).
Statistical methods.
All results are reported either as means ± standard deviation unless otherwise noted. For serum and whole-blood bacterial killing assays and for whole-lung cytokine levels, raw data were log10 transformed prior to analysis. Analysis of variance with Tukey post hoc comparisons or Kruskal-Wallis testing with similar nonparametric post hoc comparisons was used where indicated. All statistical calculations were performed with SAS 8.2 software.
RESULTS
Confirmation of O-antigen-deficient phenotype in a wbbO mutant.
The wbbO gene encodes a galactosyltransferase that is essential for synthesis of the d-galactan I component of the O1 antigen (9). The d-galactan II component represents the distal end of the O1 antigen and is linked to the lipid A core via a d-galactan I (12, 13, 24). Electrophoresis of the whole-cell lysate confirmed by silver staining and by specific immunoblotting for d-galactan I and d-galactan II that the wbbO mutation strategy resulted in loss of detectable O antigen (Fig. 1).
FIG. 1.
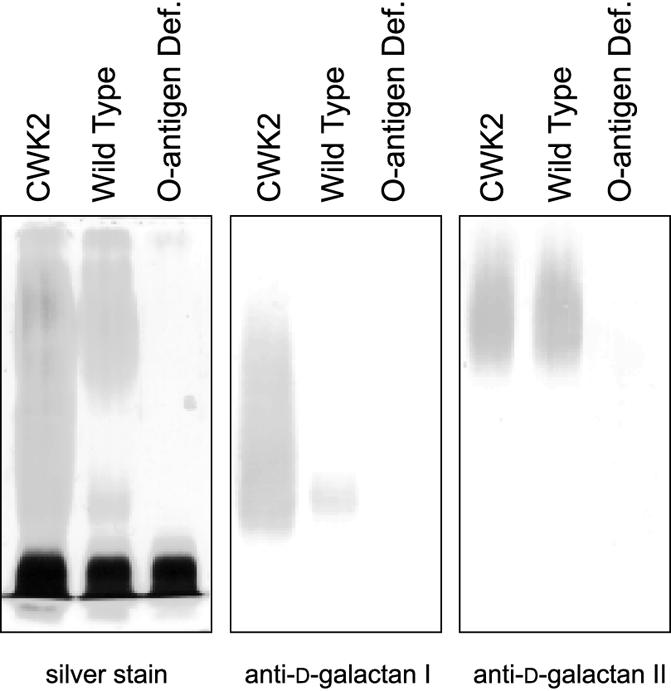
LPS phenotype from wild-type and wbbO mutant strains of K. pneumoniae O1. Periodic acid-silver stain (left panel) of LPS separated by SDS-PAGE. CWK2 is an O1:K− serotype strain used as a positive control. Compared to wild type, the wbbO mutant strain has lost its O antigen and only rough LPS remains. Immunoblots (center and right panels) of the same material developed with antibody that is specific for either the d-galactan I or d-galactan II components.
Opsonization by murine C3, and serum- and whole blood-mediated bacterial killing.
A commonly stated role for O antigen is providing defense against complement-mediated killing. Immunoassays for murine C3 on wild-type and mutant monolayers revealed no difference in EDTA-inhibitable C3 opsonization (Fig. 2). Both strains were resistant to killing by preimmune mouse serum, more than tripling their numbers during the 60-min serum exposure (Table 2). Analysis of whole-blood killing assays was comparable (Fig. 3). Both wild-type and mutant strains not only survived but continued to grow in essentially exponential fashion during the 2-h incubation with cultured whole blood.
FIG. 2.

In vitro opsonization of K. pneumoniae by murine C3. Murine serum was allowed to bind to bacterial monolayers for various amounts of time, after which an immunoassay was performed for the presence of C3. The presence or absence of O antigen did not change the magnitude of opsonization. Results are representative of at least three experiments. Abs, absorbance.
TABLE 2.
Bacterial killing by ICR-strain mouse serum
| Bacterial strain | Initial inoculum (103 CFU/100μl of NMS) | Bacterial content after incubationa (103 CFU/100 μl of NMS)
|
||
|---|---|---|---|---|
| 15 min | 30 min | 60 min | ||
| Wild type | 2.0 | 2.4 ± 0.2 | 3.7 ± 0.4 | 7.1 ± 0.2 |
| O-antigen-deficient mutant | 1.6 | 2.4 ± 0.9 | 4.2 ± 0.3 | 6.8 ± 0.5 |
Results are the means and standard deviations of three experiments each. Statistical comparisons between the wild type and the O-antigen-deficient mutant revealed no significant differences at any time point. n = 3 per group.
FIG. 3.

Susceptibility of wild-type and wbbO mutant bacteria to whole-blood-mediated killing. Various doses of both Klebsiella strains as well as the nonvirulent E. coli strain DH5a were incubated with lepirudin-anticoagulated whole blood for 2 h at 37°C. Quantitative culture results at the conclusion of the experiment were plotted against the size of the initial inocula. No statistically significant differences between wild-type and wbbO mutant strains were noted, but E. coli strain DH5α was significantly more susceptible to whole-blood bactericidal activity (P < 0.05). n = 3 per condition.
In vivo virulence and bacterial clearance from lung and blood.
Although in vitro assays of serum or whole-blood susceptibility revealed no detectable differences between wild-type and wbbO-inactivated K. pneumoniae strains, a significant difference in lethality during bacterial pneumonia was seen in vivo (Fig. 4). The 72-hour LD50 of the wild-type strain was 2 × 103 CFU, while the median lethal dose for the mutant at the same time point was 6 × 104 CFU, a 30-fold decrease in lethality associated with loss of the O antigen (P < 0.01). The difference in lethality could not be attributed to enhanced bacterial clearance from the lung alone; animals inoculated with equal numbers of wild-type or mutant organisms (i.e., isometric dosing) demonstrated similar numbers of recoverable bacteria 24 h after infection (Table 3). Instead, the difference between the two organisms at the 2 × 103 dose was the presence of bacteremia. Wild-type organisms were recovered from the blood of every infected animal at 24 h, while no organisms were detected in the blood of animals isometrically infected with 43816-01. Interestingly, when mice were infected with the isolethal mutant dose, bacteremia was observed. Taken together, these data suggest that the decrease in pathogenicity associated with loss of O-antigen expression is in part due to an increase in the threshold intratracheal dose at which bacteremia occurs and not to the extent to which bacteria are cleared from the lung early in the course of disease.
FIG. 4.

Host lethality following intratracheal infection with K. pneumoniae. Seven-day survival experiments following various doses of wild-type and O-antigen-deficient strains were performed as described in Materials and Methods. Results were used to construct logistic models of the probability of survival on any given day at any given dose of either strain. Based on these models, curves of the bacterial dose versus the probability of survival were plotted for days 2 to 7 for each Klebsiella strain. Results indicate an overall reduction of lethality of 30-fold in the O-antigen-deficient mutant strain (P < 0.01). These results were used to establish the 72-hour LD50 for the two strains (wild-type LD50, 2 × 103 CFU; mutant LD50, 6 × 104 CFU) that were used in subsequent experiments.
TABLE 3.
Lung and blood bacterial burden 24 h following inoculation with K. pneumoniae strains
| Specimen source | Median (range) CFU by straina
|
||
|---|---|---|---|
| Wild type | O-antigen-deficient mutant
|
||
| Isometric dose | Isolethal dose | ||
| Lung (entire organ) | 3 × 104 (9 × 102-1 × 105) | 5 × 104 (1 × 104-1 × 105) | 1 × 105 (3 × 103-4 × 105) |
| Blood (100 μl) | 9 × 101 (1 × 101-3 × 102) | None detected | 1 × 101 (1 × 100-5 × 101) |
n = 5 for each group.
Lethality of wild-type and mutant strains in C3-deficient mice.
To further explore the role of the complement system in regard to O-antigen-derived pathogenicity, we also examined the wild-type and O-antigen-deficient strains in C3−/− mice. Wild-type and C3−/− mice were infected with 103 CFU of wild-type or mutant bacteria and observed for survival over 96 h (Fig. 5). Survival to 96 h was approximately 25% in both mouse strains infected with the wild-type bacteria. However, among mice infected with the O-antigen-deficient strain, survival was significantly better (80%) in C3-sufficient animals (P < 0.01). The mutant bacterial strain produced a survival curve similar to that of wild-type bacteria in mice deficient in C3, suggesting that complement was contributing to host defense against the mutant, if not wild-type, bacterial challenge.
FIG. 5.

Host lethality in C3−/− mice. To examine in greater detail the in vivo role of complement in this model, C3−/− and control C3+/+ C57Bl/6J mice were intratracheally inoculated with 103 CFU of either wild-type or O-antigen-deficient K. pneumoniae and followed for 96 h for survival. In mice infected with the wild-type bacterial strain, no statistically significant differences were seen between C3+/+ and C3−/− animals, suggesting that the complement system was not significantly contributing to host defense. However, C3+/+ mice enjoyed significantly better survival compared to C3−/− animals when they were infected with the wbbO mutant (P < 0.05).
Whole-lung cytokine levels.
At 24 h after inoculation, whole-lung chemokine concentrations were elevated in animals infected with both wild-type and O-antigen-mutant K. pneumoniae strains (Fig. 6). Isometric inoculation with the wild-type and mutant strains produced comparable CC and CXC chemokine levels. However, isolethal infection with the mutant was associated with levels significantly elevated above wild-type infection (P < 0.01 for CCL3 and CXCL2). These data suggest that chemokine production in the lung during K. pneumoniae is more closely related to the size of the bacterial inoculum than to the invasiveness of the infection.
FIG. 6.

Whole-lung cytokine and chemokine content 24 h after inoculation. Homogenized whole lungs were assayed for CCL3, CXCL2, and the three cytokines associated with Th1-Th2 differentiation (IL-10, IL-12 p40, and IL-18) 24 h after bacterial inoculation, and results were expressed as picograms per milligram of total soluble lung protein. IL-10, IL-12, and IL-18 levels remained very low during the experimental period, and no statistically significant differences were seen. However, levels of both chemokines increased roughly 100-fold within 24 h of infection, with levels being comparable in mice infected with equal doses of either wild-type or mutant strains (P < 0.01). In animals infected with an isolethal rather than isometric dose of the Klebsiella mutant, chemokine levels were still higher, roughly 500 times levels seen in sham-infected animals (P < 0.01). For each group, n = 5; comparisons between groups were made with the Kruskal-Wallis test for non-normally distributed data. ND, none detected; n.s., not significant.
Unlike the chemokine response, levels of cytokines regulating cell-mediated immunity (IL-10, IL-12, and IL-18) remained very low during the first 24 h of infection in all experimental groups and no statistically significant differences were detected.
BALF leukocyte content.
Measurement of BALF leukocytes 24 h after inoculation demonstrated a pattern that paralleled the whole-lung bacterial burden and CC and CXC chemokine data. Mice infected with the wild-type and isometric mutant doses had comparable numbers of inflammatory cells in the their bronchoalveolar space, while animals receiving the isolethal dose of the mutant had significantly greater numbers of leukocytes (Fig. 7).
FIG. 7.

Leukocyte content of BALF 24 h after inoculation. Animals infected with wild-type bacteria and the isometric dose of the O-antigen mutant had comparable numbers of leukocytes (P, not significant). However, animals infected with the isolethal dose of the mutant strain had significantly greater leukocyte influx into the lung (P < 0.05 by analysis of variance). Boxes represent 25th, 50th, and 75th percentiles. Whiskers represent the total range of measured values. For wild type, n = 4; for both mutant groups, n = 5.
DISCUSSION
Gram-negative organisms display a number of surface components that contribute to their pathogenic potential, including CPS and O antigen. Although the former traditionally has been considered a defense against phagocytosis and the latter has been considered a defense against complement-mediated lysis, these distinctions have blurred as the recognized complexity of innate host defenses has grown. In the present work, we have used a conditionally replicative allelic exchange strategy to insertionally inactivate a galactosyltransferase needed by K. pneumoniae serotype O1 to initiate synthesis of O antigen. This strategy allowed selective deletion of O antigen from the bacterial surface and, as a result, significantly altered the host manifestations of acute bacterial pneumonia. Most notably, loss of O antigen from K. pneumoniae O1 resulted in a 30-fold decrease in lethality compared to wild-type bacteria and increased the magnitude of exposure required to produce bacteremia.
In a recent report, similar methods were used to compare the relative contributions of CPS and O antigen in lung infection by K. pneumoniae O1:K2 strain 52145 (6). In that work, also conducted in ICR mice, deletion of CPS was shown to render very large intratracheal doses of the pathogen nonlethal. No change in virulence was noted with the loss of O antigen. However, the dose used was very much higher than that needed to cause an inflammatory response, bacteremia, or death (107 CFU in contrast to the range of 103 to 104 CFU used in the present study) and may have overcome any subtle effects conveyed by the loss of O antigen. The range-finding results shown in Fig. 4 indicate that this polysaccharide does make a significant contribution to pathogenicity that is most evident at doses near the LD50.
As others have reported, we too were unable to demonstrate a difference in C3 binding between wild-type and O-antigen-deficient strains of Klebsiella, although to our knowledge ours is the first quantitation of mouse, rather than human, complement opsonization of this pathogen. Human complement is known to bind to both CPS and O antigen, as well as to at least one outer membrane protein (OmpK36) in Klebsiella; removal of the O antigen may have changed the sites where binding occurred without changing the overall magnitude of opsonization (1-4). In vitro susceptibility parameters were similarly unaffected by removal of O antigen (Fig. 3 and Table 2). While O antigen has been shown to convey to gram-negative organisms resistance to human serum-mediated lysis, this is not necessarily the case in mice (25).
When delivered intratracheally in a dose isometric to the 72-hour LD50 of wild-type K. pneumoniae, the wbbO mutant differed most notably in its failure to produce bacteremia despite proliferating within the lung to an extent similar to wild-type infection (Table 3). What remains to be determined is to what degree these findings are a result of decreased bacterial egress from the lung compared to enhanced clearance of the O-antigen-deficient organisms by the mononuclear phagocyte system in the liver and spleen. Regardless of which of these mechanisms is primarily responsible, increasing the size of the mutant inoculum to an isolethal dose resulted in bacteremia, further supporting the conclusion that in murine systems the lethality of K. pneumoniae is largely a result of overwhelming systemic infection rather than of the local effects of acute lung injury.
Additional work is needed to understand the role of complement in this model of gram-negative pneumonia. Although we were unable to demonstrate complement-dependent humoral or cellular killing in vitro, C3−/− mice experienced significantly greater mortality following challenge with the O-antigen mutant, indicating that a role for the complement system exists nonetheless. We hypothesize that in this instance complement may be enhancing the efficiency with which extrapulmonary bacteria are removed from the bloodstream, perhaps as a result of increased immune adherence via CR1 or through enhanced killing by splenic or hepatic macrophages (19).
We encountered two important technical challenges with K. pneumoniae 43816-01. First, we were unable to detect the second crossover event classically described with the allelic replacement strategy that we employed. The result was a plasmid cointegrant rather than a strain containing a full allelic exchange. Second, we have thus far been unable to restore the wild-type phenotype of 43816-01 by using the rfb gene containing plasmid pWQ3 as has previously been described by one of our group (5). The possibility of a second inadvertent mutation contributing to the differences of illness produced by the wild-type and plasmid cointegrant strains cannot be ruled out. However, two arguments can be made to indicate that disruption of the O-antigen synthetic pathway is responsible for the phenotypic differences we have described. First, allelic exchange is highly specific in its targeting. Second, 3′-adjacent to the rfb cluster in K. pneumoniae is the cluster for capsule production. We have found that strains 43816 and 43816-01 equally bind K2-specific anticapsular antibodies (data not shown), suggesting that the gene cluster nearest rfb is functioning appropriately.
In summary, at intratracheal doses near the LD50, the O antigen of K. pneumoniae O1:K2 significantly contributes to lethality. The most evident effect of the loss of O antigen is to prevent bacteremia early in the course of infection, rather than to limit the intrapulmonary proliferation of the pathogen. In vitro studies of serum- or whole-blood-mediated killing do not reveal obvious differences between wild-type and O-antigen-deficient strains, although an important role for the complement system is strongly suggested by differences in lethality between C3+/+ and C3−/− mice. Further work on the effect of bacterial surface carbohydrates on the propensity of K. pneumoniae to produce bacteremia and the means by which complement modulates (or fails to modulate) this phenomenon is needed.
Acknowledgments
This work was funded by federal grants HL-03817 (J.G.Y.), GM-61656 (J.G.Y. and G.A.V.), and GM-47156 (R.A.B.) and by career investigator awards from the Emergency Medicine Foundation and the American Lung Association of Michigan (J.G.Y.).
Editor: J. N. Weiser
REFERENCES
- 1.Alberti, S., D. Alvarez, S. Merino, M. Casado, F. Vivanco, J. Tomas, and V. Benedi. 1996. Analysis of complement C3 deposition and degradation on Klebsiella pneumoniae. Infect. Immun. 64:4726-4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alberti, S., G. Marques, S. Hernandez-Alles, X. Rubires, J. Tomas, F. Vivanco, and V. Benedi. 1996. Interaction between complement subcomponent C1q and the Klebsiella pneumoniae porin OmpK36. Infect. Immun. 64:4719-4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alberti, S., F. Rodriguez-Quinones, T. Schirmer, G. Rummel, J. Tomas, J. Rosenbusch, and V. Benedi. 1995. A porin from Klebsiella pneumoniae: sequence homology, three-dimensional model, and complement binding. Infect. Immun. 63:903-910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alvarez, D., S. Merino, J. Tomas, V. Benedi, and S. Alberti. 2000. Capsular polysaccharide is a major complement resistance factor in lipopolysaccharide O side chain-deficient Klebsiella pneumoniae clinical isolates. Infect. Immun. 68:953-955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clarke, B., D. Bronner, W. Keenleyside, W. Severn, J. Richards, and C. Whitfield. 1995. Role of Rfe and RfbF in the initiation of biosynthesis of d-galactan I, the lipopolysaccharide O antigen from Klebsiella pneumoniae serotype O1. J. Bacteriol. 177:5411-5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cortes, G., N. Borrell, B. de Astorza, C. Gomez, J. Sauleda, and S. Alberti. 2002. Molecular analysis of the contribution of the capsular polysaccharide and the lipopolysaccharide O side chain to the virulence of Klebsiella pneumoniae in a murine model of pneumonia. Infect. Immun. 70:2583-2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cryz, S., P. Mortimer, V. Mansfield, and R. Germanier. 1986. Seroepidemiology of Klebsiella bacteremic isolates and implications for vaccine development. J. Clin. Microbiol. 23:687-690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fung, C., B. Hu, F. Change, S. Lee, B. Kuo, M. Ho, L. Siu, and C. Lie. 2000. A 5-year study of the seroepidemiology of Klebsiella pneumoniae: high prevalence of capsular serotype K1 in Taiwan and implication for vaccine efficacy. J. Infect. Dis. 181:2075-2079. [DOI] [PubMed] [Google Scholar]
- 9.Guan, S., A. Clarke, and C. Whitfield. 2001. Functional analysis of the galactosyltransferases required for biosynthesis of d-galactan I, a component of the lipopolysaccharide O1 antigen of Klebsiella pneumoniae. J. Bacteriol. 183:3318-3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanberger, H., J. Garcia-Rodriguez, M. Gobernado, H. Goosens, L. Nilsson, and M. Struelens. 1999. Antibiotic susceptibility among aerobic gram-negative bacilli in intensive care units in 5 European countries. JAMA 281:67-71. [DOI] [PubMed] [Google Scholar]
- 11.Hitchcock, P., and T. Brown. 1983. Morphological heterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained polyacrylamide gels. J. Bacteriol. 154:269-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kol, O., J. Wieruszeski, G. Strecker, B. Fournet, R. Zalisz, and P. Smets. 1992. Structure of the O-specific polysaccharide chain of Klebsiella pneumoniae O1K2 (NCTC 5055) lipopolysaccharide. A complementary elucidation. Carbohydrate Res. 236:339-344. [DOI] [PubMed] [Google Scholar]
- 13.Kol, O., J. Wieruszeski, G. Strecker, J. Montreuil, B. Fournet, R. Zalisz, and P. Smets. 1991. Structure of the O-specific carbohydrate polysaccharide chain from Klebsiella pneumoniae O1K2 (NCTC 5055) lipopolysaccharide. Carbohydrate Res. 217:117-125. [DOI] [PubMed] [Google Scholar]
- 14.Metcalf, W., W. Jiang, L. Daniels, S. Kim, A. Haldimann, and B. Wanner. 1996. Conditionally replicative and conjugative plasmids carrying lacZa for cloning, mutagenesis, and allele replacement in bacteria. Plasmid 35:1-13. [DOI] [PubMed] [Google Scholar]
- 15.Mollnes, T. E., O. L. Brekke, M. Fung, H. Fure, D. Christiansen, G. Bergseth, V. Videm, K. T. Lappegard, J. Kohl, and J. D. Lambris 2002. Essential role of the C5a receptor in E. coli-induced oxidative burst and phagocytosis revealed by a novel lepirudin-based human whole blood model of inflammation. Blood 100:1869-1877. [PubMed] [Google Scholar]
- 16.Muller, W., W. Keppner, and I. Rasched. 1986. Versatile kanamycin-resistance cartridges for vector construction in Escherichia coli. Gene 46:131-133. [DOI] [PubMed] [Google Scholar]
- 17.Neuhauser, M., R. Weinstein, R. Rydman, L. Danziger, G. Karam, and J. Quinn. 2003. Antibiotic resistance among gram-negative bacilli in U.S. intensive care units. JAMA 289:885-888. [DOI] [PubMed] [Google Scholar]
- 18.Podschun, R., and U. Ullman. 1998. Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin. Microbiol. Rev. 11:589-603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prodinger, W., R. Wurzner, A. Erdei, and M. Dierich. 1999. Complement, p. 967-996. In W. Paul (ed.), Fundamental immunology, 4th ed. Lippincott-Raven, Philadelphia, Pa.
- 20.Soldes, O., J. Younger, and R. Hirschl. 1999. Predictors of malignancy in childhood peripheral lymphadenopathy. J. Ped. Surg. 34:1447-1452. [DOI] [PubMed] [Google Scholar]
- 21.Tsai, G., and C. E. Frasch. 1982. A sensitive silver stain for detecting lipopolysaccharides in polyacrylamide gels. Anal. Biochem. 119:115-119. [DOI] [PubMed] [Google Scholar]
- 22.Vinogradov, E., E. Frirdich, L. L. MacLean, M. B. Perry, B. O. Petersen, J. O. Duus, and C. Whitfield. 2002. Structures of lipopolysaccharides from Klebsiella pneumoniae. Elucidation of the structure of the linkage region between core and polysaccharide O chain and identification of the residues at the non-reducing termini of the O chains. J. Biol. Chem. 277:25070-25081. [DOI] [PubMed] [Google Scholar]
- 23.Wessels, M., P. Butko, M. Ma, H. Warren, A. Lage, and M. Carroll. 1995. Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc. Natl. Acad. Sci. USA 92:11490-11494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whitfield, C., J. Richards, M. Perry, B. Clarke, and L. MacLean. 1991. Expression of two structurally distinct d-galactan O antigens in the lipopolysaccharide of Klebsiella pneumoniae serotype O1. J. Bacteriol. 173:1420-1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Younger, J., S. Shankar-Sinha, M. Mickiewicz, A. Brinkman, E. Younkin, J. Sarma, T. Standiford, F. Zetoune, and P. Ward. 2003. Murine complement interactions with Pseudomonas aeruginosa and their effects during acute pneumonia. Am. J. Respir. Cell Mol. Biol. 29:432-438. [DOI] [PMC free article] [PubMed] [Google Scholar]