

SUMMARY
Neural networks that regulate an organism’s internal environment must sense perturbations, respond appropriately, and then reset. These adaptations should be reflected as changes in the efficacy of the synapses that drive the final output of these homeostatic networks. Here we show that hemorrhage, an in vivo challenge to fluid homeostasis, induces LTD at glutamate synapses onto hypothalamic magnocellular neurosecretory cells (MNCs). LTD requires the activation of postsynaptic α2-adrenoceptors and the production of endocannabinoids that act in a retrograde fashion to inhibit glutamate release. In addition, both hemorrhage and noradrenaline downregulate presynaptic group III mGluRs. This loss of mGluR function allows high-frequency activity to potentiate these synapses from their depressed state. These findings demonstrate that noradrenaline controls a form of metaplasticity that may underlie the resetting of homeostatic networks following a successful response to an acute physiological challenge.
INTRODUCTION
The survival of an organism depends upon its ability to quickly, effectively, and repeatedly defend homeostatic setpoints. This requires neural networks that regulate autonomic and neuroendocrine outputs to possess intrinsic mechanisms that allow them to respond to a challenge, adapt, and then reset. The precise nature of these mechanisms remains unresolved. Here we hypothesized that bidirectional changes in synaptic strength may be particularly useful in regulating the output of networks that must make continual adjustments in response to moment-to-moment changes in physiological input.
While persistent changes in synaptic efficacy, such as long-term potentiation (LTP) and long-term depression (LTD), are essential for information storage in neural networks (Malenka and Bear, 2004), a complementary mechanism, metaplasticity, maintains network stability and ensures that synapses operate within a functional dynamic range by modifying the expression of synaptic plasticity as a function of prior activity (Abraham, 2008). Presynaptic metabotropic glutamate receptors (mGluRs), which regulate the release of neurotransmitter at nerve terminals, have recently been implicated in a novel form of metaplasticity in the hippocampus (Pelkey et al., 2005). In this scenario, group III mGluRs undergo agonist-induced internalization, unmasking a state permissive for subsequent activity-dependent LTP of glutamate synapses. Loss of mGluR function has also been reported at glutamatergic synapses onto hypothalamic magnocellular neurosecretory cells (MNCs) in the paraventricular nucleus of the hypothalamus (PVN) (Gordon and Bains, 2003). These neurons integrate local and synaptic information regarding blood volume and make neuroendocrine adjustments to maintain body fluid homeostasis (Bourque, 2008; Poulain and Wakerley, 1982). A precipitous drop in blood volume triggers an immediate increase in the activity of these cells (Poulain et al., 1977; Wakerly et al., 1975) that is driven by the release of noradrenaline (NA) from ascending fibers (Buller et al., 1999). While this causes an immediate increase in neuronal activity through synaptic actions that increase quantal glutamate release (Boudaba et al., 2003; Gordon and Bains, 2003) and promote the postsynaptic insertion of AMPA receptors (Gordon et al., 2005), NA is also the substrate necessary for the functional inactivation of presynaptic high-affinity, group III mGluRs (Gordon and Bains, 2003).
Here we asked whether glutamate synapses onto MNCs adapt following in vivo hemorrhage and then whether these adaptations would favor an activity-dependent resetting of these synapses. Using whole-cell patch-clamp recordings from MNCs in hypothalamic brain slices, we demonstrate that in vivo hemorrhage-induced release of NA in the PVN decreases the probability of evoked glutamate release onto MNCs. This LTD is mimicked by bath application of NA to naive slices and is accompanied by a complete loss of function of group III mGluRs. Subsequent high-frequency stimulation (HFS) can cause both restoration of synaptic strength and a gain of mGluR function. These observations demonstrate that mGluRs are essential for metaplastic synaptic changes that may allow networks regulating fluid homeostasis to adapt and reset.
RESULTS
Presynaptic LTD following In Vivo Hemorrhage
To examine synaptic changes responsible for restoring body fluid volume homeostasis, we assessed synaptic efficacy in hypothalamic slices obtained from rats that had been subjected to a hypovolemic hemorrhage (removal of 25% blood volume), a stimulus that strongly activates MNCs in the PVN of the hypothalamus (Buller et al., 1999; Poulain et al., 1977; Wakerly et al., 1975). Brain slices containing the PVN were prepared 30 min after the hemorrhage to allow a sustained period of noradrenergic drive and increased neuronal activity. Glutamatergic synaptic transmission was examined in acute rat hypothalamic slices by recording from visually and electrophysiologically identified (Luther and Tasker, 2000) MNCs in the PVN. In comparison to control slices, evoked excitatory postsynaptic currents (eEPSCs) recorded from hemorrhaged rats displayed a lower probability of glutamate release (Pr) as indicated by a significantly enhanced paired-pulse ratio (PPR; EPSC2/EPSC1: control 1.51 ± 0.07; n = 57; hemorrhage 1.95 ± 0.19; n = 15; p = 0.009), more synaptic failures (failure rate, failures/30 events: control 0.04 ± 0.01; hemorrhage 0.15 ± 0.04; p = 0.0004), and an increase in the coefficient of variation (CV; standard deviation/mean; control 0.39 ± 0.02; hemorrhage 0.50 ± 0.04; p = 0.02; Figures 1A and 1B). These measures of reduced synaptic release efficacy persisted for at least 4.5 hr after slicing. No significant changes in PPR were noted if brain slices were prepared immediately (2 min) following the hemorrhage (control 1.51 ± 0.07; n = 57; 2 min hemorrhage 1.35 ± 0.13; n = 18; p = 0.28). These observations indicate that glutamate synapses onto MNCs exhibit a presynaptic LTD following hemorrhage.
Figure 1. Hemorrhage Lowers Pr at Glutamate Synapses.

(A) eEPSCs in slices obtained from hemorrhaged rats (blue) displayed an increased PPR when compared with controls (black). Current traces are averages of 30 consecutive responses from each condition. For paired-pulse recordings, synaptic stimuli were delivered 50 ms apart. Scale bars: 10 ms, 20 pA.
(B) Summary graph illustrating that hemorrhaged rats (blue; n = 15) had a reduced Pr compared to nonhemorrhaged naive rats (black; n = 57), indicated by significant increases in PPR, failure rate, and CV. *p < 0.05, **p < 0.01.
(C–E) Example of an injection site from animals microinjected with prazosin, yohimbine, or vehicle into the PVN prior to hemorrhage. The image shown is from case 5 of the yohimbine-injected group (see Figure S1). (C) Low-power view (4X) of a transmitted light image of a coronal slice of the PVN. (D) The injection site as indicated by the red fluorescence of the Fluospheres. (E) Superimposed transmitted light and epifluorescence images. 3V, third ventricle.
(F–H) Bar chart summaries of PPR, failure rate, and CV of eEPSCs obtained from control naive slices (control; n = 57) and slices from animals with yohimbine (10 nmol; n = 6), prazosin (10 nmol; n = 8), or vehicle (n = 4) microinjected into the PVN prior to hemorrhage.
Since perturbations to body fluid homeostasis sensed by peripheral receptors are relayed by noradrenergic fibers to neurons in the hypothalamus (Buller et al., 1999; Sawchenko and Swanson, 1981), we hypothesized that LTD following hemorrhage was mediated via activation of noradrenergic receptors. To test this directly we microinjected subtype selective adrenoceptor antagonists directly into the PVN prior to hemorrhaging the rat (Figures 1C–1E). Figure S1 (available online) shows the distribution of the localized injection sites for all subjects. Microinjection of the α2-adrenoceptor antagonist yohimbine (10 nmol) blocked the hemorrhage-mediated LTD (PPR 1.37 ± 0.14; n = 6; p = 0.53; failure rate 0.04 ± 0.019; p = 0.92; CV 0.38 ± 0.049; p = 0.8; Figures 1F–1H). In contrast, microinjection of the α1-adrenoceptor antagonist prazosin (10 nmol) into the PVN failed to prevent LTD following hemorrhage (PPR 2.06 ± 0.43; n = 8; p = 0.02; failure rate 0.18 ± 0.08; p = 0.0009; CV 0.58 ± 0.12; p = 0.009). Vehicle injection had no effect on hemorrhage-induced LTD (PPR 1.95 ± 0.53; n = 4; p = 0.06; failure rate 0.12 ± 0.029; p = 0.04; CV 0.58 ± 0.027; p = 0.01). These results demonstrate that hemorrhage results in LTD of glutamate synapses that requires the activation of α2, but not α1, -adrenoceptors.
α2-Adrenoceptors and Endocannabinoids Are Required for NA-Induced LTD
We next determined the mechanism through which α2-adrenoceptor activation induces LTD. In slices from naive, nonhemorrhaged rats bath application of NA (100 μM; 5 min) produced a rapid depression of eEPSCs that relaxed to a less robust, but LTD (LTDNA; eEPSC amplitude = 56.28% ± 6.67% of control; n = 17; p < 0.0001; Figures 2A and 2B). Similar to LTD observed following hemorrhage, LTDNA was also associated with an increase in PPR (control 1.32 ± 0.10; post-NA 1.69 ± 0.17; n = 7; p = 0.02; Figure 2C), an increase in the failure rate (control 0.08 ± 0.05; post-NA 0.47 ± 0.14; p = 0.02), and an increase in CV (control 0.49 ± 0.09; post-NA 0.76 ± 0.14; p = 0.01).
Figure 2. Noradrenaline Induces Presynaptic LTD.
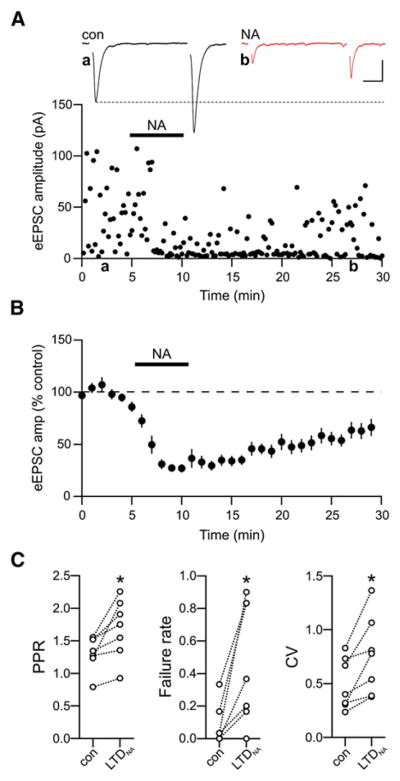
(A) Representative single eEPSC recording illustrating LTD of evoked glutamate release induced by NA (LTDNA). Traces show average paired-pulse eEPSC responses in control (black) and 20 min after wash of NA (red) at the times indicated. Scale bars: 10 ms, 20 pA.
(B) Data normalized and averaged from multiple neurons shows time course and extent of LTDNA (n = 17).
(C) LTDNA was accompanied by an increase in PPR, failure rate, and CV. *p < 0.05; n = 7.
To more carefully determine the adrenoceptor subtypes necessary for LTDNA, we conducted a number of pharmacological experiments. To test whether α1-adrenoceptors were necessary for LTDNA, we bath-applied the selective α1-adrenoceptor agonist phenylephrine (PE; 50 μM). This failed to elicit LTDNA (eEPSC amplitude = 122.78% ± 23.55% of control; n = 5; p = 0.39; Figure 3F). In addition, the specific α1-adrenoceptor antagonist prazosin (10 μM) failed to block induction of LTDNA in response to bath application of NA (eEPSC amplitude = 68.76% ± 6.27% of control; n = 5; p = 0.04; Figure 3F), a result consistent with the observed synaptic efficacy after in vivo hemorrhage. Conversely, application of the α2-adrenoceptor-subtype-selective antagonist yohimbine (20 μM) prior to and during NA application prevented induction of LTDNA (eEPSC amplitude = 93.20% ± 5.36% of control; n = 7; p = 0.22; Figures 3A and 3F). If, however, yohimbine was applied after the induction of LTDNA, synapses remained depressed (eEPSC amplitude = 59.23% ± 12.85%; n = 5; p = 0.04; Figure 3F). Application of the α2-adrenoceptor agonist clonidine (50 μM) also caused a long-lasting presynaptically expressed depression of evoked glutamate release (eEPSC amplitude = 57.07% ± 7.65% of control; n = 6; p = 0.009; PPR control 1.25 ± 0.23; PPR clonidine 2.70 ± 0.68; Figures 3B and 3F). This depression was not due to slow unbinding of clonidine from the α2-adrenoceptor because application of a yohimbine chaser after induction of LTDNA had no effect (Figure 3B).
Figure 3. LTDNA Requires Postsynaptic α2-Adre-noceptor Activation and Endocannabinoids.

(A) Average normalized eEPSC amplitude of experiments where NA was applied in the presence of the α2-adrenoceptor antagonist yohimbine (20 μM; n = 7). Inset scale bars: 10 ms, 50 pA.
(B) Average eEPSC amplitude of experiments where clonidine (50 μM; n = 6) was applied alone (open circles) and where application of clonidine was chased with yohimbine (n = 7; closed circles) at the time indicated. Inset scale bars: 10 ms, 20 pA.
(C) Effects of clonidine on eEPSCs recorded with an intracellular solution containing GDP-βs (1 mM; n = 8). Inset scale bars: 10 ms, 50 pA.
(D) Average normalized eEPSC amplitude of experiments where the CB1 receptor antagonist AM251 (1 μM) was applied at least 5 min prior to NA (n = 8). Inset scale bars: 10 ms, 200 pA.
(E) Summary of NA experiments where 10 mM BAPTA was included in the patch pipette. BAPTA was allowed to perfuse into the neuron for at least 15 min before NA was applied (n = 12). Inset scale bars: 10 ms, 100 pA.
(F) Summary data showing the percent change in eEPSC amplitude after 20 min wash of NA (100 μM; n = 17), phenylephrine (PE; 50 μM; n = 5), NA in prazosin (praz + NA; 10 μM; n = 5), NA in yohimbine (yoh + NA; 20 μM; n = 7), NA chased with yohimbine (NA + yoh chaser; n = 5), clonidine (clon; 50 μM; n = 6), clonidine followed by a yohimbine chaser (clon + yoh chaser; n = 7), clonidine in cells patched with GDP-βs included in the intracellular solution (GDP-βs + clon; n = 8), NA in the presence of AM251 (AM251 + NA), and NA in cells patched with an intracellular solution containing BAPTA (BAPTA + NA). *p < 0.05; **p < 0.01.
While the above observations strongly suggest that LTDNA requires α2-adrenoceptors and is expressed presynaptically as a reduction in glutamate Pr, they do not rule out the possibility that α2-adrenoceptors are located on the postsynaptic cell and when activated, produce a retrograde signal that depresses glutamate release. Indeed, there is compelling evidence for postsynaptic α2-adrenoceptors on MNCs in the PVN (Shirasaka et al., 2007) as well as in other brain structures (Carr et al., 2007; Cathala et al., 2002; Li and van den Pol, 2005; Liu and Alreja, 1998; Yamanaka et al., 2006), strengthening evidence that activation of postsynaptic Gi/o-coupled receptors can promote endocannabinoid (eCB) production through activation of phospholipase D (PLD) (Basavarajappa, 2007; Di Marzo et al., 1994; Jinsi-Parimoo and Deth, 1997; Senogles, 2000), and that CB1 receptors are located on nerve terminals in the hypothalamic magnocellular neurosecretory system (Di et al., 2005a; Hirasawa et al., 2004; Oliet et al., 2007). We directly tested for the involvement of postsynaptic α2-adrenoceptors by conducting experiments in which we included GDP-βs (1 mM) in our patch pipette to block postsynaptic G protein signaling. Following perfusion of the postsynaptic cell with GDP-βs for at least 10 min, application of clonidine had no effect on eEPSCs amplitude (eEPSC amplitude = 104.56% ± 8.94%; n = 8, p = 0.87; Figure 3C). This indicates that α2-adrenoceptors are located on the postsynaptic membrane. Since other Gi/o-coupled receptors act to depress transmitter release through the recruitment of eCBs (Giuffrida et al., 1999; Kreitzer and Malenka, 2007; Melis et al., 2004; Yin and Lovinger, 2006), we conducted additional experiments in which NA was bath applied in the presence of a CB1 receptor antagonist N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methly-1 H-pyrazole-3-carboximide (AM251; 1 μM). The induction of LTDNA was prevented under these experimental conditions (eEPSC amplitude = 116.34% ± 12.07% of control; n = 8; p = 0.30; Figure 3D).
It is not clear whether eCB production following activation of Gi/o-coupled receptors is Ca2+ dependent. There is evidence to support a direct, Ca2+-independent action of PLD (Jin et al., 2007), but PLD also exhibits, if not a requirement for Ca2+, then at least a reliance on Ca2+ (Kano et al., 2009; Ueda et al., 2001). To determine whether α2-induced eCB production relies on postsynaptic Ca2+, we loaded the postsynaptic cell with the Ca2+ chelator BAPTA (10 mM) (Gerdeman et al., 2002; Robbe et al., 2002). Under these conditions, NA failed to induce LTDNA (eEPSC amplitude = 95.64% ± 14.88% of control; n = 12; p = 0.22; Figure 3E). These observations show that LTDNA requires the activation of postsynaptic α2-adrenoceptors, which recruits, in a Ca2+-dependent fashion, the production of eCBs that act on the presynaptic nerve terminal to decrease glutamate release.
LTD Is Accompanied by an α1-Mediated Functional Downregulation of Group III mGluRs
In addition to activating α2-adrenoceptors, the release of NA during hemorrhage will also stimulate α1-adrenoceptors. These receptors are located on both the presynaptic nerve terminal and neighboring astrocytes (Gordon and Bains, 2005). The activation of presynaptic α1-adrenoceptors functionally inactivates high-affinity group III mGluRs at synapses onto MNCs in the PVN (Gordon and Bains, 2003). Since group III mGluRs gate metaplasticity in the hippocampus (Pelkey et al., 2005, 2008), we hypothesized that NA released during hemorrhage or bath applied in vitro to induce LTDNA may inactivate group III mGluRs in the PVN, which would serve as a molecular switch for the induction of activity-dependent synaptic potentiation.
To investigate the role of mGluRs at glutamatergic synapses onto MNCs under basal conditions, we bath applied the group III selective agonist L-2-amino-4-phosphonobutyric acid (L-AP4; 25 μM), which at this dose activates the high-affinity mGluR4 and 8 subtypes. Following a stable baseline period of recording (5–10 min), L-AP4 elicited a reversible decrease in the amplitude of eEPSCs (73.26% ± 7.48% of control; n = 9; p = 0.03; Figure 4A). This was accompanied by increases in PPR (control 1.60 ± 0.14; L-AP4 2.04 ± 0.30; p = 0.04; Figure 4B), failure rate (control 0.07 ± 0.04; L-AP4 0.13 ± 0.05; p = 0.11), and CV (control 0.34 ± 0.03; L-AP4 0.44 ± 0.05; p = 0.04), indicating a reduction in the Pr of glutamate, consistent with group III mGluR inhibition of quantal (action-potential-independent) glutamate release (Oliet et al., 2001; Panatier et al., 2004; Schrader and Tasker, 1997). In addition, application of the selective group III mGluR antagonist (RS)-a-methylserine-O-phosphate (MSOP; 100 μM) resulted in an increase in the amplitude of eEPSCs (119.28% ± 10.95% of control; n = 7; p = 0.04; Figure S4) and a decrease in the PPR (83.70% ± 5.77% of control, p = 0.03), suggesting that mGluRs are tonically active at glutamate synapses onto MNCs (Schrader and Tasker, 1997).
Figure 4. Hemorrhage and In Vitro NA Inactivates Presynaptic Group III mGluRs.

(A) Representative experiment showing that the group III mGluR agonist (L-AP4, 25 μM) depresses eEPSCs obtained from MNCs in the PVN from a naive rat. Inset shows average eEPSCs before (black) and after (green) L-AP4. Scale bars: 10 ms, 50 pA.
(B) L-AP4 depressed eEPSCs presynaptically as indicated by increases in PPR, failure rate, and CV (n = 9).
(C) Representative experiment illustrating that L-AP4 failed to depress eEPSCs in rats hemorrhaged in vivo 30 min prior to slicing. Inset shows sample eEPSCs from a hemorrhaged rat in control (blue) and after L-AP4 (green). Scale bars: 5 ms, 50 pA.
(D) In slices obtained from rats that were microinjected with yohimbine into the PVN in vivo prior to hemorrhaging, L-AP4 was not effective in depressing evoked glutamate release. Inset shows eEPSCs in control (hem + yoh; microinjected with yohimbine and then hemorrhaged) and after application of L-AP4 (green). Scale bars: 20 ms, 50 pA.
(E) After induction of LTDNA subsequent application of L-AP4 failed to depress glutamate synapses further. Inset shows sample eEPSCs after LTDNA induction (red) and subsequent application of L-AP4 (green). Scale bars: 10 ms, 20 pA.
(F) Representative recording showing the effect of L-AP4 application after clonidine. Group III mGluRs are not inactivated following induction of LTD with clonidine. Inset shows average eEPSCs after wash of clonidine (orange) and following subsequent application of L-AP4 (green). Scale bars: 20 ms, 20 pA.
(G and H) Summary bar graphs illustrating the effect of L-AP4 on the amplitude of evoked glutamate release (G) and PPR (H) in slices obtained from control naive rats (con; black), after induction of LTDNA (NA; red; n = 5), after clonidine LTD (clon; orange; n = 8), after in vivo hemorrhage (hem; blue; n = 5), and after micro-injection of yohimbine into the PVN followed by hemorrhage (hem + yoh; n = 5). *p < 0.05.
Since α1-adrenoceptor activation is coupled to a downregulation of presynaptic group III mGluRs (Gordon and Bains, 2003), we hypothesized that the effects of L-AP4 would be mitigated following either hemorrhage or NA application. Consistent with this idea, L-AP4 perfusion had no effect on eEPSCs in slices from hemorrhaged rats (98.73% ± 4.68% of control amplitude; n = 5; p = 0.95; PPR 102.48% ± 8.37%; p = 0.96; Figures 4C, 4G, and 4H). Moreover, L-AP4 failed to depress synapses from animals that were microinjected with the α2-adrenoceptor antagonist yohimbine into the PVN prior to hemorrhaging and slicing (105.76% ± 4.66% control; n = 5; Figure 4D). Thus, the in vivo yohimbine microinjection prevented induction of α2-adrenoceptor-mediated LTD (Figures 1F–1H), but did not prevent the concomitant α1-adrenoceptor-mediated inactivation of group III mGluRs (Figures 4D, 4G, and 4H).
To confirmthe role of NA in the inactivation of mGluRs, we tested the effects of L-AP4 on evoked transmission at synapses that had undergone LTDNA. Under these conditions, L-AP4 had no further effect on eEPSCs (105.21% ± 7.88% of LTDNA values; n = 5; p = 0.43; Figures 4E, 4G, and 4H), indicating that after induction of LTDNA, high-affinity group III mGluRs are inactivated/internalized. To be certain that the lack of effect of L-AP4 on synaptic transmission is due to inactivation of mGluRs and not a floor effect, we applied L-AP4 to slices that had undergone clonidine-mediated LTD. Here, mGluRs should still be active and in these experiments, L-AP4 caused a depression of eEPSC amplitude (77.50% ± 10.91% of LTD; n = 8; p = 0.04, Figure 4F). These findings indicate that native group III mGluRs function to depress glutamate release onto MNCs, but that the function of these receptors is compromised by hemorrhage-induced release of NA.
HFS Induces Potentiation following LTD
Since the inactivation/internalization of presynaptic group III mGluRs in the hippocampus unmasks a metaplastic state (Pelkey et al., 2005, 2008), we hypothesized that LTDNA, when accompanied by an α1-adrenoceptor-mediated loss of mGluR signaling, would provide the ideal conditions for activity-induced plasticity. Consistent with this hypothesis, we observed a rapid and persistent potentiation of eEPSCs in slices from hemorrhaged rats following HFS (3 × 100 Hz for 1 s, 10 s interval; 132.01% ± 8.29% of control 10–15 min post-HFS; n = 15; p = 0.0002; Figures 5A and 5B). During HFS the postsynaptic membrane potential was held constant at a hyperpolarized potential (Vhold = −80 mV). This potentiation was accompanied by decreases in PPR (control 1.80 ± 0.19; post-HFS 1.49 ± 0.12; n = 15; p = 0.02), failure rate (control 0.14 ± 0.03; post-HFS 0.03 ± 0.01; p = 0.005), and CV (control 0.54 ± 0.03; post-HFS 0.45 ± 0.03; p = 0.0008; Figure 5C). The same HFS applied to slices from control, nonhemorrhaged rats did not elicit synaptic potentiation (amplitude 98.73% ± 3.14% of control post-HFS, n = 11, p = 0.35; PPR 100.10% ± 3.47% of control post-HFS, p = 0.77; ketamine-anesthetized controls and naive rats pooled; Figures 5D and 5F). Taken together, these observations strongly suggest that an in vivo challenge to body fluid homeostasis reduces Pr at glutamatergic synapses and leads to a state where the synapses have the potential for undergoing subsequent activity-dependent, presynaptic plasticity. Interestingly, prazosin microinjection into the PVN prior to hemorrhage prevented the synapses from undergoing activity-dependent potentiation following HFS (104.32% ± 11.47% of control post-HFS; n = 5; p = 0.89; Figures 5E and 5F). Thus, acute hypovolemic hemorrhage not only causes an α2-adrenoceptor-mediated decrease in glutamatergic synaptic efficacy, but also reveals an α1-adrenoceptor-mediated unmasking of activity-dependent presynaptic potentiation in response to HFS.
Figure 5. Hemorrhage Unmasks Activity-Dependent Plasticity.

(A) Sample experiment showing HFS-induced (3 × 100 Hz for 1 s, 10 s interval) LTP in a rat hemorrhaged in vivo. Horizontal black lines indicate the average eEPSC amplitude before and after HFS. Inset shows eEPSCs from a hemorrhaged rat before (blue) and after (purple) HFS. Scale bars: 10 ms, 20 pA.
(B) Summary of experiments illustrating the response to HFS in hemorrhaged rats (n = 15).
(C) LTP in hemorrhaged rats was accompanied by a reduced PPR, failure rate, and CV. *p < 0.05; **p < 0.01.
(D) Summary of experiments showing that HFS applied to naive slices failed to increase synaptic strength (n = 11). Inset shows sample eEPSCs in control and after HFS. Scale bars: 20 ms, 50 pA.
(E) Normalized group data showing the effects of HFS on eEPSCs in slices from animals microinjected with prazosin in vivo prior to hemorrhage (n = 5). Inset shows sample eEPSCs before (hem + praz) and after HFS. Scale bars: 20 ms, 20 pA.
(F) Bar graph summarizing the group data of the changes in eEPSC amplitude and PPR after HFS in naive slices, following hemorrhage, and in animals with prazosin microinjected into the PVN prior to hemorrhage. *p < 0.05; **p < 0.01.
To determine if induction of LTDNA also unmasks a state permissible for subsequent synaptic potentiation similar to that observed following hemorrhage, we applied HFS to synapses that had undergone LTDNA while holding the postsynaptic membrane potential constant (Vhold = −80 mV). HFS resulted in rapid repotentiation/de-depression of glutamatergic transmission that persisted for the duration of the recording (>20 min; HFS post-LTDNA 96.91% ± 8.10% of pre-NA control values; n = 11; p = 0.50; Figures 6A and 6B). At these synapses, the potentiation was sufficient to completely reverse the changes in PPR (control 1.23 ± 0.13; LTDNA 1.85 ± 0.10; p = 0.008; post-HFS 1.25 ± 0.13; n = 5; p = 0.99), failure rate (control 0.08 ± 0.05; LTDNA 0.46 ± 0.11; p = 0.003; post-HFS 0.09 ± 0.04; p = 0.90), and CV (control 0.43 ± 0.06; LTDNA 0.72 ± 0.03; p = 0.01; post-HFS 0.46 ± 0.04; p = 0.99) observed during LTDNA (Figure 6C). These results indicate that LTP also has a presynaptic locus of expression and functionally represents the reverse process of LTDNA. The potentiation observed following LTDNA contrasts sharply with the absence of HFS-induced potentiation in naive (NA untreated) slices (Figure 5D), indicating that this plasticity depends upon previous activity of the network.
Figure 6. Noradrenaline-Mediated LTD Unmasks Metaplasticity.

(A) Representative experiment showing HFS-induced LTP following LTDNA induction. Sample traces are paired-pulse eEPSCs in control (black), during LTDNA (red), and after HFS (purple). Scale bars: 10 ms, 50 pA.
(B) Summary of experiments showing the effect of HFS on eEPSC amplitude after induction of LTDNA (closed black). The average time course of LTDNA in the absence of HFS is replotted from Figure 2B for comparison (open gray).
(C) HFS restored PPR, failure rate, and CV to pre-LTDNA values. **p < 0.01.
Since HFS potentiation, or metaplasticity, is observed when synapses are depressed and mGluRs are inactive, we conducted additional experiments to determine the necessity of individual component parts. First we considered the possibility that HFS-induced potentiation following LTDNA occurs because low Pr at synapses increases the “ceiling” of the synapses and makes them more amenable to potentiation in comparison to synapses with higher Pr (as in control) (Bi and Poo, 1998). To address this possibility, we carried out experiments in which Pr was reduced in naive slices by perfusion of artificial cerebrospinal fluid (ACSF) containing high Mg2+ (3 mM) and low Ca2+ (1 mM). In high-Mg2+/low-Ca2+-treated slices, eEPSC amplitude was reduced (35.29% ± 4.32% of control values) and PPR increased (control 1.79 ± 0.27; high Mg2+/low Ca2+ 2.55 ± 0.56; n = 5). When HFS was applied, these synapses failed to show potentiation (Figure S2A). Thus, we conclude that a reduction of Pr alone is not sufficient for metaplasticity at these synapses. Next, to confirm that α1- and α2-adrenoceptors specifically are necessary for metaplasticity, we applied another G protein-coupled receptor agonist baclofen (30 μM), which activates GABAB receptors and lowers Pr at glutamate synapses (Kombian et al., 1996). Baclofen depressed eEPSCs, but subsequent HFS had no effect on synaptic strength (Figure S2B). We conducted additional experiments to determine the relative contributions of LTD and the mGluRs to the metaplasticity. We observed no potentiation in response to HFS at synapses that had undergone prior LTD with clonidine (62.18% ± 3.50% of control; n = 6; Figure S3A). Finally, we asked whether inhibition of group III mGluRs was sufficient for activity-induced potentiation of synapses. In the presence of MSOP, HFS induced a pronounced posttetanic potentiation followed by a weak, longer lasting potentiation of eEPSCs that was not as robust as the potentiation observed after either LTDNA or hemorrhage (amplitude 118.23% ± 6.86% of control; n = 10; p = 0.02; PPR 85.056% ± 6.203% of control; p = 0.05; Figure S4). On the other hand, when BAPTA was included in the patch pipette to block induction of LTDNA and synapses were subsequently stimulated with HFS, no synaptic potentiation was observed (Figure S3B). Collectively, these results confirm that both LTD and mGluR inactivation are necessary for metaplasticity.
Finally, if the HFS-dependent synaptic potentiation is a resetting of the synapses back to the naive state, then LTP should also be accompanied by a recovery of function at high-affinity group III mGluRs. To test this idea, we challenged synapses that were potentiated by HFS (following LTDNA) with L-AP4. This caused a reduction in eEPSC amplitude and an increase in PPR equivalent to that observed in L-AP4-treated naive slices (73.05% ± 8.38% of post-HFS amplitude; n = 6; p = 0.04; Figure 7). These results indicate that HFS returns the synapses to a state that is functionally similar to the naive state (PPR return to control values, mGluRs are active).
Figure 7. Functional Reactivation of Group III mGluR Signaling after HFS.

(A) Average time course showing that mGluR function recovers following post-LTDNA HFS-induced LTP (n = 6). Inset shows sample eEPSCs in control, after LTDNA induction (red), after HFS (purple), and after subsequent application of L-AP4 (green). Scale bars: 5 ms, 20 pA.
(B and C) Summary graphs illustrating the percent change in eEPSC amplitude and PPR in response to L-AP4 when it’s perfused on naive slices (green), on slices after the induction of LTDNA (red; replotted from Figure 4), or on slices after LTDNA induction with subsequent HFS (purple). Each bar represents an individual experiment. *p < 0.05.
DISCUSSION
The data presented here demonstrate that glutamate synapses in circuits regulating body fluid homeostasis are metaplastic. This metaplasticity requires the functional inactivation of presynaptic group III mGluRs and is evident both after the exposure of naive slices to NA and in slices prepared from animals subjected to an acute and precipitous loss of blood volume. In vitro application of NA or in vivo hemorrhage results in a persistent reduction in evoked glutamate release onto MNCs in the PVN. The reduction in Pr after NA (LTDNA) requires the activation of postsynaptic α2-adrenoceptors, and the liberation of a retrograde signal that targets presynaptic CB1 receptors. Finally, the inactivation of mGluRs via α1-adrenoceptor stimulation primes the synapses, allowing a rapid, persistent potentiation of synaptic transmission in response to HFS of glutamatergic afferents. This LTP is accompanied by a recovery of mGluR function. Our results further support the hypothesis that mGluRs are critical molecular switches for the induction of metaplasticity and provide a link between the in vivo recruitment of neural circuits and the ability of the synapses within this circuit to undergo this powerful form of activity-dependent regulation.
An intriguing finding in this study is that presynaptic CB1 receptors are critical for the induction of LTDNA. Previous studies in the hypothalamus have shown that eCBs can modulate synaptic transmission transiently following either presynaptic HFS or postsynaptic depolarization (Di et al., 2005a; Hirasawa et al., 2004), via oxytocin receptor (Hirasawa et al., 2004; Oliet et al., 2007) or glucocorticoid receptor (Di et al., 2005b, 2009; Malcher-Lopes et al., 2006) stimulation. Our finding, however, demonstrates that eCBs contribute to an enduring plasticity in the hypothalamus and adds to the growing number of brain structures wherein eCBs mediate long-term synaptic plasticity (Chevaleyre et al., 2006; Lovinger, 2008). The activation of α2-adrenoceptors is necessary for the induction of LTDNA and is consistent with observations that activation of G protein-coupled receptors recruits eCBs (Chevaleyre and Castillo, 2003; Kreitzer and Malenka, 2005; Robbe et al., 2002; Ronesi and Lovinger, 2005; Sung et al., 2001). Interestingly, α2-adrenoceptors have largely been assumed to be located on presynaptic nerve terminals, but aside from a few elegant demonstrations clearly showing a role for presynaptic α2-adrenoceptors in synaptic depression at glutamate synapses (Carey and Regehr, 2009; Delaney et al., 2007), few studies have explicitly ruled out the possible involvement of a retrograde messenger. There are numerous observations in support of functional postsynaptic α2-adrenoceptors (Carr et al., 2007; Cathala et al., 2002; Li and van den Pol, 2005; Liu and Alreja, 1998; Shirasaka et al., 2007; Yamanaka et al., 2006). Since activation of these Gi/o-coupled receptors does not increase intracellular Ca2+, it is thought unlikely that they would participate in the production of retrograde signals. Recently, however, the D2-receptor, another Gi/o-coupled receptor, has been shown to enhance eCB release in the striatum and ventral tegmental area (Giuffrida et al., 1999; Melis et al., 2004) and to be critical for eliciting eCB-LTD in the striatum (Kreitzer and Malenka, 2005, 2007). In addition, these receptors can couple to PLD, a key enzyme in the biosynthetic pathway for eCBs (Basavarajappa, 2007). Although it’s not clear if PLD activation is important for production of eCBs in MNCs, the idea that PLD is at least partly reliant on and can be enhanced by intracellular Ca2+ (Kano et al., 2009) is supported by our data demonstrating that long-term α2-mediated inhibition is blocked by inclusion of BAPTA in our patch pipette. Although the final molecular steps mediating LTDNA remain to be determined, there is compelling evidence in other systems that it may involve the persistent inhibition of presynaptic P/Q-type or N-type Ca2+ channels (Brown et al., 2004; Hoffman and Lupica, 2000; Kreitzer and Regehr, 2001) and/or interference with release machinery (Chevaleyre et al., 2007; Gerdeman and Lovinger, 2001; Robbe et al., 2001). Our experiments, however, argue against the latter assertion because LTDNA of eEPSCs was accompanied by a robust increase in the frequency of spontaneous EPSCs (sEPSCs) (Gordon and Bains, 2003, 2005).
Consistent with our previous investigation showing that activation of α1-adrenergic receptors inactivated presynaptic group III mGluRs (Gordon and Bains, 2003), L-AP4 treatment failed to depress eEPSCs further after induction of LTDNA. This loss of mGluR function is mediated by increased PKC activity (Gordon and Bains, 2003; Macek et al., 1999), but it is not clear whether mGluRs are internalized. Interestingly, internalization of mGluR7 is critical for the expression of metaplasticity at mossy fiber-stratum lucidum interneuron (MF-SLIN) synapses in the hippocampus (Pelkey et al., 2005). The internalization of mGluR7 is triggered by exposure to agonist (Pelkey et al., 2005, 2007), whereas stable surface expression is dependent on PKC phosphorylation and receptor binding to the scaffolding protein PICK1 (Suh et al., 2008). There are, however, two important distinctions between metaplasticity at MF-SLIN synapses and that described here. First, it appears that a different subtype of group III mGluRs is involved. At glutamate synapses onto MNCs, the high-affinity group III mGluRs (mGluR4 and 8) regulate neurotransmitter release, with little contribution from the low-affinity mGluR7 subtype (Panatier et al., 2004) (although see Schrader and Tasker, 1997). Our investigations using a concentration of L-AP4 (25 μM) that targets only high-affinity group III mGluRs are consistent with these observations. Second, at MF-SLIN synapses, mGluR inactivation is dependent on prolonged binding of agonist (glutamate) to the receptors. By contrast, inactivation of mGluRs in our study depends on the activation of a second, independent chemical signal, NA. Whether mGluRs actually undergo internalization following NA is unresolved. Recent work demonstrating internalization and desensitization of mGluR4 following activation of PKC by phorbol ester treatment or Gαq-linked G protein-coupled receptor activation (Mathiesen and Ramirez, 2006) offers support for the internalization hypothesis in our scenario. Since the internalization of mGluR7 is critical to the expression of metaplasticity at MF-SLIN synapses (Pelkey et al., 2005), it seems plausible that high-affinity group III mGluR internalization is also necessary for metaplasticity at MNC glutamate synapses.
Based on our paired-pulse, CV, and failure analyses, both LTDNA and HFS potentiation have a presynaptic locus of expression, indicating that potentiation functionally represents the reverse process of LTD. Importantly, HFS not only restored presynaptic release probability, but caused a gain of mGluR function, suggesting that synapses return to a state that is functionally identical to pre-LTDNA induction. This presynaptic potentiation following HFS occurs in the absence of postsynaptic depolarization and is fundamentally different from NMDA-receptor-dependent plasticity described previously at glutamate synapses onto MNCs (Panatier et al., 2006a, 2006b).
Our work adds to a growing metaplasticity field that highlights the requirement for regulatory mechanisms to maintain synaptic efficacy within a functional dynamic range (Abraham, 2008; Clem et al., 2008). Furthermore, it provides a critical proof that recent demonstrations of presynaptic mGluRs as gatekeepers for a novel form of metaplasticity (Pelkey et al., 2005, 2006, 2008) can be extended to suggest that presynaptic mGluRs are also a mechanism to alter synaptic strength in response to a physiologically relevant in vivo challenge. How might mGluR-mediated metaplasticity in the hypothalamus contribute to restoration of body fluid homeostasis? One possibility is that the metaplasticity we describe here represents transitions of glutamate synapses through multiple functional states that tightly regulate the activity of MNCs and therefore the appropriate release of hormones from the posterior pituitary. In other words, internal homeostasis is maintained by responding immediately and effectively to a challenge, operating at a new (transient) setpoint, and then resetting the system. This includes an increase in Pr (Gordon and Bains, 2003, 2005) and an NA recruitment of astrocytes that promotes AMPA receptor insertion (Gordon et al., 2005) that initially drives MNCs, followed by an eCB-mediated persistent reduction in Pr as the system begins to adapt to the challenge, and finally, an activity-dependent resetting that is only possible if presynaptic mGluRs are first inactivated.
EXPERIMENTAL PROCEDURES
Brain Slice Preparation
All experiments were performed according to protocols approved by the University of Calgary Animal Care and Use Committee in accordance with Canadian Council on Animal Care guidelines. Male Sprague-Dawley rats (postnatal day 21 [P21]–P40) were anaesthetized using isoflurane and then decapitated. The brain was quickly removed and placed in ice-cold slicing solution containing (in mM) NaCl 87, KCl 2.5, NaHCO3 25, MgCl2 7, NaH2PO4 1.25, glucose 25, and sucrose 75 saturated with 95%/5% O2/CO2. The brain was blocked, mounted on a vibrating slicer (Leica, Nussloch, Germany), and submerged in ice-cold slicing solution. Coronal slices (250 μm) of the hypothalamus containing the PVN were cut and transferred to an incubating chamber containing ACSF at 32°C for 45 min. The ACSF contained (in mM) NaCl 126, KCl 2.5, NaHCO3 25, MgCl2 1.2, NaH2PO4 1.2, glucose 11, CaCl2 2.4, and ascorbic acid 1 saturated with 95%/5% O2/CO2. Slices were maintained for a minimum of 45 min at room temperature (21°C–24°C) before recording.
Electrophysiology
Hypothalamic slices were transferred to a recording chamber and superfused with 32°C–34°C ACSF at a flow rate of 1–2 ml/min. Whole-cell recordings were obtained from MNCs visualized with an AxioskopII FS Plus (Zeiss, Oberkochen, Germany) upright microscope fitted with infrared differential interference contrast optics. MNCs were identified based on their morphology and well-defined electrophysiological characteristics (Luther and Tasker, 2000). Recordings were obtained using borosilicate glass microelectrodes (tip resistance 3–5 MΩ) filled with a solution containing (in mM) K-gluconate 123, NaCl 8, EGTA 1, HEPES 16, MgCl2 2, K2ATP 0.3, and Na3GTP 0.3. For experiments where BAPTA was included in the pipette solution, a minimum of 15 min was allowed for intracellular diffusion. Recordings were accepted for analysis if changes in access resistance were <15%. Cells were voltage clamped at −80 mV and the perfusate always contained picrotoxin (100 μM; Sigma-Aldrich, St. Louis, MO) to block GABAA-mediated synaptic currents. Glutamatergic fibers were stimulated with an extracellular patch pipette filled with ACSF that was always positioned lateral to the PVN in approximately the same location. EPSCs were evoked at a rate of 0.1 Hz and paired-pulse responses were obtained by applying a pair of synaptic stimuli 50 ms apart. Only synapses that displayed a high degree of synchronous release 0–5 ms after the stimulation were analyzed. For high HFS, afferents were stimulated at 100 Hz for 1 s, repeated three times 10 s apart.
Signals were amplified using the Multiclamp 700B amplifier (Molecular Devices, Union City, CA), low-pass filtered at 1 kHz, and digitized at 10 kHz using the Digidata 1322 (Molecular Devices). Data were collected (pClamp 9.0, Molecular Devices) and stored on a computer for offline analysis. Evoked currents were analyzed using Clampfit 9 (Molecular Devices). The amplitude of the synaptic current was calculated from the baseline (current before evoked response) to the peak of each evoked. For clarity, the stimulus artifacts have been removed digitally from the traces depicted. PPRs (50 ms interstimulus interval), CV, and failure rates were measured over 5 min epochs of 30 eEPSCs each. To exclude possible contributions from AMPA receptor insertion in response to NA application (Gordon et al., 2005), recordings for the HFS experiments in Figure 6 were performed with an intracellular solution containing EGTA (10 mM). This concentration of EGTA is sufficient to block postsynaptic AMPA receptor insertion, but is not adequate to block the induction of LTDNA. Results are expressed as means ± SEM. Significance was determined using a Student’s t test or a one-way ANOVA with Tukey’s post hoc comparison where appropriate with significance level of p < 0.05.
Hemorrhage
Rats were anaesthetized with ketamine-xylazine (100 mg/kg). Approximately 25% of blood volume (Lee and Blaufox, 1985) was removed by cardiac puncture with a heparinized syringe over approximately 1 min. Brain slices were prepared after either 2 or 30 min after hemorrhage, or an equal time anesthetized without hemorrhage.
Microinjection of Chemicals into the PVN
Under ketamine-xylazine (100 mg/kg) anesthesia, rats were microinjected with prazosin (10 nmol), yohimbine (10 nmol), or vehicle (25% ACSF, 75% DMSO; Blevins et al., 2002). Injections into the magnocellular subnucleus of the PVN were performed using a stereotaxically fixed 10 μl Hamilton microsyringe. The stereotaxic coordinates for the PVN were as follows: 1.1–1.3 mm posterior to Bregma, 1.6–1.7 mm from the midline at a 10° angle (to avoid bleeding from the superior sagittal sinus), and 7.7–8.0 mm below the surface of the cortex (Paxinos and Watson, 2005). A volume of 1 μl was delivered over 1–2 min into the PVN to allow diffusion of the chemical. Each animal was injected unilaterally and either the right or left hemisphere was chosen randomly. Rats were hemorrhaged approximately 2–4 min after the injections. Injection sites were visually identified in hypothalamic slices by imaging FluoSpheres (0.1 μm, red fluorescent; Invitrogen, Carlsbad, CA) that were included with the vehicle at a dilution of 1:100. Injection sites were imaged using a Zeiss AxioskopII FS Plus (4X objective lens) and epifluorescence images were captured using a Retiga Exi camera (Q Imaging, Surrey, BC, Canada) with QCapture software.
Supplementary Material
Acknowledgments
We thank Drs. K. Lukowiak, S.H.R. Oliet, S. Sangha, and members of the Bains and Pittman labs for constructive comments and fruitful discussions. Q.J.P. is an Alberta Heritage Foundation for Medical Research (AHFMR) Medical Scientist. J.S.B. is an AHFMR Senior Scholar. This work was supported by operating grants from the Canadian Institutes of Health Research (J.S.B. and Q.J.P.) and the Heart and Stroke Foundation of Alberta, Yukon and NWT (J.S.B.).
Footnotes
Supplemental data for this article include four Supplemental Figures and can be found at http://www.cell.com/neuron/supplemental/S0896-6273(09)00430-9.
References
- Abraham WC. Metaplasticity: tuning synapses and networks for plasticity. Nat Rev Neurosci. 2008;9:387–399. doi: 10.1038/nrn2356. [DOI] [PubMed] [Google Scholar]
- Basavarajappa BS. Critical enzymes involved in endocannabinoid metabolism. Protein Pept Lett. 2007;14:237–246. doi: 10.2174/092986607780090829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi GQ, Poo MM. Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci. 1998;18:10464–10472. doi: 10.1523/JNEUROSCI.18-24-10464.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blevins JE, Stanley BG, Reidelberger RD. DMSO as a vehicle for central injections: tests with feeding elicited by norepinephrine injected into the paraventricular nucleus. Pharmacol Biochem Behav. 2002;71:277–282. doi: 10.1016/s0091-3057(01)00659-1. [DOI] [PubMed] [Google Scholar]
- Boudaba C, Di S, Tasker JG. Presynaptic noradrenergic regulation of glutamate inputs to hypothalamic magnocellular neurones. J Neuroendocrinol. 2003;15:803–810. doi: 10.1046/j.1365-2826.2003.01063.x. [DOI] [PubMed] [Google Scholar]
- Bourque CW. Central mechanisms of osmosensation and systemic osmoregulation. Nat Rev Neurosci. 2008;9:519–531. doi: 10.1038/nrn2400. [DOI] [PubMed] [Google Scholar]
- Brown SP, Safo PK, Regehr WG. Endocannabinoids inhibit transmission at granule cell to Purkinje cell synapses by modulating three types of presynaptic calcium channels. J Neurosci. 2004;24:5623–5631. doi: 10.1523/JNEUROSCI.0918-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buller KM, Smith DW, Day TA. Differential recruitment of hypothalamic neuroendocrine and ventrolateral medulla catecholamine cells by non-hypotensive and hypotensive hemorrhages. Brain Res. 1999;834:42–54. doi: 10.1016/s0006-8993(99)01539-5. [DOI] [PubMed] [Google Scholar]
- Carey MR, Regehr WG. Noradrenergic Control of Associative Synaptic Plasticity by Selective Modulation of Instructive Signals. Neuron. 2009;62:112–122. doi: 10.1016/j.neuron.2009.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DB, Andrews GD, Glen WB, Lavin A. alpha 2-Noradrenergic receptors activation enhances excitability and synaptic integration in rat prefrontal cortex pyramidal neurons via inhibition of HCN currents. J Physiol. 2007;584:437–450. doi: 10.1113/jphysiol.2007.141671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathala L, Guyon A, Eugene D, Paupardin-Tritsch D. alpha(2)-adrenoceptor activation increases a cationic conductance and spontaneous gabaergic synaptic activity in dopaminergic neurones of the rat substantia nigra. Neuroscience. 2002;115:1059–1065. doi: 10.1016/s0306-4522(02)00542-0. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Castillo PE. Heterosynaptic LTD of hippocampal GABAergic synapses: A novel role of endocannabinoids in regulating excitability. Neuron. 2003;38:461–472. doi: 10.1016/s0896-6273(03)00235-6. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Takahashi KA, Castillo PE. Endocannabinoid-mediated synaptic plasticity in the CNS. Annu Rev Neurosci. 2006;29:37–76. doi: 10.1146/annurev.neuro.29.051605.112834. [DOI] [PubMed] [Google Scholar]
- Chevaleyre V, Heifets BD, Kaeser PS, Sudhof TC, Purpura DP, Castillo PE. Endocannabinoid-mediated long-term plasticity requires cAMP/PKA signaling and RIM1 alpha. Neuron. 2007;54:801–812. doi: 10.1016/j.neuron.2007.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clem RL, Celikel T, Barth AL. Ongoing in vivo experience triggers synaptic metaplasticity in the neocortex. Science. 2008;319:101–104. doi: 10.1126/science.1143808. [DOI] [PubMed] [Google Scholar]
- Delaney AJ, Crane JW, Sah P. Noradrenaline modulates transmission at a central synapse by a presynaptic mechanism. Neuron. 2007;56:880–892. doi: 10.1016/j.neuron.2007.10.022. [DOI] [PubMed] [Google Scholar]
- Di S, Boudaba C, Popescu IR, Weng FJ, Harris C, Marcheselli VL, Bazan NG, Tasker JG. Activity-dependent release and actions of endocannabinoids in the rat hypothalamic supraoptic nucleus. J Physiol. 2005a;569:751–760. doi: 10.1113/jphysiol.2005.097477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di S, Malcher-Lopes R, Marcheselli VL, Bazan NG, Tasker JG. Rapid glucocorticoid-mediated endocannabinoid release and opposing regulation of glutamate and gamma-aminobutyric acid inputs to hypothalamic magnocellular neurons. Endocrinology. 2005b;146:4292–4301. doi: 10.1210/en.2005-0610. [DOI] [PubMed] [Google Scholar]
- Di S, Maxson MM, Franco A, Tasker JG. Glucocorticoids regulate glutamate and GABA synapse-specific retrograde transmission via divergent nongenomic signaling pathways. J Neurosci. 2009;29:393–401. doi: 10.1523/JNEUROSCI.4546-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Marzo V, Fontana A, Cadas H, Schinelli S, Cimino G, Schwartz JC, Piomelli D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- Gerdeman G, Lovinger DM. CB1 cannabinoid receptor inhibits synaptic release of glutamate in rat dorsolateral striatum. J Neurophysiol. 2001;85:468–471. doi: 10.1152/jn.2001.85.1.468. [DOI] [PubMed] [Google Scholar]
- Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- Giuffrida A, Parsons LH, Kerr TM, Rodriguez de Fonseca F, Navarro M, Piomelli D. Dopamine activation of endogenous cannabinoid signaling in dorsal striatum. Nat Neurosci. 1999;2:358–363. doi: 10.1038/7268. [DOI] [PubMed] [Google Scholar]
- Gordon GRJ, Bains JS. Priming of excitatory synapses by alpha(1) adrenoceptor-mediated inhibition of group III metabotropic glutamate receptors. J Neurosci. 2003;23:6223–6231. doi: 10.1523/JNEUROSCI.23-15-06223.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon GRJ, Bains JS. Noradrenaline triggers multivesicular release at glutamatergic synapses in the hypothalamus. J Neurosci. 2005;25:11385–11395. doi: 10.1523/JNEUROSCI.2378-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon GRJ, Baimoukhametova DV, Hewitt SA, Rajapaksha WRAK, Fisher TE, Bains JS. Norepinephrine triggers release of glial ATP to increase postsynaptic efficacy. Nat Neurosci. 2005;8:1078–1086. doi: 10.1038/nn1498. [DOI] [PubMed] [Google Scholar]
- Hirasawa M, Schwab Y, Natah S, Hillard CJ, Mackie K, Sharkey KA, Pittman QJ. Dendritically released transmitters cooperate via autocrine and retrograde actions to inhibit afferent excitation in rat brain. J Physiol. 2004;559:611–624. doi: 10.1113/jphysiol.2004.066159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman AF, Lupica CR. Mechanisms of cannabinoid inhibition of GABA(A) synaptic transmission in the hippocampus. J Neurosci. 2000;20:2470–2479. doi: 10.1523/JNEUROSCI.20-07-02470.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin XH, Okamoto Y, Morishita J, Tsuboi K, Tonai T, Ueda N. Discovery and characterization of a Ca2+-independent phosphatidylethanolamine N-acyltransferase generating the anandamide precursor and its congeners. J Biol Chem. 2007;282:3614–3623. doi: 10.1074/jbc.M606369200. [DOI] [PubMed] [Google Scholar]
- Jinsi-Parimoo A, Deth RC. Reconstitution of alpha 2D-adrenergic receptor coupling to phospholipase D in a PC12 cell lysate. J Biol Chem. 1997;272:14556–14561. doi: 10.1074/jbc.272.23.14556. [DOI] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Kombian SB, Zidichouski JA, Pittman QJ. GABA(B) receptors presynaptically modulate excitatory synaptic transmission in the rat supraoptic nucleus in vitro. J Neurophysiol. 1996;76:1166–1179. doi: 10.1152/jn.1996.76.2.1166. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto Purkinje cells. Neuron. 2001;29:717–727. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Dopamine modulation of state-dependent endocannabinoid release and long-term depression in the striatum. J Neurosci. 2005;25:10537–10545. doi: 10.1523/JNEUROSCI.2959-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- Lee HB, Blaufox MD. Blood-Volume in the Rat. J Nucl Med. 1985;26:72–76. [PubMed] [Google Scholar]
- Li Y, van den Pol AN. Direct and indirect inhibition by catecholamines of hypocretin/orexin neurons. J Neurosci. 2005;25:173–183. doi: 10.1523/JNEUROSCI.4015-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Alreja M. Norepinephrine inhibits neurons of the intermediate subnucleus of the lateral septum via alpha(2)-adrenoceptors. Brain Res. 1998;806:36–54. doi: 10.1016/s0006-8993(98)00728-8. [DOI] [PubMed] [Google Scholar]
- Lovinger DM. Presynaptic modulation by endocannabinoids. Handb Exp Pharmacol. 2008;184:435–477. doi: 10.1007/978-3-540-74805-2_14. [DOI] [PubMed] [Google Scholar]
- Luther JA, Tasker JG. Voltage-gated currents distinguish parvocellular from magnocellular neurones in the rat hypothalamic paraventricular nucleus. J Physiol. 2000;523:193–209. doi: 10.1111/j.1469-7793.2000.t01-1-00193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macek TA, Schaffhauser H, Conn PJ. Activation of PKC disrupts presynaptic inhibition by group II and group in metabotropic glutamate receptors and uncouples the receptor from GTP-binding proteins. Ann NY Acad Sci. 1999;868:554–557. doi: 10.1111/j.1749-6632.1999.tb11327.x. [DOI] [PubMed] [Google Scholar]
- Malcher-Lopes R, Di S, Marcheselli VS, Weng FJ, Stuart CT, Bazan NG, Tasker JG. Opposing crosstalk between leptin and glucocorticoids rapidly modulates synaptic excitation via endocannabinoid release. J Neurosci. 2006;26:6643–6650. doi: 10.1523/JNEUROSCI.5126-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: An embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Mathiesen JM, Ramirez MT. The metabotropic glutamate receptor 4 is internalized and desensitized upon protein kinase C activation. Br J Pharmacol. 2006;148:279–290. doi: 10.1038/sj.bjp.0706733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melis M, Pistis M, Perra S, Muntoni AL, Pillolla G, Gessa GL. Endocannabinoids mediate presynaptic inhibition of glutamatergic transmission in rat ventral tegmental area dopamine neurons through activation of CB1 receptors. J Neurosci. 2004;24:53–62. doi: 10.1523/JNEUROSCI.4503-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SHR, Piet R, Poulain DA. Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science. 2001;292:923–926. doi: 10.1126/science.1059162. [DOI] [PubMed] [Google Scholar]
- Oliet SHR, Baimoukhametova DV, Piet R, Bains JS. Retrograde regulation of GABA transmission by the tonic release of oxytocin and endocannabinoids governs postsynaptic firing. J Neurosci. 2007;27:1325–1333. doi: 10.1523/JNEUROSCI.2676-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panatier A, Poulain DA, Oliet SHR. Regulation of transmitter release by high-affinity group III mGluRs in the supraoptic nucleus of the rat hypothalamus. Neuropharmacology. 2004;47:333–341. doi: 10.1016/j.neuropharm.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Panatier A, Gentles SJ, Bourque CW, Oliet SHR. Activity-dependent synaptic plasticity in the supraoptic nucleus of the rat hypothalamus. J Physiol. 2006a;573:711–721. doi: 10.1113/jphysiol.2006.109447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, Poulain DA, Oliet SHR. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell. 2006b;125:775–784. doi: 10.1016/j.cell.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. Amsterdam: Elsevier/Academic Press; 2005. [Google Scholar]
- Pelkey KA, Lavezzari G, Racca C, Roche KW, McBain CJ. mGluR7 is a metaplastic switch controlling bidirectional plasticity of feedforward inhibition. Neuron. 2005;46:89–102. doi: 10.1016/j.neuron.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Topolnik L, Lacaille JC, McBain CJ. Compartmentalized Ca2+ channel regulation at divergent mossy-fiber release sites underlies target cell-dependent plasticity. Neuron. 2006;52:497–510. doi: 10.1016/j.neuron.2006.08.032. [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Yuan XQ, Lavezzari G, Roche KW, McBain CJ. mGluR7 undergoes rapid internalization in response to activation by the allosteric agonist AMN082. Neuropharmacology. 2007;52:108–117. doi: 10.1016/j.neuropharm.2006.07.020. [DOI] [PubMed] [Google Scholar]
- Pelkey KA, Topolnik L, Yuan XQ, Lacaille JC, McBain CJ. State-dependent cAMP sensitivity of presynaptic function underlies metaplasticity in a hippocampal feedforward inhibitory circuit. Neuron. 2008;60:980–987. doi: 10.1016/j.neuron.2008.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulain DA, Wakerley JB. Electrophysiology of hypothalamic magnocellular neurones secreting oxytocin and vasopressin. Neuroscience. 1982;7:773–808. doi: 10.1016/0306-4522(82)90044-6. [DOI] [PubMed] [Google Scholar]
- Poulain DA, Wakerley JB, Dyball REJ. Electrophysiological differentiation of oxytocin- and vasopressin-secreting neurones. Proc R Soc Lond B Biol Sci. 1977;196:367–384. doi: 10.1098/rspb.1977.0046. [DOI] [PubMed] [Google Scholar]
- Robbe D, Alonso G, Duchamp F, Bockaert J, Manzoni OJ. Localization and mechanisms of action of cannabinoid receptors at the glutamatergic synapses of the mouse nucleus accumbens. J Neurosci. 2001;21:109–116. doi: 10.1523/JNEUROSCI.21-01-00109.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbe D, Kopf M, Remaury A, Bockaert J, Manzoni OJ. Endogenous cannabinoids mediate long-term synaptic depression in the nucleus accumbens. Proc Natl Acad Sci USA. 2002;99:8384–8388. doi: 10.1073/pnas.122149199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronesi J, Lovinger DM. Induction of striatal long-term synaptic depression by moderate frequency activation of cortical afferents in rat. J Physiol. 2005;562:245–256. doi: 10.1113/jphysiol.2004.068460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawchenko PE, Swanson LW. Central noradrenergic pathways for the integration of hypothalamic neuroendocrine and autonomic responses. Science. 1981;214:685–687. doi: 10.1126/science.7292008. [DOI] [PubMed] [Google Scholar]
- Schrader LA, Tasker JG. Presynaptic modulation by metabotropic glutamate receptors of excitatory and inhibitory synaptic inputs to hypothalamic magnocellular neurons. J Neurophysiol. 1997;77:527–536. doi: 10.1152/jn.1997.77.2.527. [DOI] [PubMed] [Google Scholar]
- Senogles SE. The D2s dopamine receptor stimulates phospholipase D activity: A novel signaling pathway for dopamine. Mol Pharmacol. 2000;58:455–462. doi: 10.1124/mol.58.2.455. [DOI] [PubMed] [Google Scholar]
- Shirasaka T, Kannan H, Takasaki M. Activation of a G protein-coupled inwardly rectifying K+ current and suppression of I-h contribute to dexmedetomidine-induced inhibition of rat hypothalamic paraventricular nucleus neurons. Anesthesiology. 2007;107:605–615. doi: 10.1097/01.anes.0000281916.65365.4e. [DOI] [PubMed] [Google Scholar]
- Suh YH, Pelkey KA, Lavezzari G, Roche PA, Huganir RL, McBain CJ, Roche KW. Corequirement of PICK1 binding and PKC phosphorylation for stable surface expression of the metabotropic glutamate receptor mGluR7. Neuron. 2008;58:736–748. doi: 10.1016/j.neuron.2008.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung KW, Choi S, Lovinger DM. Activation of group I mGluRs is necessary for induction of long-term depression at Striatal synapses. J Neurophysiol. 2001;86:2405–2412. doi: 10.1152/jn.2001.86.5.2405. [DOI] [PubMed] [Google Scholar]
- Ueda N, Liu Q, Yamanaka K. Marked activation of the N-acylphosphatidylethanolamine-hydrolyzing phosphodiesterase by divalent cations. Biochim Biophys Acta. 2001;1532:121–127. doi: 10.1016/s1388-1981(01)00120-2. [DOI] [PubMed] [Google Scholar]
- Wakerly JB, Poulain DA, Dyball REJ, Cross BA. Activity of phasic neurosecretory cells during hemorrhage. Nature. 1975;258:82–84. doi: 10.1038/258082a0. [DOI] [PubMed] [Google Scholar]
- Yamanaka A, Muraki Y, Ichiki K, Tsujino N, Kilduff TS, Goto K, Sakurai T. Orexin neurons are directly and indirectly regulated by catecholamines in a complex manner. J Neurophysiol. 2006;96:284–298. doi: 10.1152/jn.01361.2005. [DOI] [PubMed] [Google Scholar]
- Yin HH, Lovinger DM. Frequency-specific and D2 receptor-mediated inhibition of glutamate release by retrograde endocannabinoid signaling. Proc Natl Acad Sci USA. 2006;103:8251–8256. doi: 10.1073/pnas.0510797103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.