

Abstract
Clinicopathologic correlation studies are critically important for the field of Alzheimer disease (AD) research. Studies on human subjects with autopsy confirmation entail numerous potential biases that affect both their general applicability and the validity of the correlations. Many sources of data variability can weaken the apparent correlation between cognitive status and AD neuropathologic changes. Indeed, most persons in advanced old age have significant non-AD brain lesions that may alter cognition independently of AD. Worldwide research efforts have evaluated thousands of human subjects to assess the causes of cognitive impairment in the elderly, and these studies have been interpreted in different ways. We review the literature focusing on the correlation of AD neuropathologic changes (i.e. β-amyloid plaques and neurofibrillary tangles) with cognitive impairment. We discuss the various patterns of brain changes that have been observed in elderly individuals to provide a perspective for understanding AD clinicopathologic correlation and conclude that evidence from many independent research centers strongly supports the existence of a specific disease, as defined by the presence of Aβ plaques and neurofibrillary tangles. Although Aβ plaques may play a key role in AD pathogenesis, the severity of cognitive impairment correlates best with the burden of neocortical neurofibrillary tangles.
Keywords: Aging, Alzheimer disease, Amyloid, Dementia, Epidemiology, Neuropathology, MAPT, Neurofibrillary tangles
INTRODUCTION
Decades of research worldwide have generated a large body of clinicopathologic correlation (CPC) scholarship related to Alzheimer disease (AD). Here we review the literature from the perspective of neuropathologists and clinicians performing research in this field. The review is designed for a broad target audience. We discuss the pathognomonic features of AD (1-3), highlight some of the challenges in CPC studies, discuss the specificity of AD neuropathologic changes, and, mindful of the controversies in this field of research, describe particular combinations of concomitant lesions that are found in human brains. We then review CPC studies that have documented observations on patients along the spectrum from intact cognition to end-stage dementia linked with AD-type neuropathologic changes. Pertinent reviews of historical background are available (4-10). Terms used in this review are defined in Table 1.
TABLE 1.
Definition of Terms
Pathologic terms:
|
Anatomic terms:
|
OVERVIEW OF CHALLENGES TO CPC STUDIES IN AD
There are many challenges related to the study of the neuropathologic correlates of cognitive impairment in the elderly. Sources of potential bias in AD CPC studies are summarized in Table 2; some of these are virtually impossible to avoid in the design of a research study. Autopsy is required for definitive AD diagnosis, yet autopsy rates are generally low and autopsy inevitably confers a selection bias. Furthermore, CPC studies rarely are a random sample of the population and clinic- and hospital-based CPCs are subject to other potential biases. For example, because persons with behavioral problems are more likely to require help than those with isolated memory problems, the prevalence and types of Lewy body disease derived from a clinic are likely to be different from those from a broader community (11). Most prior studies have been performed in countries with the most advantageous socioeconomic conditions and have concentrated on middle- and upper-income whites. Thus, overall, ideal conditions have not yet been achieved for comprehensive, population-level epidemiologic study in which information from detailed, longitudinal neurocognitive assessments can be combined with that from state-of-the-art postmortem examinations. Another challenge of CPC studies is that the molecular, anatomic, and clinical changes of AD may not progress in a uniform fashion (7, 12-15). This problem may not be easily solved using statistical models because the progression of both the clinical and the pathologic disease are not necessarily parametrically distributed. The tendency to dichotomize the disease (i.e. “demented vs nondemented”) and overreliance on ordinal variables may also obscure important relationships. Variation in the elapsed time between final clinical evaluation and autopsy and competing mortality risks add more uncertainty. Studies on CPC are additionally complicated by the high prevalence and high morbidity of concurrent diseases (16, 17) (discussed in more detail below). Some researchers tend to focus on unusual or extreme cases with atypical presentations, whereas others describe the distribution of outcomes in larger and perhaps more representative samples. Both approaches can provide insights; CPC data must be reconciled or at least better understood with reference to multiple variables and potential confounders. In sum, both clinical and pathologic assessments are imperfect, variably applied, and constantly evolving. These complexities should be kept in mind as we assess the sometimes-controversial CPC literature.
TABLE 2.
Sources of Potential Bias for Clinicopathologic Correlation Studies of Alzheimer Disease
| Patient Characteristics | Clinical Workup | Study Design | Disease Heterogeneity | Pathologic Workup |
|---|---|---|---|---|
| >Baseline “cognitive reserve” and education-linked factors | >Quantification of “cognition”: nonparametric cognitive changes | >Recruitment, inclusion, and exclusion criteria | >Different genetic risk factors at play | >Evaluation and quantification of other pathologies |
| >Varied access to high-quality health care (diagnostics and therapeutics) | >Quantification of non-AD changes such as cerebrovascular disease | >Cross-sectional vs longitudinal assessments | >Some “atypical” forms of disease | >Focus on complete brain or mainly hippocampus |
| >Non-AD structural brain comorbidities (cerebrovascular, neurotrauma, etc) | >Cognitive assessment instruments used | >Focus on rare cases or attempting to understand “epidemiological” perspective | >Unknown effects of environmental factors | >Multiple methods to detect AβPs and NFTs |
| >Emotional and mood disorders | >Individual clinician “thresholds” | >Bias in terms of autopsy rates | >Overlap and interplay between different diseases | >Skew toward end-stage disease at autopsy |
| >Systemic diseases that affect cognition (metabolic, hormonal, neoplastic, etc.) | >Variation among clinician practices | >Age of individuals in cohort at death | >Specificity of clinical, biomarker, and pathologic features | >Individual pathologist “thresholds”? |
| >Environmental and behavioral (substance abuse) | >Evolution in assessment methodology over time | >Definitions: “case” and “control” and other terms | >Variation among pathologist practices | |
| >Cohort effects | >Use of biomarkers | >Interval between final clinic evaluation and death | >Accentuation nonhallmark lesions (acetylcholine, synapses) | |
| >Use of semiquantitative or ordinal variables | >Biostatistical methodology | >Quantitative or ordinal variables |
AβP, amyloid β-containing plaque; AD, Alzheimer disease; CPC, clinicopathologic correlations; NFT, neurofibrillary tangle.
AβPS AND NEUROFIBRILLARY TANGLES: THE HISTOPATHOLOGIC HALLMARKS LINKED TO AD
The neuropathologic hallmarks of AD are neurofibrillary tangles (NFTs; including pretangles) and AβPs (including diffuse and neuritic plaques, which may also be referred to as “senile plaques” [2, 18, 19]; Fig. 1 and Table 1). Additional changes that may also occur in the brains of AD patients include amyloid angiopathy, age-related brain atrophy, synaptic pathology, white matter rarefaction, granulovacuolar degeneration, neuron loss, TDP-43 proteinopathy, and neuroinflammation (20-26), which may contribute to cognitive impairment; however, these are not considered pathognomonic features of AD (3). Therefore, we focus this review only on the defining features of AD neuropathologic changes: AβPs and NFTs (3). Excellent reviews are available about the molecular characteristics of NFTs and AβPs (27, 28).
FIGURE 1.
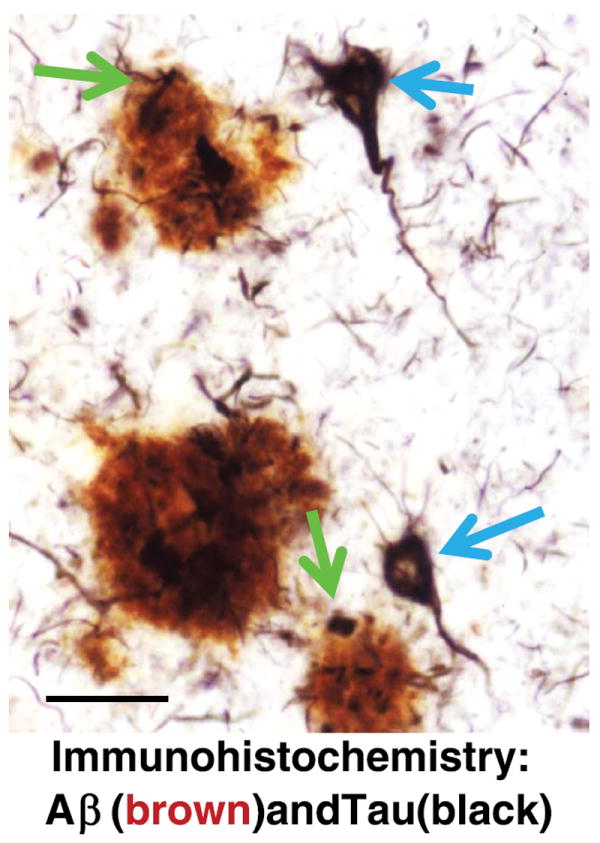
Photomicrograph of a section from the cerebral neocortex of an Alzheimer disease brain stained using double-label immunohistochemistry for β-amyloid (Aβ, reddish brown) and microtubule-associated protein tau (black). Aβ plaques (AβPs; blue arrows) are roughly spherical and extracellular, whereas neurofibrillary tangles (NFTs; green arrows) develop within neurons. Note that some of the dystrophic neurites in the AβPs contain aberrant tau protein pathology (black), which is biochemically identical to that seen in intracellular NFTs. These AβPs have been described to be “neuritic plaques.” Scale bar = 50 μm.
Neurofibrillary tangles are not specific for AD (29-32); indeed, they are found in almost every class of brain disease and are universal (yet topographically restricted) in normal aging subjects (33). Neurofibrillary tangles are found in the brains of individuals who experienced frontotemporal lobar degeneration with tauopathy (FTLD-MAPT), focal cortical dysplasia, myotonic dystrophy, prion diseases, metabolic/storage diseases, some brain tumors, chronic traumatic encephalopathy, viral encephalitis, and other brain diseases (34-37) (Table 3). This suggests that NFTs are, at least under some conditions, a secondary response to injury. On the other hand, tau gene mutations can produce clinical dementia with NFTs; this indicates that, under some conditions, NFTs may be directly linked to the primary or at least proximal neurodegenerative changes (29, 31, 37).
TABLE 3.
Alzheimer DiseaseYLinked Pathologic Features and Their Presence in Conditions That Have Pathologic Overlap With Alzheimer Disease
| Neuropathologic Observation | |||||
|---|---|---|---|---|---|
|
| |||||
| Clinical Condition | Diffuse AβPs | AβPs With Tau Neurites | Hippocampal NFTs | Neocortical NFTs | References |
| Alzheimer disease | +++ | +++ | +++ | +++ | This review |
| “Oldest old” without dementia | ± | − − | +−++ | − − | (172, 324, 325) |
| Tangle-only dementia | +++ | + | (116, 326) | ||
| Dementia with Lewy bodies | +−++ | − − | + | − − | (327, 328) |
| Chronic traumatic encephalopathy | +−+++ | ± | +/++ | +−+++ | (329-331) |
| Niemann-Pick Type C | ± | ± | + | + | (332, 333) |
| Guamanian amyotrophic lateral sclerosis parkinsonism/dementia syndrome | ++ | − − | ++ | ++ | (334-338) |
| Tauopathies: PSP, CBD, Pick disease, FTLD-MAPT | − − | − − | ++ | ++ | (37, 339, 340) |
AβP, neuritic and diffuse amyloid plaques containing Aβ peptide; FTLD-MAPT, frontotemporal lobar degeneration with mutation in MAPT; CBD, corticobasal degeneration; NFT, neurofibrillary tangle; PSP, progressive supranuclear palsy.
− −, no disease-specific pathologic feature; ±, scattered or inconsistent pathologic features; +, low level of pathologic features; ++, moderate level of pathologic features; +++, high level of pathologic features.
Both the density and the neuroanatomic localization of NFTs are important parameters in AD neuropathology. The findings reported by Tomlinson et al (38) in 1970 have been replicated many times: “[NFTs are] found in both controls and dements [demented subjects] in the hippocampus, but elsewhere in the cortex was only severe or widespread in the dements; severe generalized neurofibrillary change in the cortex was not seen in any control; it seems possible that its occurrence always indicates dementia.” This passage underscores the key point that may be relevant to the study of tau biomarkers, that is, a modest number of medial temporal lobe NFTs are universally present in subjects older than 70 years (33), even in persons who have intact cognition. Recently, it has been reported that NFTs are also very common in certain brainstem nuclei in subjects without dementia (39-44). As such, neurofibrillary degeneration restricted to subcortical sites is often subclinical, whereas widespread neocortical NFTs are almost always associated with severe cognitive impairment in more than 1 disease state.
In contrast to NFTs, AβPs are extracellular (45, 46) and are found in a high proportion of all elderly persons but are not universal (44, 47, 48). A particularly important subtype of AβPs are “neuritic plaques” (NPs), as they are more likely to be associated with cognitive impairment than “diffuse plaques” (49-51). Neuritic plaques are AβPs surrounded by degenerating axons and dendrites that often contain hyperphosphorylated tau aggregates. This subset of AβPs is a hallmark of the current diagnostic criteria for AD, although there is no universally accepted definition of a NP. Indeed, AβPs that lack degenerating tau-positive neurites may have dystrophic neurites that are detected using other methods, including the Bielschowsky silver method, thioflavine S, or immunohistochemistry that detects p62, ubiquitin, phosphorylated neurofilament proteins, or amyloid precursor protein (APP). Some of the variability may be due to the evolution of the NP as part of the disease process (52-54). Because non-AD tauopathies usually lack NPs, they are not an inevitable cellular response to tau pathology (Table 2).
AβPs alone do not seem to be a sufficient substrate for advanced clinical AD dementia (ADD). However, in contrast to neurofibrillary (tau) pathology, there seems to be a strong association between AD genetics and Aβ plaque formation. All high-penetrance AD genetic risk alleles (i.e. APOE ε4 allele, Down syndrome, APP mutations and duplications, PSEN1 and PSEN2 mutations) have been linked in various experimental systems with increased Aβ deposition and increased formation of putative toxic Aβ peptide species (46, 55-57). These data have strong mechanistic implications with an epidemiological scope because genetic factors confer approximately 70% of an individual’s risk for AD (58, 59).
The concordance of genetics and Aβ deposition is aligned with CPC studies that indicate that AβPs may be a temporally “upstream” feature of the neocortical disease, with the caveats that brainstem and medial temporal lobe pretangles and NFTs are seen in subjects without Aβ deposition in all age categories (33, 39, 44). Thus, AβPs and NFTs are the hallmark features of AD but do not develop in the human brain according to the same temporal or anatomic pattern (60, 61). These nuanced observations, which have been gathered and analyzed by many researchers, provide the bases to address some of the controversies that have existed in the field for decades.
CONTROVERSIES IN AD CPC RESEARCH
A basic goal of AD CPC research has been to assess critically whether CPC data support the hypothesis that AD neuropathologic changes (AβPs and NFTs) correlate with clinical dementia. We note that there are publications contending that CPC data argue against a deleterious role for AD pathologic changes (i.e. that they may be an “epiphenomenon” of aging) and that the disease should not necessarily be defined by their presence (62-68). This controversy has been ongoing for many years (38). There are 4 separate assertions that seem to have gained traction in the field, thereby resulting in uncertainty:
Assertion 1. One sometimes observes persons without dementia with advanced AD pathologic changes at autopsy.
Assertion 2. One sometimes observes persons with cognitive impairment who clinically to have AD, but who lack AD pathologic changes at autopsy.
Assertion 3. Clinical trials aimed at reducing AD pathologic changes have failed.
Assertion 4. AβPs and NFTs may be neuroprotective rather than neurotoxic.
Assertions 1 and 2 are addressed directly by extensive data from AD CPC studies that support 3 points. The definition of “advanced AD pathologic changes” is critical. Some groups may consider widespread diffuse and/or NPs as advanced AD pathologic diagnosis regardless of the numbers and distributions of NFTs. It is clear, as mentioned previously, that AβPs, in the absence of any other neurodegenerative disease lesions or other pathologies, are not a sufficient substrate for severe dementia and thus do not constitute “advanced AD pathologic changes.” In contrast, dense and extensive neocortical NFTs are very consistently associated with dementia and thus, according to new diagnostic criteria, these are required to constitute high burden of AD neuropathologic change (3). As for Assertion 2, it is true that dementia, even one that may be clinically similar to probable AD, may be present without AD pathologic changes; this is simply not AD, but presumably one among many other diseases that cause dementia (e.g. hippocampal sclerosis or vascular dementia). These points will be considered in the context of the AD CPC literature.
Assertion 3 relates to what clinical trials data tell us about the impact of AD pathologic changes. There have been cases in which individuals with mid- to late-stage clinical AD were administered anti-Aβ immunotherapy that partially cleared AβPs, but this clearance did not seem to mitigate NFT formation or the inexorable disease course (69-71). There are multiple reasons why these trials may have failed, including the late timing of the therapy. Thus, conclusions drawn from these trials may be premature (72, 73). In fact, there have been hints of some beneficial effects (clinical and biochemical) linked to human anti-Aβ immunotherapies (74-79). As yet, there are no agents that are capable of reducing tangle density. In sum, clinical trials have not provided definitive answers as to the direct role of AβPs or tangles in the cognitive impairment associated with AD pathologic diagnosis (80, 81).
Assertion 4 is that AβPs and NFTs are actually neuroprotective rather than toxic. Because autopsies provide only cross-sectional data, they cannot prove mechanism. Since both tau phosphorylation and Aβ peptide generation are seen in “normal” brains, some contend that there are probably beneficial and adaptive aspects to these molecular processes. This is important to consider when developing therapies that seek to inhibit those pathways (62). Yet, there are strong arguments in favor of the hypotheses that the molecular processes that underlie tau and Aβ deposition in NFTs and AβPs are indeed pathologic. Abundant evidence from many independent studies indicates that Aβ peptides and other plaque substances (in their various molecular forms) may be neurotoxic, both directly and through secondary responses to stress, injury, and inflammation (46, 82, 83). Both gain-of-toxic-function and loss-of-normal-function deleterious effects also have been described for phospho-tau (84-89). The toxicity of oligomeric forms of tau and Aβ peptides could be connected with biochemical changes that also produce NPs and NFTs; thus, the oligomeric species may be directly related to the neuropathology, although they are not exactly correlated (90-95). It must be acknowledged that a simplistic conceptualization of “plaques and tangles” does not adequately reflect the complexity of the biochemical changes in the brains of patients with AD. Thus, peptides and higher-order aggregates derived from products of APP and MAPT genes can potentially lead to combinations of neuroprotective and toxic attributes. Here, we focus on data and analyses from particular human autopsy studies to address the specific question, “are there strong correlations between antemortem cognitive impairment and the ‘defining’ hallmarks of the disease?”
We consider it to be an important clue that most brains from elderly human subjects show abnormalities that fall somewhere along particular continua of pathologic severity (60, 96-100). These patterns of brain changes indicate specific features that can be correlated with clinical parameters, modeled to understand disease mechanisms, and targeted for therapeutics. Before addressing the “classic” AD pathologic changes and their clinical correlates, we discuss in the following section some of the less common pathologic findings in human brains, that is, the “exceptions that help test the rules.”
LESS-PREVALENT PATTERNS OF CLINICAL AND PATHOLOGIC FEATURES FOUND IN HUMAN BRAINS
“Plaque-Only” Dementia
In 1987, Terry et al (101) described what has become known as “plaque-only dementia.” There were 58 subjects with dementia and AD changes, of which 40 had 2 or more neocortical tangles per high-magnification field in frontal, temporal, and parietal lobes; 18 had no neocortical NFTs. The cases did not differ in hippocampal tangle densities and had no significant differences in brain weights, neocortical thickness, or neocortical neuron counts. Despite this, both groups were demented and did not differ significantly on the Blessed Dementia Scale. The existence of this “plaque-only” dementia category has been controversial because some subjects with pathologic findings similar to the plaque-only group described by Terry et al have dementia and some do not. The current revision of the National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of AD stipulate that subjects with moderately dense neuritic AβPs may be regarded as having an intermediate level of AD neuropathologic change and that other potential contributors to cognitive impairment and dementia should be sought (3). Notably, prior studies necessarily included brains from persons with diseases (e.g. neocortical α-synucleinopathy or TDP-43 pathology) that were not at that time well characterized, but which can coexist with variable neocortical amyloid loads. Indeed, Hansen et al subsequently published an article titled “Plaque-only Alzheimer disease is usually the Lewy body variant, and vice versa” (102). When modern pathologic methods are used, severe cognitive impairment associated only with diffuse AβPs in the brain at autopsy is rare and has an unknown relationship to AD. Subtle cognitive impairment may be associated with diffuse AβPs, indicating that these lesions may play a direct role in incipient AD (54), although a direct role of diffuse Aβ plaques without tau pathology has not been proven. Furthermore, diffuse AβPs may be associated with cerebral amyloid angiopathy, which could produce impairments separately (103, 104), although see Nelson et al (105). As the field moves toward identifying subtle (i.e. early) cognitive impairment, more work will be required to characterize any deleterious influence of diffuse AβPs on cognition and clinical manifestations in cases of presumed incipient AD. On the other hand, the density of tau-positive NPs does correlate directly and statistically independently with some degree of cognitive impairment (105-107) (Fig. 2).
FIGURE 2.
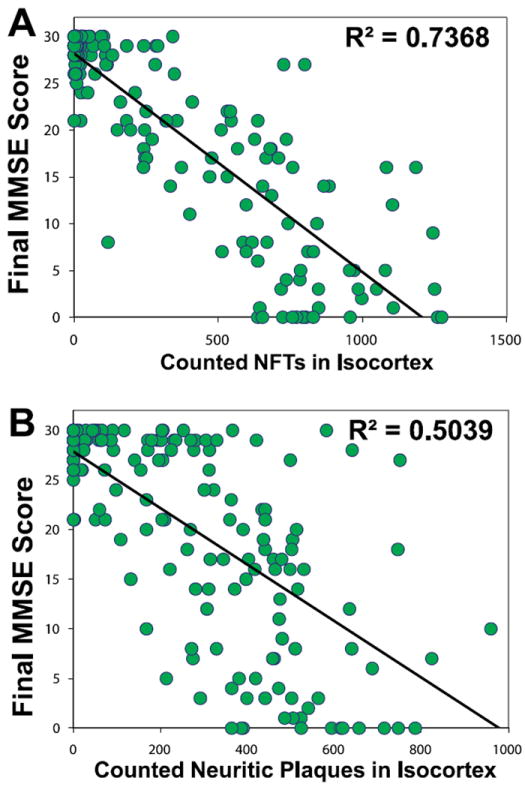
(A, B) Correlations between antemortem cognitive status (final Mini-Mental State Examination [MMSE] scores), counted neocortical neurofibrillary tangles (NFTs; A), and neuritic β-amyloid plaques (NPs; B) for 178 patients lacking concomitant neuropathologic findings (189). Each circle represents data from a single individual. Neurofibrillary tangles and NPs were counted and summed from 4 different portions of cerebral neocortex: Brodmann areas 21/22, 18/19, 9, and 35, as described (189). Data are reprinted with permission from the Journal of Neuropathology and Experimental Neurology (2007;66:1136–46). Copyright 2007, American Association of Neuropathologists. The correlation between final MMSE scores and neocortical NFT counts is stronger than that between MMSE scores and NP counts.
“Tangle-Only” or “Tangle-Predominant” Dementia and Other Non-AD Tauopathies
In most community-based autopsy series, non-AD tauopathies constitute approximately 1% of dementia cases (108-115) (but see Noda et al [116]). In autopsy series associated with dementia clinics, a higher proportion of non-AD tauopathies are seen, presumably due to recruitment bias (111). Among well-recognized forms of tauopathies are FTLD-MAPT, progressive supranuclear palsy, corticobasal degeneration, Pick disease, argyrophilic grain disease, and “tangle-only dementia” (116-125). There are also some individuals with intact or subtly diminished cognitive abilities whose brains harbor hippocampal and/or amygdala NFTs (Braak stages III–IV) with few or no AβPs; these represent approximately 5% of cases in some large autopsy series (41). In this condition, the NFTs comprise tau isoforms similar to those of classic AD, but these subjects do not have the same distribution of APOE genotypes as subjects with AD (118). It remains to be determined whether these individuals died early in the course of an idiosyncratic subtype of AD (12) or instead reflect a completely different pathogenesis.
AβPs and NFTs But No Dementia
Confusion persists about individuals lacking documented cognitive impairment yet whose brains reveal neuropathologic findings of AD at autopsy. This is an important issue because it challenges the principle that the processes that underlie AD neuropathologic changes are the sole cause of cognitive impairment. To address this issue, longitudinal studies of participants who are enrolled while cognitively normal and followed for several decades can be used to study the sequential clinical and corresponding molecular pathologic changes in AD. Rigorous analysis requires preliminary modeling of what is expected at a population level based on what is known about the disease.
The prevalence of dementia due to AD, as judged by clinicians, doubles every half-decade after age 65; thus, approximately 30% to 40% of living 95-year-old patients receive the diagnosis of ADD (126-128). The interval between initial ADD clinical diagnosis and death is approximately 8 years (129), but AβPs and NFTs are present well over a decade before death (130-138). New terminology has been developed to delineate stages of preclinical AD (48, 139, 140). The median life expectancy in Western countries is approximately 79 years (141). Thus, using these data, we can deduce confidently that an “average” person dying at age 79 has an approximately 15% likelihood of having been diagnosed clinically with ADD, but an approximately 30% to 40% chance of harboring significant AD neuropathologic changes (otherwise, the prevalence of ADD could not reach approximately 30%–40% by age 95). Indeed, this is precisely the observation reported by one of the largest autopsy series to date (142) and is well supported by AD biomarker studies (143-145).
The existence of persons without dementia with some AD neuropathologic changes is therefore not problematic. It is expected that many persons die with brains that exhibit AD neuropathologic change in the preclinical phase of AD; indeed, data support the modeled expectations (44, 142). One would expect a normally distributed population-based model of AD to contain individuals with early or moderate AD who are classified dichotomously (and falsely) as lacking dementia. Pathologic changes that are present before clinical manifestations are a well-accepted concept in other chronic diseases such as cancer, atherosclerosis, and others (146, 147) but are equally relevant to discussions of AD. Significantly, the severity and distribution of AD neuropathologic changes are much different (lower) in preclinical cases than in cases with clinically severe ADD (48, 108, 148-150). We are aware of no reported case of truly intact cognition despite extremely severe AD pathologic changes (i.e. widespread neocortical NFTs) measured with quantitative pathologic analysis, although many persons with intact cognition have been assessed in autopsy series (33, 42, 54, 133, 142, 148, 151-166).
Dementia Cases Lacking AβPs, NFTs, or Other Pathologic Substrates
The literature does not indicate large numbers of patients with advanced dementia whose brains lack any pathologic changes when up-to-date neuropathologic methods have been applied to their brains. Older studies that reported cases with dementia and no other known pathologic substrate lacked insights into more recently defined pathologies, such as those with aberrant TDP-43, valosin-containing protein, and fused in sarcoma inclusions (37, 167). Moreover, some prior methods (e.g. Bielschowsky impregnation) were less sensitive to some tauopathies than Gallyas for NFTs, Campbell-Switzer for AβPs, and immunohistochemistry. On the basis of the discovery of these and other novel neurodegenerative disease lesions in the past 20 years, we view reports that some individuals in old age may have moderate cognitive impairment without advanced AD pathologic diagnosis as a challenge to the research community to elucidate the novel mechanisms underlying cognitive impairments in those subjects (168-173). A notable example of this is schizophrenia, which, in elderly patients, is associated with dementia, although the underlying basis of this dementia is enigmatic (174-176). Normal pressure hydrocephalus is another poorly understood cause of dementia that may lack AD pathologic changes (177, 178). Understanding these phenomena and how they may contribute to cognitive impairment in a broader population will require more thorough study of non-AD brain diseases.
AD-Related Brain Pathology in Individuals Older Than 90 Years
There have been reports of dissociation between AD neuropathologic changes and cognitive status in extreme old age, but these data merit careful analysis. The incidence of dementia increases continually up to (179-183) and above (139, 184) 90 years of age. In contrast to the increased prevalence of clinical dementia with advanced age, the prevalence of cases with high NP and high NFT pathologic diagnosis at autopsy seems to level off in the oldest old according to multiple autopsy series (154, 168-172). However, not all studies are in agreement on this point (185, 186); these “survivors” represent a small fraction of the human population with underrepresentation of APOE ε4 allele (187). Even in extreme old age, the presence of many neocortical NFTs correlates with antemortem cognitive decline (105, 188-191). Thus, no “dissociation” exists between the AD neuropathologic change and cognitive impairment. The enigma relates to cognitively impaired individuals of advanced age whose brains lack substantial AD pathologic diagnosis at autopsy.
Cognitive impairment is associated with many diseases. Cerebrovascular disease (CVD) is the most prevalent non-AD pathologic diagnosis in the brains of the advanced aged and is directly relevant to any CPC study related to dementia. The difficulties introduced by this multifaceted and unpredictable disease category in aged individuals have been discussed previously (17, 192-204); 75% to 90% of patients older than 90 years have some degree of CVD pathologic diagnosis (189, 205-207). There is no universally applied rubric for CVD pathologic diagnosis, although the recent National Institute on Aging–Alzheimer’s Association criteria attempted to systematize neuropathologic assessment of this complex form of brain injury (3). Nevertheless, the profound impact of CVD on studies pertinent to cognition in the elderly seems to be underappreciated in dementia research (208-210).
In addition to AD and CVD, there are many widespread contributors to cognitive impairment in the elderly. Examples of these include hippocampal sclerosis (a prevalent disease that plays a strong deleterious role in extreme old age and is distinct from the disease linked to epilepsy in younger individuals [211]), α-synucleinopathies, hematomas, argyrophilic grain disease, neuropsychiatric disorders and their therapies, failure of other organ systems, diabetes, hypertension, chemotherapy, and other adverse effects of medications (157, 212-220).
Thus, in the oldest old, the prevalence of concomitant non-AD brain diseases, including CVD and hippocampal sclerosis, begins to mimic the effects of AD pathologic diagnosis (154, 221, 222). Together, these and perhaps other uncharacterized changes may weaken the apparent association between AD pathologic diagnosis and cognitive status (189, 195, 198). An analogy can be made to heart disease: it would be difficult to study the clinicopathologic correlation between coronary atherosclerosis and cardiac function if two thirds of cases had alular dysfunction, infectious endocarditis, or severe arrhythmias. Despite all the potential pitfalls, many high-quality studies related to the correlations between cognitive status near the time of death and AD-type pathologic burden at autopsy have been performed.
CLINICOPATHOLOGIC CORRELATION IN AD
Correlation Between AβPs and Cognitive Status
More than 40 CPC studies have assessed the correlation between AβPs and severity of antemortem cognitive impairment (38, 98, 103, 105-108, 158, 162, 171, 185, 189, 191, 223-260). These studies vary with respect to many factors including the research cohort characteristics, anatomic areas examined, pathologic methods, AβP subcategories and counting techniques, metrics for cognition, the range and domains of cognitive decline tested, and the rigor with which concomitant pathologic findings were evaluated and factored into the study.
Although the study designs were diverse in CPC studies involving AβPs, most studies confirm a significant correlation between antemortem cognitive impairment and neocortical AβPs, as demonstrated by Blessed et al (254) (Fig. 3). These workers used the von Braunmühl silver stain, which could not differentiate between diffuse and neuritic AβPs. This, and some other earlier studies, incorporated a bias related to the inclusion of many individuals at opposite ends of the AD continuum thereby masking the poorer correlation between plaque load and dementia severity among the impaired subjects. Subsequent studies have established that the densities of NPs correlate with antemortem cognitive impairment better than the densities of diffuse AβPs (45, 98, 106, 154, 189, 261).
FIGURE 3.
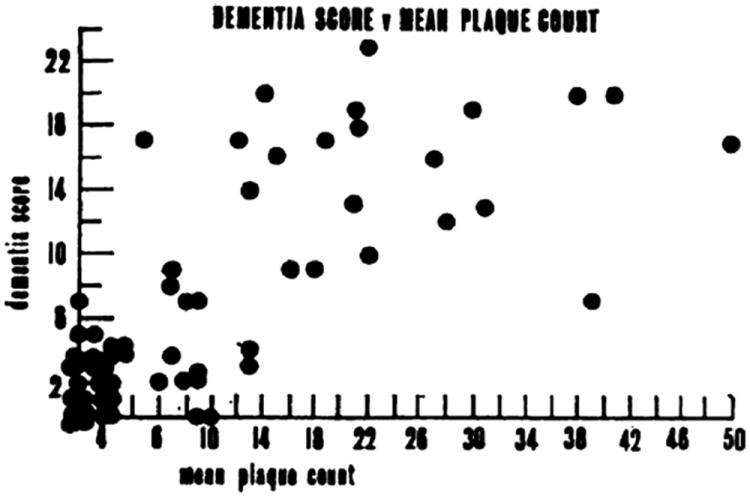
Correlations between antemortem cognitive status (“dementia scores”) and counted amyloid plaques from the 1968 article by Blessed et al (254). Dementia scores were derived from “psychological tests of orientation, remote memory, recent memory, and concentration.” Amyloid plaques were visualized using the von Braunmühl silver stain. Each circle represents data from a single individual. There is reasonable correlation between the dementia scores and the number of plaques, although this work antedated the era of neocortical synucleinopathy, TDP-43, and other factors now known to both clinicians and neuropathologists. This figure was reproduced with permission from The British Journal of Psychiatry (1968;114:797–811). Copyright 1968, The Royal College of Psychiatrists.
It is important to note, however, that many prior studies have only considered limbic and neocortical plaques. The distribution of AβP pathology may occur throughout the entire neuraxis in advanced disease. Elderly without dementia show early stages of this hierarchical process. The progression of AβP pathology seems to begin in the neocortex with a few diffuse plaques and then sequentially progress to also include 1) the hippocampus, entorhinal cortex, cingulate cortex, and amygdala; 2) basal ganglia and diencephalon; 3) midbrain and medulla oblongata; and 4) pons and cerebellum (262). A recent study with large numbers of subjects assessed with standardized antemortem cognitive testing found that the amyloid stage that has progressed to involve the striatum is highly predictive of dementia (263). Correlations between amyloid stages (Thal phases), NFT burden, and cognitive impairment are shown in Figure 4.
FIGURE 4.
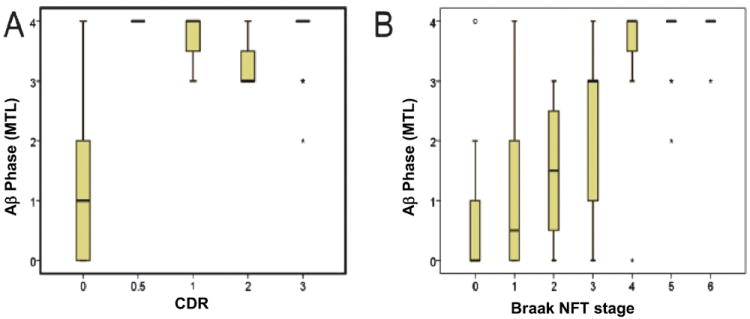
β-Amyloid (Aβ) phase showing the relationship between cognition, as represented by the retrospectively determined clinical dementia rating scale (CDR) score, and the phase of Aβ deposition determined in the medial temporal lobe (MTL) in 202 cases (103, 262). Non–Alzheimer disease (AD) dementia cases were excluded from this analysis. (A) Almost all demented cases exhibited end phases of the expansion of Aβ deposition without major differences; cases without dementia had early-phase Aβ pathology (partial correlation controlled for age and sex for all cases: r = 0.582, p < 0.001; only for AD cases: r = 0.086, p = 0.605). (B) Relationship between Braak neurofibrillary tangle (NFT) stage and Aβ phase in 201 AD and control cases. With increasing Braak ? NFT stage, the distribution of Aβ plaque deposition expanded to end-phase Aβ deposition reached with Braak NFT stage IV (partial correlation controlled for age and sex for all cases: r = 0.621, p < 0.001; only for AD cases: r = 0.07, p = 0.671).
A critical point supporting the importance of plaques is that widespread neocortical NFTs are virtually nonexistent without the presence of widespread AβPs, except in the minority of cases that show a clear-cut non-AD tauopathy (e.g. progressive supranuclear palsy). Owing in part to the scarcity of neocortical NFTs in the absence of AβPs, and the relatively modest apparent direct impact of AβPs on cognition, it has been suggested that the pathogenetic effect of AβP-related substances may be mediated by “seeding” neurofibrillary pathology (46, 88, 189, 264, 265). The current clinical trial literature is compatible with the idea that AβP-related substances kindle a self-propagating process related to neocortical NFTs, as has been hypothesized (266-269). Regarding AβPs in AD, an analogy can be made to the established role of high blood cholesterol in heart disease. Hypercholesterolemia promotes atherosclerosis that over time can cause myocardial infarction. Therefore, high blood cholesterol is considered a causative risk factor for heart disease, although it does not correlate with clinical heart disease in all groups (many people live with hypercholesterolemia for years without experiencing myocardial infarction). In the same way, AβPs may be a key pathologic factor in AD without equating with the clinical features of ADD.
Often neglected is the question of lesion turnover (i.e. AβPs being possibly reabsorbed during life so that they would not be “counted” at autopsy), which seems to occur in some subjects (70, 270). For this reason, the number of AβPs might plateau (or at least the rate of increase flatten out), leading to a “ceiling effect” relatively early in disease process that confounds linear correlations between plaque number and severity of cognitive change (253). Aβ vaccine trials also attest to the ability of the immune system to clear diffuse AβPs (69, 70, 271), but it is not known whether toxic APP metabolites or other plaque components remain after clearance of fibrillar Aβ-peptides (73, 272). The active “turnover” during life could reduce the correlation between the lesion numbers observed at autopsy and clinical features of the disease.
Neuritic AβP (NP): A Pathologic Entity With Mechanistic Implications
AβPs ringed by degenerating neurites that usually contain abundant PHF-tau, that is, NPs (Table 1), deserve special consideration. In histologic sections, NPs are characterized by nearby abnormal nerve cell processes (Fig. 1). The abnormal NP-contacting dendrites and axons contain pathologic tau fibrils identical to those seen in NFTs (244, 273), with loss of adjacent synapses (274, 275). Dystrophic axons from completely different afferent sources, expressing distinct neurotransmitters, have been shown to be present in particular NPs (276, 277), which strongly implies that toxicity is directed from the (extracellular) plaque to the radially arranged intracellular neurites. The particular toxic substance(s) within NPs still have not been completely characterized (90, 103, 278), but NPs represent a nidus in which extracellular plaque substance(s) seem to induce intracellular degenerative changes with tau pathology (18, 50, 279-281). This process may be synergistic or identical to the stimuli that could promote NFTs in human brains. Neuritic plaques seem to develop after diffuse AβPs and NFTs (44). Using data from the National Alzheimer’s Coordinating Center with inclusion and exclusion criteria, as described previously (282), NPs and NFTs tend to coexist (Table 4); this corroborates earlier observations (283). The characteristic anatomic distribution of NPs and NFTs supports additional hypotheses in relation to their formation that are outside the scope of this review (33, 39, 284-286).
TABLE 4.
Distribution of Cases According to Braak Stages and Neuritic Amyloid Plaque Densities
| Neuritic Aβ Plaque Density
|
||||
|---|---|---|---|---|
| None | Sparse | Moderate | Frequent | |
| Braak staging (NFTs) | ||||
| 0 | 52 | 14 | 10 | 3 |
| I | 79 | 24 | 31 | 7 |
| II | 68 | 59 | 48 | 17 |
| III | 48 | 59 | 108 | 64 |
| IV | 22 | 41 | 160 | 158 |
| V | 0 | 25 | 134 | 553 |
| VI | 3 | 14 | 72 | 1,035 |
Data are from the National Alzheimer’s Coordinating Center (n = 2,903 included, as described previously [282]) to show distribution of cases according to Braak stages (0 to VI) of neurofibrillary tangles (NFTs) (60) and neuritic amyloid (Aβ) plaque densities, graded according to Consortium to Establish A Registry for Alzheimer’s Disease criteria (51).
Correlation Between NFTs and Cognitive Status
Numerous CPC studies have assessed the association between NFTs and cognitive status (38, 98, 154, 158, 162, 185, 188, 189, 191, 223-232, 235-240, 244, 250-252, 258, 261, 264, 287-296). As with studies on AβPs, those of NFTs have used diverse study designs. Regardless of methodology, the correlation between neocortical NFTs (but not necessarily allocortical or subcortical NFTs) and antemortem cognitive status is strong in studies that span the spectrum of cognitive impairment (provided there is not a strong influence by other diseases that can cause cognitive impairment). Results from the University of Kentucky Alzheimer’s Disease Center (189) corroborate previous findings (Fig. 2). Duyckaerts et al (287) and Sabbagh et al (252) independently arrived at the conclusion that the density of NFTs in selected cerebral cortical fields significantly (p < 0.01) correlated with Blessed or Mini-Mental State Examination scores (297). Likewise, Dickson et al (251) described a strong correlation between cortical NFT counts or phospho-tau ELISA measurements and Blessed Information, Concentration, and Memory test scores. Similar results were obtained by Bennett et al (264) who found a strong correlation between global cognitive status and densities of neocortical tau-positive tangles.
On the basis of the review of an enormous sample of cross-sectional autopsy data, the pattern of NFT development across the clinical spectrum of disease has been integrated in Braak neuropathologic staging (60, 298, 299). Neurofibrillary pathology is observed in the human brain long before Aβ plaques develop (39, 44, 300). At 40 years, all individuals studied by Braak and Del Tredici (39) and Braak et al (44) exhibited at least initial neurofibrillary pathology in the brainstem (Fig. 5). The relationship between the near-universal tau pathology seen in the locus coeruleus of middle-aged individuals and AD is not known. As AD progresses, the NFTs become numerous in locus coeruleus which may suggest the same pathogenetic process is becoming more severe (301).
FIGURE 5.
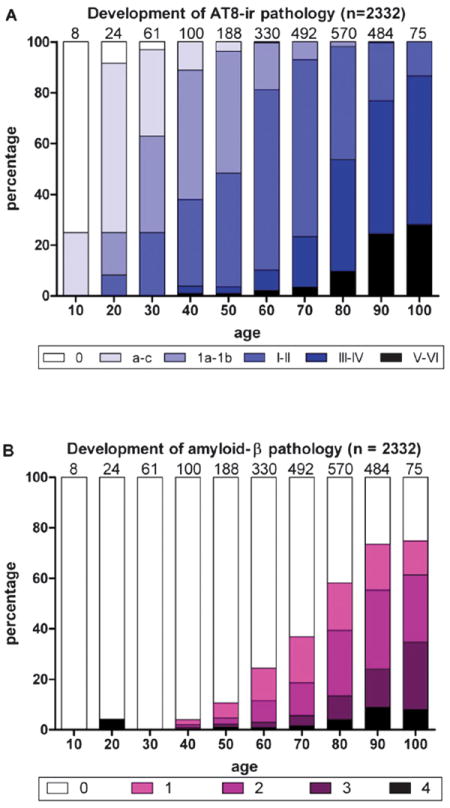
Development of phospho-tau (AT8)-immunoreactivity (ir) versus β-amyloid (Aβ) pathologic findings. (A) White columns indicate the relative frequency of 2,332 nonselected autopsy cases devoid of any abnormal intraneuronal tau deposits. Columns in shades of blue indicate the relative frequency of cases with all types of intraneuronal lesions (Braak NFT stages). (B) Development of extracellular Aβ deposits. Purple areas within the columns indicate subgroups of cases showing plaque-like Aβ-amyloid deposits in temporal neocortex (Phase 1, light purple), allocortex and neocortical association areas (Phases 2 and 3, middle purple and dark purple), or in virtually all cerebral cortical regions (Phase 4, black). Note the relatively late appearance of Aβ plaques in comparison to subcortical neurofibrillary tangles. This figure is reproduced with permission from the Journal of Neuropathology and Experimental Neurology (2011;70:960–99) (44). Copyright 2011, American Association of Neuropathologists.
In more advanced disease, the neuroanatomic distribution of NFTs correlates both with the location at which neurons die (302-305) and with the cognitive domains affected in patients with ADD. For example, early AD symptoms tend to relate to memory, when the anatomic substrates of memory in the medial temporal lobe are selectively affected by NFTs (Braak stages III–V). The cognitive domains affected in mid-and late-stage ADD expand to include areas of executive function, visuospatial capacities, and speech. These manifestations occur in synchrony with the development of NFTs in the neocortical areas responsible for those functions (Braak stages V–VI). Braak staging provides a useful ordinal system for describing the topographical distribution of pretangles and NFTs in human brain. Nevertheless, Braak staging also masks the fact that marked variability in pathologic lesion density across cases exists within a given Braak stage (7, 12) and that not all AD cases fall within the same spatial continuum with regard to brain NFT distributions (12, 15, 151).
A key question in this regard is whether a heavy neocortical NFT load is present in individuals with intact cognition. In a review of 11 different studies comprising 555 subjects without dementia, a total of 12 brains were assessed as Braak stage V (2.2%); 3 were assessed as Braak stage VI (0.5%) (142). However, not all Braak stage VI brains were the same; there was significant variation in NFT burden in these cases (142, 151). In the Braak stage VI cases studied where neocortical NFTs were most abundant, cognitive impairment was found on testing (151). It is important to emphasize that, among thousands of cases from dozens of CPC studies worldwide, there never has been a report of a thoroughly documented individual with truly “end-stage” neocortical NFT pathology who lacked antemortem cognitive impairment proximal to death (142, 151). Even the single case report “outlier,” a Braak neuropathologic stage VI case without full-blown dementia, had some degree of cognitive impairment within expectations for late preclinical AD (306).
As with AβPs, NFTs are quantified with certainty only at autopsy. Therefore, it is necessary to know whether some NFTs are removed (or reabsorbed) from the brain before death. Some, but not all, studies have indicated that more neurons disappear in brains of demented subjects than can be explained directly by the number of NFTs and “ghost tangles” observed at autopsy (285, 304, 307-310) (but see [305]). Thus, some evidence exists for NFT “turnover,” neuronal shrinkage, and/or non-NFT cell death mechanism(s).
Overview of Specific Large Autopsy Series
A subset of the CPC studies is highlighted in Table 5. These studies were chosen because they represent large autopsy series with detailed clinical and pathologic assessments. The inclusion criteria for Table 5 were as follows: number of patients more than 40, neuropathologic assessments that included study of both NFTs and AβPs in the cerebral neocortex (not just the hippocampus), subjects with a broad range of pathologic severities, and nondichotomous correlation with cognitive status (i.e. not demented vs nondemented). These inclusion criteria do not minimize the contributions of studies with smaller sample sizes, for example, the important CPC study by Arriagada et al (293) that assessed 10 patients. Nonetheless, a detailed description of all smaller autopsy series would comprise many dozens of additional studies along with a commentary regarding each of their specific attributes. It should also be mentioned that some of the important autopsy series of AD CPC are excluded from this list because they only assessed AβPs and not NFTs, assessed only the hippocampus, or viewed cognitive status as a dichotomous variable. Studies not shown in Table 5 include the seminal articles of Blessed et al (254), Roth et al (255), and Tomlinson et al (38, 256), which found a correlation between dementia severity and amyloid plaques counted in cerebral cortex (with the caveat described previously), and previous studies that had also helped to establish the connection between AβPs, NFTs, and antemortem cognitive impairment (311-320).
TABLE 5.
Large Clinicopathologic Studies of Alzheimer Disease
| Study Institution | References* | Year | N | Avg Int | Methodological Considerations† | Main Finding(s) Correlating AD Pathology to CI |
|---|---|---|---|---|---|---|
| Harvard Medical School | (261) | 1991 | 56 | NR | C-S, QP | NFT > total amyloid plaques correlate with dementia severity |
| Oxford Project to Investigate Memory and Aging | (296, 341) | 1995 | 86 | NR | L, BP | NFTs > NPs > total amyloid plaques correlate with dementia severity |
| Mount Sinai ADRC | (291, 342, 343) | 1995 | 70 | NR | C-S, QP, cognitive status based on chart review | NFTs ≫ AβPs correlate strongly with CI |
| Albert Einstein | (154, 155, 251) | 1995 | 45 | 11.7 | L, QP | NFTs > AβPs correlate strongly with CI |
| Vienna Longitudinal Study on Dementia | (98) | 1996 | 122 | 6.8 | L, QP | NFTs > AβPs correlate strongly with cognitive status |
| Washington University ADRC | (225, 344) | 1998 | 199 | NR | L, QP | Correlation to CI: NFTS > “cored senile plaques” > total senile plaques |
| Nun Study | (109, 151, 288) | 1999 | 495 | 9.6 | L, CC, QP, only (female) nuns | NFTs correlate best with CI; all cases above threshold demented |
| Cambridge, UK Group | (345) | 2000 | 48 | <12 | L, BP | NFTs ~ NPs >total amyloid plaques correlate with global “clinical severity” of AD |
| Mayo Clinic ADC | (346) | 2002 | 67 | 0.7 | L, BP | Significant correlation between Braak stage and CI |
| Rush University | (190, 264, 347, 348) | 2004 | 652 | 8.2 | L, CC, QP | NFTs > NPS correlate with CI |
| University of California, San Diego, ADRC | (106) | 2004 | 131 | 8.5 | L,QP | NFTs correlate best with late AD CI, but NPs correlate better in early disease |
| Honolulu-Asia Aging Study | (349) | 2005 | 333 | 16.0 | L, CC, QP, only males | NFTs and NPs far more dense in subjects with than those without dementia Caveat: many concomitant pathologies |
| Adult Changes in Thought | (108, 350) | 2007 | 211 | <24 | L, BP, CC | NFTS (Braak stage) correlates with CI much stronger than AβPs |
| University of Kentucky ADC | (105, 189) | 2007 | 334 | 13.1 | L, QP | NFTs correlate best with cognitive status; all cases above threshold demented |
| Combined ADC project | (54) | 2009 | 97 | 0.7 | Various, QP, Subjects without dementia only | Presence of diffuse amyloid plaques correlates with subtle CI |
| Banner-Sun Health Research Institute | (252, 351) (258) | 2010 | 150 | 12.5 | L, QP, BP | NFTs > AβPs correlate strongly with CI |
| Baltimore Longitudinal Study on Aging | (161, 188) | 2010 | 209 | 8.8 | L, BP | NFTs in neocortex the key correlate of CI across the aging spectrum including past 90 y |
| 90+ Study | (191) | 2011 | 108 | 5 | L, QP, BP | Neocortical NFTs and hippocampal sclerosis key CI correlates in aged group |
References refer to the most relevant articles. See text for inclusion and exclusion criteria.
Methodological considerations: AβP, neuritic and diffuse amyloid plaques containing Aβ peptide; AD, Alzheimer disease; ADC, Alzheimer Disease Center; ADRC, Alzheimer Disease Research Center; Avg Int, average documented interval between death and autopsy in months; BP, pathology assessed using Braak stages (60); CC, community-based cohort; C-S, cross-sectional; CI, cognitive impairment; L, longitudinal; N, number of subjects in most relevant and recent article; NFT, neurofibrillary tangle; NP, neuritic plaque; NR, not recorded; QP, quantitative pathology (lesion counts).
Diversely designed studies from at least 18 different research centers have produced high-quality data with some common conclusions, notably that the density of neocortical NFTs is the pathologic feature that best correlates with antemortem cognitive status (Table 5). The correlation is less strong for neuritic AβPs, and less still for diffuse AβPs. Hippocampal pathology is nearly universal in AD cases, yet AD neuropathologic changes in hippocampus do not correlate as well as neocortical pathology with cognitive status at any stage of the disease due to floor and ceiling effects. Finally, it is critically important to take into account concomitant pathologies that contribute substantial “noise” to the system.
There are increasingly detailed and insightful assessments of both clinical and neuropathologic parameters, combined with more optimal subject recruitment, improved testing for new disease entities, and well-powered sample sizes. All of these factors help us to perform more valid analyses and generate consistent data. The overwhelming conclusion from many academic centers around the world is that AD neuropathologic changes, especially in the advanced stages of the disease (i.e. with abundant neocortical diffuse and neuritic AβPs and NFTs), correlate with the severity of antemortem cognitive impairment.
EVOLVING CONCEPTS AND UNANSWERED QUESTIONS
The field of AD research has made progress with respect to in vivo diagnostic methods to track the progression of AD and to identify patients who may benefit from candidate therapies. For the most part, these methods, which include neuroimaging, cerebrospinal fluid, and blood markers, are beyond the scope of the current review. Nonetheless, the strong focus on neuroimaging and biomarkers has raised important issues about the basic concept and definition of AD. There may be a future method that does not involve brain autopsy and that is optimized for diagnosing specifically the devastating illness that we refer to as AD. However, until such a method becomes available, neuropathologic examination remains the criterion standard for disease definition. Because cognitive impairment in aging may be attributed to many different conditions other than AD, current biomarkers for AD are limited to predicting which patients may become cognitively impaired with neocortical AβPs and NFTs, rather than just predicting cognitive impairment alone. For example, hippocampal atrophy is visualized on magnetic resonance imaging in both AD and hippocampal sclerosis patients and is therefore not a distinct AD biomarker. This is an important consideration because up to 20% of individuals in advanced age are affected by hippocampal sclerosis that correlates with cognitive impairment independently of AD pathologic diagnosis (105, 211, 321-323). A patient lacking AD pathologic diagnosis at autopsy, despite “biomarker positivity,” is not an AD case and suggests the biomarker has imperfect specificity. Conversely, AD pathologic diagnosis at autopsy in a biomarker-negative case implies imperfect sensitivity. Moreover, it is important to remember that AD is often mixed with other pathologies in the aging brain; thus, the finding of a positive biomarker for AD will not eliminate the need for autopsy studies. Indeed, there are still also no recognized biomarker-based criterion standards for a large number of other neurodegenerative tauopathies, synucleinopathies, and vascular brain changes.
There are limitations to CPC studies and significant questions remain unanswered at this time. Pathologic assessments cannot prove molecular mechanisms; instead, they identify and describe deviations from what is recognized as “normal.” We try to understand these changes by using an array of immunohistochemical and experimental methods. Although the density of neocortical NFTs and neuritic AβPs on autopsy correlate with the severity of cognitive dysfunction, and the observed results align with a plausible hypothesis, this does not prove that these pathologic hallmarks are proximate to the ultimate neurotoxic species. Moreover, we still seek answers to the following critical questions: What biochemical changes are upstream of the formation of NFTs and AbPs What exactly is the role of pretangles in the pathologic process? What processes occur in parallel with “classic” neuropathologic lesions? And, why are some cell populations differentially vulnerable?
SUMMARY AND CONCLUSIONS
Summary points are presented in Table 6. Neuropathology of aging-related brain disease must take into account diverse medical, technical, biochemical, and anatomic considerations. Concomitant pathologies are very common in aging brains. These diseases and many other factors collectively are formidable obstacles to aligning pathology and cognition along strictly linear scales. Although copathologies (CVD, hippocampal sclerosis, synucleinopathies, etc.) contribute to cognitive impairment, the universal observation of a strong independent correlation between plaques and tangles with dementia severity (despite the “background noise”) means that AD neuropathologic changes are likely significant. An extensive literature on CPC correlations related to AD dementia, a body of research-based on work performed around the world with many thousands of patients, indicates a sequence that may begin with specific genetic and environmental factors that increase risk of widespread AβPs. Neuritic plaques combine extracellular Aβ deposition and intracellular neurofibrillary (tau protein) pathology. Cross-sectional data indicate that pretangles and NFTs develop first in the brainstem and medial temporal lobe structures. Among patients with measurable clinical disease and lacking comorbidities, the extent of cognitive impairment parallels the severity of neocortical NFT pathology. In sum, there is a complex but predictable correlation between AD pathologic hallmarks and cognitive impairment.
TABLE 6.
Summary Points
|
AD, Alzheimer disease; CPC, clinicopathologic correlation; NFT, neurofibrillary tangle.
Acknowledgments
The authors thank the many research volunteers and wonderful colleagues around the world who participated in research efforts to understand AD and related diseases. We thank Dr Nina Silverberg for reviewing the manuscript.
Supported by Grants AG05136, AG02219, AG13854, AG05138, AG06647, AG10124, AG016976, AG035071, AG08051, AG008017, AG10161, AG15819, AG028383, and AG05681 from the National Institutes of Health/National Alzheimer’s Coordinating Center. Additional grants were from the Deutsche Forschungsgemeinschaft, Alzheimer Forschung Initiative, Michael J. Fox Foundation for Parkinson’s Research, Medical Research Council (UK), Alzheimer’s Research UK, Alzheimer’s Society, Alzheimer’s Association and BRACE.
References
- 1.Alzheimer A. Über eigenartige Krankheitsfälle des späteren Lebensalters. Z Ges Neurol. 1911;4:356–85. [Google Scholar]
- 2.Consensus recommendations for the postmortem diagnosis of Alzheimer’s disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer’s Disease. Neurobiol Aging. 1997;18:S1–2. [PubMed] [Google Scholar]
- 3.Montine TJ, Phelps CH, Beach TG, et al. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathol. 2012;123:1–11. doi: 10.1007/s00401-011-0910-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beach TG. The history of Alzheimer’s disease: Three debates. J Hist Med Allied Sci. 1987;42:327–49. doi: 10.1093/jhmas/42.3.327. [DOI] [PubMed] [Google Scholar]
- 5.Boller F, Bick K, Duyckaerts C. They have shaped Alzheimer disease: The protagonists, well known and less well known. Cortex. 2007;43:565–69. doi: 10.1016/s0010-9452(08)70251-x. [DOI] [PubMed] [Google Scholar]
- 6.Berchtold NC, Cotman CW. Evolution in the conceptualization of dementia and Alzheimer’s disease: Greco-Roman period to the 1960s. Neurobiol Aging. 1998;19:173–89. doi: 10.1016/s0197-4580(98)00052-9. [DOI] [PubMed] [Google Scholar]
- 7.von Gunten A, Bouras C, Kövari E, et al. Neural substrates of cognitive and behavioral deficits in atypical Alzheimer’s disease. Brain Res Rev. 2006;51:176–211. doi: 10.1016/j.brainresrev.2005.11.003. [DOI] [PubMed] [Google Scholar]
- 8.Berrios GE. Alzheimer’s disease: A conceptual history. Int J Geriatr Psychiatry. 1990;5:355–65. [Google Scholar]
- 9.Goedert M. Oskar Fischer and the study of dementia. Brain. 2009;132:1102–11. doi: 10.1093/brain/awn256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maurer K, Volk S, Gerbaldo H. Auguste D and Alzheimer’s disease. Lancet. 1997;349:1546–49. doi: 10.1016/S0140-6736(96)10203-8. [DOI] [PubMed] [Google Scholar]
- 11.Schneider JA, Aggarwal NT, Barnes L, et al. The neuropathology of older persons with and without dementia from community versus clinic cohorts. J Alzheimers Dis. 2009;18:691–701. doi: 10.3233/JAD-2009-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murray ME, Graff-Radford NR, Ross OA, et al. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: A retrospective study. Lancet Neurol. 2011;10:785–96. doi: 10.1016/S1474-4422(11)70156-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Han L, Cole M, Bellavance F, et al. Tracking cognitive decline in Alzheimer’s disease using the Mini-Mental State Examination: A meta-analysis. Int Psychogeriatr. 2000;12:231–47. doi: 10.1017/s1041610200006359. [DOI] [PubMed] [Google Scholar]
- 14.Clark CM, Sheppard L, Fillenbaum GG, et al. Variability in annual Mini-Mental State Examination score in patients with probable Alzheimer disease: A clinical perspective of data from the Consortium to Establish a Registry for Alzheimer’s Disease. Arch Neurol. 1999;56:857–62. doi: 10.1001/archneur.56.7.857. [DOI] [PubMed] [Google Scholar]
- 15.Hof PR, Vogt BA, Bouras C, et al. Atypical form of Alzheimer’s disease with prominent posterior cortical atrophy: A review of lesion distribution and circuit disconnection in cortical visual pathways. Vision Res. 1997;37:3609–25. doi: 10.1016/S0042-6989(96)00240-4. [DOI] [PubMed] [Google Scholar]
- 16.Jellinger KA. Criteria for the neuropathological diagnosis of dementing disorders: Routes out of the swamp? Acta Neuropathol. 2009;117:101–10. doi: 10.1007/s00401-008-0466-z. [DOI] [PubMed] [Google Scholar]
- 17.Schneider JA, Arvanitakis Z, Bang W, et al. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69:2197–204. doi: 10.1212/01.wnl.0000271090.28148.24. [DOI] [PubMed] [Google Scholar]
- 18.Wisniewski HM, Ghetti B, Terry RD. Neuritic (senile) plaques and filamentous changes in aged rhesus monkeys. J Neuropathol Exp Neurol. 1973;32:566–84. doi: 10.1097/00005072-197310000-00007. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki K, Terry RD. Fine structural localization of acid phosphatase in senile plaques in Alzheimer’s presenile dementia. Acta Neuropathol. 1967;8:276–84. doi: 10.1007/BF00688828. [DOI] [PubMed] [Google Scholar]
- 20.Hirano A, Dembitzer HM, Kurland LT, et al. The fine structure of some intraganglionic alterations. Neurofibrillary tangles, granulovacuolar bodies and “rod-like” structures as seen in Guam amyotrophic lateral sclerosis and parkinsonism-dementia complex. J Neuropathol Exp Neurol. 1968;27:167–82. [PubMed] [Google Scholar]
- 21.Thal DR, Del Tredici K, Ludolph AC, et al. Stages of granulovacuolar degeneration: Their relation to Alzheimer’s disease and chronic stress response. Acta Neuropathol. 2011;122:577–89. doi: 10.1007/s00401-011-0871-6. [DOI] [PubMed] [Google Scholar]
- 22.Masliah E. Mechanisms of synaptic dysfunction in Alzheimer’s disease. Histol Histopathol. 1995;10:509–19. [PubMed] [Google Scholar]
- 23.Masliah E, Mallory M, Hansen L, et al. Quantitative synaptic alterations in the human neocortex during normal aging. Neurology. 1993;43:192–97. doi: 10.1212/wnl.43.1_part_1.192. [DOI] [PubMed] [Google Scholar]
- 24.Nunomura A, Castellani RJ, Zhu X, et al. Involvement of oxidative stress in Alzheimer disease. J Neuropathol Exp Neurol. 2006;65:631–41. doi: 10.1097/01.jnen.0000228136.58062.bf. [DOI] [PubMed] [Google Scholar]
- 25.Butterfield DA, Perluigi M, Sultana R. Oxidative stress in Alzheimer’s disease brain: New insights from redox proteomics. Eur J Pharmacol. 2006;545:39–50. doi: 10.1016/j.ejphar.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 26.Scheff SW, Price DA. Alzheimer’s disease–related alterations in synaptic density: Neocortex and hippocampus. J Alzheimers Dis. 2006;9:101–15. doi: 10.3233/jad-2006-9s312. [DOI] [PubMed] [Google Scholar]
- 27.Gomez-Isla T, Spires T, De Calignon A, et al. Neuropathology of Alzheimer’s disease. Handb Clin Neurol. 2008;89:233–43. doi: 10.1016/S0072-9752(07)01222-5. [DOI] [PubMed] [Google Scholar]
- 28.Duyckaerts C, Delatour B, Potier MC. Classification and basic pathology of Alzheimer disease. Acta Neuropathol. 2009;118:5–36. doi: 10.1007/s00401-009-0532-1. [DOI] [PubMed] [Google Scholar]
- 29.Goedert M. Tau protein and neurodegeneration. Semin Cell Dev Biol. 2004;15:45–49. doi: 10.1016/j.semcdb.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 30.Arai T, Ikeda K, Akiyama H, et al. Distinct isoforms of tau aggregated in neurons and glial cells in brains of patients with Pick’s disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. 2001;101:167–73. doi: 10.1007/s004010000283. [DOI] [PubMed] [Google Scholar]
- 31.Buée L, Bussière T, Buée-Scherrer V, et al. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 2000;33:95–130. doi: 10.1016/s0165-0173(00)00019-9. [DOI] [PubMed] [Google Scholar]
- 32.Brat DJ, Gearing M, Goldthwaite PT, et al. Tau-associated neuropathology in ganglion cell tumours increases with patient age but appears unrelated to ApoE genotype. Neuropathol Appl Neurobiol. 2001;27:197–205. doi: 10.1046/j.1365-2990.2001.00311.x. [DOI] [PubMed] [Google Scholar]
- 33.Bouras C, Hof PR, Giannakopoulos P, et al. Regional distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of elderly patients: A quantitative evaluation of a one-year autopsy population from a geriatric hospital. Cereb Cortex. 1994;4:138–50. doi: 10.1093/cercor/4.2.138. [DOI] [PubMed] [Google Scholar]
- 34.Bancher C, Leitner H, Jellinger K, et al. On the relationship between measles virus and Alzheimer neurofibrillary tangles in subacute sclerosing panencephalitis. Neurobiol Aging. 1996;17:527–33. doi: 10.1016/0197-4580(96)00069-3. [DOI] [PubMed] [Google Scholar]
- 35.Tabaton M, Mandybur TI, Perry G, et al. The widespread alteration of neurites in Alzheimer’s disease may be unrelated to amyloid deposition. Ann Neurol. 1989;26:771–78. doi: 10.1002/ana.410260614. [DOI] [PubMed] [Google Scholar]
- 36.Wang XF, Dong CF, Zhang J, et al. Human tau protein forms complex with PrP and some GSS- and fCJD-related PrP mutants possess stronger binding activities with tau in vitro. Mol Cell Biochem. 2008;310:49–55. doi: 10.1007/s11010-007-9664-6. [DOI] [PubMed] [Google Scholar]
- 37.Cairns NJ, Bigio EH, Mackenzie IR, et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: Consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol (Berl) 2007;114:5–22. doi: 10.1007/s00401-007-0237-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tomlinson BE, Blessed G, Roth M. Observations on the brains of demented old people. J Neurol Sci. 1970;11:205–42. doi: 10.1016/0022-510x(70)90063-8. [DOI] [PubMed] [Google Scholar]
- 39.Braak H, Del Tredici K. The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol. 2011;121:171–81. doi: 10.1007/s00401-010-0789-4. [DOI] [PubMed] [Google Scholar]
- 40.Del Tredici K, Braak H. Neurofibrillary changes of the Alzheimer type in very elderly individuals: Neither inevitable nor benign: Commentary on “No disease in the brain of a 115-year-old woman”. Neurobiol Aging. 2008;29:1133–36. doi: 10.1016/j.neurobiolaging.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 41.Nelson PT, Abner EL, Schmitt FA, et al. Brains with medial temporal lobe neurofibrillary tangles but no neuritic amyloid plaques are a diagnostic dilemma but may have pathogenetic aspects distinct from Alzheimer disease. J Neuropathol Exp Neurol. 2009;68:774–84. doi: 10.1097/NEN.0b013e3181aacbe9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hof PR, Glannakopoulos P, Bouras C. The neuropathological changes associated with normal brain aging. Histol Histopathol. 1996;11:1075–88. [PubMed] [Google Scholar]
- 43.Simic G, Stanic G, Mladinov M, et al. Does Alzheimer’s disease begin in the brainstem? Neuropathol Appl Neurobiol. 2009;35:532–54. doi: 10.1111/j.1365-2990.2009.01038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Braak H, Thal DR, Ghebremedhin E, et al. Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J Neuropathol Exp Neurol. 2011;70:960–69. doi: 10.1097/NEN.0b013e318232a379. [DOI] [PubMed] [Google Scholar]
- 45.Dickson DW. Neuropathological diagnosis of Alzheimer’s disease: A perspective from longitudinal clinicopathological studies. Neurobiol Aging. 1997;18:S21–26. doi: 10.1016/s0197-4580(97)00065-1. [DOI] [PubMed] [Google Scholar]
- 46.Selkoe DJ. Biochemistry and molecular biology of amyloid β-protein and the mechanism of Alzheimer’s disease. Handb Clin Neurol. 2008;89:245–60. doi: 10.1016/S0072-9752(07)01223-7. [DOI] [PubMed] [Google Scholar]
- 47.Davies L, Wolska B, Hilbich C, et al. A4 amyloid protein deposition and the diagnosis of Alzheimer’s disease: Prevalence in aged brains determined by immunocytochemistry compared with conventional neuropathologic techniques. Neurology. 1988;38:1688–93. doi: 10.1212/wnl.38.11.1688. [DOI] [PubMed] [Google Scholar]
- 48.Jicha GA, Abner EL, Schmitt FA, et al. Preclinical AD Workgroup staging: Pathological correlates and potential challenges. Neurobiol Aging. 2012;33:662.e1–662.e16. doi: 10.1016/j.neurobiolaging.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Terry RD, Masliah E, Hansen LA. Structural basis of the cognitive alterations in Alzheimer disease. In: Terry RD, Katzman R, Bick KL, editors. Alzheimer Disease. New York, NY: Raven Press; 1994. pp. 179–96. [Google Scholar]
- 50.Wisniewski HM, Vorbrodt AW, Moretz RC, et al. Pathogenesis of neuritic (senile) and amyloid plaque formation. Exp Brain Res. 1982;(Suppl 5):3–9. doi: 10.1007/978-3-642-68507-1_1. [DOI] [PubMed] [Google Scholar]
- 51.Mirra SS. The CERAD neuropathology protocol and consensus recommendations for the postmortem diagnosis of Alzheimer’s disease: A commentary. Neurobiol Aging. 1997;18:S91–94. doi: 10.1016/s0197-4580(97)00058-4. [DOI] [PubMed] [Google Scholar]
- 52.Thal DR, Rüb U, Schultz C, et al. Sequence of Aβ-protein deposition in the human medial temporal lobe. J Neuropathol Exp Neurol. 2000;59:733–48. doi: 10.1093/jnen/59.8.733. [DOI] [PubMed] [Google Scholar]
- 53.Thal DR, Capetillo-Zarate E, Del Tredici K, et al. The development of amyloid β protein deposits in the aged brain. Sci Aging Knowledge Environ. 2006;2006:re1. doi: 10.1126/sageke.2006.6.re1. [DOI] [PubMed] [Google Scholar]
- 54.Price JL, McKeel DW, Jr, Buckles VD, et al. Neuropathology of non-demented aging: Presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging. 2009;30:1026–36. doi: 10.1016/j.neurobiolaging.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hardy J. Alzheimer’s disease: The amyloid cascade hypothesis: An update and reappraisal. J Alzheimers Dis. 2006;9:151–53. doi: 10.3233/jad-2006-9s317. [DOI] [PubMed] [Google Scholar]
- 56.Mayeux R, Hyslop PS. Alzheimer’s disease: Advances in trafficking. Lancet Neurol. 2008;7:2–3. doi: 10.1016/S1474-4422(07)70298-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reitz C, Mayeux R. Use of genetic variation as biomarkers for Alzheimer’s disease. Ann N Y Acad Sci. 2009;1180:75–96. doi: 10.1111/j.1749-6632.2009.04945.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bertram L, Lill CM, Tanzi RE. The genetics of Alzheimer disease: Back to the future. Neuron. 2010;68:270–81. doi: 10.1016/j.neuron.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 59.Gatz M, Reynolds CA, Fratiglioni L, et al. Role of genes and environments for explaining Alzheimer disease. Arch Gen Psychiatry. 2006;63:168–74. doi: 10.1001/archpsyc.63.2.168. [DOI] [PubMed] [Google Scholar]
- 60.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–59. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 61.Ohm TG, Müller H, Braak H, et al. Close-meshed prevalence rates of different stages as a tool to uncover the rate of Alzheimer’s disease–related neurofibrillary changes. Neuroscience. 1995;64:209–17. doi: 10.1016/0306-4522(95)90397-p. [DOI] [PubMed] [Google Scholar]
- 62.Castellani RJ, Lee HG, Zhu X, et al. Alzheimer disease pathology as a host response. J Neuropathol Exp Neurol. 2008;67:523–31. doi: 10.1097/NEN.0b013e318177eaf4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lee HG, Castellani RJ, Zhu X, et al. Amyloid-β in Alzheimer’s disease: The horse or the cart? Pathogenic or protective? Int J Exp Pathol. 2005;86:133–38. doi: 10.1111/j.0959-9673.2005.00429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Whitehouse PJ, George DR, D’Alton S. Describing the dying days of “Alzheimer’s disease”. J Alzheimers Dis. 2011;24:11–13. doi: 10.3233/JAD-2010-101639. [DOI] [PubMed] [Google Scholar]
- 65.Chen M, Maleski JJ, Sawmiller DR. Scientific truth or false hope? Understanding Alzheimer’s disease from an aging perspective. J Alzheimers Dis. 2011;24:3–10. doi: 10.3233/JAD-2010-101638. [DOI] [PubMed] [Google Scholar]
- 66.Herrup K. Reimagining Alzheimer’s disease—an age-based hypothesis. J Neurosci. 2010;30:16755–62. doi: 10.1523/JNEUROSCI.4521-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mondragon-Rodriguez S, Basurto-Islas G, Lee HG, et al. Causes versus effects: The increasing complexities of Alzheimer’s disease pathogenesis. Expert Rev Neurother. 2010;10:683–91. doi: 10.1586/ern.10.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Maltsev AV, Bystryak S, Galzitskaya OV. The role of β-amyloid peptide in neurodegenerative diseases. Ageing Res Rev. 2011;10:440–52. doi: 10.1016/j.arr.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 69.Nicoll JA, Barton E, Boche D, et al. Aβ species removal after Aβ42 immunization. J Neuropathol Exp Neurol. 2006;65:1040–48. doi: 10.1097/01.jnen.0000240466.10758.ce. [DOI] [PubMed] [Google Scholar]
- 70.Holmes C, Boche D, Wilkinson D, et al. Long-term effects of Aβ42 immunisation in Alzheimer’s disease: Follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–23. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- 71.Gilman S, Koller M, Black RS, et al. Clinical effects of Aβ immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553–62. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- 72.Boche D, Denham N, Holmes C, et al. Neuropathology after active Aβ42 immunotherapy: Implications for Alzheimer’s disease pathogenesis. Acta Neuropathol. 2010;120:369–84. doi: 10.1007/s00401-010-0719-5. [DOI] [PubMed] [Google Scholar]
- 73.Roher AE, Maarouf CL, Daugs ID, et al. Neuropathology and amyloid-β spectrum in a bapineuzumab immunotherapy recipient. J Alzheimers Dis. 2011;24:315–25. doi: 10.3233/JAD-2011-101809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Serrano-Pozo A, William CM, Ferrer I, et al. Beneficial effect of human anti–amyloid-β active immunization on neurite morphology and tau pathology. Brain. 2010;133:1312–27. doi: 10.1093/brain/awq056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Black RS, Sperling RA, Safirstein B, et al. A single ascending dose study of bapineuzumab in patients with Alzheimer disease. Alzheimer Dis Assoc Disord. 2010;24:198–203. doi: 10.1097/WAD.0b013e3181c53b00. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Salloway S, Sperling R, Gilman S, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009;73:2061–70. doi: 10.1212/WNL.0b013e3181c67808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Boche D, Donald J, Love S, et al. Reduction of aggregated tau in neuronal processes but not in the cell bodies after Aβ42 immunisation in Alzheimer’s disease. Acta Neuropathol. 2010;120:13–20. doi: 10.1007/s00401-010-0705-y. [DOI] [PubMed] [Google Scholar]
- 78.Wisniewski T. AD vaccines: Conclusions and future directions. CNS Neurol Disord Drug Targets. 2009;8:160–66. doi: 10.2174/187152709787847289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Selkoe DJ. Resolving controversies on the path to Alzheimer’s therapeutics. Nat Med. 2011;17:1521. doi: 10.1038/nm.2460. [DOI] [PubMed] [Google Scholar]
- 80.Wisniewski T, Konietzko U. Amyloid-β immunisation for Alzheimer’s disease. Lancet Neurol. 2008;7:805–11. doi: 10.1016/S1474-4422(08)70170-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer’s disease: An appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10:698–712. doi: 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- 82.Selkoe DJ. Soluble oligomers of the amyloid β-protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106–13. doi: 10.1016/j.bbr.2008.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–39. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Pritchard SM, Dolan PJ, Vitkus A, et al. The toxicity of tau in Alzheimer disease: Turnover, targets and potential therapeutics. J Cell Mol Med. 2011;15:1621–35. doi: 10.1111/j.1582-4934.2011.01273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Takashima A. Hyperphosphorylated tau is a cause of neuronal dysfunction in tauopathy. J Alzheimers Dis. 2008;14:371–75. doi: 10.3233/jad-2008-14403. [DOI] [PubMed] [Google Scholar]
- 86.Pei JJ, Sjogren M, Winblad B. Neurofibrillary degeneration in Alzheimer’s disease: From molecular mechanisms to identification of drug targets. Curr Opin Psychiatry. 2008;21:555–61. doi: 10.1097/YCO.0b013e328314b78b. [DOI] [PubMed] [Google Scholar]
- 87.Delacourte A. Tau pathology and neurodegeneration: An obvious but misunderstood link. J Alzheimers Dis. 2008;14:437–40. doi: 10.3233/jad-2008-14412. [DOI] [PubMed] [Google Scholar]
- 88.De Felice FG, Wu D, Lambert MP, et al. Alzheimer’s disease–type neuronal tau hyperphosphorylation induced by A β oligomers. Neurobiol Aging. 2008;29:1334–47. doi: 10.1016/j.neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Johnson GV, Stoothoff WH. Tau phosphorylation in neuronal cell function and dysfunction. J Cell Sci. 2004;117:5721–29. doi: 10.1242/jcs.01558. [DOI] [PubMed] [Google Scholar]
- 90.Shankar GM, Li S, Mehta TH, et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–42. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jin M, Shepardson N, Yang T, et al. Soluble amyloid {β}-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A. 2011;108:5819–24. doi: 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gadad BS, Britton GB, Ks R. Targeting oligomers in neurodegenerative disorders: lessons from alpha-synuclein, tau, and amyloid-β peptide. J Alzheimers Dis. 2011;24(Suppl 2):223–32. doi: 10.3233/JAD-2011-110182. [DOI] [PubMed] [Google Scholar]
- 93.Wu HY, Hudry E, Hashimoto T, et al. Amyloid β induces the morphological neurodegenerative triad of spine loss, dendritic simplification, and neuritic dystrophies through calcineurin activation. J Neurosci. 2010;30:2636–49. doi: 10.1523/JNEUROSCI.4456-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Darocha-Souto B, Scotton TC, Coma M, et al. Brain oligomeric β-amyloid but not total amyloid plaque burden correlates with neuronal loss and astrocyte inflammatory response in amyloid precursor protein/tau transgenic mice. J Neuropathol Exp Neurol. 2011;70:360–76. doi: 10.1097/NEN.0b013e318217a118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.van Helmond Z, Miners JS, Kehoe PG, et al. Higher soluble amyloid β concentration in frontal cortex of young adults than in normal elderly or Alzheimer’s disease. Brain Pathol. 2010;20:787–93. doi: 10.1111/j.1750-3639.2010.00374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Alafuzoff I, Arzberger T, Al-Sarraj S, et al. Staging of neurofibrillary pathology in Alzheimer’s disease: A study of the BrainNet Europe Consortium. Brain Pathol. 2008;18:484–96. doi: 10.1111/j.1750-3639.2008.00147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Archer T, Kostrzewa RM, Beninger RJ, et al. Staging neurodegenerative disorders: Structural, regional, biomarker, and functional progressions. Neurotox Res. 2011;19:211–34. doi: 10.1007/s12640-010-9190-2. [DOI] [PubMed] [Google Scholar]
- 98.Bancher C, Jellinger K, Lassmann H, et al. Correlations between mental state and quantitative neuropathology in the Vienna Longitudinal Study on Dementia. Eur Arch Psychiatry Clin Neurosci. 1996;246:137–46. doi: 10.1007/BF02189115. [DOI] [PubMed] [Google Scholar]
- 99.Braak H, Alafuzoff I, Arzberger T, et al. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Arnold SE, Hyman BT, Flory J, et al. The topographical and neuroanatomical distribution of neurofibrillary tangles and neuritic plaques in the cerebral cortex of patients with Alzheimer’s disease. Cereb Cortex. 1991;1:103–16. doi: 10.1093/cercor/1.1.103. [DOI] [PubMed] [Google Scholar]
- 101.Terry RD, Hansen LA, DeTeresa R, et al. Senile dementia of the Alzheimer type without neocortical neurofibrillary tangles. J Neuropathol Exp Neurol. 1987;46:262–68. doi: 10.1097/00005072-198705000-00003. [DOI] [PubMed] [Google Scholar]
- 102.Hansen LA, Masliah E, Galasko D, et al. Plaque-only Alzheimer disease is usually the Lewy body variant, and vice versa. J Neuropathol Exp Neurol. 1993;52:648–54. doi: 10.1097/00005072-199311000-00012. [DOI] [PubMed] [Google Scholar]
- 103.Thal DR, Griffin WS, Braak H. Parenchymal and vascular Aβ-deposition and its effects on the degeneration of neurons and cognition in Alzheimer’s disease. J Cell Mol Med. 2008;12:1848–62. doi: 10.1111/j.1582-4934.2008.00411.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Arvanitakis Z, Leurgans SE, Wang Z, et al. Cerebral amyloid angiopathy pathology and cognitive domains in older persons. Ann Neurol. 2011;69:320–27. doi: 10.1002/ana.22112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nelson PT, Abner EL, Schmitt FA, et al. Modeling the association between 43 different clinical and pathological variables and the severity of cognitive impairment in a large autopsy cohort of elderly persons. Brain Pathol. 2010;20:66–79. doi: 10.1111/j.1750-3639.2008.00244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tiraboschi P, Hansen LA, Thal LJ, et al. The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology. 2004;62:1984–89. doi: 10.1212/01.wnl.0000129697.01779.0a. [DOI] [PubMed] [Google Scholar]
- 107.Matthews FE, Brayne C, Lowe J, et al. Epidemiological pathology of dementia: Attributable-risks at death in the Medical Research Council Cognitive Function and Ageing Study. PLoS Med. 2009;6:e1000180. doi: 10.1371/journal.pmed.1000180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sonnen JA, Larson EB, Crane PK, et al. Pathological correlates of dementia in a longitudinal, population-based sample of aging. Ann Neurol. 2007;62:406–13. doi: 10.1002/ana.21208. [DOI] [PubMed] [Google Scholar]
- 109.Wolf DS, Gearing M, Snowdon DA, et al. Progression of regional neuropathology in Alzheimer disease and normal elderly: Findings from the Nun Study. Alzheimer Dis Assoc Disord. 1999;13:226–31. doi: 10.1097/00002093-199910000-00009. [DOI] [PubMed] [Google Scholar]
- 110.Tschanz JT, Treiber K, Norton MC, et al. A population study of Alzheimer’s disease: Findings from the Cache County Study on Memory, Health, and Aging. Care Manag J. 2005;6:107–14. doi: 10.1891/cmaj.6.2.107. [DOI] [PubMed] [Google Scholar]
- 111.Schneider JA, Aggarwal NT, Barnes L, et al. The neuropathology of older persons with and without dementia from community versus clinic cohorts. J Alzheimers Dis. 2009;18:691–701. doi: 10.3233/JAD-2009-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lim A, Tsuang D, Kukull W, et al. Clinico-neuropathological correlation of Alzheimer’s disease in a community-based case series. J Am Geriatr Soc. 1999;47:564–69. doi: 10.1111/j.1532-5415.1999.tb02571.x. [DOI] [PubMed] [Google Scholar]
- 113.Kaivorinne AL, Kruger J, Kuivaniemi K, et al. Role of MAPT mutations and haplotype in frontotemporal lobar degeneration in Northern Finland. BMC Neurol. 2008;8:48. doi: 10.1186/1471-2377-8-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rosso SM, Donker Kaat L, Baks T, et al. Frontotemporal dementia in the Netherlands: Patient characteristics and prevalence estimates from a population-based study. Brain. 2003;126:2016–22. doi: 10.1093/brain/awg204. [DOI] [PubMed] [Google Scholar]
- 115.Tyas SL, Snowdon DA, Desrosiers MF, et al. Healthy ageing in the Nun Study: Definition and neuropathologic correlates. Age Ageing. 2007;36:650–55. doi: 10.1093/ageing/afm120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Noda K, Sasaki K, Fujimi K, et al. Quantitative analysis of neurofibrillary pathology in a general population to reappraise neuropathological criteria for senile dementia of the neurofibrillary tangle type (tangle-only dementia): The Hisayama Study. Neuropathology. 2006;26:508–18. doi: 10.1111/j.1440-1789.2006.00722.x. [DOI] [PubMed] [Google Scholar]
- 117.Ulrich J, Spillantini MG, Goedert M, et al. Abundant neurofibrillary tangles without senile plaques in a subset of patients with senile dementia. Neurodegeneration. 1992:257–84. [Google Scholar]
- 118.Jellinger KA, Attems J. Neurofibrillary tangle-predominant dementia: Comparison with classical Alzheimer disease. Acta Neuropathol. 2007;113:107–17. doi: 10.1007/s00401-006-0156-7. [DOI] [PubMed] [Google Scholar]
- 119.Feany MB, Dickson DW. Neurodegenerative disorders with extensive tau pathology: A comparative study and review. Ann Neurol. 1996;40:139–48. doi: 10.1002/ana.410400204. [DOI] [PubMed] [Google Scholar]
- 120.Yamada M, Itoh Y, Sodeyama N, et al. Senile dementia of the neurofibrillary tangle type: A comparison with Alzheimer’s disease. Dement Geriatr Cogn Disord. 2001;12:117–26. doi: 10.1159/000051245. [DOI] [PubMed] [Google Scholar]
- 121.Winton MJ, Joyce S, Zhukareva V, et al. Characterization of tau pathologies in gray and white matter of Guam parkinsonism-dementia complex. Acta Neuropathol. 2006;111:401–12. doi: 10.1007/s00401-006-0053-0. [DOI] [PubMed] [Google Scholar]
- 122.Matsumoto S, Hirano A, Goto S. Spinal cord neurofibrillary tangles of Guamanian amyotrophic lateral sclerosis and parkinsonism-dementia complex: An immunohistochemical study. Neurology. 1990;40:975–79. doi: 10.1212/wnl.40.6.975. [DOI] [PubMed] [Google Scholar]
- 123.Joachim CL, Morris JH, Kosik KS, et al. Tau antisera recognize neurofibrillary tangles in a range of neurodegenerative disorders. Ann Neurol. 1987;22:514–20. doi: 10.1002/ana.410220411. [DOI] [PubMed] [Google Scholar]
- 124.Wong KT, Allen IV, McQuaid S, et al. An immunohistochemical study of neurofibrillary tangle formation in post-encephalitic Parkinsonism. Clin Neuropathol. 1996;15:22–25. [PubMed] [Google Scholar]
- 125.Jellinger KA, Bancher C. Senile dementia with tangles (tangle predominant form of senile dementia) Brain Pathol. 1998;8:367–76. doi: 10.1111/j.1750-3639.1998.tb00160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hebert LE, Scherr PA, Bienias JL, et al. Alzheimer disease in the US population: Prevalence estimates using the 2000 census. Arch Neurol. 2003;60:1119–22. doi: 10.1001/archneur.60.8.1119. [DOI] [PubMed] [Google Scholar]
- 127.Aronson MK, Ooi WL, Geva DL, et al. Dementia. Age-dependent incidence, prevalence, and mortality in the old old. Arch Intern Med. 1991;151:989–92. doi: 10.1001/archinte.151.5.989. [DOI] [PubMed] [Google Scholar]
- 128.Katzman R, Kawas C. The epidemiology of dementia and Alzheimer disease. In: Terry RD, editor. Alzheimer Disease. New York, NY: Raven Press, Ltd; 1994. pp. 105–22. [Google Scholar]
- 129.Williams MM, Xiong C, Morris JC, et al. Survival and mortality differences between dementia with Lewy bodies vs Alzheimer disease. Neurology. 2006;67:1935–41. doi: 10.1212/01.wnl.0000247041.63081.98. [DOI] [PubMed] [Google Scholar]
- 130.Scarmeas N, Habeck CG, Hilton J, et al. APOE related alterations in cerebral activation even at college age. J Neurol Neurosurg Psychiatry. 2005;76:1440–44. doi: 10.1136/jnnp.2004.053645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Small BJ, Rosnick CB, Fratiglioni L, et al. Apolipoprotein E and cognitive performance: A meta-analysis. Psychol Aging. 2004;19:592–600. doi: 10.1037/0882-7974.19.4.592. [DOI] [PubMed] [Google Scholar]
- 132.Smith CD, Chebrolu H, Wekstein DR, et al. Brain structural alterations before mild cognitive impairment. Neurology. 2007;68:1268–73. doi: 10.1212/01.wnl.0000259542.54830.34. [DOI] [PubMed] [Google Scholar]
- 133.Thal DR, Del Tredici K, Braak H. Neurodegeneration in normal brain aging and disease. Sci Aging Knowledge Environ. 2004;2004:pe26. doi: 10.1126/sageke.2004.23.pe26. [DOI] [PubMed] [Google Scholar]
- 134.Galvin JE, Powlishta KK, Wilkins K, et al. Predictors of preclinical Alzheimer disease and dementia: A clinicopathologic study. Arch Neurol. 2005;62:758–65. doi: 10.1001/archneur.62.5.758. [DOI] [PubMed] [Google Scholar]
- 135.Mosconi L, De Santi S, Rusinek H, et al. Magnetic resonance and PET studies in the early diagnosis of Alzheimer’s disease. Expert Rev Neurother. 2004;4:831–49. doi: 10.1586/14737175.4.5.831. [DOI] [PubMed] [Google Scholar]
- 136.Rusinek H, De Santi S, Frid D, et al. Regional brain atrophy rate predicts future cognitive decline: 6-year longitudinal MR imaging study of normal aging. Radiology. 2003;229:691–96. doi: 10.1148/radiol.2293021299. [DOI] [PubMed] [Google Scholar]
- 137.Backman L, Jones S, Berger AK, et al. Cognitive impairment in preclinical Alzheimer’s disease: A meta-analysis. Neuropsychology. 2005;19:520–31. doi: 10.1037/0894-4105.19.4.520. [DOI] [PubMed] [Google Scholar]
- 138.Backman L, Small BJ, Fratiglioni L. Stability of the preclinical episodic memory deficit in Alzheimer’s disease. Brain. 2001;124:96–102. doi: 10.1093/brain/124.1.96. [DOI] [PubMed] [Google Scholar]
- 139.Seshadri S, Beiser A, Au R, et al. Operationalizing diagnostic criteria for Alzheimer’s disease and other age-related cognitive impairment—Part 2. Alzheimers Dement. 2011;7:35–52. doi: 10.1016/j.jalz.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Jack CR, Jr, Albert MS, Knopman DS, et al. Introduction to the recommendations from the National Institute on Aging–Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:257–62. doi: 10.1016/j.jalz.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Central Intelligence Agency. The World Factbook. Washington, DC: Central Intelligence Agency; 2008. [Google Scholar]
- 142.Nelson PT, Braak H, Markesbery WR. Neuropathology and cognitive impairment in Alzheimer disease: A complex but coherent relationship. J Neuropathol Exp Neurol. 2009;68:1–14. doi: 10.1097/NEN.0b013e3181919a48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Leow AD, Yanovsky I, Parikshak N, et al. Alzheimer’s disease neuroimaging initiative: A one-year follow up study using tensor-based morphometry correlating degenerative rates, biomarkers and cognition. Neuroimage. 2009;45:645–55. doi: 10.1016/j.neuroimage.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Jack CR, Jr, Bernstein MA, Fox NC, et al. The Alzheimer’s Disease Neuroimaging Initiative (ADNI): MRI methods. J Magn Reson Imaging. 2008;27:685–91. doi: 10.1002/jmri.21049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Sonnen JA, Montine KS, Quinn JF, et al. Cerebrospinal fluid biomarkers in mild cognitive impairment and dementia. J Alzheimers Dis. 2010;19:301–9. doi: 10.3233/JAD-2010-1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Burton EC, Troxclair DA, Newman WP., 3rd Autopsy diagnoses of malignant neoplasms: How often are clinical diagnoses incorrect? JAMA. 1998;280:1245–48. doi: 10.1001/jama.280.14.1245. [DOI] [PubMed] [Google Scholar]
- 147.Avgerinos DV, Bjornsson J. Malignant neoplasms: Discordance between clinical diagnoses and autopsy findings in 3,118 cases. APMIS. 2001;109:774–80. doi: 10.1034/j.1600-0463.2001.d01-145.x. [DOI] [PubMed] [Google Scholar]
- 148.Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology. 2006;66:1837–44. doi: 10.1212/01.wnl.0000219668.47116.e6. [DOI] [PubMed] [Google Scholar]
- 149.Santacruz KS, Sonnen JA, Pezhouh MK, et al. Alzheimer disease pathology in subjects without dementia in 2 studies of aging: The Nun study and the Adult Changes in Thought Study. J Neuropathol Exp Neurol. 2011;70:832–40. doi: 10.1097/NEN.0b013e31822e8ae9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Sonnen JA, Santa Cruz K, Hemmy LS, et al. Ecology of the aging human brain. Arch Neurol. 2011;68:1049–56. doi: 10.1001/archneurol.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Abner EL, Kryscio RJ, Schmitt FA, et al. “End-stage” neurofibrillary tangle pathology in preclinical Alzheimer’s disease: fact or fiction? J Alzheimers Dis. 2011;25:445–53. doi: 10.3233/JAD-2011-101980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Arriagada PV, Marzloff K, Hyman BT. Distribution of Alzheimer-type pathologic changes in nondemented elderly individuals matches the pattern in Alzheimer’s disease. Neurology. 1992;42:1681–88. doi: 10.1212/wnl.42.9.1681. [DOI] [PubMed] [Google Scholar]
- 153.Berg L, McKeel DW, Jr, Miller JP, et al. Neuropathological indexes of Alzheimer’s disease in demented and nondemented persons aged 80 years and older. Arch Neurol. 1993;50:349–58. doi: 10.1001/archneur.1993.00540040011008. [DOI] [PubMed] [Google Scholar]
- 154.Crystal HA, Dickson DW, Sliwinski MJ, et al. Pathological markers associated with normal aging and dementia in the elderly. Ann Neurol. 1993;34:566–73. doi: 10.1002/ana.410340410. [DOI] [PubMed] [Google Scholar]
- 155.Dickson DW, Crystal HA, Mattiace LA, et al. Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging. 1992;13:179–89. doi: 10.1016/0197-4580(92)90027-u. [DOI] [PubMed] [Google Scholar]
- 156.Driscoll I, Resnick SM, Troncoso JC, et al. Impact of Alzheimer’s pathology on cognitive trajectories in nondemented elderly. Ann Neurol. 2006;60:688–95. doi: 10.1002/ana.21031. [DOI] [PubMed] [Google Scholar]
- 157.Erkinjuntti T, Sulkava R, Kovanen J, et al. Suspected dementia: Evaluation of 323 consecutive referrals. Acta Neurol Scand. 1987;76:359–64. doi: 10.1111/j.1600-0404.1987.tb03594.x. [DOI] [PubMed] [Google Scholar]
- 158.Haroutunian V, Purohit DP, Perl DP, et al. Neurofibrillary tangles in nondemented elderly subjects and mild Alzheimer disease. Arch Neurol. 1999;56:713–18. doi: 10.1001/archneur.56.6.713. [DOI] [PubMed] [Google Scholar]
- 159.Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol. 1999;45:358–68. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 160.Schmitt FA, Davis DG, Wekstein DR, et al. “Preclinical” AD revisited: Neuropathology of cognitively normal older adults. Neurology. 2000;55:370–76. doi: 10.1212/wnl.55.3.370. [DOI] [PubMed] [Google Scholar]
- 161.Troncoso JC, Cataldo AM, Nixon RA, et al. Neuropathology of preclinical and clinical late-onset Alzheimer’s disease. Ann Neurol. 1998;43:673–76. doi: 10.1002/ana.410430519. [DOI] [PubMed] [Google Scholar]
- 162.Xuereb JH, Brayne C, Dufouil C, et al. Neuropathological findings in the very old. Results from the first 101 brains of a population-based longitudinal study of dementing disorders. Ann N Y Acad Sci. 2000;903:490–96. doi: 10.1111/j.1749-6632.2000.tb06404.x. [DOI] [PubMed] [Google Scholar]
- 163.Knopman DS, Parisi JE, Salviati A, et al. Neuropathology of cognitively normal elderly. J Neuropathol Exp Neurol. 2003;62:1087–95. doi: 10.1093/jnen/62.11.1087. [DOI] [PubMed] [Google Scholar]
- 164.Iseki E, Tsunoda S, Suzuki K, et al. Regional quantitative analysis of NFT in brains of non-demented elderly persons: Comparisons with findings in brains of late-onset Alzheimer’s disease and limbic NFT dementia. Neuropathology. 2002;22:34–39. doi: 10.1046/j.0919-6544.2001.00425.x. [DOI] [PubMed] [Google Scholar]
- 165.Hulette CM, Welsh-Bohmer KA, Murray MG, et al. Neuropathological and neuropsychological changes in “normal” aging: Evidence for preclinical Alzheimer disease in cognitively normal individuals. J Neuropathol Exp Neurol. 1998;57:1168–74. doi: 10.1097/00005072-199812000-00009. [DOI] [PubMed] [Google Scholar]
- 166.Jellinger KA, Attems J. Neuropathology and general autopsy findings in nondemented aged subjects. Clin Neuropathol. 2012;31:87–98. doi: 10.5414/np300418. [DOI] [PubMed] [Google Scholar]
- 167.Mackenzie IR, Neumann M, Bigio EH, et al. Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: An update. Acta Neuropathol. 2010;119:1–4. doi: 10.1007/s00401-009-0612-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Crystal HA, Dickson D, Davies P, et al. The relative frequency of “dementia of unknown etiology” increases with age and is nearly 50% in nonagenarians. Arch Neurol. 2000;57:713–19. doi: 10.1001/archneur.57.5.713. [DOI] [PubMed] [Google Scholar]
- 169.Neuropathology Group. Medical Research Council Cognitive Function and Ageing Study. Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS) Lancet. 2001;357:169–75. doi: 10.1016/s0140-6736(00)03589-3. [DOI] [PubMed] [Google Scholar]
- 170.Haroutunian V, Schnaider-Beeri M, Schmeidler J, et al. Role of the neuropathology of Alzheimer disease in dementia in the oldest-old. Arch Neurol. 2008;65:1211–17. doi: 10.1001/archneur.65.9.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Savva GM, Wharton SB, Ince PG, et al. Age, neuropathology, and dementia. N Engl J Med. 2009;360:2302–9. doi: 10.1056/NEJMoa0806142. [DOI] [PubMed] [Google Scholar]
- 172.von Gunten A, Ebbing K, Imhof A, et al. Brain aging in the oldest-old. Curr Gerontol Geriatr Res. doi: 10.1155/2010/358531. published online ahead of print July 25, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Marshall GA, Fairbanks LA, Tekin S, et al. Early-onset Alzheimer’s disease is associated with greater pathologic burden. J Geriatr Psychiatry Neurol. 2007;20:29–33. doi: 10.1177/0891988706297086. [DOI] [PubMed] [Google Scholar]
- 174.Arnold SE, Trojanowski JQ. Recent advances in defining the neuropathology of schizophrenia. Acta Neuropathol. 1996;92:217–31. doi: 10.1007/s004010050512. [DOI] [PubMed] [Google Scholar]
- 175.Arnold SE, Trojanowski JQ, Gur RE, et al. Absence of neurodegeneration and neural injury in the cerebral cortex in a sample of elderly patients with schizophrenia. Arch Gen Psychiatry. 1998;55:225–32. doi: 10.1001/archpsyc.55.3.225. [DOI] [PubMed] [Google Scholar]
- 176.Jellinger KA, Gabriel E. No increased incidence of Alzheimer’s disease in elderly schizophrenics. Acta Neuropathol. 1999;97:165–69. doi: 10.1007/s004010050969. [DOI] [PubMed] [Google Scholar]
- 177.Leinonen V, Koivisto AM, Savolainen S, et al. Post-mortem findings in 10 patients with presumed normal pressure hydrocephalus and review of the literature. Neuropathol Appl Neurobiol. doi: 10.1111/j.1365-2990.2011.01195.x. published online ahead of print February 10, 2012. [DOI] [PubMed] [Google Scholar]
- 178.Savolainen S, Hurskainen H, Paljarvi L, et al. Five-year outcome of normal pressure hydrocephalus with or without a shunt: Predictive value of the clinical signs, neuropsychological evaluation and infusion test. Acta Neurochir (Wien) 2002;144:515–23. doi: 10.1007/s00701-002-0936-3. discussion 23. [DOI] [PubMed] [Google Scholar]
- 179.Kukull WA, Higdon R, Bowen JD, et al. Dementia and Alzheimer disease incidence: A prospective cohort study. Arch Neurol. 2002;59:1737–46. doi: 10.1001/archneur.59.11.1737. [DOI] [PubMed] [Google Scholar]
- 180.Rocca WA, Cha RH, Waring SC, et al. Incidence of dementia and Alzheimer’s disease: A reanalysis of data from Rochester, Minnesota, 1975-1984. Am J Epidemiol. 1998;148:51–62. doi: 10.1093/oxfordjournals.aje.a009560. [DOI] [PubMed] [Google Scholar]
- 181.Hall CB, Verghese J, Sliwinski M, et al. Dementia incidence may increase more slowly after age 90: Results from the Bronx Aging Study. Neurology. 2005;65:882–86. doi: 10.1212/01.wnl.0000176053.98907.3f. [DOI] [PubMed] [Google Scholar]
- 182.Rocca WA, Petersen RC, Knopman DS, et al. Trends in the incidence and prevalence of Alzheimer’s disease, dementia, and cognitive impairment in the United States. Alzheimers Dement. 2011;7:80–93. doi: 10.1016/j.jalz.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Launer LJ. Counting dementia: There is no one “best” way. Alzheimers Dement. 2011;7:10–14. doi: 10.1016/j.jalz.2010.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Corrada MM, Brookmeyer R, Paganini-Hill A, et al. Dementia incidence continues to increase with age in the oldest old: The 90+ study. Ann Neurol. 2010;67:114–21. doi: 10.1002/ana.21915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Imhof A, Kovari E, von Gunten A, et al. Morphological substrates of cognitive decline in nonagenarians and centenarians: A new paradigm? J Neurol Sci. 2007;257:72–9. doi: 10.1016/j.jns.2007.01.025. [DOI] [PubMed] [Google Scholar]
- 186.Jellinger KA, Attems J. Prevalence of dementia disorders in the oldest-old: An autopsy study. Acta Neuropathol. 2010;119:421–33. doi: 10.1007/s00401-010-0654-5. [DOI] [PubMed] [Google Scholar]
- 187.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993;261:921–23. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 188.Dolan D, Troncoso J, Resnick SM, et al. Age, Alzheimer’s disease and dementia in the Baltimore Longitudinal Study of Ageing. Brain. 2010;133:2225–31. doi: 10.1093/brain/awq141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Nelson PT, Jicha GA, Schmitt FA, et al. Clinicopathologic correlations in a large Alzheimer disease center autopsy cohort: Neuritic plaques and neurofibrillary tangles “do count” when staging disease severity. J Neuropathol Exp Neurol. 2007;66:1136–46. doi: 10.1097/nen.0b013e31815c5efb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Wilson RS, Leurgans SE, Boyle PA, et al. Neurodegenerative basis of age-related cognitive decline. Neurology. 2010;75:1070–78. doi: 10.1212/WNL.0b013e3181f39adc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Robinson JL, Geser F, Corrada MM, et al. Neocortical and hippocampal amyloid-β and tau measures associate with dementia in the oldest-old. Brain. 2011;134(Pt 12):3708–15. doi: 10.1093/brain/awr308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Bowler JV, Munoz DG, Merskey H, et al. Fallacies in the pathological confirmation of the diagnosis of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 1998;64:18–24. doi: 10.1136/jnnp.64.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Fernando MS, Ince PG. Vascular pathologies and cognition in a population-based cohort of elderly people. J Neurol Sci. 2004;226:13–17. doi: 10.1016/j.jns.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 194.Jellinger KA, Attems J. Neuropathological evaluation of mixed dementia. J Neurol Sci. 2007;257:80–87. doi: 10.1016/j.jns.2007.01.045. [DOI] [PubMed] [Google Scholar]
- 195.Schneider JA, Boyle PA, Arvanitakis Z, et al. Subcortical infarcts, Alzheimer’s disease pathology, and memory function in older persons. Ann Neurol. 2007;62:59–66. doi: 10.1002/ana.21142. [DOI] [PubMed] [Google Scholar]
- 196.Schneider JA, Wilson RS, Bienias JL, et al. Cerebral infarctions and the likelihood of dementia from Alzheimer disease pathology. Neurology. 2004;62:1148–55. doi: 10.1212/01.wnl.0000118211.78503.f5. [DOI] [PubMed] [Google Scholar]
- 197.Todorov AB, Go RC, Constantinidis J, et al. Specificity of the clinical diagnosis of dementia. J Neurol Sci. 1975;26:81–98. doi: 10.1016/0022-510x(75)90116-1. [DOI] [PubMed] [Google Scholar]
- 198.Chui HC, Zarow C, Mack WJ, et al. Cognitive impact of subcortical vascular and Alzheimer’s disease pathology. Ann Neurol. 2006;60:677–87. doi: 10.1002/ana.21009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Jicha GA, Parisi JE, Dickson DW, et al. Neuropathologic outcome of mild cognitive impairment following progression to clinical dementia. Arch Neurol. 2006;63:674–81. doi: 10.1001/archneur.63.5.674. [DOI] [PubMed] [Google Scholar]
- 200.Rockwood K. Mixed dementia: Alzheimer’s and cerebrovascular disease. Int Psychogeriatr. 2003;15:39–46. doi: 10.1017/S1041610203008949. [DOI] [PubMed] [Google Scholar]
- 201.Kövari E, Gold G, Herrmann FR, et al. Cortical microinfarcts and demyelination affect cognition in cases at high risk for dementia. Neurology. 2007;68:927–31. doi: 10.1212/01.wnl.0000257094.10655.9a. [DOI] [PubMed] [Google Scholar]
- 202.Gold G, Giannakopoulos P, Herrmann FR, et al. Identification of Alzheimer and vascular lesion thresholds for mixed dementia. Brain. 2007;130:2830–36. doi: 10.1093/brain/awm228. [DOI] [PubMed] [Google Scholar]
- 203.Murray ME, Knopman DS, Dickson DW. Vascular dementia: Clinical, neuroradiologic and neuropathologic aspects. Panminerva Med. 2007;49:197–207. [PubMed] [Google Scholar]
- 204.Jellinger K, Danielczyk W, Fischer P, et al. Clinicopathological analysis of dementia disorders in the elderly. J Neurol Sci. 1990;95:239–58. doi: 10.1016/0022-510x(90)90072-u. [DOI] [PubMed] [Google Scholar]
- 205.White L, Petrovitch H, Hardman J, et al. Cerebrovascular pathology and dementia in autopsied Honolulu-Asia Aging Study participants. Ann N Y Acad Sci. 2002;977:9–23. doi: 10.1111/j.1749-6632.2002.tb04794.x. [DOI] [PubMed] [Google Scholar]
- 206.White L, Small BJ, Petrovitch H, et al. Recent clinical-pathologic research on the causes of dementia in late life: Update from the Honolulu-Asia Aging Study. J Geriatr Psychiatry Neurol. 2005;18:224–27. doi: 10.1177/0891988705281872. [DOI] [PubMed] [Google Scholar]
- 207.Polvikoski TM, van Straaten EC, Barkhof F, et al. Frontal lobe white matter hyperintensities and neurofibrillary pathology in the oldest old. Neurology. 2010;75:2071–78. doi: 10.1212/WNL.0b013e318200d6f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Korczyn AD. Mixed dementia—the most common cause of dementia. Ann N Y Acad Sci. 2002;977:129–34. doi: 10.1111/j.1749-6632.2002.tb04807.x. [DOI] [PubMed] [Google Scholar]
- 209.Thal DR, Ghebremedhin E, Orantes M, et al. Vascular pathology in Alzheimer disease: Correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol. 2003;62:1287–301. doi: 10.1093/jnen/62.12.1287. [DOI] [PubMed] [Google Scholar]
- 210.Cordonnier C, van der Flier WM. Brain microbleeds and Alzheimer’s disease: Innocent observation or key player? Brain. 2011;134:335–44. doi: 10.1093/brain/awq321. [DOI] [PubMed] [Google Scholar]
- 211.Nelson PT, Schmitt FA, Lin Y, et al. Hippocampal sclerosis in advanced age: Clinical and pathological features. Brain. 2011;134:1506–18. doi: 10.1093/brain/awr053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Amador-Ortiz C, Ahmed Z, Zehr C, et al. Hippocampal sclerosis dementia differs from hippocampal sclerosis in frontal lobe degeneration. Acta Neuropathol (Berl) 2007;113:245–52. doi: 10.1007/s00401-006-0183-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Arai T, Hasegawa M, Akiyama H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–11. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- 214.Corey-Bloom J, Sabbagh MN, Bondi MW, et al. Hippocampal sclerosis contributes to dementia in the elderly. Neurology. 1997;48:154–60. doi: 10.1212/wnl.48.1.154. [DOI] [PubMed] [Google Scholar]
- 215.Miller LA, Munoz DG, Finmore M. Hippocampal sclerosis and human memory. Arch Neurol. 1993;50:391–94. doi: 10.1001/archneur.1993.00540040051014. [DOI] [PubMed] [Google Scholar]
- 216.Knopman DS, Petersen RC, Cha RH, et al. Incidence and causes of nondegenerative nonvascular dementia: A population-based study. Arch Neurol. 2006;63:218–21. doi: 10.1001/archneur.63.2.218. [DOI] [PubMed] [Google Scholar]
- 217.Pao WC, Dickson DW, Crook JE, et al. Hippocampal sclerosis in the elderly: Genetic and pathologic findings, some mimicking Alzheimer disease clinically. Alzheimer Dis Assoc Disord. 2011;25:364–8. doi: 10.1097/WAD.0b013e31820f8f50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Nelson PT, Schmitt FA, Lin Y, et al. Hippocampal sclerosis in advanced age: Clinical and pathological features in 106 cases. Brain. 2011;134(Pt 5):1506–18. doi: 10.1093/brain/awr053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Jagust W. Dementia: Finding the signals in the noise. Lancet Neurol. 2005;4:10–11. doi: 10.1016/S1474-4422(04)00949-4. [DOI] [PubMed] [Google Scholar]
- 220.Thal DR, Schultz C, Botez G, et al. The impact of argyrophilic grain disease on the development of dementia and its relationship to concurrent Alzheimer’s disease–related pathology. Neuropathol Appl Neurobiol. 2005;31:270–79. doi: 10.1111/j.1365-2990.2005.00635.x. [DOI] [PubMed] [Google Scholar]
- 221.Brumback-Peltz C, Balasubramanian AB, Corrada MM, et al. Diagnosing dementia in the oldest-old. Maturitas. 2011;70:164–8. doi: 10.1016/j.maturitas.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Nelson PT, Head E, Schmitt FA, et al. Alzheimer’s disease is not “brain aging”: Neuropathological, genetic, and epidemiological human studies. Acta Neuropathol. 2011;121:571–87. doi: 10.1007/s00401-011-0826-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Guillozet AL, Weintraub S, Mash DC, et al. Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment. Arch Neurol. 2003;60:729–36. doi: 10.1001/archneur.60.5.729. [DOI] [PubMed] [Google Scholar]
- 224.Markesbery WR, Schmitt FA, Kryscio RJ, et al. Neuropathologic substrate of mild cognitive impairment. Arch Neurol. 2006;63:38–46. doi: 10.1001/archneur.63.1.38. [DOI] [PubMed] [Google Scholar]
- 225.Berg L, McKeel DW, Jr, Miller JP, et al. Clinicopathologic studies in cognitively healthy aging and Alzheimer’s disease: Relation of histologic markers to dementia severity, age, sex, and apolipoprotein E genotype. Arch Neurol. 1998;55:326–35. doi: 10.1001/archneur.55.3.326. [DOI] [PubMed] [Google Scholar]
- 226.Cummings BJ, Pike CJ, Shankle R, et al. Beta-amyloid deposition and other measures of neuropathology predict cognitive status in Alzheimer’s disease. Neurobiol Aging. 1996;17:921–33. doi: 10.1016/s0197-4580(96)00170-4. [DOI] [PubMed] [Google Scholar]
- 227.Davis PC, Gearing M, Gray L, et al. The CERAD experience, Part VIII: Neuroimaging-neuropathology correlates of temporal lobe changes in Alzheimer’s disease. Neurology. 1995;45:178–79. doi: 10.1212/wnl.45.1.178. [DOI] [PubMed] [Google Scholar]
- 228.Holtzer R, Irizarry MC, Sanders J, et al. Relation of quantitative indexes of concurrent alpha-synuclein abnormalities to clinical outcome in autopsy-proven Alzheimer disease. Arch Neurol. 2006;63:226–30. doi: 10.1001/archneur.63.2.226. [DOI] [PubMed] [Google Scholar]
- 229.Koepsell TD, Kurland BF, Harel O, et al. Education, cognitive function, and severity of neuropathology in Alzheimer disease. Neurology. 2008;70(19 Pt 2):1732–9. doi: 10.1212/01.wnl.0000284603.85621.aa. [DOI] [PubMed] [Google Scholar]
- 230.Sabbagh MN, Corey-Bloom J, Tiraboschi P, et al. Neurochemical markers do not correlate with cognitive decline in the Lewy body variant of Alzheimer disease. Arch Neurol. 1999;56:1458–61. doi: 10.1001/archneur.56.12.1458. [DOI] [PubMed] [Google Scholar]
- 231.Sze CI, Troncoso JC, Kawas C, et al. Loss of the presynaptic vesicle protein synaptophysin in hippocampus correlates with cognitive decline in Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:933–44. doi: 10.1097/00005072-199708000-00011. [DOI] [PubMed] [Google Scholar]
- 232.Tiraboschi P, Hansen LA, Alford M, et al. Early and widespread cholinergic losses differentiate dementia with Lewy bodies from Alzheimer disease. Arch Gen Psychiatry. 2002;59:946–51. doi: 10.1001/archpsyc.59.10.946. [DOI] [PubMed] [Google Scholar]
- 233.Wang DS, Bennett DA, Mufson E, et al. Decreases in soluble alpha-synuclein in frontal cortex correlate with cognitive decline in the elderly. Neurosci Lett. 2004;359:104–8. doi: 10.1016/j.neulet.2004.01.044. [DOI] [PubMed] [Google Scholar]
- 234.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer’s disease: Correlation with cognitive severity. Ann Neurol. 1990;27:457–64. doi: 10.1002/ana.410270502. [DOI] [PubMed] [Google Scholar]
- 235.Green MS, Kaye JA, Ball MJ. The Oregon brain aging study: Neuropathology accompanying healthy aging in the oldest old. Neurology. 2000;54:105–13. doi: 10.1212/wnl.54.1.105. [DOI] [PubMed] [Google Scholar]
- 236.Kraybill ML, Larson EB, Tsuang DW, et al. Cognitive differences in dementia patients with autopsy-verified AD, Lewy body pathology, or both. Neurology. 2005;64:2069–73. doi: 10.1212/01.WNL.0000165987.89198.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Mungas D, Reed BR, Ellis WG, et al. The effects of age on rate of progression of Alzheimer disease and dementia with associated cerebrovascular disease. Arch Neurol. 2001;58:1243–47. doi: 10.1001/archneur.58.8.1243. [DOI] [PubMed] [Google Scholar]
- 238.Gold G, Bouras C, Kovari E, et al. Clinical validity of Braak neuropathological staging in the oldest-old. Acta Neuropathol (Berl) 2000;99:579–82. doi: 10.1007/s004010051163. discussion 83-84. [DOI] [PubMed] [Google Scholar]
- 239.Silver MH, Newell K, Brady C, et al. Distinguishing between neurodegenerative disease and disease-free aging: Correlating neuropsychological evaluations and neuropathological studies in centenarians. Psychosom Med. 2002;64:493–501. doi: 10.1097/00006842-200205000-00014. [DOI] [PubMed] [Google Scholar]
- 240.Duyckaerts C, Delaère P, Hauw JJ, et al. Rating of the lesions in senile dementia of the Alzheimer type: Concordance between laboratories. A European multicenter study under the auspices of EURAGE. J Neurol Sci. 1990;97:295–323. doi: 10.1016/0022-510x(90)90226-d. [DOI] [PubMed] [Google Scholar]
- 241.Yasha TC, Shankar L, Santosh V, et al. Histopathological & immunohistochemical evaluation of ageing changes in normal human brain. Indian J Med Res. 1997;105:141–50. [PubMed] [Google Scholar]
- 242.Nelson PT, Abner EL, Scheff SW, et al. Alzheimer’s-type neuropathology in the precuneus is not increased relative to other areas of neocortex across a range of cognitive impairment. Neurosci Lett. 2009;450:336–39. doi: 10.1016/j.neulet.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Caramelli P, Robitaille Y, Laroche-Cholette A, et al. Structural correlates of cognitive deficits in a selected group of patients with Alzheimer’s disease. Neuropsychiatry Neuropsychol Behav Neurol. 1998;11:184–90. [PubMed] [Google Scholar]
- 244.Barcikowska M, Wisniewski HM, Bancher C, et al. About the presence of paired helical filaments in dystrophic neurites participating in the plaque formation. Acta Neuropathol. 1989;78:225–31. doi: 10.1007/BF00687751. [DOI] [PubMed] [Google Scholar]
- 245.Delaère P, Duyckaerts C, Brion JP, et al. Tau, paired helical filaments and amyloid in the neocortex: A morphometric study of 15 cases with graded intellectual status in aging and senile dementia of Alzheimer type. Acta Neuropathol. 1989;77:645–53. doi: 10.1007/BF00687893. [DOI] [PubMed] [Google Scholar]
- 246.McKeel DW, Jr, Price JL, Miller JP, et al. Neuropathologic criteria for diagnosing Alzheimer disease in persons with pure dementia of Alzheimer type. J Neuropathol Exp Neurol. 2004;63:1028–37. doi: 10.1093/jnen/63.10.1028. [DOI] [PubMed] [Google Scholar]
- 247.Gold G, Kövari E, Corte G, et al. Clinical validity of A β-protein deposition staging in brain aging and Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:946–52. doi: 10.1093/jnen/60.10.946. [DOI] [PubMed] [Google Scholar]
- 248.Masliah E, Terry RD, Mallory M, et al. Diffuse plaques do not accentuate synapse loss in Alzheimer’s disease. Am J Pathol. 1990;137:1293–97. [PMC free article] [PubMed] [Google Scholar]
- 249.Wang D, Munoz DG. Qualitative and quantitative differences in senile plaque dystrophic neurites of Alzheimer’s disease and normal aged brain. J Neuropathol Exp Neurol. 1995;54:548–56. doi: 10.1097/00005072-199507000-00009. [DOI] [PubMed] [Google Scholar]
- 250.Duyckaerts C, Colle MA, Dessi F, et al. Progression of Alzheimer histopathological changes. Acta Neurol Belg. 1998;98:180–85. [PubMed] [Google Scholar]
- 251.Dickson DW, Crystal HA, Bevona C, et al. Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol Aging. 1995;16:285–98. doi: 10.1016/0197-4580(95)00013-5. discussion 298-304. [DOI] [PubMed] [Google Scholar]
- 252.Sabbagh MN, Cooper K, DeLange J, et al. Functional, global and cognitive decline correlates to accumulation of Alzheimer’s pathology in MCI and AD. Curr Alzheimer Res. 2010;7:280–86. doi: 10.2174/156720510791162340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Ingelsson M, Fukumoto H, Newell KL, et al. Early Aβ accumulation and progressive synaptic loss, gliosis, and tangle formation in AD brain. Neurology. 2004;62:925–31. doi: 10.1212/01.wnl.0000115115.98960.37. [DOI] [PubMed] [Google Scholar]
- 254.Blessed G, Tomlinson BE, Roth M. The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects. Br J Psychiatry. 1968;114:797–811. doi: 10.1192/bjp.114.512.797. [DOI] [PubMed] [Google Scholar]
- 255.Roth M, Tomlinson BE, Blessed G. Correlation between scores for dementia and counts of ‘senile plaques’ in cerebral grey matter of elderly subjects. Nature. 1966;209:109–10. doi: 10.1038/209109a0. [DOI] [PubMed] [Google Scholar]
- 256.Tomlinson BE, Blessed G, Roth M. Observations on the brains of non-demented old people. J Neurol Sci. 1968;7:331–56. doi: 10.1016/0022-510x(68)90154-8. [DOI] [PubMed] [Google Scholar]
- 257.Potter PE, Rauschkolb PK, Pandya Y, et al. Pre- and post-synaptic cortical cholinergic deficits are proportional to amyloid plaque presence and density at preclinical stages of Alzheimer’s disease. Acta Neuropathol. 2011;122:49–60. doi: 10.1007/s00401-011-0831-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Beach TG, Adler CH, Lue L, et al. Unified staging system for Lewy body disorders: Correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. 2009;117:613–34. doi: 10.1007/s00401-009-0538-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Bussière T, Friend PD, Sadeghi N, et al. Stereologic assessment of the total cortical volume occupied by amyloid deposits and its relationship with cognitive status in aging and Alzheimer’s disease. Neuroscience. 2002;112:75–91. doi: 10.1016/s0306-4522(02)00056-8. [DOI] [PubMed] [Google Scholar]
- 260.Josephs KA, Whitwell JL, Knopman DS, et al. Abnormal TDP-43 immunoreactivity in AD modifies clinicopathologic and radiologic phenotype. Neurology. 2008;70:1850–57. doi: 10.1212/01.wnl.0000304041.09418.b1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.McKee AC, Kosik KS, Kowall NW. Neuritic pathology and dementia in Alzheimer’s disease. Ann Neurol. 1991;30:156–65. doi: 10.1002/ana.410300206. [DOI] [PubMed] [Google Scholar]
- 262.Thal DR, Rüb U, Orantes M, et al. Phases of A β-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–800. doi: 10.1212/wnl.58.12.1791. [DOI] [PubMed] [Google Scholar]
- 263.Beach TG, Sue LI, Walker DG, et al. Striatal amyloid plaque density predicts braak neurofibrillary stage and clinicopathological Alzheimer’s disease: Implications for amyloid imaging. J Alzheimers Dis. 2012;28:869–76. doi: 10.3233/JAD-2011-111340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Bennett DA, Schneider JA, Wilson RS, et al. Neurofibrillary tangles mediate the association of amyloid load with clinical Alzheimer disease and level of cognitive function. Arch Neurol. 2004;61:378–84. doi: 10.1001/archneur.61.3.378. [DOI] [PubMed] [Google Scholar]
- 265.Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12:383–88. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 266.de Calignon A, Fox LM, Pitstick R, et al. Caspase activation precedes and leads to tangles. Nature. 2010;464:1201–4. doi: 10.1038/nature08890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284:12845–52. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Clavaguera F, Bolmont T, Crowther RA, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909–13. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Guo JL, Lee VM. Seeding of normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem. 2011;286:15317–31. doi: 10.1074/jbc.M110.209296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Bennett DA, Cochran EJ, Saper CB, et al. Pathological changes in frontal cortex from biopsy to autopsy in Alzheimer’s disease. Neurobiol Aging. 1993;14:589–96. doi: 10.1016/0197-4580(93)90043-b. [DOI] [PubMed] [Google Scholar]
- 271.Nicoll JA, Wilkinson D, Holmes C, et al. Neuropathology of human Alzheimer disease after immunization with amyloid-β peptide: A case report. Nat Med. 2003;9:448–52. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 272.Miners JS, Barua N, Kehoe PG, et al. Aβ-degrading enzymes: Potential for treatment of Alzheimer disease. J Neuropathol Exp Neurol. 2011;70:944–59. doi: 10.1097/NEN.0b013e3182345e46. [DOI] [PubMed] [Google Scholar]
- 273.Masliah E, Mallory M, Deerinck T, et al. Re-evaluation of the structural organization of neuritic plaques in Alzheimer’s disease. J Neuropathol Exp Neurol. 1993;52:619–32. doi: 10.1097/00005072-199311000-00009. [DOI] [PubMed] [Google Scholar]
- 274.Spires TL, Hyman BT. Neuronal structure is altered by amyloid plaques. Rev Neurosci. 2004;15:267–78. doi: 10.1515/revneuro.2004.15.4.267. [DOI] [PubMed] [Google Scholar]
- 275.Koffie RM, Meyer-Luehmann M, Hashimoto T, et al. Oligomeric amyloid β associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A. 2009;106:4012–17. doi: 10.1073/pnas.0811698106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Benzing WC, Mufson EJ, Armstrong DM. Immunocytochemical distribution of peptidergic and cholinergic fibers in the human amygdala: Their depletion in Alzheimer’s disease and morphologic alteration in non-demented elderly with numerous senile plaques. Brain Res. 1993;625:125–38. doi: 10.1016/0006-8993(93)90145-d. [DOI] [PubMed] [Google Scholar]
- 277.Armstrong DM, Benzing WC, Evans J, et al. Substance P and somatostatin coexist within neuritic plaques: Implications for the pathogenesis of Alzheimer’s disease. Neuroscience. 1989;31:663–71. doi: 10.1016/0306-4522(89)90431-4. [DOI] [PubMed] [Google Scholar]
- 278.Klein WL, Krafft GA, Finch CE. Targeting small Aβ oligomers: The solution to an Alzheimer’s disease conundrum? Trends Neurosci. 2001;24:219–24. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- 279.Benzing WC, Ikonomovic MD, Brady DR, et al. Evidence that transmitter-containing dystrophic neurites precede paired helical filament and Alz-50 formation within senile plaques in the amygdala of non-demented elderly and patients with Alzheimer’s disease. J Comp Neurol. 1993;334:176–91. doi: 10.1002/cne.903340203. [DOI] [PubMed] [Google Scholar]
- 280.Duyckaerts C, Delaère P, Poulain V, et al. Does amyloid precede paired helical filaments in the senile plaque? A study of 15 cases with graded intellectual status in aging and Alzheimer disease. Neurosci Lett. 1988;91:354–59. doi: 10.1016/0304-3940(88)90706-9. [DOI] [PubMed] [Google Scholar]
- 281.Lenders MB, Peers MC, Tramu G, et al. Dystrophic peptidergic neurites in senile plaques of Alzheimer’s disease hippocampus precede formation of paired helical filaments. Brain Res. 1989;481:344–49. doi: 10.1016/0006-8993(89)90812-3. [DOI] [PubMed] [Google Scholar]
- 282.Nelson PT, Jicha GA, Kryscio RJ, et al. Low sensitivity in clinical diagnoses of dementia with Lewy bodies. J Neurol. 2010;257:359–66. doi: 10.1007/s00415-009-5324-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Probst A, Anderton BH, Brion JP, et al. Senile plaque neurites fail to demonstrate anti-paired helical filament and anti-microtubule-associated protein-tau immunoreactive proteins in the absence of neurofibrillary tangles in the neocortex. Acta Neuropathol. 1989;77:430–36. doi: 10.1007/BF00687379. [DOI] [PubMed] [Google Scholar]
- 284.Morrison JH, Hof PR, Bouras C. An anatomic substrate for visual disconnection in Alzheimer’s disease. Ann N Y Acad Sci. 1991;640:36–43. doi: 10.1111/j.1749-6632.1991.tb00187.x. [DOI] [PubMed] [Google Scholar]
- 285.Bussière T, Giannakopoulos P, Bouras C, et al. Progressive degeneration of nonphosphorylated neurofilament protein-enriched pyramidal neurons predicts cognitive impairment in Alzheimer’s disease: Stereologic analysis of prefrontal cortex area 9. J Comp Neurol. 2003;463:281–302. doi: 10.1002/cne.10760. [DOI] [PubMed] [Google Scholar]
- 286.Lewis DA, Campbell MJ, Terry RD, et al. Laminar and regional distributions of neurofibrillary tangles and neuritic plaques in Alzheimer’s disease: A quantitative study of visual and auditory cortices. J Neurosci. 1987;7:1799–808. doi: 10.1523/JNEUROSCI.07-06-01799.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Duyckaerts C, Bennecib M, Grignon Y, et al. Modeling the relation between neurofibrillary tangles and intellectual status. Neurobiol Aging. 1997;18:267–73. doi: 10.1016/s0197-4580(97)80306-5. [DOI] [PubMed] [Google Scholar]
- 288.Riley KP, Snowdon DA, Markesbery WR. Alzheimer’s neurofibrillary pathology and the spectrum of cognitive function: Findings from the Nun Study. Ann Neurol. 2002;51:567–77. doi: 10.1002/ana.10161. [DOI] [PubMed] [Google Scholar]
- 289.Giannakopoulos P, Kövari E, Gold G, et al. Pathological substrates of cognitive decline in Alzheimer’s disease. Front Neurol Neurosci. 2009;24:20–29. doi: 10.1159/000197881. [DOI] [PubMed] [Google Scholar]
- 290.Whitwell JL, Josephs KA, Murray ME, et al. MRI correlates of neurofibrillary tangle pathology at autopsy: A voxel-based morphometry study. Neurology. 2008;71:743–49. doi: 10.1212/01.wnl.0000324924.91351.7d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Bierer LM, Hof PR, Purohit DP, et al. Neocortical neurofibrillary tangles correlate with dementia severity in Alzheimer’s disease. Arch Neurol. 1995;52:81–88. doi: 10.1001/archneur.1995.00540250089017. [DOI] [PubMed] [Google Scholar]
- 292.Brayne C, Richardson K, Matthews FE, et al. Neuropathological correlates of dementia in over-80-year-old brain donors from the population-based Cambridge city over-75s cohort (CC75C) study. J Alzheimers Dis. 2009;18:645–58. doi: 10.3233/JAD-2009-1182. [DOI] [PubMed] [Google Scholar]
- 293.Arriagada PV, Growdon JH, Hedley-Whyte ET, et al. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42:631–39. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 294.Grober E, Dickson D, Sliwinski MJ, et al. Memory and mental status correlates of modified Braak staging. Neurobiol Aging. 1999;20:573–79. doi: 10.1016/s0197-4580(99)00063-9. [DOI] [PubMed] [Google Scholar]
- 295.Nagy Z, Esiri MM, Jobst KA, et al. Relative roles of plaques and tangles in the dementia of Alzheimer’s disease: Correlations using three sets of neuropathological criteria. Dementia. 1995;6:21–31. doi: 10.1159/000106918. [DOI] [PubMed] [Google Scholar]
- 296.Nagy Z, Hindley NJ, Braak H, et al. The progression of Alzheimer’s disease from limbic regions to the neocortex: Clinical, radiological and pathological relationships. Dement Geriatr Cogn Disord. 1999;10:115–20. doi: 10.1159/000017111. [DOI] [PubMed] [Google Scholar]
- 297.Folstein MF, Folstein SE, McHugh PR. “Mini-Mental State.” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–98. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 298.Braak H, Braak E. Staging of Alzheimer’s disease–related neurofibrillary changes. Neurobiol Aging. 1995;16:271–78. doi: 10.1016/0197-4580(95)00021-6. discussion 278-84. [DOI] [PubMed] [Google Scholar]
- 299.Braak H, Braak E, Bohl J. Staging of Alzheimer-related cortical destruction. Eur Neurol. 1993;33:403–8. doi: 10.1159/000116984. [DOI] [PubMed] [Google Scholar]
- 300.Duyckaerts C, Hauw JJ. Prevalence, incidence and duration of Braak’s stages in the general population: Can we know? Neurobiol Aging. 1997;18:362–69. doi: 10.1016/s0197-4580(97)00047-x. discussion 389-92. [DOI] [PubMed] [Google Scholar]
- 301.Grudzien A, Shaw P, Weintraub S, et al. Locus coeruleus neurofibrillary degeneration in aging, mild cognitive impairment and early Alzheimer’s disease. Neurobiol Aging. 2007;28:327–35. doi: 10.1016/j.neurobiolaging.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 302.Bobinski M, Wegiel J, Tarnawski M, et al. Relationships between regional neuronal loss and neurofibrillary changes in the hippocampal formation and duration and severity of Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:414–20. doi: 10.1097/00005072-199704000-00010. [DOI] [PubMed] [Google Scholar]
- 303.Rössler M, Zarski R, Bohl J, et al. Stage-dependent and sector-specific neuronal loss in hippocampus during Alzheimer’s disease. Acta Neuropathol. 2002;103:363–69. doi: 10.1007/s00401-001-0475-7. [DOI] [PubMed] [Google Scholar]
- 304.Gomez-Isla T, Hollister R, West H, et al. Neuronal loss correlates with but exceeds neurofibrillary tangles in Alzheimer’s disease. Ann Neurol. 1997;41:17–24. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- 305.Fukutani Y, Kobayashi K, Nakamura I, et al. Neurons, intracellular and extracellular neurofibrillary tangles in subdivisions of the hippocampal cortex in normal ageing and Alzheimer’s disease. Neurosci Lett. 1995;200:57–60. doi: 10.1016/0304-3940(95)12083-g. [DOI] [PubMed] [Google Scholar]
- 306.Berlau DJ, Kahle-Wrobleski K, Head E, et al. Dissociation of neuropathologic findings and cognition: Case report of an apolipoprotein E epsilon2/epsilon2 genotype. Arch Neurol. 2007;64:1193–96. doi: 10.1001/archneur.64.8.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Kril JJ, Patel S, Harding AJ, et al. Neuron loss from the hippocampus of Alzheimer’s disease exceeds extracellular neurofibrillary tangle formation. Acta Neuropathol. 2002;103:370–76. doi: 10.1007/s00401-001-0477-5. [DOI] [PubMed] [Google Scholar]
- 308.Vogt BA, Vogt LJ, Vrana KE, et al. Multivariate analysis of laminar patterns of neurodegeneration in posterior cingulate cortex in Alzheimer’s disease. Exp Neurol. 1998;153:8–22. doi: 10.1006/exnr.1998.6852. [DOI] [PubMed] [Google Scholar]
- 309.Bussi¨re T, Gold G, Kövari E, et al. Stereologic analysis of neurofibrillary tangle formation in prefrontal cortex area 9 in aging and Alzheimer’s disease. Neuroscience. 2003;117:577–92. doi: 10.1016/s0306-4522(02)00942-9. [DOI] [PubMed] [Google Scholar]
- 310.Hof PR, Morrison JH. The aging brain: Morphomolecular senescence of cortical circuits. Trends Neurosci. 2004;27:607–13. doi: 10.1016/j.tins.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 311.Mann DM, Yates PO, Marcyniuk B. Correlation between senile plaque and neurofibrillary tangle counts in cerebral cortex and neuronal counts in cortex and subcortical structures in Alzheimer’s disease. Neurosci Lett. 1985;56:51–55. doi: 10.1016/0304-3940(85)90439-2. [DOI] [PubMed] [Google Scholar]
- 312.Mann DM, Yates PO, Marcyniuk B. Changes in Alzheimer’s disease in the magnocellular neurones of the supraoptic and paraventricular nuclei of the hypothalamus and their relationship to the noradrenergic deficit. Clin Neuropathol. 1985;4:127–34. [PubMed] [Google Scholar]
- 313.Dayan AD. Quantitative histological studies on the aged human brain. II. Senile plaques and neurofibrillary tangles in senile dementia (with an appendix on their occurrence in cases of carcinoma. Acta Neuropathol. 1970;16:95–102. doi: 10.1007/BF00687664. [DOI] [PubMed] [Google Scholar]
- 314.Dayan AD. Quantitative histological studies on the aged human brain. I. Senile plaques and neurofibrillary tangles in “normal” patients. Acta Neuropathol. 1970;16:85–94. doi: 10.1007/BF00687663. [DOI] [PubMed] [Google Scholar]
- 315.Wisniewski H, Terry RD. Further studies on experimental neurofibrillary tangles. J Neuropathol Exp Neurol. 1968;27:149–50. [PubMed] [Google Scholar]
- 316.Terry RD. The fine structure of neurofibrillary tangles in Alzheimer’s disease. J Neuropathol Exp Neurol. 1963;22:629–42. doi: 10.1097/00005072-196310000-00005. [DOI] [PubMed] [Google Scholar]
- 317.Ball MJ. Neuronal loss, neurofibrillary tangles and granulovacuolar degeneration in the hippocampus with ageing and dementia. A quantitative study Acta Neuropathol. 1977;37:111–18. doi: 10.1007/BF00692056. [DOI] [PubMed] [Google Scholar]
- 318.Hirano A, Zimmerman HM. Alzheimer’s neurofibrillary changes. A topographic study. Arch Neurol. 1962;7:227–42. doi: 10.1001/archneur.1962.04210030065009. [DOI] [PubMed] [Google Scholar]
- 319.Wilcock GK, Esiri MM. Plaques, tangles and dementia. A quantitative study. J Neurol Sci. 1982;56:343–56. doi: 10.1016/0022-510x(82)90155-1. [DOI] [PubMed] [Google Scholar]
- 320.Wilcock GK, Esiri MM, Bowen DM, et al. Alzheimer’s disease. Correlation of cortical choline acetyltransferase activity with the severity of dementia and histological abnormalities. J Neurol Sci. 1982;57:407–17. doi: 10.1016/0022-510x(82)90045-4. [DOI] [PubMed] [Google Scholar]
- 321.Robinson JL, Geser F, Corrada MM, et al. Neocortical and hippocampal amyloid-β and tau measures associate with dementia in the oldest-old. Brain. 2011;134(Pt 12):3708–15. doi: 10.1093/brain/awr308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Rauramaa T, Pikkarainen M, Englund E, et al. TAR-DNA binding protein-43 and alterations in the hippocampus. J Neural Transm. 2011;118:683–9. doi: 10.1007/s00702-010-0574-5. [DOI] [PubMed] [Google Scholar]
- 323.Dickson DW, Davies P, Bevona C, et al. Hippocampal sclerosis: A common pathological feature of dementia in very old (> or = 80 years of age) humans. Acta Neuropathol. 1994;88:212–21. doi: 10.1007/BF00293396. [DOI] [PubMed] [Google Scholar]
- 324.Jellinger KA. Clinical validity of Braak staging in the oldest-old. Acta Neuropathol (Berl) 2000;99:583–84. doi: 10.1007/s004010051163. [DOI] [PubMed] [Google Scholar]
- 325.Giannakopoulos P, Hof PR, Giannakopoulos AS, et al. Dementia in the oldest-old: Quantitative analysis of 12 cases from a psychiatric hospital. Dementia. 1994;5:348–56. doi: 10.1159/000106745. [DOI] [PubMed] [Google Scholar]
- 326.Bancher C, Jellinger KA. Neurofibrillary tangle predominant form of senile dementia of Alzheimer type: A rare subtype in very old subjects. Acta Neuropathol. 1994;88:565–70. doi: 10.1007/BF00296494. [DOI] [PubMed] [Google Scholar]
- 327.Fujishiro H, Ferman TJ, Boeve BF, et al. Validation of the neuropathologic criteria of the third consortium for dementia with Lewy bodies for prospectively diagnosed cases. J Neuropathol Exp Neurol. 2008;67:649–56. doi: 10.1097/NEN.0b013e31817d7a1d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328.Lowe J, Dickson D. Pathological diagnostic criteria for dementia associated with cortical Lewy bodies: Review and proposal for a descriptive approach. J Neural Transm Suppl. 1997;51:111–20. doi: 10.1007/978-3-7091-6846-2_9. [DOI] [PubMed] [Google Scholar]
- 329.McKee AC, Cantu RC, Nowinski CJ, et al. Chronic traumatic encephalopathy in athletes: Progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68:709–35. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Hof PR, Bouras C, Buée L, et al. Differential distribution of neurofibrillary tangles in the cerebral cortex of dementia pugilistica and Alzheimer’s disease cases. Acta Neuropathol. 1992;85:23–30. doi: 10.1007/BF00304630. [DOI] [PubMed] [Google Scholar]
- 331.Roberts GW, Allsop D, Bruton C. The occult aftermath of boxing. J Neurol Neurosurg Psychiatry. 1990;53:373–78. doi: 10.1136/jnnp.53.5.373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Auer IA, Schmidt ML, Lee VM, et al. Paired helical filament tau (PHFtau) in Niemann-Pick Type C disease is similar to PHFtau in Alzheimer’s disease. Acta Neuropathol. 1995;90:547–51. doi: 10.1007/BF00318566. [DOI] [PubMed] [Google Scholar]
- 333.Mattsson N, Zetterberg H, Bianconi S, et al. gamma-Secretase–dependent amyloid-β is increased in Niemann-Pick Type C: A cross-sectional study. Neurology. 2011;76:366–72. doi: 10.1212/WNL.0b013e318208f4ab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Schmidt ML, Lee VM, Saido T, et al. Amyloid plaques in Guam amyotrophic lateral sclerosis/parkinsonism-dementia complex contain species of A β similar to those found in the amyloid plaques of Alzheimer’s disease and pathological aging. Acta Neuropathol. 1998;95:117–22. doi: 10.1007/s004010050774. [DOI] [PubMed] [Google Scholar]
- 335.Schmidt ML, Zhukareva V, Perl DP, et al. Spinal cord neurofibrillary pathology in Alzheimer disease and Guam Parkinsonism-dementia complex. J Neuropathol Exp Neurol. 2001;60:1075–86. doi: 10.1093/jnen/60.11.1075. [DOI] [PubMed] [Google Scholar]
- 336.Hirano A, Malamud N, Kurland LT. Parkinsonism-dementia complex, an endemic disease on the island of Guam. II. Pathological features. Brain. 1961;84:662–79. doi: 10.1093/brain/84.4.662. [DOI] [PubMed] [Google Scholar]
- 337.Hof PR, Perl DP, Loerzel AJ, et al. Neurofibrillary tangle distribution in the cerebral cortex of parkinsonism-dementia cases from Guam: Differences with Alzheimer’s disease. Brain Res. 1991;564:306–13. doi: 10.1016/0006-8993(91)91467-f. [DOI] [PubMed] [Google Scholar]
- 338.Hof PR, Perl DP, Loerzel AJ, et al. Amyotrophic lateral sclerosis and parkinsonism-dementia from Guam: Differences in neurofibrillary tangle distribution and density in the hippocampal formation and neocortex. Brain Res. 1994;650:107–16. doi: 10.1016/0006-8993(94)90212-7. [DOI] [PubMed] [Google Scholar]
- 339.Williams DR. Tauopathies: Classification and clinical update on neurodegenerative diseases associated with microtubule-associated protein tau. Intern Med J. 2006;36:652–60. doi: 10.1111/j.1445-5994.2006.01153.x. [DOI] [PubMed] [Google Scholar]
- 340.Dickson DW, Kouri N, Murray ME, et al. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-Tau) J Mol Neurosci. 2011;45:384–9. doi: 10.1007/s12031-011-9589-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Nagy Z, Esiri MM, Jobst KA, et al. Influence of the apolipoprotein E genotype on amyloid deposition and neurofibrillary tangle formation in Alzheimer’s disease. Neuroscience. 1995;69:757–61. doi: 10.1016/0306-4522(95)00331-c. [DOI] [PubMed] [Google Scholar]
- 342.Haroutunian V, Hoffman LB, Beeri MS. Is there a neuropathology difference between mild cognitive impairment and dementia? Dialogues Clin Neurosci. 2009;11:171–79. doi: 10.31887/DCNS.2009.11.2/vharoutunian. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Hof PR, Archin N, Osmand AP, et al. Posterior cortical atrophy in Alzheimer’s disease: Analysis of a new case and re-evaluation of a historical report. Acta Neuropathol. 1993;86:215–23. doi: 10.1007/BF00304135. [DOI] [PubMed] [Google Scholar]
- 344.Nagy Z, Esiri MM, Hindley NJ, et al. Accuracy of clinical operational diagnostic criteria for Alzheimer’s disease in relation to different pathological diagnostic protocols. Dement Geriatr Cogn Disord. 1998;9:219–26. doi: 10.1159/000017050. [DOI] [PubMed] [Google Scholar]
- 345.Mukaetova-Ladinska EB, Garcia-Siera F, Hurt J, et al. Staging of cytoskeletal and β-amyloid changes in human isocortex reveals biphasic synaptic protein response during progression of Alzheimer’s disease. Am J Pathol. 2000;157:623–36. doi: 10.1016/s0002-9440(10)64573-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Jack CR, Jr, Dickson DW, Parisi JE, et al. Antemortem MRI findings correlate with hippocampal neuropathology in typical aging and dementia. Neurology. 2002;58:750–57. doi: 10.1212/wnl.58.5.750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Bennett DA, Schneider JA, Bienias JL, et al. Mild cognitive impairment is related to Alzheimer disease pathology and cerebral infarctions. Neurology. 2005;64:834–41. doi: 10.1212/01.WNL.0000152982.47274.9E. [DOI] [PubMed] [Google Scholar]
- 348.Dowling NM, Tomaszewski Farias S, Reed BR, et al. Neuropathological associates of multiple cognitive functions in two community-based cohorts of older adults. J Int Neuropsychol Soc. 2010:1–13. doi: 10.1017/S1355617710001426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Petrovitch H, Ross GW, Steinhorn SC, et al. AD lesions and infarcts in demented and non-demented Japanese-American men. Ann Neurol. 2005;57:98–103. doi: 10.1002/ana.20318. [DOI] [PubMed] [Google Scholar]
- 350.Haneuse S, Larson E, Walker R, et al. Neuropathology-based risk scoring for dementia diagnosis in the elderly. J Alzheimers Dis. 2009;17:875–85. doi: 10.3233/JAD-2009-1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Sabbagh MN, Sandhu SS, Farlow MR, et al. Correlation of clinical features with argyrophilic grains at autopsy. Alzheimer Dis Assoc Disord. 2009;23:229–33. doi: 10.1097/WAD.0b013e318199d833. [DOI] [PMC free article] [PubMed] [Google Scholar]