

Abstract
Nucelosides such as adenosine (Ado) influence nearly every aspect of physiology and pathophysiology. Extracellular nucleotides liberated at local sites of inflammation are metabolized through regulated phosphohydrolysis by a series of ecto-nucleotidases including ectonucleoside triphosphate diphosphohydrolase-1 (CD39) and ecto-5′-nucleotidase (CD73), found on the surface of a variety of cell types. Once generated, Ado is made available to bind and activate one of four G-protein-coupled Ado receptors. Recent in vitro and in vivo studies implicate Ado in a broad array of tissue protective mechanisms that provide new insight into adenosine actions. Studies in cultured cells and murine tissues have indicated that Ado receptors couple to novel post-translational protein modifications, including Cullin deneddylation, as a new anti-inflammatory mechanism. Studies in Ado receptor-null mice have been revealing and indicate a particularly important role for the Ado A2B receptor in animal models of intestinal inflammation. Here, we review contributions of Ado to cell and tissue stress responses, with a particular emphasis on the gastrointestinal mucosa.
Keywords: mucosa, inflammation, colitis, neutrophil, epithelium, endothelium, murine model
I. Introduction
Circulating or locally released nucleotides are rapidly metabolized by ecto-enzymes localized to the cell surface. Ecto-5′-nucleotidase (CD73) is a glycosyl phosphatidylinositol (GPI)-linked, membrane-bound glycoprotein which hydrolyzes extracellular nucleoside monophosphates into bioactive nucleoside intermediates[1]. Surface-bound CD73 metabolizes adenosine 5′-monophosphate (AMP) to adenosine, which when released can activate one of four types of G-protein coupled, seven transmembrane spanning adenosine receptors (AdoR) or can be internalized through dipyridamole-sensitive carriers[2]. Adenosine receptors are expressed on a wide variety of cells, and many cell types have been shown to express more than one isoform of the receptor. Likewise, activation of surface AdoR has been shown to regulate diverse physiologic endpoints. In the recent years, our understanding of nucleotide metabolic pathways has benefited from the development of genetically manipulated animals, particularly mice genetically deficient in the various adenosine receptors. Here, we review relevant studies addressing physiologic influences of Ado within the gastrointestinal mucosa utilizing a variety of model systems.
II. Functional Mucosal Responses Mediated by Adenosine
The gastrointestinal tract is lined by functionally differentiated epithelial cells. Primary physiologic functions of the epithelium include barrier function, absorption and mucosal hydration. Mucosal hydration is accomplished through a coordinated series of ion transport events. As part of a tissue adaptive response, a number of purine nucleotide metabolites, including adenosine, have been implicated to influence epithelial electrogenic chloride secretion, the transport event responsible for mucosal hydration. This aspect of epithelial function has been studied in detail utilizing models of intact epithelial cell layers coupled with electrophysiologic strategies. Studies by Madara et al. examining biological properties of soluble mediators derived from activated inflammatory cells (e.g. neutrophils and eosinophils) identified a small, protease-resistant fraction originally termed neutrophil-derived secretagogue (NDS)[3, 4]. When incubated with epithelia, NDS potently activated electrogenic chloride secretion and associated fluid transport in cultured intestinal epithelial cells. Subsequent biochemical analysis of NDS identified this molecule to be 5-AMP[4]. With no known AMP receptor, studies turned toward defining potential 5′-AMP metabolic pathways. These studies revealed the expression of CD73 as a critical control point for the metabolism of 5′-AMP to Ado[5]. CD73 was shown to be expressed at a very high level on the apical aspect on intestinal epithelial cells. The only known function of CD73 is the phosphohydrolysis of AMP to Ado. From such observations, biochemical and pharmacologic studies demonstrated the expression of the Ado A2B receptor (A2BAR) isoform of adenosine receptors on cultured and primary intestinal epithelial cells and in human tissue as the primary determinant for mucosal hydration[6]. A mechanism of such fluid transport mediated by Ado is shown in Figure 1.
Figure 1. Coordinated activation of mucosal hydration during inflammation.

During active inflammation, PMN transmigration (in conjunction with platelets, see text) migrate across the apical side of intestinal epithelial cells. PMN and platelet-derived ATP is metabolized to adenosine by a two-step enzymatic reaction involving ecto-apyrase (CD39 and CD39-like enzymes) and ecto-nucleotidase (CD73). Adenosine binding to epithelial adenosine A2B receptors results in activation of electrogenic Cl secretion and the paracellular movement of water. Such platelet/PMN - epithelial crosstalk pathway may serve as a defensive response by which mucosal surfaces are flushed from bacteria and bacterial products under inflammatory conditions.
A number of studies have also implicated adenosine receptors in the control of tissue barrier function. For example, A2BAR, which couples via G proteins Gs and possibly Gq[7, 8], has been demonstrated to be critical for the restitution of epithelial and endothelial monolayers following transmigration. Successful transmigration of leukocytes across endothelial and epithelial cells is accomplished by temporary PMN self-deformation with localized widening of the inter-junctional spaces[9, 10], a process with the potential to disturb endothelial and epithelial barrier function. Original studies by Lennon et al. revealed that the prominent signaling pathway for closing inter-endothelial gaps during transmigration involved adenosine-stimulated “resealing” of the barrier[11]. Subsequently, it was shown that protein kinase A signaling through the A2BAR recruits the vasodilator-stimulated phosphoprotein (VASP), a protein originally identified in platelets implicated in actin-binding and cross-linking functions[12]. This work demonstrated that VASP is phosphorylated via a PKA-dependent process in conditions that enhance barrier recovery following epithelial [13] and endothelial [14] disruption. Confocal microscopy studies revealed that VASP localizes with ZO-1 at epithelial tight junctions and at cell-cell borders and that phosphorylated VASP appears at the junction during epithelial and endothelial restitution. Subsequent transfection studies utilizing epithelial cells expressing truncated forms of VASP abnormal in oligomerization or actin-binding activity revealed a functional diminution of barrier recovery [13, 14] (see Figure 2).
Figure 2. Model of adenosine-mediated epithelial restitution following PMN migration.

Adenosine-mediated signaling increases levels of cAMP in epithelial cells and results in the activation of protein kinase A (PKA), and subsequent phosphorylation of VASP. In this model, phospho-VASP interactions with actin and tight junction proteins contribute to diminishing paracellular permeability through relaxation of actin cytoskeletal tension. Such a mechanism contributes to the resealing of epithelial monolayers following PMN transmigration.
III. Mucosal Sources of Adenosine
In ongoing inflammation, a number of cell types actively release adenine nucleotides, primarily in the form of ADP and ATP [15-17]. Inflammatory cells can be a rich source of nucleotides, and given the association of neutrophils with adenine-nucleotide/nucleoside signaling in the inflammatory milieu[4, 11], it was hypothesized that PMNs could represent a potential source of extracellular ATP [18, 19]. Initial studies utilizing luminometric ATP detection assays from supernatant fractions revealed that PMNs release ATP in an activation-dependent manner. Pharmacological strategies identified a mechanism involving connexin 43 (Cx43) hemichannels as the membrane pore for ATP release. Cx43 molecules assemble as hexameric “connexons” that form junctional complexes between different cell types[20] and various leukocytes have been shown to express Cx43[21]. More recent studies implicated Cx43 connexons as intercellular signaling channels via the release of ATP [20, 22]. PMNs from induced Cx43-/- mice, whereby activated PMNs release less than 15% of ATP relative to littermate controls and Cx43 heterozygote PMNs were intermediate in their capacity for ATP release. This study implicated Cx43 in activated PMN ATP release, therein contributing to the innate metabolic control of the inflammatory milieu [18]. Subsequent studies by others revealed that human neutrophils release ATP predominantly from the leading edge of their cell surface as a mechanism to amplify chemotactic signals and direct cell orientation by feedback through P2Y2 nucleotide receptors [23, 24].
Activated platelets are also known to release nucleotides at high concentration [25]. In this context, Weissmuller et. al. highlighted the interaction between PMN and platelets in regulating intestinal inflammation and fluid transport via nucleotide release [26]. Mucosal diseases are often characterized by a mixed inflammatory infiltrate that includes PMNs and platelets. These studies showed that platelets migrate across intestinal epithelial cells in a PMN-dependent manner. Furthermore, platelet-PMN co-migration was observed in intestinal tissue derived from human patients with inflammatory bowel disease (IBD). The translocated platelets were found to release large quantities of ATP, which was metabolized to adenosine via a 2-step enzymatic reaction involving CD73 and CD39-like molecules expressed on intestinal epithelial cells (IEC). Subsequent studies revealed a mechanism involving Ado-mediated activation of electrogenic chloride secretion, with concomitant water movement into the intestinal lumen[4]. Together, these studies demonstrated that ecto-NTDases are expressed on IEC and interact with platelet-derived nucleotides through a mechanism involving platelets that “piggy back” across mucosal barriers while attached to the surface of PMN [26] (Figure 1).
IV. Mechanisms of Adenosine Action
It is long appreciated that G protein-coupled receptors, particularly those which elevate intracellular cAMP (i.e A2AAR and A2BAR), can serve as sensors of inflammation and promote tissue protective responses[27]. Less is known about post-receptor events and molecular mechanisms of action [28]. It is recently appreciated that Ado receptor signaling is linked to a post-translational modification termed neddylation, the reversible conjugation of the ubiquitin-like NEDD8 (Neural precursor cell expressed, developmentally down-regulated 8) [29] moiety to proteins [30]. Neddylation and deneddylation responses are highly conserved and exist in a wide variety of cell types [31] and species [32-35]. Activation of the Nedd8-precursor through cleavage a carboxy-terminal glycine residue by UCH-L3 (also called SENP8) enables conjugation to the E1 UBA3-APPBP1 heterodimer, also known as Nedd8-Activating-Enzyme (NAE) [36-39]. Subsequently NEDD8 is conjugated to its specific E2 Ubc12 (ubiquitin conjugating enzyme) [40] and afterwards linked to the E3 complex [41, 42] (Figure 3). Neddylation plays an essential role in the post-translational modification of Cullin-RING-ligases [43] involved in the ubiquitin pathway. Cullins function as scaffolding proteins and are essential for the assembly of the ubiquitin E3 ligase complex conjugating ubiquitin to target proteins and thus marking them for proteasomal degradation, including IκB, which leads to translocation of the NFκB p50/p65 heterodimer to the nucleus [44, 45]. Of note, the ubiquitin E3 ligase complex integrating Cullin-2 is central to the regulation of hypoxia-inducible factor[31], a molecule which has gained significant attention as a protective factor in various models of mucosal inflammation[46].
Figure 3. Molecular mechanism of anti-inflammation by adenosine.
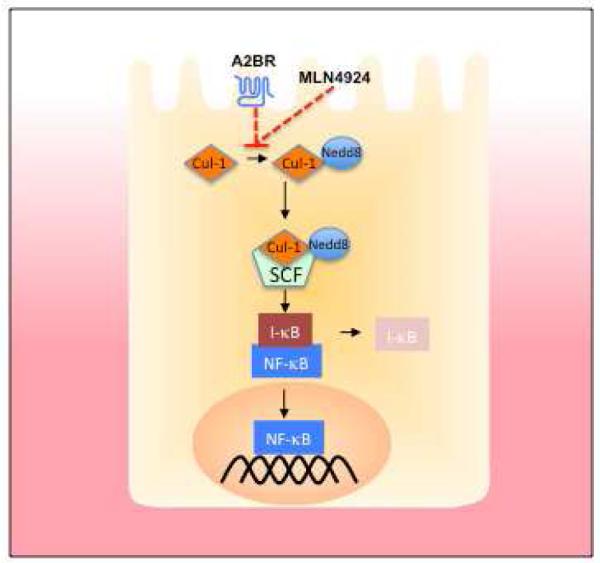
The canonical NFκB pathway is activated through the phosphorylation of IκB. This phosphorylation allows for its recognition by the neddylated Skp-Cullin-F-Box (SCF) complex, polyubiquitination and subsequent proteasomal degradation. Neddylation of Cul-1 is achieved through a multienzyme process, conjugating a NEDD8 moiety to the target protein (see text). Adenosine receptor activation, as well as the neddylation inhibitor MLN4924, inhibit Cullin neddylation or promote deneddylation of Cullin-proteins, thereby inhibiting the overall function of the SCF complex.
Ado potently inhibits the neddylation of Cullin-1 in endothelial and epithelial cells [47]. Insights into potential roles for Cullin-deneddylation in inflammation were provided by Neish et al. who demonstrated that commensal bacteria-associated attenuation of NFκB is Cullin-de-neddylation-dependent in epithelial cells [48]. Furthermore, Kumar et al. demonstrated that commensal bacteria influence the neddylation status of Cullin-1 (Cul-1) through generation of reactive oxygen species (ROS)[49]. Initially it was demonstrated that epithelial cells elicit increases in ROS when co-cultured with commensal bacteria. This resulted in a transient and reversible deneddylation of Cul-1 and subsequent decrease of NFκB pathway end products. Interestingly, they were able to show that different commensal bacterial strains differ in the amount of ROS they generate. Since an altered microbiota is a prevalent observation in patients with IBD[50] differing amounts of ROS in individual locations within the intestine may altering the inflammatory response in IBD [51].
One particularly intriguing mechanism suggests that Ado inhibits NF-κB through actions on proteasomal degradation of IκB proteins [47]. These findings were based on studies addressing adenosine signaling mechanisms which revealed that adenosine and adenosine analogs display a dose-dependent deneddylation of Cul-1 with rank order of specificity A2BAR >A1AR>>A2AAR = A3AR [47]. Current understanding indicates that deneddylation reactions on Cullin targets via CSN-associated proteolysis is increasingly implicated as a central point for Cullin-mediated E3 ubiquitylation [43]. Notably, other pathways for deneddylation have been reported. For example, signaling elicited by adrenomedullin, which is induced in LPS-associated inflammation[52], deneddylated Cul-1 with beneficial influences in a murine model of IBD [53]. Furthermore, the identification of the Nedd8-specific protease SENP8 has provided new insight into this emerging field. SENP8 appears to contain isopeptidase activity capable of directly deneddylating Cullin targets [54, 55]. Moreover, recent studies indicate that SENP8 is central for inflammatory responses at the vascular interface [56]. Pharmacological targeting of the neddylation pathway, more specifically the NAE, using MLN4924[57], a structural homolog of 5′-AMP, significantly abrogated NF-κB responses and reduced secretion of LPS-elicited and TNF-α-elicited pro-inflammatory cytokines in human endothelial cells[56] as well as in human and murine macrophage cells lines [58]. Likewise, MLN4924 abrogated pro-inflammatory responses to LPS in vivo [56]. These studies identify SENP8 as a proximal regulator of Cullin neddylation and provide an important role for SENP8 in fine-tuning of the inflammatory response (see Figure 3).
IV. The role of individual Ado receptors (ARs) in GI inflammation in vivo
The rather broad expression pattern of individual AdoR in the GI tract have engendered significant interest as therapeutic targets in GI inflammation. A number of studies have addressed the contribution of individual ARs (A1AR, A2AAR, A2BAR, or A3AR) to gastrointestinal inflammation in murine models. Specifically, the availability of AdoR-null mice and selective agonists and antagonists have revealed important insight into the potential contributions of Ado to GI inflammation.
While less is known about A1AR in GI disease models, the A2A and A2BAR receptors have been studied in some detail [59-61]. The A2AAR is critical for A2AAR signaling in T cell-mediated regulation of colitis. Moreover, treatment with a specific A2AAR agonist attenuated the production of pro-inflammatory cytokines and attenuation of colitis [62]. In addition, recent studies have performed a “head-on” comparisons between A2AAR-/- and A2BAR-/- in murine colitis models [63]. Previous studies had revealed that neither A2AAR-/- nor A2BAR-/- animals manifest outward immunological defects when housed in specific pathogen-free conditions[64]. Herein, A2AAR-/- or A2BAR-/- mice were subjected to DSS protocols and compared their responses to littermate wild-type controls. Initial studies using 3.5% DSS showed that A2BAR-/- mice became profoundly ill within 3 days of induction, associated with high mortality which required lowering the DSS concentration to 2.5%. A2AAR-/- animals remained healthy at 2.5% DSS with no significant differences observed in weight loss curves between A2AAR-/- mice and littermate controls. Notably, A2AAR-/- mice showed increased susceptibility to higher concentrations of DSS. Indeed, when DSS concentrations were increased from 2.5% to 4.5%, A2AAR-/- showed significantly increased weight loss on days 2 – 6 and increased colon contraction compared to wild-type controls. Thus, it would appear that both A2AAR-/- and A2BAR-/- mice have increased susceptibility to DSS colitis, but that this phenotype is likely more severe in A2AAR-/-[63]. Likewise, enteral administration of the selective A2BAR inhibitor PSB1115 resulted in a similar increase in severity of DSS colitis.[63] Together, these studies indicate a central regulatory role for the A2BAR in modulating the acute inflammatory phase of DSS colitis [63]. These studies are consistent with other studies that found a more severe phenotype of A2BAR-/- mice during intestinal ischemia-reperfusion injury[65] and implicate the A2BAR agonist BAY 60-6583 in the treatment of this condition[65]. Moreover, other studies from our laboratory have shown attenuation of mucosal inflammation during acute lung injury[61] or during intestinal hypoxia exposure[60, 64] mediated by the A2BAR. Similarly, A2BAR signaling is protective during renal[66] or myocardial ischemia[67, 68]. This is also consistent with several studies from other investigators, indicating A2BAR signaling in protection from vascular inflammation[69, 70], inflammation during organ transplantation[71] or attenuation of pulmonary inflammation during hypoxia[47].
By contrast, some studies have suggested a pathological role of A2BAR signaling in murine colitis [72, 73] and in Clostridium difficile intoxication[74]. Exposure of mice to DSS induced colitis have revealed that A2BAR-/- are protected and mucosal inflammation is attenuated. Such observations would be consistent with previous studies in cultured epithelial indicating more a pathological role for A2BAR signaling, including the observations that TNF strongly induces A2BAR expression [75]and the findings that A2BAR signaling enhances IL-6 and other pro-inflammatory cytokines [76-78].
Why the results from this study[73] might be different from our own [63, 65] is not readily apparent. In contrast to the study by Kolachala et al.,[73] several previous studies support an anti-inflammatory and tissue protective role of A2BAR signaling in different organ systems. As outlined above, gene-targeted mice for the A2BAR show enhanced vascular inflammation when exposed to endotoxin[70] or during acute vascular injury.[69] Similarly, gene-targeted deletion of the A2BAR is associated with enhanced vascular leak and inflammation during hypoxia.[60, 64, 79] Potential explanations why the studies of Kolachala et al.[72, 73] found a detrimental role of A2BAR signaling during murine colitis could include details in the colitis protocol, differences in murine strains with genetic deletion of the A2BAR or differences of the animal facilities resulting in microbial flora that influence disease outcome. In this regard, it is well documented differences in the microflora can fundamentally change the course of disease in various mouse models of colitis[80]. It is important to note that in some of their studies, A2BAR-/- mice showed a strongly a pro-inflammatory phenotype[73]. For example, A2BAR-/- mice showed increased susceptibility to systemic Salmonella infection, where >90% of A2BAR-/- mice died within 10 days compared with 20% of wild-type mice following orally administered S typhimurium. Consistent with the mortality data, A2BAR-/- mice also showed signs of weight loss earlier than WT mice[73]. In this regard, it is also possible that the lack of A2BAR signaling in A2BAR-/- could fundamentally influence innate immune cell function. For example, it has been shown that A2BAR contributes significantly to the differentiation of dendritic cells (DC)[81]. Ado-differentiated DC resulted in a pro-angiogenic, pro-inflamamtory and impaired allostimulatory phenotype. Thus, at the present time, it remains unclear exactly how the A2BAR functions in complex mucosal inflammatory models. Likely, additional comparisons between the individual mouse strains may be necessary to rectify some of these discrepancies.
Finally, studies in A3AR-/- mice have proven quite interesting and potentially revealed a specific role for A3AR in the resolution of GI inflammation. For example, Butler et al. and Ren et al. have recently shown that AA3R-/- show overall decreased pathology in acute DSS colitis[82], but these animals failed to resolve inflammation-associated with chronic inflammation in this model[82]. Likewise, the adenosine A3 receptor agonist, N6-(3-iodobenzyl)-adenosine-5′-N-methyluronamide, has proven to be protective in at least two murine models of colitis.[83] In humans, A3AR expression negatively correlates with acute inflammatory score, Crohn’s Disease Activity Index (CDAI) and disease chronicity[84], strongly implicating a need for more studies with this receptor subtype.
Conclusions
The gastrointestinal mucosa represents a unique environment for which to study nucleotide metabolism. The dynamic nature of multiple cell types in a highly proliferative area provides unique opportunities to define novel molecular mechanisms. Studies in animal models of IBD have revealed key seminal observations to the contribution of Ado receptors in mucosal immunology. More recent work has identified new molecular mechanisms of Ado receptor activation, particularly those related to post-translational modificaton of key effector molecules. Such findings provide a promising template for both drug discovery as well as novel endogenous adaptive pathways that reveal windows into the relevance of metabolism to disease processes.
Acknowledgments
This work was supported by grants from the National Institutes of Health (NIH; HL60569, DK50189, and DK095491), the Deutsche Forschungsgemeinschaft (DFG EH 371/1-1) and a grant from the Crohn’s and Colitis Foundation of America.
Footnotes
Disclosure
The authors declare that they have no conflict of interests.
The authors declare no financial interests in any of the work submitted here.
References
- 1.Zimmermann H, Braun N. Ecto-nucleotidases--molecular structures, catalytic properties, and functional roles in the nervous system. Prog Brain Res. 1999;120:371–385. [PubMed] [Google Scholar]
- 2.Linden J. Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annu Rev Pharmacol Toxicol. 2001;41:775–787. doi: 10.1146/annurev.pharmtox.41.1.775. [DOI] [PubMed] [Google Scholar]
- 3.Madara JL, Parkos CA, Colgan SP, MacLeod RJ, Nash S, Matthews J, Delp C, Lencer WS. Cl-secretion in a model intestinal epithelium induced by a neutrophil-derived secretagogue. J. Clin. Invest. 1992;89:1938–1944. doi: 10.1172/JCI115800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Madara JL, Patapoff TW, Gillece-Castro B, Colgan SP, Parkos CA, Delp C, Mrsny RJ. 5′-adenosine monophosphate is the neutrophil-derived paracrine factor that elicits chloride secretion from T84 intestinal epithelial cell monolayers. J. Clin. Invest. 1993;91:2320–2325. doi: 10.1172/JCI116462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Strohmeier GR, Lencer WI, Patapoff TW, Thompson LF, Carlson SL, Moe SJ, Mrsny RJ, Madara JL. Surface expression, polarization, and functional significance of CD73 in human intestinal epithelia. J. Clin. Invest. 1997;99:2588–2601. doi: 10.1172/JCI119447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strohmeier GR, Reppert SM, Lencer WI, Madara JL. The A2b adenosine receptor mediates cAMP responses to adenosine receptor agonists in human intestinal epithelia. J. Biol. Chem. 1995;270:2387–2394. doi: 10.1074/jbc.270.5.2387. [DOI] [PubMed] [Google Scholar]
- 7.Feoktistov I, Goldstein AE, Ryzhov S, Zeng D, Belardinelli L, Voyno-Yasenetskaya T, Biaggioni I. Differential expression of adenosine receptors in human endothelial cells: role of A2B receptors in angiogenic factor regulation. Circ Res. 2002;90:531–538. doi: 10.1161/01.res.0000012203.21416.14. [DOI] [PubMed] [Google Scholar]
- 8.Ryzhov S, Goldstein AE, Biaggioni I, Feoktistov I. Cross-talk between G(s)- and G(q)-coupled pathways in regulation of interleukin-4 by A(2B) adenosine receptors in human mast cells. Mol Pharmacol. 2006;70:727–735. doi: 10.1124/mol.106.022780. Epub 2006 May 2017. [DOI] [PubMed] [Google Scholar]
- 9.Chin AC, Parkos CA. Pathobiology of neutrophil transepithelial migration: implications in mediating epithelial injury. Annu Rev Pathol. 2007;2:111–143. doi: 10.1146/annurev.pathol.2.010506.091944. [DOI] [PubMed] [Google Scholar]
- 10.Ley K. Plugging the leaks. Nat Med. 2001;7:1105–1106. doi: 10.1038/nm1001-1105. [DOI] [PubMed] [Google Scholar]
- 11.Lennon PF, Taylor CT, Stahl GL, Colgan SP. Neutrophil-derived 5′-adenosine monophosphate promotes endothelial barrier function via CD73-mediated conversion to adenosine and endothelial A2B receptor activation. J Exp Med. 1998;188:1433–1443. doi: 10.1084/jem.188.8.1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reinhard M, Halbrugge M, Scheer U, Wiegand C, Jockusch BM, Walter U. The 46/50 kDa phosphoprotein VASP purified from human platelets is a novel protein associated with actin filaments and focal contacts. Embo J. 1992;11:2063–2070. doi: 10.1002/j.1460-2075.1992.tb05264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lawrence DW, Comerford KM, Colgan SP. Role of VASP in reestablishment of epithelial tight junction assembly after Ca2+ switch. Am J Physiol Cell Physiol. 2002;282:C1235–1245. doi: 10.1152/ajpcell.00288.2001. [DOI] [PubMed] [Google Scholar]
- 14.Comerford KM, Lawrence DW, Synnestvedt K, Levi BP, Colgan SP. Role of vasodilator-stimulated phosphoprotein in PKA-induced changes in endothelial junctional permeability. Faseb J. 2002;16:583–585. doi: 10.1096/fj.01-0739fje. [DOI] [PubMed] [Google Scholar]
- 15.Aherne CM, Kewley EM, Eltzschig HK. The resurgence of A2B adenosine receptor signaling. Biochim Biophys Acta. 2010 doi: 10.1016/j.bbamem.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eckle T, Koeppen M, Eltzschig HK. Role of extracellular adenosine in acute lung injury. Physiology (Bethesda) 2009;24:298–306. doi: 10.1152/physiol.00022.2009. [DOI] [PubMed] [Google Scholar]
- 17.Eltzschig HK. Adenosine: an old drug newly discovered. Anesthesiology. 2009;111:904–915. doi: 10.1097/ALN.0b013e3181b060f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eltzschig HK, Eckle T, Mager A, Kuper N, Karcher C, Weissmuller T, Boengler K, Schulz R, Robson SC, Colgan SP. ATP release from activated neutrophils occurs via connexin 43 and modulates adenosine-dependent endothelial cell function. Circ Res. 2006;99:1100–1108. doi: 10.1161/01.RES.0000250174.31269.70. [DOI] [PubMed] [Google Scholar]
- 19.Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, Robson SC, Colgan SP. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med. 2003;198:783–796. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodenough DA, Paul DL. Beyond the gap: functions of unpaired connexon channels. Nat Rev Mol Cell Biol. 2003;4:285–294. doi: 10.1038/nrm1072. [DOI] [PubMed] [Google Scholar]
- 21.Jara PI, Boric MP, Saez JC. Leukocytes express connexin 43 after activation with lipopolysaccharide and appear to from gap junctions with endothelial cells after ischemia-reperfusion. Proc Nat Acad Sci (USA) 1995;92:7011–7015. doi: 10.1073/pnas.92.15.7011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Novak I. ATP as a Signaling Molecule: the Exocrine Focus. News Physiol Sci. 2003;18:12–17. doi: 10.1152/nips.01409.2002. [DOI] [PubMed] [Google Scholar]
- 23.Chen Y, Corriden R, Inoue Y, Yip L, Hashiguchi N, Zinkernagel A, Nizet V, Insel PA, Junger WG. ATP release guides neutrophil chemotaxis via P2Y2 and A3 receptors. Science. 2006;314:1792–1795. doi: 10.1126/science.1132559. [DOI] [PubMed] [Google Scholar]
- 24.Linden J. Purinergic Chemotaxis. Science. 2006;314:1689–1690. doi: 10.1126/science.1137190. [DOI] [PubMed] [Google Scholar]
- 25.Gordon JL. Extracellular ATP: effects, sources and fate. Biochem J. 1986;233:309–319. doi: 10.1042/bj2330309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weissmuller T, Campbell EL, Rosenberger P, Scully M, Beck PL, Furuta GT, Colgan SP. PMNs facilitate translocation of platelets across human and mouse epithelium and together alter fluid homeostasis via epithelial cell-expressed ecto-NTPDases. J Clin Invest. 2008;118:3682–3692. doi: 10.1172/JCI35874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sitkovsky MV, Lukashev D, Apasov S, Kojima H, Koshiba M, Caldwell C, Ohta A, Thiel M. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annu Rev Immunol. 2004;22:657–682. doi: 10.1146/annurev.immunol.22.012703.104731. [DOI] [PubMed] [Google Scholar]
- 28.Eltzschig HK, Rivera-Nieves J, Colgan SP. Targeting the A2B adenosine receptor during gastrointestinal ischemia and inflammation. Expert Opin Ther Targets. 2009;13:1267–1277. doi: 10.1517/14728220903241666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar S, Tomooka Y, Noda M. Identification of a set of genes with developmentally down-regulated expression in the mouse brain. Biochem Biophys Res Commun. 1992;185:1155–1161. doi: 10.1016/0006-291x(92)91747-e. [DOI] [PubMed] [Google Scholar]
- 30.Kamitani T, Kito K, Nguyen HP, Yeh ET. Characterization of NEDD8, a developmentally down-regulated ubiquitin-like protein. J Biol Chem. 1997;272:28557–28562. doi: 10.1074/jbc.272.45.28557. [DOI] [PubMed] [Google Scholar]
- 31.Mikus P, Zundel W. COPing with hypoxia. Semin Cell Dev Biol. 2005;16:462–473. doi: 10.1016/j.semcdb.2005.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones D, Crowe E, Stevens TA, Candido EP. Functional and phylogenetic analysis of the ubiquitylation system in Caenorhabditis elegans: ubiquitin-conjugating enzymes, ubiquitin-activating enzymes, and ubiquitin-like proteins. Genome Biol. 2002;3 doi: 10.1186/gb-2001-3-1-research0002. RESEARCH0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Osaka F, Saeki M, Katayama S, Aida N, Toh EA, Kominami K, Toda T, Suzuki T, Chiba T, Tanaka K, et al. Covalent modifier NEDD8 is essential for SCF ubiquitin-ligase in fission yeast. EMBO J. 2000;19:3475–3484. doi: 10.1093/emboj/19.13.3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tateishi K, Omata M, Tanaka K, Chiba T. The NEDD8 system is essential for cell cycle progression and morphogenetic pathway in mice. J Cell Biol. 2001;155:571–579. doi: 10.1083/jcb.200104035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ou CY, Lin YF, Chen YJ, Chien CT. Distinct protein degradation mechanisms mediated by Cul1 and Cul3 controlling Ci stability in Drosophila eye development. Genes Dev. 2002;16:2403–2414. doi: 10.1101/gad.1011402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wada H, Kito K, Caskey LS, Yeh ET, Kamitani T. Cleavage of the C-terminus of NEDD8 by UCH-L3. Biochem Biophys Res Commun. 1998;251:688–692. doi: 10.1006/bbrc.1998.9532. [DOI] [PubMed] [Google Scholar]
- 37.Huang DT, Miller DW, Mathew R, Cassell R, Holton JM, Roussel MF, Schulman BA. A unique E1-E2 interaction required for optimal conjugation of the ubiquitin-like protein NEDD8. Nat Struct Mol Biol. 2004;11:927–935. doi: 10.1038/nsmb826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mendoza HM, Shen LN, Botting C, Lewis A, Chen J, Ink B, Hay RT. NEDP1, a highly conserved cysteine protease that deNEDDylates Cullins. J Biol Chem. 2003;278:25637–25643. doi: 10.1074/jbc.M212948200. [DOI] [PubMed] [Google Scholar]
- 39.Wu K, Yamoah K, Dolios G, Gan-Erdene T, Tan P, Chen A, Lee CG, Wei N, Wilkinson KD, Wang R, et al. DEN1 is a dual function protease capable of processing the C terminus of Nedd8 and deconjugating hyper-neddylated CUL1. J Biol Chem. 2003;278:28882–28891. doi: 10.1074/jbc.M302888200. [DOI] [PubMed] [Google Scholar]
- 40.Liakopoulos D, Doenges G, Matuschewski K, Jentsch S. A novel protein modification pathway related to the ubiquitin system. EMBO J. 1998;17:2208–2214. doi: 10.1093/emboj/17.8.2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hori T, Osaka F, Chiba T, Miyamoto C, Okabayashi K, Shimbara N, Kato S, Tanaka K. Covalent modification of all members of human cullin family proteins by NEDD8. Oncogene. 1999;18:6829–6834. doi: 10.1038/sj.onc.1203093. [DOI] [PubMed] [Google Scholar]
- 42.Jones J, Wu K, Yang Y, Guerrero C, Nillegoda N, Pan ZQ, Huang L. A targeted proteomic analysis of the ubiquitin-like modifier nedd8 and associated proteins. J Proteome Res. 2008;7:1274–1287. doi: 10.1021/pr700749v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parry G, Estelle M. Regulation of cullin-based ubiquitin ligases by the Nedd8/RUB ubiquitin-like proteins. Semin Cell Dev Biol. 2004;15:221–229. doi: 10.1016/j.semcdb.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 44.Amir RE, Iwai K, Ciechanover A. The NEDD8 pathway is essential for SCF(beta -TrCP)-mediated ubiquitination and processing of the NF-kappa B precursor p105. J Biol Chem. 2002;277:23253–23259. doi: 10.1074/jbc.M200967200. [DOI] [PubMed] [Google Scholar]
- 45.Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 46.Colgan SP, Taylor CT. Hypoxia: an alarm signal during intestinal inflammation. Nat Rev Gastroenterol Hepatol. 2010;7:281–287. doi: 10.1038/nrgastro.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Khoury J, Ibla JC, Neish AS, Colgan SP. Antiinflammatory adaptation to hypoxia through adenosine-mediated cullin-1 deneddylation. J Clin Invest. 2007;117:703–711. doi: 10.1172/JCI30049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Neish AS, Gewirtz AT, Zeng H, Young AN, Hobert ME, Karmali V, Rao AS, Madara JL. Prokaryotic regulation of epithelial responses by inhibition of IkappaB-alpha ubiquitination. Science. 2000;289:1560–1563. doi: 10.1126/science.289.5484.1560. [DOI] [PubMed] [Google Scholar]
- 49.Kumar A, Wu H, Collier-Hyams LS, Kwon YM, Hanson JM, Neish AS. The bacterial fermentation product butyrate influences epithelial signaling via reactive oxygen species-mediated changes in cullin-1 neddylation. J Immunol. 2009;182:538–546. doi: 10.4049/jimmunol.182.1.538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. Epub 12007 Aug 13715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kumar A, Wu H, Collier-Hyams LS, Hansen JM, Li T, Yamoah K, Pan ZQ, Jones DP, Neish AS. Commensal bacteria modulate cullin-dependent signaling via generation of reactive oxygen species. EMBO J. 2007;26:4457–4466. doi: 10.1038/sj.emboj.7601867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ehrentraut S, Frede S, Stapel H, Mengden T, Grohe C, Fandrey J, Meyer R, Baumgarten G. Antagonism of lipopolysaccharide-induced blood pressure attenuation and vascular contractility. Arterioscler Thromb Vasc Biol. 2007;27:2170–2176. doi: 10.1161/ATVBAHA.107.146100. Epub 2007 Jul 2126. [DOI] [PubMed] [Google Scholar]
- 53.MacManus CF, Campbell EL, Keely S, Burgess A, Kominsky DJ, Colgan SP. Anti-inflammatory actions of adrenomedullin through fine tuning of HIF stabilization. FASEB J. 2011;25:1856–1864. doi: 10.1096/fj.10-170316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mendoza HM, Shen LN, Botting C, Lewis A, Chen J, Ink B, Hay RT. NEDP1, a highly conserved cysteine protease that deNEDDylates Cullins. J Biol Chem. 2003;278:25637–25643. doi: 10.1074/jbc.M212948200. [DOI] [PubMed] [Google Scholar]
- 55.Wu K, Yamoah K, Dolios G, Gan-Erdene T, Tan P, Chen A, Lee CG, Wei N, Wilkinson KD, Wang R, et al. DEN1 is a dual function protease capable of processing the C terminus of Nedd8 and deconjugating hyper-neddylated CUL1. J Biol Chem. 2003;278:28882–28891. doi: 10.1074/jbc.M302888200. [DOI] [PubMed] [Google Scholar]
- 56.Ehrentraut SF, Kominsky DJ, Glover LE, Campbell EL, Kelly CJ, Bowers BE, Bayless AJ, Colgan SP. Central role for endothelial human deneddylase-1/SENP8 in fine-tuning the vascular inflammatory response. J. Immunol. 2013 doi: 10.4049/jimmunol.1202041. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 58.Chang FM, Reyna SM, Granados JC, Wei SJ, Innis-Whitehouse W, Maffi SK, Rodriguez E, Slaga TJ, Short JD. Inhibition of Neddylation Represses Lipopolysaccharide-induced Proinflammatory Cytokine Production in Macrophage Cells. J Biol Chem. 2012;287:35756–35767. doi: 10.1074/jbc.M112.397703. doi: 35710.31074/jbc.M35112.397703. Epub 392012 Aug 397727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414:916–920. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- 60.Rosenberger P, Schwab JM, Mirakaj V, Masekowsky E, Mager A, Morote-Garcia JC, Unertl K, Eltzschig HK. Hypoxia-inducible factor-dependent induction of netrin-1 dampens inflammation caused by hypoxia. Nat Immunol. 2009;10:195–202. doi: 10.1038/ni.1683. [DOI] [PubMed] [Google Scholar]
- 61.Eckle T, Grenz A, Laucher S, Eltzschig HK. A2B adenosine receptor signaling attenuates acute lung injury by enhancing alveolar fluid clearance in mice. J Clin Invest. 2008;118:3301–3315. doi: 10.1172/JCI34203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Naganuma M, Wiznerowicz EB, Lappas CM, Linden J, Worthington MT, Ernst PB. Cutting edge: Critical role for A2A adenosine receptors in the T cell-mediated regulation of colitis. J Immunol. 2006;177:2765–2769. doi: 10.4049/jimmunol.177.5.2765. [DOI] [PubMed] [Google Scholar]
- 63.Frick JS, MacManus CF, Scully M, Glover LE, Eltzschig HK, Colgan SP. Contribution of Adenosine A2B Receptors to Inflammatory Parameters of Experimental Colitis. J Immunol. 2009 doi: 10.4049/jimmunol.0801324. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eckle T, Faigle M, Grenz A, Laucher S, Thompson LF, Eltzschig HK. A2B adenosine receptor dampens hypoxia-induced vascular leak. Blood. 2008;111:2024–2035. doi: 10.1182/blood-2007-10-117044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hart M, Jacobi B, Schittenhelm J, Henn M, Eltzschig HK. A2B Adenosine Receptor Signaling Provides Potent Protection during Intestinal Ischemia/Reperfusion Injury. J Immunol. 2009 doi: 10.4049/jimmunol.0802193. in press. [DOI] [PubMed] [Google Scholar]
- 66.Grenz A, Osswald H, Eckle T, Yang D, Zhang H, Tran ZV, Klingel K, Ravid K, Eltzschig HK. The Reno-Vascular A2B Adenosine Receptor Protects the Kidney from Ischemia. PLoS Medicine. 2008;5:e137. doi: 10.1371/journal.pmed.0050137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Eckle T, Kohler D, Lehmann R, El Kasmi KC, Eltzschig HK. Hypoxia-Inducible Factor-1 Is Central to Cardioprotection: A New Paradigm for Ischemic Preconditioning. Circulation. 2008;118:166–175. doi: 10.1161/CIRCULATIONAHA.107.758516. [DOI] [PubMed] [Google Scholar]
- 68.Eckle T, Krahn T, Grenz A, Kohler D, Mittelbronn M, Ledent C, Jacobson MA, Osswald H, Thompson LF, Unertl K, et al. Cardioprotection by ecto-5′-nucleotidase (CD73) and A2B adenosine receptors. Circulation. 2007;115:1581–1590. doi: 10.1161/CIRCULATIONAHA.106.669697. [DOI] [PubMed] [Google Scholar]
- 69.Yang D, Koupenova M, McCrann DJ, Kopeikina KJ, Kagan HM, Schreiber BM, Ravid K. The A2b adenosine receptor protects against vascular injury. Proc Natl Acad Sci U S A. 2008;105:792–796. doi: 10.1073/pnas.0705563105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yang D, Zhang Y, Nguyen HG, Koupenova M, Chauhan AK, Makitalo M, Jones MR, St Hilaire C, Seldin DC, Toselli P, et al. The A2B adenosine receptor protects against inflammation and excessive vascular adhesion. J Clin Invest. 2006;116:1913–1923. doi: 10.1172/JCI27933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hasegawa T, Bouis D, Liao H, Visovatti SH, Pinsky DJ. Ecto-5′ nucleotidase (CD73)-mediated adenosine generation and signaling in murine cardiac allograft vasculopathy. Circ Res. 2008;103:1410–1421. doi: 10.1161/CIRCRESAHA.108.180059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kolachala V, Ruble B, Vijay-Kumar M, Wang L, Mwangi S, Figler H, Figler R, Srinivasan S, Gewirtz A, Linden J, et al. Blockade of adenosine A2B receptors ameliorates murine colitis. Br J Pharmacol. 2008;155:127–137. doi: 10.1038/bjp.2008.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kolachala VL, Vijay-Kumar M, Dalmasso G, Yang D, Linden J, Wang L, Gewirtz A, Ravid K, Merlin D, Sitaraman SV. A2B Adenosine Receptor Gene Deletion Attenuates Murine Colitis. Gastroenterology. 2008 doi: 10.1053/j.gastro.2008.05.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Warren CA, Li Y, Calabrese GM, Freire RS, Zaja-Milatovic S, van Opstal E, Figler RA, Linden J, Guerrant RL. Contribution of Adenosine A2B Receptors in Clostridium difficile Intoxication and Infection. Infect Immun. 2012;8:8. doi: 10.1128/IAI.00782-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kolachala V, Asamoah V, Wang L, Obertone TS, Ziegler TR, Merlin D, Sitaraman SV. TNF-alpha upregulates adenosine 2b (A2b) receptor expression and signaling in intestinal epithelial cells: a basis for A2bR overexpression in colitis. Cell Mol Life Sci. 2005;62:2647–2657. doi: 10.1007/s00018-005-5328-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ryzhov S, Goldstein AE, Novitskiy SV, Blackburn MR, Biaggioni I, Feoktistov I. Role of A2B adenosine receptors in regulation of paracrine functions of stem cell antigen 1-positive cardiac stromal cells. J Pharmacol Exp Ther. 2012;341:764–774. doi: 10.1124/jpet.111.190835. Epub 2012 Mar 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ryzhov S, Zaynagetdinov R, Goldstein AE, Novitskiy SV, Blackburn MR, Biaggioni I, Feoktistov I. Effect of A2B adenosine receptor gene ablation on adenosine-dependent regulation of proinflammatory cytokines. J Pharmacol Exp Ther. 2008;324:694–700. doi: 10.1124/jpet.107.131540. Epub 2007 Oct 2026. [DOI] [PubMed] [Google Scholar]
- 78.Sitaraman SV, Merlin D, Wang L, Wong M, Gewirtz AT, Si-Tahar M, Madara JL. Neutrophil-epithelial crosstalk at the intestinal lumenal surface mediated by reciprocal secretion of adenosine and IL-6. J Clin Invest. 2001;107:861–869. doi: 10.1172/JCI11783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thompson LF, Eltzschig HK, Ibla JC, Van De Wiele CJ, Resta R, Morote-Garcia JC, Colgan SP. Crucial role for ecto-5′-nucleotidase (CD73) in vascular leakage during hypoxia. J. Exp. Med. 2004;200:1395–1405. doi: 10.1084/jem.20040915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Blumberg RS, Saubermann LJ, Strober W. Animal models of mucosal inflammation and their relation to human inflammatory bowel disease. Curr Opin Immunol. 1999;11:648–656. doi: 10.1016/s0952-7915(99)00032-1. [DOI] [PubMed] [Google Scholar]
- 81.Novitskiy SV, Ryzhov S, Zaynagetdinov R, Goldstein AE, Huang Y, Tikhomirov OY, Blackburn MR, Biaggioni I, Carbone DP, Feoktistov I, et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood. 2008;112:1822–1831. doi: 10.1182/blood-2008-02-136325. Epub 2008 Jun 1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Butler M, Sanmugalingam D, Burton VJ, Wilson T, Pearson R, Watson RP, Smith P, Parkinson SJ. Impairment of adenosine a3 receptor activity disrupts neutrophil migratory capacity and impacts innate immune function in vivo. Eur J Immunol. 2012;2:201242655. doi: 10.1002/eji.201242655. [DOI] [PubMed] [Google Scholar]
- 83.Mabley J, Soriano F, Pacher P, Hasko G, Marton A, Wallace R, Salzman A, Szabo C. The adenosine A3 receptor agonist, N6-(3-iodobenzyl)-adenosine-5′-N-methyluronamide, is protective in two murine models of colitis. Eur J Pharmacol. 2003;466:323–329. doi: 10.1016/s0014-2999(03)01570-x. [DOI] [PubMed] [Google Scholar]
- 84.Rybaczyk L, Rozmiarek A, Circle K, Grants I, Needleman B, Wunderlich JE, Huang K, Christofi FL. New bioinformatics approach to analyze gene expressions and signaling pathways reveals unique purine gene dysregulation profiles that distinguish between CD and UC. Inflamm Bowel Dis. 2009;15:971–984. doi: 10.1002/ibd.20893. [DOI] [PMC free article] [PubMed] [Google Scholar]