

INTRODUCTION
Glutamate is the predominant fast excitatory neurotransmitter in the brain (Greenamyre, 1986). However, it also possesses toxic effects to the nervous system under certain circumstances through a process known as excitotoxicity. During acute insults like status epilepticus, mechanical trauma or ischemia, the concentrations of extra-neuronal glutamate is elevated as a result of cellular leakage or depolarization-induced exocytosis (Lau and Tymianski, 2010). The raise of the glutamate concentration leads to an intensive activation of ionotropic glutamate receptors [N-Methyl-D-aspartic acid receptor (NMDAR), 2-amino-3-(5-methyl-3-oxo-1,2- oxazol-4-yl)propanoic acid receptor (AMPAR) and kainate receptors], causing an excessive calcium influx to the neurons and triggering apoptosis (Szydlowska and Tymianski, 2010). Yet the precise mechanism of glutamate-induced cell death is still elusive, it is reported that the process involves several pathways including activation of nitric oxide synthase, calcium-sensitive proteases, caspases and mitochondrial damage (Wang and Qin, 2010). In addition, accumulated evidence have suggested that neuronal apoptosis was also involved in chronic disorders such as Parkinson's disease and Alzheimer's disease (Dong et al., 2009). Thus, modulating the glutamate-provoked excitotoxicity through pharmacological manipulations or activation of the intrinsic protective mechanisms has a great implication to attenuate neuronal damage during the pathological conditions.
Studies over the past decades have identified phosphoinositide 3-kinase (PI3K)/Akt cascade as the central pathway to protect neurons from apoptosis (Dudek et al., 1997). For example, hypoxia-induced apoptosis in primary hippocampal neurons are blocked by the activation of PI3K/Akt signaling (Yamaguchi et al., 2001). Yet the importance of PI3K/Akt pathway in neuroprotection has long been recognized, our understanding on the regulations of these kinases during excitotoxic insults are limited. We have previously identified a novel class of GTPases, the phosphoinositide 3-kinase enhancer (PIKE), which enhances the activity of PI3K/Akt through direct interactions (Ahn et al., 2004, Rong et al., 2003, Ye et al., 2000). There are three isoforms of PIKE (PIKE-L, PIKE-S and PIKE-A) generated from alternative splicing of the gene CENTG1 (Chan and Ye, 2007). In hippocampal neurons, PIKE-L complexes with Homer and metabotropic glutamate receptor 1 (mGlu1) when the receptor is activated, which is essential for mGlu1-dependent PI3K initiation (Rong, Ahn, 2003). We have further demonstrated that PIKE-L is essential for netrins to execute the cell survival functions (Tang et al., 2008). PIKE-L also associates with the DNase inhibitor SET and protects it from proteolytic cleavage by asparagine endopeptidase (AEP) during acidosis-provoked neuronal death (Liu et al., 2008). Recently, we have generated the PIKE knockout (PIKE −/−) mice and found that PIKE-L is a downstream component of brain derived neurotrophic factor (BDNF) signaling to maintain normal cortical development (Chan et al., 2011d). It is also a novel interaction partner of AMPAR GluA2 subunit to regulate the receptor trafficking and long-term potentiation formation (Chan et al., 2011a). However, it remains unexplored if PIKE processes any significant role in protecting the brain cells from apoptosis during excitotoxic insults.
We report here that PIKE −/− mice are hypersensitive to kainic acid (KA)-induced seizure. Moreover, a higher degree of neurodegeneration is observed in PIKE −/− brain after KA injection. Biochemical analysis reveals that the apoptotic pathway is elevated in the absence of PIKE and electrophysiological study shows that PIKE-null neurons are more susceptible to KA-induced Ca2+ entry, leading to a higher sensitivity to Ca2+-provoked apoptosis. Thus, our results demonstrate that the integrity of PIKE signaling is essential for protecting the neurons from excitotoxicity-stimulated damage both in vitro and in vivo.
MATERIALS AND METHODS
Animals
PIKE −/− mice with a targeted deletion of exons 3 to 6 of CENTG1 were generated as previously reported (Chan et al., 2010b). KA (Sigma-Aldrich, USA) was dissolved in PBS and administrated into the animals (2 to 3-months-old) by intraperitoneal injection. All animal experiments were performed according to the care of experimental animal guidelines from Emory University.
Primary neuron culture
Cortical primary neurons were dissected from E18 embryo and cultured as described (Chan et al., 2011b). DIV 7 neurons were infected with various adenoviruses as indicated. Two days after infection, the neurons were treated with either PBS or KA (100 μM) for 24 h.
Caspase activities assay
Cell lysates from cultured neurons or whole mouse brain were collected and homogenized in lysis buffer 50 mm Tris, pH 7.4, 40 mm NaCl, 1 mm EDTA, 0.5% Triton X-100, 1.5 mm Na3VO4, 50 mm NaF, 10 mm sodium pyrophosphate, 10 mm sodium β-glycerophosphate, 1 mm phenylmethylsulfonyl fluoride (PMSF), 5 mg/ml aprotinin, 1 mg/ml leupeptin, and 1 mg/ml pepstatin A). KA-induced caspases activities in cultured neurons were assayed by Caspase-Glo 3/7 Assay Kit (Promega, USA). Total caspases activity in PIKE −/− brain was determined by CaspACE (Colorimetric) Assay Kit (Promega, USA).
Immunohistochemical staining
Brain tissues were fixed in 4% paraformaldehyde, paraffin embedded and sectioned (8 μm) in standard procedures. After serial rehydration and permeabilization in 0.1% TBST, sections were immunostained using specific antibodies as indicated and counterstained with hematoxylin using Zymed Histostain SP kit (Invitrogen, USA) as previously described (Chan et al., 2010a).
Recording of hippocampal slices/neurons
Hippocampal slices were prepared as described previously (Woo et al., 2007). In brief, mice (4–8 weeks old, male) were decapitated after being anaesthetized. Transverse hippocampal slices (400 μm) were prepared using a Vibroslice (Leica VT 1000S) in an ice-cold artificial cerebrospinal fluid (ACSF) containing (in mM): 126 NaCl, 26 NaHCO3, 3.0 KCl, 1.2 NaH2PO4, 2.0 CaCl2, 1.0 MgSO4, 10 glucose, 0.02 bicuculline or 0.1 picrotoxin. CA1 neurons were visualized with infrared optics using an upright microscope equipped with a ×40 water-immersion lens (Olympus, BX51WI) and infrared-sensitive CCD camera. NMDA-EPSC was determined by subtracting the traces obtained in 100 μM D-L AP5 at + 40 mV from those obtained before. The isolated AMPA-EPSC was measured at −60 mV in the presence of 100 μM D-L AP5. Internal recording solution contained (in mM): 140 CsF, 35 CsOH, 10 HEPES, 2 MgCl2, 11 EGTA, 2 TEA, 1 CaCl2, 2 Mg-ATP (pH 7.3, 300 mosM) whereas the extracellular solution contained (in mM): 140 NaCl, 0.2 CaCl2 5.4 KCl, 25 HEPES, 33 glucose, 0.001 TTX (pH 7.4, 320–335 mosM). Whole-cell currents were recorded by ramping the membrane potential from −60 mV to 60 mV at a rate of 50 mV/s in the absence and presence of 200 μM kainate. I–V relations of AMAP receptor-mediated responses were constructed by subtracting I–V curves in the absence of kainate from those obtained in the presence of kainate. Rectification index (RI) was calculated by using the equation RI = [I+40/(40 − Erev)]/[I−60/(−60 − Erev)] where I+40 and I−60 were the amplitudes of kainate-induced currents at +40 and −60 mV, respectively, and Erev was the reversal potential (Iino et al., 1994, Ozawa et al., 1991)
To characterize Ca2+ permeability of AMPA receptor channels, neurons were recorded in the large nucleated outside-out configuration at room temperature (Sather et al., 1992). Hippocampal neurons were mechanically isolated in 1 mg/ml papain from CA1 slices of 4–5 weeks old mice as described previously (Wang and MacDonald, 1995). The intracellular solution was the same as that containing 140 mM CsF; but the extracellular solution contained (in mM) 100 CaCl2, 10 glucose, 5 HEPES, and 24 N-methylglucamine (pH7.4). Relative permeability ratios were determined from the Erev recorded in NaC1-free solution using the constant field equation: PCa /PCS = [Cs+]i/[Ca2+]0 exp (EF/RT) [exp (EF/RT) + 1]/4 where E is the reversal potential, and F, R, and T have their conventional meaning. PCa and PCs represent the permeability coefficients to Ca2+ and Cs+.
Electroencephalograph (EEG)
Electroencephalograph was performed as described (Martin et al., 2007). Four stainless-steel electrodes were placed subdurally (1st electrode: +2.00 anterior to bregma suture and +1.2 from the midline; 2nd electrode: −1.5 posterior to bregma and +1.2 from the midline; 3rd electrode: −0.5 posterior to bregma and −2.2 from the midline; and 4th electrode: −3.5 from bregma and −2.2 from midline) and a pair of fine wires (Cooner AS 632, USA) were inserted in the nuchal muscle for movement detection (EMG). After 7 days of recovery, each animal was placed into a Plexiglas box (15 cm × 15 cm × 15 cm) and was attached to an amplifier (Stellate, Canada) via a small counterbalanced commutator (Dragonfly Research, USA). Amplified ECoG and EMG signals were collected and analyzed with the Stellate Harmonie Software (version 6.1c EN-0, Canada). After 2 hours of baseline recording, the animals were injected with KA (15 mg/Kg) and ECoG and EMG data were collected for 3 hours. The numbers of spike discharges in 5 s samples of ECoG recording at every 5 minute interval over a period of 180 minutes were measured.
Statistical analysis
Results were expressed as mean ± S.E.M. and were considered significant when P<0.05. Statistic analysis of the data was performed using either Student's t-test or two-way ANOVA followed by Bonferroni post-tests by the computer program GraphPad Prism (GraphPad Software, USA).
RESULTS AND DISCUSSIONS
To study the neuroprotective role of PIKE during excitotoxic insults, we exposed wild-type (WT) and PIKE-ablated (PIKE −/−) cortical neurons (DIV 7) to KA (100 μM). KA is a non-selective agonist of AMPAR/kainate receptor, which causes neuronal depolarization and excessive Ca2+ influx (Murphy et al., 1987, Robinson and Deadwyler, 1981). It is also a well-known excitotoxic agent to induce neuronal apoptosis when administrated in vivo and in vitro (Filipkowski et al., 1994, Simonian et al., 1996). Caspase activity assay revealed that KA stimulation caused cell death in both wild-type and PIKE −/− neurons. However, the extent of apoptosis was significantly higher in PIKE −/− neurons after KA-stimulation, suggesting neurons with PIKE-ablation are more susceptible to KA-induced cell death (Fig 1A).
fig 1. Kainic acid triggers more cell death in PIKE ablated neurons.

A. Enhanced apoptosis in PIKE-ablated neurons. Cortical neurons from wild-type (+/+) and PIKE knockout (−/−) mice were cultured (DIV 7) and stimulated with KA (100 μM, 24 h). The cell lysates were then collected and the total activities of caspase 3 and 7 were determined (*: P<0.05, **: P<0.01, ***: P<0.001, Student's t-test, n=3).
B. Reintroduction of the PIKE isoforms in null cells reduces KA-induced neuronal cell death. Cortical neurons from wild-type (+/+) and PIKE knockout (−/−) mice were cultured (DIV 7) and infected with either control adenovirus (Ad Control), adenovirus ovexpressing PIKE-A (Ad PIKE-A) or PIKE-L (Ad PIKE-L) for 48 hr. The infected cells were then treated with KA (100 μM, 24 h). The cell lysates were then collected and the total activities of caspase 3 and 7 were determined (**: P<0.01, ***: P<0.001, Student's t-test, n=3).
We further examined the isoforms-specific role of PIKE in protecting neuronal apoptosis. We infected PIKE −/− neurons with control adenovirus or adenovirus overexpressing PIKE-L (Ad PIKE-L) or PIKE-A (Ad PIKE-A) and treated the infected cells with KA. As shown in Fig 1B, overexpression of either PIKE isoforms inhibited the basal and KA-induced caspase activities, indicating both PIKE isoforms are indispensable in neuroprotection.
The above in vitro studies, together with our previous findings, supported that PIKE GTPases are crucial for promoting neuronal survival in cultured neurons (Liu, Jang, 2008, Rong, Ahn, 2003, Tang, Jang, 2008). However, whether PIKE proteins possess the same protective role against neurotoxic insults in vivo has not been explored. Therefore, we examined the degree of neuronal damage in PIKE −/− brains after KA injection. In basal condition, total caspase activity in PIKE −/− brain was significantly higher (Fig 2A). The caspase activity in both wild-type and PIKE −/− brains was augmented after KA injection, but the increment in PIKE −/− mice was significantly higher, indicating that PIKE is essential to inhibit caspases activation under basal and neuroexcitotoxic conditions. Immunohistochemical analysis also revealed higher caspase 3 activation in the dentate gyrus of the PIKE −/− brain after KA injection (Fig 2B, lower panels). However, no active caspase-3 was detected in the dentate gyrus of both genotypes under basal condition (Fig 2B, upper panels). Nevertheless, active caspase 3 could be observed in some cortical neurons in PIKE −/− mice but not in control mice under physiological condition (Fig 2B, upper insets), which may account for the elevated caspase activity shown in Fig 2A. In agreement with the staining, enhanced cleavage of caspase was observed in PIKE −/− brain compared to the control (Fig 2C, lower panel). Notably, PIKE-L was also cleaved in wild-type brain after KA treatment (Fig 2C, upper panel), which confirms our previous finding that PIKE is a substrate of caspases during apoptosis (Tang and Ye, 2006).
fig 2. Kainic acid induces severer neuronal death in PIKE −/− mice.

A. PIKE −/− brain displayed higher caspase activity under basal or KA-induced condition. Four days after intraperitoneal injection of saline or 20 mg/kg KA, the brains from wild-type and PIKE −/− mice were collected and the total caspase activities were determined using colorimetric assay (**: P<0.01; ***: P<0.001, Student's t-test, n=4).
B. KA injection caused more potent apoptosis in dentate gyrus of PIKE −/− mice. Five days after saline or 20 mg/kg KA challenge, the mouse brains from wild-type or PIKE −/− mice were fixed, sectioned and stained with specific antibody against active caspase 3. Neurons with positive signal were indicated with arrows. Caspase activity was also detected in cortical regions of PIKE −/− animals but not the wild-type control as shown in the box of the corresponding upper panels. Scale bar: 100 μm. Inserts in the upper panels are cortical neurons from the same animal, whereas the inserts in lower panels are magnified view of hippocampal neurons showing positive signals.
C. Ablation of PIKE in brain enhanced cell death. Wild-type and PIKE −/− mice were treated with 20 mg/kg KA or saline via intraperitoneal injection. Four days after injection, the mice were scarified and the apoptotic markers in the brain were detected by immunoblotting. Increased apoptotic degradation of caspase 3 was detected in PIKE knockout brain (lower panel). The expression of PIKE was confirmed using anti-PIKE-L N-terminus (PIKE (N)) antibody (top panel).
In addition to inducing neurodegeneration, KA also elicits seizures in rodents by activating the ionotropic glutamate receptors directly and increasing the release of excitatory amino acids from nerve terminals (Ferkany et al., 1984, Zaczek et al., 1981). To further demonstrate that PIKE −/− mice are more vulnerable to KA stimulation, we treated wild-type and PIKE −/− mice with epileptogenic dose of KA intraperitoneally and scored the degrees of induced seizure (1: Immobility, 2: Forelimb and/or tail extension, rigid posture; 3: Repetitive movements, head bobbing; 4: Rearing and falling; 5: Continuous rearing and falling; 6: Severe tonic-clonic seizure) (Zhang et al., 2002). While 30 mg/kg KA induced abrupt tonic-clonic seizure in both mice group within 10 min (data not shown) and persisted for about an hour, 20 mg/kg KA induced seizure in both genotypes with progressively increased severity. However, PIKE −/− mice showed much higher degree of seizure (Fig 3A). Significant statistical difference of the induced seizure was observed 25 min after KA injection and persisted till the end of the experiment. To test if the elevated severity of KA-triggered seizure in PIKE −/− mice was caused by increased neuronal excitability, we studied the electroencephalogram (EEG) patterns in the cortex of PIKE −/− mice after KA injection (Fig 3B). Under basal condition, there was no significant difference in the amplitudes of spike wave-discharge between wild-type and PIKE −/− mice (data not shown). After KA (15 mg/kg) injection, the amplitude of spike-wave was increased to a similar extend in both wild-type and PIKE −/− mice. However, the frequency of discharge tripled in PIKE −/− mice (Fig 3C). These results indicate that the increased seizure in PIKE −/− mice may be caused by enhanced excitability induced by KA.
fig 3. PIKE −/− mice are more susceptible to KA-induced seizure.

A. PIKE depletion increases seizure scores. Adult (2 to 3-month-old) wild-type (□) and PIKE −/− (■) mice were injected intraperitoneally with KA (20 mg/kg). Induced seizure after KA injection was then scored (*: P<>0.01, two-way ANOVA, n=6).
B. Representative EEG recording before and after KA (15 mg/kg) injection in wild-type animals.
C. KA-provoked more spike discharge in PIKE −/− mice. Number of spike-wave discharges that were higher than the basal levels during the EEG examination in the wild-type and PIKE −/− mice after 15 mg/kg KA injection was quantified (*: P<0.05, Student's t-test, n=4).
We have reported that the PIKE-L is involved in controlling the cell surface expression of AMPAR subunit GluA2 in neurons (Chan, Chen, 2011a). Among the AMPAR subunits, GluA2 is impermeable to Ca2+ (Hollmann et al., 1991). When GluA2 is unable to be recruited into the synapse, the composition of AMPAR at the excitatory synapses may be altered, leading to the formation of Ca2+ permeable AMPARs through increasing the amount of aberrant receptor complexes composed of GluA1 and GluA3 (Sans et al., 2003). Moreover, it has been reported that mice without GluA2-containing AMPAR were hypersensitive to KA challenge (Jia et al., 1996). Indeed, the loss of surface GluA2 is an important contributing factor to neuronal dysfunction and cell death in excitotoxicity (Bell et al., 2009). Thus, PIKE −/− neurons might have a higher permeability to Ca2+ influx after KA stimulation. To test this hypothesis, KA (200 μM)-evoked whole-cell currents from PIKE −/− hippocampal neurons were recorded by ramping the membrane potential from -60 mV to 60 mV at a rate of 50 mV/s (Fig 4A). Wild-type CA1 pyramidal neurons exhibited little or no rectification, suggesting that most AMPARs in CA1 neurons contained the GluA2 subunit and have a low permeability to Ca2+. In contrast, neurons of PIKE −/− mice demonstrated enhanced inward rectification, an effect similar to that observed in neurons lacking the GluA2 subunit (Burnashev et al., 1995, Jia, Agopyan, 1996, Jonas et al., 1994) (Fig 4B). Rectification index (RI) of PIKE −/− neurons was significantly lower (Fig 4C), further suggesting an increased Ca2+ permeability in the absence of PIKE. Nevertheless, the reversal potentials were closed to 0 mV in both PIKE −/− and wild-type neurons.
fig 4. Higher calcium influx in PIKE −/− neurons after KA stimulation.

A. Representative traces showing AMPA receptor medicated currents in response to synaptic stimulation in presence of the NMDA receptor blocker (d-AP5, 100 μM). Traces were obtained at holding currents of −60 mV to +40 mV, in 20 mV steps. The scales represent 100 pA and 30 ms respectively (n=6).
B. Peak I/V relationship of wild-type (◯) and PIKE −/− (●) neurons.
C. Bar graph showing the rectification index in (B). The rectification index was calculated by the traces at the holding potentials of −60, 0, +40 mV using the following formula: RI = [(I+40 − I0)/ (I0 − I−70)] × 3/2 (**: P<0.01, Student's t-test, n=6).
To further determine the Ca2+ permeability when PIKE is ablated, neurons were recorded in a large nucleated outside-out configuration (Sather, Dieudonne, 1992) and in Na+ free extracellular solution that contained 100 mM Ca2+. The I/V curves of kainate-activated currents indicated a higher reversal potentials (Erev) in PIKE −/− neurons, confirming the increased Ca2+ permeability in PIKE −/− neurons (Fig. 4A). Moreover, PIKE −/− neurons has a higher PCa2+ to PCs+ ratio, an indicator of Ca2+ permeability (Burnashev, Zhou, 1995), than wild-type neurons (Fig 5B). Thus, the high Ca2+ permeability in PIKE −/− neurons may be responsible for their hypersensitivity to KA-induced seizure and cell death.
fig 5. Large nucleated outside-out recordings of PIKE −/− neurons.
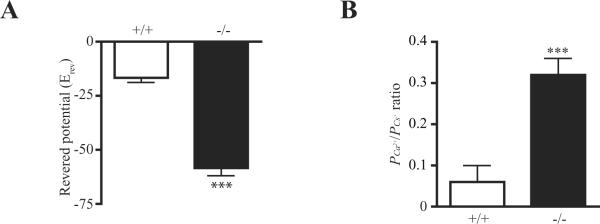
A. Reversed potential of neurons after KA stimulation (***: P<0.001, Student's t-test, n=5).
B. PCa2+ to PCs+ ratio of neurons after KA stimulation (***: P<0.001, Student's t-test, n=5).
In this report, we have demonstrated that PIKE GTPases are indispensable components of the defensive mechanism against neurotoxic insults in neurons. We have shown previously that PIKE ablation in neurons results in decreased PI3K/Akt signaling (Chan, Liu, 2011d). Since PI3K/Akt is the vital signaling cascade to maintain cell survival, the reduced PI3K activity in PIKE −/− brain thus make the neurons more susceptible to cell death induced by various stimuli. In addition to KA challenge, we have also shown that more cells were committed to apoptosis in PIKE −/− brain after middle cerebral artery occlusion (MCAO)-induced stroke, another animal model of glutamate-related excitotoxicity (Matsumoto et al., 1993). Moreover, we have shown that PIKE-L is critical for controlling the surface expression of GluA2 subunit of AMPA receptor in hippocampal and cortical neurons. Since a reduction of surface GluA2 increases the calcium influx in mouse brain (Jia, Agopyan, 1996, Sans, Vissel, 2003), it is reasonable to observe that PIKE −/− brains, where the amount of cell surface GluA2 is reduced, are more permeable to Ca2+ entry after KA stimulation. Therefore, PIKE GTPases have a dual role in resisting apoptosis: they maintain the PI3K/Akt pathway on one hand and limit the amount of Ca2+-permeable AMPA receptor on cell surface on the other hand (Fig 6).
fig 6. Proposed role of PIKE in neuroprotection.

PIKE GTPases protect the neuron from excitotoxicity-induced dysfunction and cell death by dual mechanisms: they limit the ampount of Ca2+-impermeable AMPA-R (GluA2) on the cell surface to prevent excessive Ca2+ influx and activate the PI3K/Akt pathway to block the death signaling induced by prolonged elevation of intracellular Ca2+ concentration ([Ca2+]i).
What is the upstream factor to initiate the PIKE-dependent defensive mechanism when the neurons are confronted with neurotoxic insults? We have shown that PIKE is a downstream factor of BDNF signaling (Chan, Liu, 2011d). Given that neurotrophins are prominent factors in preventing neuronal cell death under various devastating injuries like stroke and hypoxia (Almeida et al., 2005), it is thus reasonable to suggest that BDNF is the upstream activator of PIKE to initiate the neuroprotection. Indeed, studies have reported that BDNF protected glutamate-induced apoptotic cell death in the central nervous system via the PI3K/Akt pathway (Almeida, Manadas, 2005, Gratacos et al., 2001). We have also reported that PIKE-A is a novel insulin receptor (IR) activator during insulin stimulation (Chan et al., 2011c). We observed a partial insulin resistance in the PIKE −/− brain after insulin injection (i.p.) that insulin triggered less IR activation in the PIKE −/− brain (data not shown). Since PI3K and Akt are the major members of the IR signaling, it is not surprising to find that insulin can protect neuronal apoptosis caused by serum deprivation or AMPAR overactivation via PI3K/Akt (Kim and Han, 2005, Ryu et al., 1999). Moreover, insulin has an anti-seizure activity against KA-induced myoclonic twitches and tonic-clonic convulsions (Uysal et al., 1996). Although further experimental data are needed before a solid conclusion is made, it is tempting to suggest that PIKE may serve as common regulators of BDNF and insulin signaling to execute its protective functions in neurons.
Although BDNF is a prominent neuroprotective factor against KA-induced cell death, it is also a significant contributor to seizure pathogenesis. It has been reported that BDNF heterozygous null mice have a higher threshold for both electrically and chemically induced seizure, suggesting BDNF promotes epileptogenesis (Barton and Shannon, 2005). The studies in neuronal-specific deletion of TrkB further support the essential role of BDNF in seizure induction as the TrkB −/− mice are resistant to kindling epileptogenesis (He et al., 2004). Further, transgenic overexpression of BDNF results in spontaneous seizure and enhanced sensitivity to KA-evolved seizures (Croll et al., 1999). If PIKE is a downstream effector of BDNF to induce spontaneous seizure, the threshold for seizure induction in PIKE ablated animals should be heightened. However, PIKE −/− mice are more vulnerable to KA-induced seizure work, which suggests a discrepant role of BDNF/PIKE signaling in KA-induced neuronal death and epileptogenesis. Further experiments are thus warranted to clarify the precise role of BDNF/PIKE signaling during excitotoxicity-induced apoptosis and seizure.
In all, we have demonstrated that PIKE GTPases are essential components of the intrinsic neuroprotective mechanism against neurotoxic insults in mice, which also provided the in vivo data to support our previous in vitro observations. Thus, maintaining the integrity of PIKE activities is beneficial for preserving neuronal survival under pathological conditions.
SUMMARY
Phosphoinositide 3-kinase enhancer (PIKE) is a group of novel GTPases that regulate the PI3K/Akt activity through direct interactions. Previous in vitro studies suggest that PIKE are involved in numerous mechanisms to prevent apoptosis in neurons but their roles in intact animal remain unexplored. Here, we show that neurons with PIKE ablation (PIKE −/−) are hypersensitive to neuroexcitotoxic stimulation as higher Ca2+ permeability and cell death are detected in kainic acid (KA)-treated PIKE −/− neurons. We further examined the neuroprotective role of PIKE in vivo by administrating KA into the knockout animals via intraperitoneal injection. Immunohistological and biochemical analysis reveal that KA injection causes severer neuronal damage in PIKE> −/− brains. The discharge frequency of electroencephalogram is also higher in PIKE −/− mice after KA administration, results in a stronger epileptogenic behavior in the mutant line. Therefore, our data provide compelling evidence on the neuroprotective role of PIKE GTPases in animals that PIKE proteins are essential for protecting against neurotoxic insults.
ACKNOWLEDGEMENT
We would like to thank the Histology and EM Laboratory of Yerkes National Primate Research Center, Emory University for the preparation of the tissue sections. This work is supported by grant R01-NS045627 from NIH to K. Ye and the URC grant 00016337 from Emory University to C.B.Chan.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ahn JY, Rong R, Kroll TG, Van Meir EG, Snyder SH, Ye K. PIKE (phosphatidylinositol 3-kinase enhancer)-A GTPase stimulates Akt activity and mediates cellular invasion. J Biol Chem. 2004;279:16441–51. doi: 10.1074/jbc.M312175200. [DOI] [PubMed] [Google Scholar]
- Almeida RD, Manadas BJ, Melo CV, Gomes JR, Mendes CS, Graos MM, et al. Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ. 2005;12:1329–43. doi: 10.1038/sj.cdd.4401662. [DOI] [PubMed] [Google Scholar]
- Barton ME, Shannon HE. The seizure-related phenotype of brain-derived neurotrophic factor knockdown mice. Neuroscience. 2005;136:563–9. doi: 10.1016/j.neuroscience.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Bell JD, Park E, Ai J, Baker AJ. PICK1-mediated GluR2 endocytosis contributes to cellular injury after neuronal trauma. Cell Death Differ. 2009;16:1665–80. doi: 10.1038/cdd.2009.106. [DOI] [PubMed] [Google Scholar]
- Burnashev N, Zhou Z, Neher E, Sakmann B. Fractional calcium currents through recombinant GluR channels of the NMDA, AMPA and kainate receptor subtypes. J Physiol. 1995;485(Pt 2):403–18. doi: 10.1113/jphysiol.1995.sp020738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CB, Chen Y, Liu X, Tang X, Lee CW, Mei L, et al. PIKE-mediated PI3-kinase activity is required for AMPA receptor surface expression. EMBO J. 2011a doi: 10.1038/emboj.2011.281. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CB, Chen Y, Liu X, Tang X, Lee CW, Mei L, et al. PIKE-mediated PI3-kinase activity is required for AMPA receptor surface expression. EMBO J. 2011b doi: 10.1038/emboj.2011.281. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CB, Liu X, Ensslin MA, Dillehay DL, Ormandy CJ, Sohn P, et al. PIKE-A is required for prolactin-mediated STAT5a activation in mammary gland development. EMBO J. 2010a;29:956–68. doi: 10.1038/emboj.2009.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CB, Liu X, He K, Qi Q, Jung DY, Kim JK, et al. The association of phosphoinositide 3-kinase enhancer A with hepatic insulin receptor enhances its kinase activity. EMBO Rep. 2011c doi: 10.1038/embor.2011.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CB, Liu X, Jung DY, Jun JY, Luo HR, Kim JK, et al. Deficiency of phosphoinositide 3-kinase enhancer protects mice from diet-induced obesity and insulin resistance. Diabetes. 2010b;59:883–93. doi: 10.2337/db09-1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CB, Liu X, Pradoldej S, Hao C, An J, Yepes M, et al. Phosphoinositide 3-kinase enhancer regulates neuronal dendritogenesis and survival in neocortex. J Neurosci. 2011d;31:8083–92. doi: 10.1523/JNEUROSCI.1129-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CB, Ye K. PIKE GTPase are phosphoinositide-3-kinase enhancers, suppressing programmed cell death. J Cell Mol Med. 2007;11:39–53. doi: 10.1111/j.1582-4934.2007.00014.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croll SD, Chesnutt CR, Greene NA, Lindsay RM, Wiegand SJ. Peptide immunoreactivity in aged rat cortex and hippocampus as a function of memory and BDNF infusion. Pharmacol Biochem Behav. 1999;64:625–35. doi: 10.1016/s0091-3057(99)00122-7. [DOI] [PubMed] [Google Scholar]
- Dong XX, Wang Y, Qin ZH. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin. 2009;30:379–87. doi: 10.1038/aps.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, et al. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–5. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- Ferkany JW, Zaczek R, Coyle JT. The mechanism of kainic acid neurotoxicity. Nature. 1984;308:561–2. doi: 10.1038/308561a0. [DOI] [PubMed] [Google Scholar]
- Filipkowski RK, Hetman M, Kaminska B, Kaczmarek L. DNA fragmentation in rat brain after intraperitoneal administration of kainate. Neuroreport. 1994;5:1538–40. doi: 10.1097/00001756-199407000-00032. [DOI] [PubMed] [Google Scholar]
- Gratacos E, Perez-Navarro E, Tolosa E, Arenas E, Alberch J. Neuroprotection of striatal neurons against kainate excitotoxicity by neurotrophins and GDNF family members. J Neurochem. 2001;78:1287–96. doi: 10.1046/j.1471-4159.2001.00538.x. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT. The role of glutamate in neurotransmission and in neurologic disease. Arch Neurol. 1986;43:1058–63. doi: 10.1001/archneur.1986.00520100062016. [DOI] [PubMed] [Google Scholar]
- He XP, Kotloski R, Nef S, Luikart BW, Parada LF, McNamara JO. Conditional deletion of TrkB but not BDNF prevents epileptogenesis in the kindling model. Neuron. 2004;43:31–42. doi: 10.1016/j.neuron.2004.06.019. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Hartley M, Heinemann S. Ca2+ permeability of KA-AMPA--gated glutamate receptor channels depends on subunit composition. Science. 1991;252:851–3. doi: 10.1126/science.1709304. [DOI] [PubMed] [Google Scholar]
- Iino M, Mochizuki S, Ozawa S. Relationship between calcium permeability and rectification properties of AMPA receptors in cultured rat hippocampal neurons. Neurosci Lett. 1994;173:14–6. doi: 10.1016/0304-3940(94)90139-2. [DOI] [PubMed] [Google Scholar]
- Jia Z, Agopyan N, Miu P, Xiong Z, Henderson J, Gerlai R, et al. Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron. 1996;17:945–56. doi: 10.1016/s0896-6273(00)80225-1. [DOI] [PubMed] [Google Scholar]
- Jonas P, Racca C, Sakmann B, Seeburg PH, Monyer H. Differences in Ca2+ permeability of AMPA-type glutamate receptor channels in neocortical neurons caused by differential GluR-B subunit expression. Neuron. 1994;12:1281–9. doi: 10.1016/0896-6273(94)90444-8. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Han Y. Insulin inhibits AMPA-induced neuronal damage via stimulation of protein kinase B (Akt) J Neural Transm. 2005;112:179–91. doi: 10.1007/s00702-004-0163-6. [DOI] [PubMed] [Google Scholar]
- Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010;460:525–42. doi: 10.1007/s00424-010-0809-1. [DOI] [PubMed] [Google Scholar]
- Liu Z, Jang SW, Liu X, Cheng D, Peng J, Yepes M, et al. Neuroprotective actions of PIKE-L by inhibition of SET proteolytic degradation by asparagine endopeptidase. Mol Cell. 2008;29:665–78. doi: 10.1016/j.molcel.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin MS, Tang B, Papale LA, Yu FH, Catterall WA, Escayg A. The voltage-gated sodium channel Scn8a is a genetic modifier of severe myoclonic epilepsy of infancy. Hum Mol Genet. 2007;16:2892–9. doi: 10.1093/hmg/ddm248. [DOI] [PubMed] [Google Scholar]
- Matsumoto K, Graf R, Rosner G, Taguchi J, Heiss WD. Elevation of neuroactive substances in the cortex of cats during prolonged focal ischemia. J Cereb Blood Flow Metab. 1993;13:586–94. doi: 10.1038/jcbfm.1993.76. [DOI] [PubMed] [Google Scholar]
- Murphy SN, Thayer SA, Miller RJ. The effects of excitatory amino acids on intracellular calcium in single mouse striatal neurons in vitro. J Neurosci. 1987;7:4145–58. doi: 10.1523/JNEUROSCI.07-12-04145.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozawa S, Iino M, Abe M. Excitatory synapse in the rat hippocampus in tissue culture and effects of aniracetam. Neurosci Res. 1991;12:72–82. doi: 10.1016/0168-0102(91)90101-4. [DOI] [PubMed] [Google Scholar]
- Robinson JH, Deadwyler SA. Kainic acid produces depolarization of CA3 pyramidal cells in the vitro hippocampal slice. Brain Res. 1981;221:117–27. doi: 10.1016/0006-8993(81)91067-2. [DOI] [PubMed] [Google Scholar]
- Rong R, Ahn JY, Huang H, Nagata E, Kalman D, Kapp JA, et al. PI3 kinase enhancer-Homer complex couples mGluRI to PI3 kinase, preventing neuronal apoptosis. Nat Neurosci. 2003;6:1153–61. doi: 10.1038/nn1134. [DOI] [PubMed] [Google Scholar]
- Ryu BR, Ko HW, Jou I, Noh JS, Gwag BJ. Phosphatidylinositol 3-kinase-mediated regulation of neuronal apoptosis and necrosis by insulin and IGF-I. J Neurobiol. 1999;39:536–46. [PubMed] [Google Scholar]
- Sans N, Vissel B, Petralia RS, Wang YX, Chang K, Royle GA, et al. Aberrant formation of glutamate receptor complexes in hippocampal neurons of mice lacking the GluR2 AMPA receptor subunit. J Neurosci. 2003;23:9367–73. doi: 10.1523/JNEUROSCI.23-28-09367.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sather W, Dieudonne S, MacDonald JF, Ascher P. Activation and desensitization of N-methyl-D-aspartate receptors in nucleated outside-out patches from mouse neurones. J Physiol. 1992;450:643–72. doi: 10.1113/jphysiol.1992.sp019148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonian NA, Getz RL, Leveque JC, Konradi C, Coyle JT. Kainic acid induces apoptosis in neurons. Neuroscience. 1996;75:1047–55. doi: 10.1016/0306-4522(96)00326-0. [DOI] [PubMed] [Google Scholar]
- Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium. 2010;47:122–9. doi: 10.1016/j.ceca.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Tang X, Jang SW, Okada M, Chan CB, Feng Y, Liu Y, et al. Netrin-1 mediates neuronal survival through PIKE-L interaction with the dependence receptor UNC5B. Nat Cell Biol. 2008;10:698–706. doi: 10.1038/ncb1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Ye K. Pike tyrosine phosphorylation regulates its apoptotic cleavage during programmed cell death. Adv Enzyme Regul. 2006;46:289–300. doi: 10.1016/j.advenzreg.2006.01.017. [DOI] [PubMed] [Google Scholar]
- Uysal H, Kuli P, Caglar S, Inan LE, Akarsu ES, Palaoglu O, et al. Antiseizure activity of insulin: insulin inhibits pentylenetetrazole, penicillin and kainic acid-induced seizures in rats. Epilepsy Res. 1996;25:185–90. doi: 10.1016/s0920-1211(96)00078-2. [DOI] [PubMed] [Google Scholar]
- Wang LY, MacDonald JF. Modulation by magnesium of the affinity of NMDA receptors for glycine in murine hippocampal neurones. J Physiol. 1995;486(Pt 1):83–95. doi: 10.1113/jphysiol.1995.sp020792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Qin ZH. Molecular and cellular mechanisms of excitotoxic neuronal death. Apoptosis. 2010;15:1382–402. doi: 10.1007/s10495-010-0481-0. [DOI] [PubMed] [Google Scholar]
- Woo RS, Li XM, Tao Y, Carpenter-Hyland E, Huang YZ, Weber J, et al. Neuregulin-1 enhances depolarization-induced GABA release. Neuron. 2007;54:599–610. doi: 10.1016/j.neuron.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Yamaguchi A, Tamatani M, Matsuzaki H, Namikawa K, Kiyama H, Vitek MP, et al. Akt activation protects hippocampal neurons from apoptosis by inhibiting transcriptional activity of p53. J Biol Chem. 2001;276:5256–64. doi: 10.1074/jbc.M008552200. [DOI] [PubMed] [Google Scholar]
- Ye K, Hurt KJ, Wu FY, Fang M, Luo HR, Hong JJ, et al. Pike. A nuclear gtpase that enhances PI3kinase activity and is regulated by protein 4.1N. Cell. 2000;103:919–30. doi: 10.1016/s0092-8674(00)00195-1. [DOI] [PubMed] [Google Scholar]
- Zaczek R, Nelson M, Coyle JT. Kainic acid neurotoxicity and seizures. Neuropharmacology. 1981;20:183–9. doi: 10.1016/0028-3908(81)90202-1. [DOI] [PubMed] [Google Scholar]
- Zhang J, Zhang D, McQuade JS, Behbehani M, Tsien JZ, Xu M. c-fos regulates neuronal excitability and survival. Nat Genet. 2002;30:416–20. doi: 10.1038/ng859. [DOI] [PubMed] [Google Scholar]