

Abstract
Objective
To identify the genetic and epigenetic defects in patients presenting with a facioscapulohumeral (FSHD) clinical phenotype without D4Z4 contractions on chromosome 4q35 tested by linear gel electrophoresis (LGE) and Southern blot analysis.
Design and patients
We studied 16 patients displaying an FSHD-like phenotype, with normal cardiovascular and respiratory function, a myopathic pattern on EMG, and a muscle biopsy being normal or displaying only mild and a specific dystrophic changes. We sequenced the genes calpain 3 (CAPN3), valosin containing protein (VCP) and four and a half LIM domains protein 1 (FHL1) and we analyzed the D4Z4 repeat arrays by extensive genotyping and DNA methylation analysis.
Results
We identified one patient carrying a complex rearrangement in the FSHD locus that masked the D4Z4 contraction associated with FSHD1 in standard genetic testing, one patient with somatic mosaicism for the D4Z4 4q35 contraction, six patients that were diagnosed with FSHD2, four patients with CAPN3 mutations, two patients with a VCP mutation, No mutations were detected in FHL1, and in two patients we could not identify the genetic defect.
Conclusions
In patients presenting an FSHD-like clinical phenotype with a negative molecular testing for FSHD consider: 1) detailed genetic testing including D4Z4 contraction of permissive hybrid D4Z4 repeat arrays, p13E-11 probe deletions, D4Z4 hypomethylation in absence of repeat contraction as observed in FSHD2, 2) mutations in CAPN3 even in the absence of protein deficiency on western blot analysis 3) VCP mutations even in the absence cognitive impairment, Paget disease and typical inclusion in muscle biopsy.
INTRODUCTION
Autosomal dominant facioscapulohumeral muscular dystrophy (FSHD) is characterized by facial and shoulder girdle muscle weakness, spreading, with the progression of the disease, to anterior foreleg, abdominal and pelvic girdle muscles.
Facial involvement frequently includes orbicularis oculi and oris muscles resulting in impaired palpebral occlusion and transverse smile, determining the typical facial appearance of these patients[1]. Asymmetry of muscle involvement is considered as typical feature, but is not always present[2].
The genetic lesions in the majority of FSHD cases are linked to the subtelomeric region of chromosome 4q (FSHD1)[3]. This region contains a polymorphic repeat array consisting of 3.3 kb repeated D4Z4 units which can vary in the general population between 11–100 units. In FSHD1 patients, at least one 4q35 residing D4Z4 repeat array is contracted to 1–10 units. Standard molecular diagnosis is based on Southern blot analysis of restriction enzyme-digested DNA fragments after linear gel electrophoresis (LGE)[4] using the probe p13E-11, which hybridizes proximally to the D4Z4 region [3]. This technique allows for a correct molecular diagnosis in most of the patients displaying classical clinical picture of FSHD; nevertheless, in a limited number of patients, it yields false positive or false negative results [5]. Indeed, two distinct 4q chromosomes (4qA and 4qB) were identified, differing essentially for the presence of an 0,3 kb pLAM fragment and an 6.2kb beta satellite DNA fragment immediately distal to the D4Z4 locus on 4qA chromosomes. Only D4Z4 repeat contractions on 4qA alleles are pathogenic[6, 7]. In addition, as a result of an ancient duplication, the subtelomere of chromosome 10q also contains a repeat array highly homologous to 4q D4Z4, but repeat contractions on this chromosome are not associated with FSHD[4]. The pLAM fragment contains the 3′ untranslated region and the polyadenylation (poly(A)) signal of the DUX4 gene which is located in each D4Z4 unit[8, 9]. Contractions of D4Z4 in FSHD1 lead to local chromatin relaxation, with loss of DNA methylation, and transcriptional derepression of the DUX4 gene[9–12].
Multiple haplotypes of chromosome 4q have been distinguished on the basis of sequence variations in the FSHD locus, yet contractions in only few of these cause FSHD. More specifically, contractions of D4Z4 on 4A166 chromosomes appear to be non-pathogenic, or at least much less pathogenic, as evidenced by the identification of multiple healthy family members in two Dutch families carrying contracted D4Z4 repeat arrays on a 4A166 chromosome[13]. A detailed description of the structure of these alleles has been reported elsewhere[9, 13, 14].
Consequently, to avoid false positives, exact determination of the genetic background of chromosome 4q is necessary to confirm FSHD, in sporadic cases and in patients presenting with an atypical clinical phenotypes[5, 13].
Some FSHD1 patients may harbor a contraction of repeats with non-canonical D4Z4 composition consisting of both 4q-type and 10q-type D4Z4 units[14]. These arrays may appear as chromosome 10q-derived repeats and complicate FSHD molecular diagnosis generating false negatives results[15]. Alternatively, patients may carry a proximal deletion including the p13E-11 probe region that cannot be detected by the standard approach [16]. A specific subgroup of FSHD patients present with chromatin relaxation and DNA hypomethylation on non-contracted D4Z4 repeat arrays in the presence of at least one permissive chromosome. These patients have been referred to as FSHD2 patients and no causative mutation in a specific gene has been found yet[17–19].
Mutations in other genes may also mimic FSHD. For example, calpain 3 (CAPN3) mutations have been associated with autosomal recessive limb girdle muscular dystrophy type 2A (LGMD2A), that, in a subgroup of patients, may present with scapulohumeral muscle weakness resembling FSHD, a milder phenotype compared to the classic form, with later onset and no contractures [20]. Diagnosis of LGMD2A is currently based on western blot analysis of a muscle biopsy, but genetically confirmed patients with normal protein expression levels have been reported[21, 22], and, interestingly, some of them displayed a scapulohumeral phenotype[20].
Mutations in valosin containing protein (VCP) have been associated with an autosomal dominant progressive disorder comprising inclusion body myopathy, Paget’s disease of bone and fronto-temporal dementia. VCP patients may display isolated muscular weakness with scapuloperoneal distribution resembling FSHD[23]. Finally, X-linked dominant scapulo-peroneal syndrome has been reported in association with mutations encoding four-and-a-half-LIM protein 1 (FHL1)[24].
PATIENTS AND METHODS
Patients
Inclusion criteria for the study were: an FSHD clinical phenotype defined as the presence of at least three of the following features: 1) evidence of autosomal dominant inheritance; weakness in 2) facial muscles; 3) shoulder girdle muscles; 4) anterior foreleg muscles and 5) asymmetric involvement. In addition, all patients had 1) normal serum or plasma biochemical parameters (with the exclusion of CK) including erythrocyte sedimentation rate, electrolytes, thyroid and parathyroid hormones; 2) normal electrocardiogram, echocardiography and respiratory pulmonary function; 3) EMG with myogenic pattern in affected territories without evidence of myotonic discharges; and 4) a muscle biopsy showing no abnormalities or only mild and unspecific dystrophic changes. Muscle samples were processed using standard procedures[25].
For each patient included in this study, data concerning age, sex, family history, distribution of weakness, and creatine kinase (CK) blood levels were collected (Table 1) after informed consent. None of these patients displayed contractures or involvement of ocular muscles. Clinical data collection, extensive D4Z4 genotyping and D4Z4 methylation analysis were also carried out in all FSHD2 patient family members that were available after informed consent.
Table 1.
Clinical data
| Patient | Molecular findings | Age of onset/age at clinical examination (years) | Sex | Family history | CK values | Facial weakness | Scapular fixator weakness | Humeral muscles weakness | Abdominal muscle weakness | Pelvic girdle weakness | Anterior forelegs weakness | Asymmetry |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| P1 | FSHD1 (Mosaic) | 35/44 | F | Sporadic case | N | No | Yes | Yes | Yes | No | Yes | Yes |
| P2 | FSHD1 (Hybrid allele) | 15/48 | F | Daughter affected | N | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| P3 | FSHD2 | 25/43 | M | Sporadic case | x1 | Yes | Yes | No | No | No | Yes | Yes |
| P4 | FSHD2 | 37/60 | M | Sporadic case | x1 | Yes | Yes | Yes | Yes | No | Yes | No |
| P5 | FSHD2 | 30/45 | M | Sporadic case | x2 | Yes | Yes | Yes | Yes | No | No | Yes |
| P6 | FSHD2 | 32/42 | M | Sporadic case | x2 | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| P7 | FSHD2 | 25/51 | M | Sporadic case | x2 | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| P8 | FSHD2 | 35/41 | F | Sporadic case | x2 | Yes | Yes | Yes | Yes | Yes | Yes | Yes |
| P9 | LGMD2A | 25/42 | F | Sporadic case | x3 | Yes | Yes | Yes | Yes | Yes | Yes | No |
| P10 | LGMD2A | 25/37 | M | Sporadic case | x4 | Yes | Yes | Yes | Yes | Yes | Yes | No |
| P11 | LGMD2A | 40/60 | F | Sporadic case | x2 | Yes | Yes | Yes | No | Yes | Yes | No |
| P12 | LGMD2A | 20/51 | F | Sporadic case | x2 | Yes | Yes | Yes | No | Yes | Yes | No |
| P13 | VCP gene mutation | 30/55 | M | Father affected | x3 | No | Yes | No | Yes | Yes | Yes | Yes |
| P14 | VCP gene mutation | 35/59 | M | ? | x4 | Yes | Yes | Yes | No | Yes | Yes | No |
| P15 | Unknown | 25/46 | M | Sporadic case | x4 | Yes | Yes | Yes | Yes | No | No | Yes |
| P16 | Unknown | 30/52 | M | Sporadic | x6 | Yes | Yes | Yes | No | No | Yes | Yes |
N: Normal CK values (90–200 IU/L); Clinical features corresponding to inclusion criteria are highlighted in bold.
Molecular analyses
Molecular analysis of CAPN3, VCP and FHL1 was performed using reported protocols[24, 26, 27].
Single sequence length polymorphism (SSLP), repeat length and distal variation determination was performed as described[14] (se also the Fields Center for FSHD Research Website http://www.urmc.rochester.edu/fields-center/).
Methylation levels in the proximal D4Z4 repeat units of chromosomes 4q and 10q was performed as reported[10, 19].
RESULTS
Sixteen patients (P1–16) fitted the inclusion criteria described above, ten men and six women. The mean age at clinical evaluation was 48,5±7,2 and the mean age at onset of the disease reported by patients was 29±6,6. Clinical characteristics of the patients are reported in Table 1. Genomic DNA of all patients was subjected to CAPN3, VCP and FHL1 mutation analysis by direct exon sequencing, to extensive D4Z4 genotyping by PFGE analysis, and to DNA methylation studies of the D4Z4 repeat by methylation-sensitive Southern blot analysis. The results of these studies are summarized in Table 2 and Table 3.
Table 2.
Chromosome 4q and 10q D4Z4 genotype and chromosome 4q D4Z4 methylation status
| Patient | Molecular findings | 4q repeat (1) | 4q repeat (2) | 10q repeat (1) | 10q repeat (2) | BsaAI | FseI |
|---|---|---|---|---|---|---|---|
| P1 | FSHD1 Mosaic | 11kb 4A161 (55%)/78kb 4A161 (45%) | 110kb 4A163 | 14kb 10A166 | 26kb 10A166 | 53 | 53 |
| P2 | FSHD1 Hybrid | 11H kb 4A161 | 45kb 4B163 | 25kb 10A166 | 54kb 10A166 | Not possible | Not possible |
| P3 | FSHD2 | 51kb 4A161 | 123kb 4A161 | 46kb 10A176 | 69kb 10A166 | 25 | 10 |
| P4 | FSHD2 | 45kb 4qA161 | 99kb 4B163 | 94kb 10A166 | 141kb 10A166 (75%)/120kb 10A166 (25%) | 32 | 17 |
| P5 | FSHD2 | 65kb 4A161 | 160kb 4A161 | 60kb 10A166 | 220kb 10A166 | 30 | 13 |
| P6 | FSHD2 | 62kb 4B163 | 95kb 4A161 | 23kb 10A166 | 245kb 10A164 | 21 | 12 |
| P7 | FSHD2 | 50kb 4A161 | 90kb 4A166 | 160kb 10A166 | 170kb 10A166 | 23 | 17 |
| P8 | FSHD2 | 45kb 4A161 | 55kb 4B163 | 52kb 10A166 | 55kb 10A166 | 22 | 13 |
| P9 | LGMD1A | 105kb 4B168 | 180kb 4A161 | 93kb 10A166 | 95kb 10A166 | 63 | 66 |
| P10 | LGMD1A | 130Hkb 4A166 | 134kb 4A161 | 21kb 10A166 | 144kb 10A166 | 58 | 63 |
| P11 | LGMD1A | 60kb 4B* | 70kb 4A* | 90kb 10B* | 100kb 10A* | ND | ND |
| P12 | LGMD1A | 80kb 4B* | 100kb 4A* | 125kb 4A* | 130kb 4A* | ND | ND |
| P13 | VCP gene mutation | 78kb 4A161 | 155kb 4A161 | 48kb 4A166 | 125kb 4A166 | 66 | 75 |
| P14 | VCP gene mutation | 45kb 4A161 | 100kb 4A161 | 45kb 10A166 | 45kb 10A166 | 50 | 47 |
| P15 | Unknown | 137kb 4A161 | 137kb 4B168 | 22kb 10A166 | 24kb 10A166 | 51 | 46 |
| P16 | Unknown | 76H kb 4A166H | 105kb 4B163 | 20kb 10A166 | 51kb 10A166 | 52 | 46 |
The number reported on BsaAI and FseI columns indicate the percentage of methylation of the BsaAI and FseI restriction sites in the most proximal D4Z4 unit of the repeat array.
ND: not done.
No 4q and 10q haplotype determination available
Table 3.
Molecular data of patients with CAPN3 or VCP mutations
| Patient | Gene | Nucleotide change | Amino acid change | Reference |
|---|---|---|---|---|
| P9 | CAPN3 | c.1469 G>A | p.R490Q | Richard et al.1995 |
| c.1469 G>A | p.R490Q | Richard et al.1995 | ||
| P10 | CAPN3 | c.1469 G>A | p.R490Q | Richard et al.1995 |
| c.550 del A | p.T184RfsX25 | Richard et al.1995 | ||
| P11 | CAPN3 | c.1319 G>A | p.R440Q | Piluso et al. 2005 |
| c.2362_2363delinsTCATCT | p.R788SfsX14 | Blazquez et al. 2008 | ||
| P12 | CAPN3 | c.2362_2363delinsTCATCT | p.R788SfsX14 | Blazquez et al. 2008 |
| c.2362_2363delinsTCATCT | p.R788SfsX14 | Blazquez et al. 2008 | ||
| P13 | VCP | c.572 G>A | p.R191Q | Stojkovic et al. 2009 |
| P14 | VCP | c.572 G>A | p.R191Q | Stojkovic et al. 2009 |
Patient 1: Somatic mosaicism for D4Z4 contraction: FSHD1
Patient P1 is a sporadic case and was found by PFGE analysis to be mosaic for a contracted D4Z4 repeat on a 4A161 allele of 11kb in ~55% of her blood cells, corresponding to 1 D4Z4 repeat unit. Reported disease onset was at age 35 years: she showed a typical FSHD clinical phenotype including asymmetric facial weakness, shoulder girdle muscle involvement, abdominal and anterior foreleg muscle weakness. The milder phenotype displayed by P1, still ambulant at the age of 44, which was not expected based on the small residual repeat size (1 repeat unit), may be explained by the mosaic nature of the contraction.
Patient 2: Hybrid D4Z4 repeat: FSHD1
This patient, a 48 years-old woman, presented at age 5 years with a typical FSHD phenotype including facial, scapulohumeral and anterior forelegs asymmetric muscular weakness. At age 45 abdominal and pelvic girdle muscle were also involved.
Her 27 year old daughter was also affected presenting mild facial and shoulder girdle involvement, right dorsiflexor muscle weakness. She carried the same genetic defect, while the two younger twin sisters did not carry the pathogenic allele and were asymptomatic. Facial weakness in both patients consisted in asymmetric involvement of orbicularis oculi and orbicularis oris (Supplementary figure 1).
Genotype studies of this family have been described in a previous report (Family 3 of Lemmers et al.[9]). Briefly, the pathogenic chromosome is the result of a meiotic exchange between chromosomes 10q and 4q creating a contracted hybrid D4Z4 repeat array on a permissive chromosomal background.
Interestingly, all carriers of this contracted hybrid D4Z4 repeat are less severely affected than what was expected based on the residual number of D4Z4 units, which is one. We suggest that the composition of the array, which mainly consists of non-permissive 10q sequences, may play an important role in determining the milder phenotype. On the other hand, less severe FSHD phenotypes have been often described in females [28].
Patients 3, 4, 5, 6, 7 and 8: FSHD2 patients
These patients were all sporadic cases, displaying a typical FSHD phenotype. Interestingly, most of them complained of pain and fatigue, symptoms frequently reported by FSHD1 patients[29], and one of them (P3) had sensorineural hearing loss, a feature described in 25–65% of FSHD1 patients[30]. CK values were normal to twofold elevated, and muscle biopsy showed only mild dystrophic changes. All these patients carried a 4A161 allele of 45–95kb and marked hypomethylation of the D4Z4 locus, typical of FSHD2 (Table 2).
Pedigrees of P3, P4, P6, P7, P8 are reported in Supplementary Figure 2, while the pedigree of P5 has already been reported and discussed in detail (Family 6 in de Greef et al.[19]). Family members of these patients available have also been clinically characterized and D4Z4 genotyping and methylation studies were performed.
The mothers of P3, P6, and P7 and the father of P8 carried the same 4A161 allele as their affected offspring, but they were not hypomethylated and were asymptomatic (Supplementary Figure 1A, C, D, E). Interestingly, the 26 years-old daughter of P4 displayed significant hypomethylation but she had D4Z4 repeats on a non-permissive chromosome 4 background and is asymptomatic (Supplementary Figure 1B).
Patients 9 to 12: LGMD2A
Patients P9–P12 were sporadic cases, presenting with symmetric facial, shoulder girdle, pelvic, and anterior forelegs muscles weakness. All carried pathogenic mutations in CAPN3 (Table 3)[31–33]. In these four patients, western blots for calpain 3 protein in muscle were normal and muscle biopsies were characterized by mild dystrophic features.
Mean CK values were three and four times the upper limit of normal values in P9 and P10, which is higher then the mean CK values classically found in FSHD patients, while P11 and P12 had twice the upper limit of normal CK values. Facial involvement was present in all patients and consisted essentially of a hypotonic and hypokinetic face without involvement of orbicularis oculi muscles. Indeed, patients were unable to whistle, to blow a candle, but they could properly close their eyes and mimic a kiss.
Patients 13 and 14: VCP mutations
Patient P13 had a clinical evidence of autosomal dominant inheritance, since the father was diagnosed with a myopathy of unknown origin. He displayed a scapuloperoneal phenotype with a certain degree of asymmetry in the legs. Facial involvement was not present and CK values were three times the upper limit of normal values. The muscle biopsy was mildly dystrophic without inclusion bodies.
P14 was adopted and displayed facial involvement, shoulder and pelvic girdle weakness and anterior foreleg symmetric muscle weakness. CK values were four-fold increased. Interestingly facial involvement presented with difficulties in closing the eyes and protruding the lips, as is typically seen in FSHD patients.
The muscle biopsies of P13 and P14 showed mild dystrophic changes and no inclusion bodies. Both carried a mutation in the VCP gene reported in Table 3, previously described as pathogenic[23]. After the molecular diagnosis, they underwent an extensive neuropsychological evaluation that was normal in P14, while revealing a mild dysexecutive syndrome in P13, characterized by low scores in short term memory scales. Brain MRI, serum alkaline phosphatase activity (APA), and skeletal X ray (pelvis, hip, femur, humerus, vertebral bodies and skull) were normal in both patients.
Patients 15 and 16
P15 and P16 are both sporadic cases with a typical FSHD phenotype, including facial weakness, and no peculiar features in EMG or in muscle biopsy that may suggest a specific form of muscular dystrophy. CK values were respectively of four and six times the upper limit of normal values. No mutation was found in all genes sequenced and the genotype and methylation study of the D4Z4 locus did not show evidence for FSHD or FSHD2. This may suggest the presence of a mutation in a gene mimicking an FSHD phenotype, which has not yet been identified.
DISCUSSION
The Clinical diagnosis of FSHD is frequently not an issue for a neurologist expert in neuromuscular disorders due to the typical pattern of muscle involvement. Nevertheless, the classical genetic approach based on identification of 4q35 D4Z4 contracted allele after EcoRI and EcoRI/BlnI digestion, LGE, Southern blotting and p13E-11 hybridization fails to offer the molecular confirmation in about 5–10% of cases[14].
The present study was conducted to guide the clinician in the choice of additional genetic or epigenetic tests to confirm the clinical diagnosis, since several conditions can partially mimic the clinical phenotype of FSHD.
In clinical practice, when standard genetic testing does not confirm the clinical suspicion of FSHD1, EMG is required to confirm the myogenic pattern of muscle weakness and to rule out neurogenic involvement. For example, patients harboring deletions of PMP22[34], or mutations in the TRPV4 gene [35], both transmitted as autosomal dominant traits, may present with a scapuloperoneal distribution of muscular involvement of neurogenic origin.
Other inherited myopathies may present with a clinical phenotype resembling FSHD: some limb girdle muscular dystrophies (LGMD) [36], glycogenosis type II (Pompe disease) [23] and glycogenosis type V (McArdle’s disease) [37], myosin storage myopathies [38], desminopathies [39], other myofibrillar myopathies[24, 40], and mitochondrial myopathies [41].
In most of the cases, the age of onset, the disease progression rate, the pattern of inheritance, the presence of respiratory muscle or cardiac involvement, the absence of facial weakness, the presence of contractures or ocular involvement, may aid the clinician in orientating the diagnosis toward these diseases.
In all these cases, muscle biopsy is mandatory to confirm the clinical suspicion. Indeed, histological, immunohistochemical and western blot studies on muscle samples are generally adequate to distinguish among different LGMDs due to lack of sarcolemmal protein, or to identify storage, vacuolar, mitochondrial, congenital or myofibrillar myopathies.
If EMG and muscle biopsy studies are not conclusive, the use of detailed PFGE based D4Z4 genotyping on agarose-embedded DNA plugs[42] can be used to identify FSHD1 patients that could have been missed by the less detailed standard molecular diagnosis.
In our study we found two of such a patients among the 16 patients included in the study, P1 and P2. P1 was mosaic for the D4Z4 contraction. This condition results from a mitotic rearrangement of D4Z4 probably in early embryogenesis, is quite frequent in de novo cases[43, 44] and mosaicism often goes undetected[45]. Their clinical phenotype appears to be milder compared to individuals carrying the same residual repeat size in all of their cells. In P2, the pathogenic hybrid allele was identified by PFGE and the distal D4Z4 fragment was analyzed by direct sequencing to confirm the pathogenicity[9].
In all cases without a diagnosis, and with a permissive chromosome, FSHD2 should be suspected [10, 17–19]. Indeed, FSHD2 patients display: 1) a clinical phenotype identical to FSHD1 patients; 2) normal-sized repeats with at least one permissive chromosome (contrary to FSHD1 where the repeat is contracted on a permissive chromosome) and 3) reduced DNA methylation of the D4Z4 repeats on chromosomes 4q and 10q, as measured on the methylation-sensitive restriction sites FseI and BsaAI.
In our series six patients (P3 – P8) were found to meet these criteria. All of them were sporadic cases, and we studied the segregation of the permissive allele and the DNA methylation in all members available for the six families confirming that the FSHD clinical phenotype appears only in patients displaying hypomethylation on a permissive 4A161 background. Finally, when EMG studies, muscle biopsy studies and extensive D4Z4 repeat arrays genotyping and DNA methylation analysis are negative, we suggest reconsidering the possibility of a FSHD phenocopy.
In our series of patients, six out of the sixteen patients (P9 to P14) carried mutations in genes not related to FSHD, i.e. CAPN3 and VCP. These genetic defects had gone unnoticed by standard histological, immunohistochemical and Western blot studies on muscle biopsy. Actually, approximately 20% of CAPN3 mutations do not cause a reduction of the protein steady state levels, but affect its autocatalytic activity. A specific test is available to measure this activity in muscle, avoiding these misdiagnoses[22]. Unfortunately, this test is done only in a restricted number of laboratories, and in the case of the present study, was not performed because no muscle sample was left. Furthermore, it has been reported that patients harboring VCP mutation may display only minor and non-specific histopathological changes on muscle biopsies [14].
Accordingly, in P13 and P14, carrying a mutation in the VCP gene, no sign of Paget’s disease and no characteristic rimmed vacuoles on muscle biopsy were found. Nevertheless, after the molecular diagnosis, P14 underwent to a neuropsychological test demonstrating the presence of a mild dysexecutive syndrome. APA measurement, standard bone X ray (pelvis, hip, femur, humerus, vertebral bodies and skull) and adapted neuropsychological tests may help in detecting asymptomatic Paget’s disease or fronto-temporal dementia that are usually associated with a inclusion body myopathy and mutations in the VCP gene[27].
Interestingly, CK levels in four out of the six patients displaying a phenotype resembling FSHD but carrying mutations in other genes, were more elevated then in classical FSHD1 patients, and five over six patients displayed no asymmetric muscular involvement. Concerning facial weakness, P9, P10, P11 and P12 (LGMD2A) and P14 (VCP) showed transversal smile and difficulty to blow, as P4 and P6 (FSHD2) while P2 (FSHD1), P3, P5, P7 and P8 (FSHD2) showed also weakness of orbicularis oculi (Supplementary Figure 1). Altogether, these clinical features, if present, may prompt clinicians to sequence CAPN3 and VCP gene, before undergoing more complex epigenetic studies.
Mutations in FHL1 have been described to cause a scapuloperoneal phenotype mimicking FSHD, inherited in an X dominant fashion[24]. We could not find FHL1 mutations in our patients, possibly because they are less frequently associated with an FSHD-like phenotype. Nevertheless we cannot exclude that mutation in this gene may be associated to FSHD-like phenotype due to the small number of patients included in the present study.
In two out of sixteen patients (P15, P16) included in this study, no genetic or epigenetic defect could be found. In these two sporadic patients, CK values were elevated four to six times the upper limit of normal values, which may suggest the presence of other loci determining an FSHD-like phenotype. In the absence of an identified genetic or epigenetic defect, FSHD-like patients should undergo a complete clinical workout in order to identify additional signs that may be useful to orientate the diagnosis. Annual follow-up is also recommended to monitor the pattern of muscle weakness progression and, possibly, the appearance of signs of symptoms evocative of a specific muscular dystrophy. The possibility of a second muscle biopsy must be carefully discussed with the patient in case of worsening of clinical conditions.
In conclusion, in patients with a FSHD phenotype and no D4Z4 contraction on chromosome 4q35 detected by standard techniques it is important to consider these possibilities: 1) a false negative diagnosis; 2) FSHD2; and 3) a mutation in another gene resembling FSHD (especially if CK levels are consistently elevated, and there is evidence of symmetric muscle involvement); (see the Diagnostic Flowchart in Figure 1). Extensive D4Z4 genotyping by PFGE analysis and methylation studies of the D4Z4 repeat, which are currently available only in selected research laboratories, are required to diagnose the first two conditions.
Figure 1.
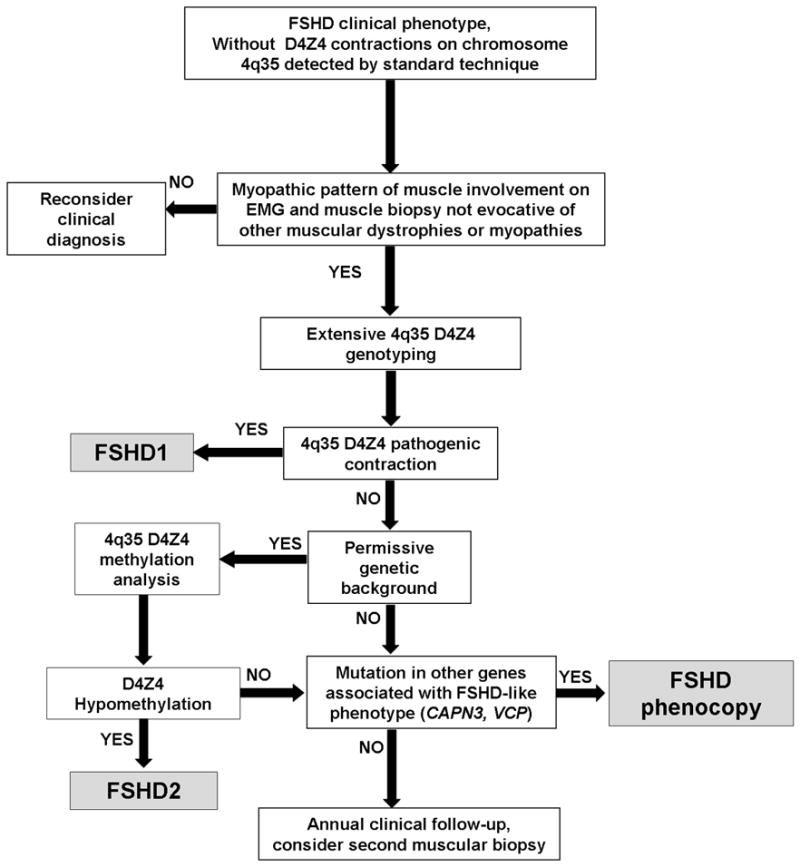
Diagnostic flowchart for patients with FSHD-like phenotype.
Novel diagnostic approaches are currently being developed to simplify the diagnosis of FSHD and of other muscle disorders. Molecular combing is a promising new strategy which is apparently simpler and faster than classical Southern blot, and allows allele determination and the detection of mosaics and of complex alleles in a single step procedure [46]. Furthermore, next-generation sequencing technology will allow to study large panels of genes for a fraction of the cost (and of the time) required by classic Sanger sequencing.
Supplementary Material
Acknowledgments
We acknowledge the patients and their families, the French Association against Myopathies (AFM) for its help in referring patients. Pilar Camaño and Adolfo Lopez de Munain Arregui are funded by the Centro Investigación Biomédica en Red para Enfermedades Neurodegenerativas (CIBERNED), the Basque Government (Fellowship grant, No 2008111011), the Instituto Carlos III, ILUNDAIN Fundazioa, the Prinses Beatrix Fonds, the Fields Center for FSHD and Neuromuscular Diseases and the National Institutes of Health (P01NS069539, AR059966) to take care of clinical evaluation and molecular studies (VCP and CAPN3 gene).
References
- 1.Padberg G. Facioscapulohumeral muscular dystrophy: a clinician’s experience. In: Upadhyaya M, Cooper DN, editors. Facioscapulohumeral muscular dystrophy. Clinical medicine and molecular cell biology. Oxon, United Kingdom: Garland Science/BIOS Scientific Publishers; 2004. [Google Scholar]
- 2.Padberg G. Facioscapulohumeral Disease. Leiden: Leiden University; 1982. [Google Scholar]
- 3.Wijmenga C, Hewitt JE, Sandkuijl LA, et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet. 1992;2:26–30. doi: 10.1038/ng0992-26. [DOI] [PubMed] [Google Scholar]
- 4.Deidda G, Cacurri S, Piazzo N, et al. Direct detection of 4q35 rearrangements implicated in facioscapulohumeral muscular dystrophy (FSHD) J Med Genet. 1996;33:361–5. doi: 10.1136/jmg.33.5.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sacconi S, Salviati L, Bourget I, et al. Diagnostic challenges in facioscapulohumeral muscular dystrophy. Neurology. 2006;67:1464–6. doi: 10.1212/01.wnl.0000240071.62540.6f. [DOI] [PubMed] [Google Scholar]
- 6.Lemmers RJ, de Kievit P, Sandkuijl L, et al. Facioscapulohumeral muscular dystrophy is uniquely associated with one of the two variants of the 4q subtelomere. Nat Genet. 2002;32:235–6. doi: 10.1038/ng999. [DOI] [PubMed] [Google Scholar]
- 7.Lemmers RJ, Wohlgemuth M, Frants RR, et al. Contractions of D4Z4 on 4qB subtelomeres do not cause facioscapulohumeral muscular dystrophy. Am J Hum Genet. 2004;75:1124–30. doi: 10.1086/426035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dixit M, Ansseau E, Tassin A, et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc Natl Acad Sci U S A. 2007;104:18157–62. doi: 10.1073/pnas.0708659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lemmers RJ, van der Vliet PJ, Klooster R, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science. 2010;329:1650–3. doi: 10.1126/science.1189044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Overveld PG, Lemmers RJ, Sandkuijl LA, et al. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat Genet. 2003;35:315–7. doi: 10.1038/ng1262. [DOI] [PubMed] [Google Scholar]
- 11.Snider L, Geng LN, Lemmers RJ, et al. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet. 2010;6:e1001181. doi: 10.1371/journal.pgen.1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pearson CE. FSHD: a repeat contraction disease finally ready to expand (our understanding of its pathogenesis) PLoS Genet. 2010;6:e1001180. doi: 10.1371/journal.pgen.1001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lemmers RJ, Wohlgemuth M, van der Gaag KJ, et al. Specific sequence variations within the 4q35 region are associated with facioscapulohumeral muscular dystrophy. Am J Hum Genet. 2007;81:884–94. doi: 10.1086/521986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lemmers RJ, van der Vliet PJ, van der Gaag KJ, et al. Worldwide population analysis of the 4q and 10q subtelomeres identifies only four discrete interchromosomal sequence transfers in human evolution. Am J Hum Genet. 2010;86:364–77. doi: 10.1016/j.ajhg.2010.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Buzhov BT, Lemmers RJ, Tournev I, et al. Genetic confirmation of facioscapulohumeral muscular dystrophy in a case with complex D4Z4 rearrangments. Hum Genet. 2005;116:262–6. doi: 10.1007/s00439-004-1237-0. [DOI] [PubMed] [Google Scholar]
- 16.Lemmers RJ, Osborn M, Haaf T, et al. D4F104S1 deletion in facioscapulohumeral muscular dystrophy: phenotype, size, and detection. Neurology. 2003;61:178–83. doi: 10.1212/01.wnl.0000078889.51444.81. [DOI] [PubMed] [Google Scholar]
- 17.de Greef JC, Wohlgemuth M, Chan OA, et al. Hypomethylation is restricted to the D4Z4 repeat array in phenotypic FSHD. Neurology. 2007;69:1018–26. doi: 10.1212/01.wnl.0000271391.44352.fe. [DOI] [PubMed] [Google Scholar]
- 18.de Greef JC, Lemmers RJ, van Engelen BG, et al. Common epigenetic changes of D4Z4 in contraction-dependent and contraction-independent FSHD. Hum Mutat. 2009;30:1449–59. doi: 10.1002/humu.21091. [DOI] [PubMed] [Google Scholar]
- 19.de Greef JC, Lemmers RJ, Camano P, et al. Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology. 2010;75:1548–54. doi: 10.1212/WNL.0b013e3181f96175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Angelini C, Nardetto L, Borsato C, et al. The clinical course of calpainopathy (LGMD2A) and dysferlinopathy (LGMD2B) Neurol Res. 2010;32:41–6. doi: 10.1179/174313209X380847. [DOI] [PubMed] [Google Scholar]
- 21.Fanin M, Nascimbeni AC, Fulizio L, et al. Loss of calpain-3 autocatalytic activity in LGMD2A patients with normal protein expression. Am J Pathol. 2003;163:1929–36. doi: 10.1016/S0002-9440(10)63551-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fanin M, Nascimbeni AC, Tasca E, et al. How to tackle the diagnosis of limb-girdle muscular dystrophy 2A. Eur J Hum Genet. 2009;17:598–603. doi: 10.1038/ejhg.2008.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stojkovic T, Hammouda el H, Richard P, et al. Clinical outcome in 19 French and Spanish patients with valosin-containing protein myopathy associated with Paget’s disease of bone and frontotemporal dementia. Neuromuscul Disord. 2009;19:316–23. doi: 10.1016/j.nmd.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 24.Quinzii CM, Vu TH, Min KC, et al. X-linked dominant scapuloperoneal myopathy is due to a mutation in the gene encoding four-and-a-half-LIM protein 1. Am J Hum Genet. 2008;82:208–13. doi: 10.1016/j.ajhg.2007.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sacconi S, Trevisson E, Salviati L, et al. Coenzyme Q10 is frequently reduced in muscle of patients with mitochondrial myopathy. Neuromuscul Disord. 2010;20:44–8. doi: 10.1016/j.nmd.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 26.Richard I, Brenguier L, Dincer P, et al. Multiple independent molecular etiology for limb-girdle muscular dystrophy type 2A patients from various geographical origins. Am J Hum Genet. 1997;60:1128–38. [PMC free article] [PubMed] [Google Scholar]
- 27.Watts GD, Wymer J, Kovach MJ, et al. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377–81. doi: 10.1038/ng1332. [DOI] [PubMed] [Google Scholar]
- 28.Tawil R, Van Der Maarel SM. Facioscapulohumeral muscular dystrophy. Muscle Nerve. 2006;34:1–15. doi: 10.1002/mus.20522. [DOI] [PubMed] [Google Scholar]
- 29.Kalkman JS, Schillings ML, Zwarts MJ, et al. The development of a model of fatigue in neuromuscular disorders: a longitudinal study. J Psychosom Res. 2007;62:571–9. doi: 10.1016/j.jpsychores.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 30.Trevisan CP, Pastorello E, Ermani M, et al. Facioscapulohumeral muscular dystrophy: a multicenter study on hearing function. Audiol Neurootol. 2008;13:1–6. doi: 10.1159/000107431. [DOI] [PubMed] [Google Scholar]
- 31.Blazquez L, Azpitarte M, Saenz A, et al. Characterization of novel CAPN3 isoforms in white blood cells: an alternative approach for limb-girdle muscular dystrophy 2A diagnosis. Neurogenetics. 2008;9:173–82. doi: 10.1007/s10048-008-0129-1. [DOI] [PubMed] [Google Scholar]
- 32.Piluso G, Politano L, Aurino S, et al. Extensive scanning of the calpain-3 gene broadens the spectrum of LGMD2A phenotypes. J Med Genet. 2005;42:686–93. doi: 10.1136/jmg.2004.028738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Richard I, Broux O, Allamand V, et al. Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell. 1995;81:27–40. doi: 10.1016/0092-8674(95)90368-2. [DOI] [PubMed] [Google Scholar]
- 34.Verma A. Neuropathic scapuloperoneal syndrome (Davidenkow’s syndrome) with chromosome 17p11.2 deletion. Muscle Nerve. 2005;32:668–71. doi: 10.1002/mus.20402. [DOI] [PubMed] [Google Scholar]
- 35.Zimon M, Baets J, Auer-Grumbach M, et al. Dominant mutations in the cation channel gene transient receptor potential vanilloid 4 cause an unusual spectrum of neuropathies. Brain. 2010;133:1798–809. doi: 10.1093/brain/awq109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benedetti S, Menditto I, Degano M, et al. Phenotypic clustering of lamin A/C mutations in neuromuscular patients. Neurology. 2007;69:1285–92. doi: 10.1212/01.wnl.0000261254.87181.80. [DOI] [PubMed] [Google Scholar]
- 37.Nadaj-Pakleza AA, Vincitorio CM, Laforet P, et al. Permanent muscle weakness in McArdle disease. Muscle Nerve. 2009;40:350–7. doi: 10.1002/mus.21351. [DOI] [PubMed] [Google Scholar]
- 38.Muelas N, Hackman P, Luque H, et al. MYH7 gene tail mutation causing myopathic profiles beyond Laing distal myopathy. Neurology. 2010;75:732–41. doi: 10.1212/WNL.0b013e3181eee4d5. [DOI] [PubMed] [Google Scholar]
- 39.Walter MC, Reilich P, Huebner A, et al. Scapuloperoneal syndrome type Kaeser and a wide phenotypic spectrum of adult-onset, dominant myopathies are associated with the desmin mutation R350P. Brain. 2007;130:1485–96. doi: 10.1093/brain/awm039. [DOI] [PubMed] [Google Scholar]
- 40.Selcen D. Myofibrillar myopathies. Neuromuscul Disord. 2011;21:161–71. doi: 10.1016/j.nmd.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rowland LP, Blake DM, Hirano M, et al. Clinical syndromes associated with ragged red fibers. Rev Neurol (Paris) 1991;147:467–73. [PubMed] [Google Scholar]
- 42.Lemmers RJ, van der Maarel SM, van Deutekom JC, et al. Inter- and intrachromosomal sub-telomeric rearrangements on 4q35: implications for facioscapulohumeral muscular dystrophy (FSHD) aetiology and diagnosis. Hum Mol Genet. 1998;7:1207–14. doi: 10.1093/hmg/7.8.1207. [DOI] [PubMed] [Google Scholar]
- 43.Zatz M, Marie SK, Passos-Bueno MR, et al. High proportion of new mutations and possible anticipation in Brazilian facioscapulohumeral muscular dystrophy families. Am J Hum Genet. 1995;56:99–105. [PMC free article] [PubMed] [Google Scholar]
- 44.van der Maarel SM, Deidda G, Lemmers RJ, et al. De novo facioscapulohumeral muscular dystrophy: frequent somatic mosaicism, sex-dependent phenotype, and the role of mitotic transchromosomal repeat interaction between chromosomes 4 and 10. Am J Hum Genet. 2000;66:26–35. doi: 10.1086/302730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lemmers RJ, van der Wielen MJ, Bakker E, et al. Somatic mosaicism in FSHD often goes undetected. Ann Neurol. 2004;55:845–50. doi: 10.1002/ana.20106. [DOI] [PubMed] [Google Scholar]
- 46.Nguyen K, Walrafen P, Bernard R, et al. Molecular combing reveals allelic combinations in facioscapulohumeral dystrophy. Ann Neurol. doi: 10.1002/ana.22513. Published Online First 7 June 2011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.