

Abstract
Background
The PR interval (PR) as measured by the resting, standard 12-lead electrocardiogram (ECG) reflects the duration of atrial/atrioventricular nodal depolarization. Substantial evidence exists for a genetic contribution to PR, including genome-wide association studies that have identified common genetic variants at nine loci influencing PR in populations of European and Asian descent. However, few studies have examined loci associated with PR in African Americans.
Methods and Results
We present results from the largest genome-wide association study to date of PR in 13,415 adults of African descent from ten cohorts. We tested for association between PR (ms) and approximately 2.8 million genotyped and imputed single nucleotide polymorphisms. Imputation was performed using HapMap 2 YRI and CEU panels. Study-specific results, adjusted for global ancestry and clinical correlates of PR, were meta-analyzed using the inverse variance method. Variation in genome-wide test statistic distributions was noted within studies (lambda range: 0.9–1.1), although not after genomic control correction was applied to the overall meta-analysis (lambda: 1.008). In addition to generalizing previously reported associations with MEIS1, SCN5A, ARHGAP24, CAV1, and TBX5 to African American populations at the genome-wide significance level (P<5.0×10−8), we also identified a novel locus: ITGA9, located in a region previously implicated in SCN5A expression. The 3p21 region harboring SCN5A also contained two additional independent secondary signals influencing PR (P<5.0×10−8).
Conclusions
This study demonstrates the ability to map novel loci in African Americans as well as the generalizability of loci associated with PR across populations of African, European and Asian descent.
Keywords: electrocardiography, epidemiology, GWAS, single nucleotide polymorphism genetics, PR interval
Introduction
The PR interval (PR) is an electrocardiographic measurement of atrial conduction spanning the onset of sinus depolarization through the atrioventricular node. PR is a predictor of incident atrial fibrillation,1 a common cardiac arrhythmia,2 and a potent risk factor for pacemaker implantation, heart failure, stroke, and all-cause mortality.1, 3 Substantial evidence exists for a genetic contribution to PR. Family-based studies have estimated the heritability of PR at approximately 34%4, 5 and rare sodium channel mutations associated with atrial cardiac conduction defects have been characterized.6, 7 Recent genome-wide association (GWA) studies performed in populations of European and Asian descent have identified common polymorphisms at nine loci that are associated with variation in PR.5, 8–11 For example, ARHGAP24, CAV1, SCN10A, and TBX5 have been reported in at least two PR GWA studies.
To date, the majority of GWA studies examining PR were performed in populations of European or Asian descent. The exception is a report by Smith and colleagues (2011),12 which generalized four previously described PR loci identified in European and Asian populations (SCN5A, SCN10A, MEIS1, and TBX5) to 6,247 African American participants from four cohorts. However, Smith and colleagues neither detected novel associations nor identified genome-wide significant associations with several previously replicated loci, including ARHGAP24, CAV1 and WNT11.9, 10 It is therefore unclear whether these loci are relevant in African Americans. Additionally, the increased genetic diversity in populations of African descent provides opportunities for the identification of novel variants influencing PR. Epidemiologic studies have also reported that PR is longer in individuals of African compared to European ancestry,13, 14 which provides additional motivation for GWA studies of PR in populations of African descent.
To further characterize genetic determinants of PR in populations of African descent, we extended the earlier efforts of Smith et al.12 by including GWA study data from six additional African American cohorts (7,168 additional participants). These results were meta-analyzed with those previously reported by Smith et al. to provide the largest GWA study of PR to date in populations of African ancestry.
Results
We performed a GWA analysis of PR in 13,415 adults of African descent from ten cohorts, including three studies from the Continental Origins and Genetic Epidemiology Network (COGENT)15 and four studies from the Candidate-gene Association Resource (CARe) consortia.16 Four of the ten studies were included in the earlier study by Smith et al.:12 the Atherosclerosis Risk in Communities (ARIC) Study, the Cleveland Family Study (CFS), the Jackson Heart Study (JHS), and the Multi-Ethnic Study of Atherosclerosis (MESA). Variation in study size was noted across cohorts (range: 191 – 4,149 participants) and the largest contributing study was composed entirely of females (Table 1). Across studies, participants were predominantly female (72%), middle-aged (overall mean age: 58 years), obese (overall mean body mass index (BMI): 31 kg/m2) and pre-hypertensive (overall mean systolic blood pressure (SBP): 130 mmHg). Modest evidence of test statistic inflation was noted for the family-based CFS (λ: 1.10) and JHS (λ: 1.08), although inflation was neither observed in the remaining studies (λ range: 0.95, 1.04) nor in the overall meta-analysis after genomic control was applied (λ: 1.008) (Supplemental Figure 1). A total of 2.8 million genotyped and imputed autosomal SNPs were available for analysis after applying genotyping and imputation quality control measures (Supplemental Table 1).
Table 1.
Characteristics of 13,415 African-American participants from ten cohort studies.a
| Variableb | ARIC n=2,391 |
BLSA n=155 |
BHS n=191 |
CFS n=267 |
CHS n=674 |
HABCc n=1,054 |
HANDLS n=945 |
JHS n=1,962 |
MESA n=1,627 |
WHIc n=4,149 |
|---|---|---|---|---|---|---|---|---|---|---|
| PR interval (ms) | 172 ± 27 | 172 ± 25 | 161 ± 23 | 169 ± 26 | 172 ± 29 | 171 ± 28 | 162 ± 25 | 171 ± 26 | 171 ± 26 | 167 ± 25 |
| RR interval (ms) | 923 ± 150 | 957 ± 130 | 896 ± 149 | 903 ± 131 | 921 ± 158 | 931 ± 154 | 907 ± 154 | 949 ± 148 | 975 ± 155 | 915 ± 146 |
| Age (years) | 53.2 ± 8.8 | 64.4 ± 11.4 | 35.7 ± 4.8 | 44.3 ± 15.2 | 72.6 ± 5.5 | 73.4 ± 2.9 | 48.6 ± 9.0 | 49.3 ± 11.7 | 62.1 ± 10.1 | 61.6 ± 6.8 |
| Female sex (%) | 1,480 (62) | 98 (63) | 127 (66) | 154 (58) | 431 (64) | 609 (58) | 527 (56) | 1,203 (61) | 887 (55) | 4,149 (100) |
| BMI (kg/m2) | 29.5 ± 6.1 | 28.3 ± 5.2 | 31.5 ± 8.7 | 34.5 ± 9.2 | 28.4 ± 5.5 | 28.5 ± 5.4 | 29.9 ± 8.1 | 32.3 ± 7.8 | 30.2 ± 5.9 | 31.6 ± 6.2 |
| Systolic BP (mmHg) | 128.1 ± 20.7 | 133.7 ± 15.6 | 124.3 ± 17.9 | 126.1 ± 14.4 | 146.2 ± 21.5 | 138.7 ± 22.0 | 120.8 ± 21.9 | 124.6 ± 17.8 | 131.6 ± 21.6 | 131.9 ± 17.3 |
| Genomic inflation factor (λ) | 1.023 | 0.969 | 0.989 | 1.099 | 1.043 | 1.014 | 0.947 | 1.079 | 1.008 | 1.010 |
| % European ancestryd | 15 (11, 22) | ND | 18 (13, 21) | 18 (13, 26) | 24 (16, 36) | 19 (12, 28) | 16 (11, 22) | 16 (12, 21) | 19 (12, 30) | 21 (13, 31) |
Sample sizes presented are the maximum number of participants with SNP data.
Data are presented as mean (standard deviation) for continuous variables and percentages for categorical variables.
The HABC and WHI studies replaced imputed data with genotyped data when available and therefore have a range of genotyped participants (HABC minimum = 939 participants; WHI minimum = 3,898 participants).
Presented as median (25th percentile, 75th percentile)
ARIC, Atherosclerosis Risk in Communities; BLSA, Baltimore Longitudinal Study on Aging; BHS, Bogalusa Heart Study; CFS, Cleveland Family Study; CHS, Cardiovascular Health Study; HABC, The Health, Aging, and Body Composition Study; HANDLS, The Healthy Aging in Neighborhoods of Diversity across the Life Span Study; JHS, Jackson Heart Study; MESA, Multi-Ethnic Study of Atherosclerosis; ND, not determined. WHI, Women’s Health Initiative.
In the meta-analysis, 90 SNPs at six loci were associated with PR at the genome-wide significance threshold of P < 5.0 × 10−8 after applying genomic control (Figure 1, Table 2). The strongest primary PR signal (P = 5.26 × 10−43, primary signals defined as the locus-specific SNP with the lowest P-value), was observed for rs3922844 in SCN5A (effect allele frequency (AF) = 0.58), and corresponded to a 4.5 ms decrease in PR per copy of the C allele (Figure 2c). We also identified two independent secondary signals at SCN5A/10A, a region characterized by low patterns of linkage disequilibrium (LD) and multiple recombination peaks (Figure 2C); one in SCN5A and a second in SCN10A, which was located 14.3 kb downstream of the SCN5A primary signal (Table 2, secondary signals defined as the locus-specific SNP with the lowest genome-wide significant P-value after conditioning on primary signals and successive secondary signals). Estimates for the eight signals (six primary; two secondary) were generally consistent across cohorts (Supplemental Table 2), and there was little evidence of among-study heterogeneity (Cochran’s Q P ≥ 0.05). The primary signals also were robust to adjustment for local ancestry (Supplemental Table 3).
Figure 1.
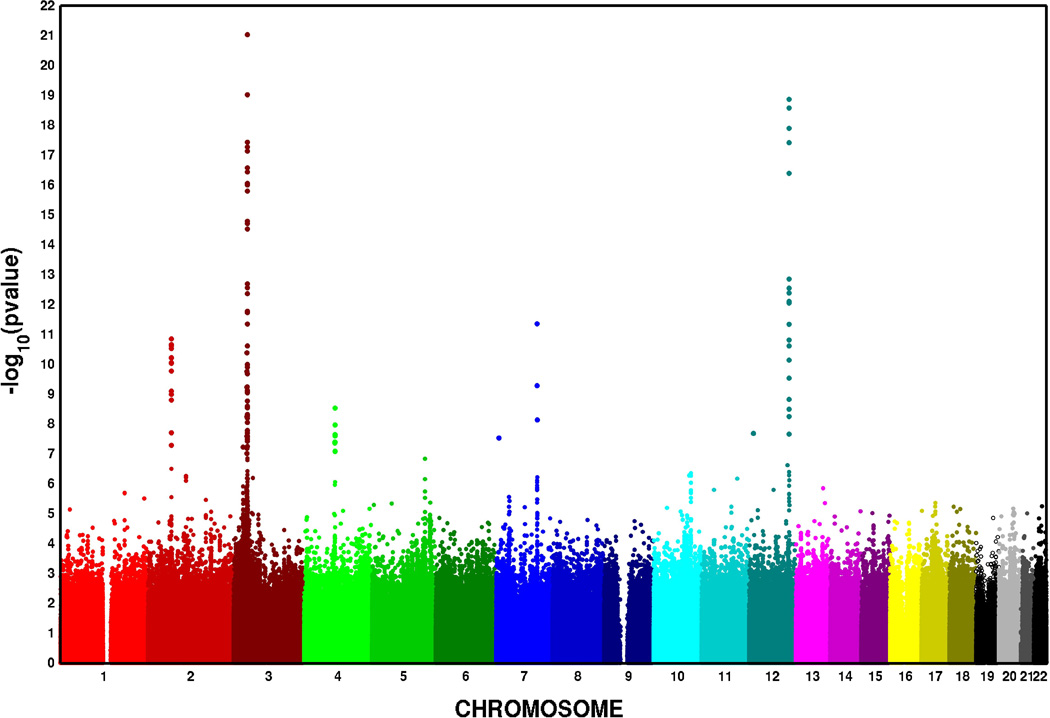
Manhattan plot of the association of SNPs with PR in a meta-analysis of ten African American cohorts. The x-axis represents the chromosomal position for each SNP, and the y-axis represents the −log10 P-value for association with PR, which is truncated at 1×10−23.
Table 2.
Summary of six primary and two secondary independent loci (P < 5.0×10−8) obtained for PR in 13,415 African-American participants from ten cohort studies.
| SNP | Gene | Chr | Position (Build 36) |
Allelesa | Effect allele Frequency |
Study-specific direction of βb |
β (se) | p | phet |
|---|---|---|---|---|---|---|---|---|---|
| Primary signalsc | |||||||||
| rs3891585 | MEIS1 | 2 | 66,610,480 | A/G | 0.43 | +−++−+++++ | 2.13 (0.31) | 1.42 × 10−11 | 0.11 |
| rs267567 | ITGA9 | 3 | 37,549,028 | A/G | 0.18 | +++−++++++ | 2.73 (0.41) | 4.14 × 10−11 | 0.54 |
| rs3922844 | SCN5A | 3 | 38,599,257 | T/C | 0.58 | −−−−−−−−−− | −4.54 (0.33) | 5.26 × 10−43 | 0.58 |
| rs11732231 |
ARHGAP2 4 |
4 | 86,902,584 | C/G | 0.23 | ++++++++++ | 2.28 (0.39) | 2.96 × 10−9 | 0.30 |
| rs11773845 | CAV1 | 7 | 115,978,537 | A/C | 0.36 | −−−−−−−−−− | −2.29 (0.33) | 4.45 × 10−12 | 0.53 |
| rs1895585 | TBX5 | 12 | 113,286,521 | A/G | 0.30 | ++−+++++++ | 3.19 (0.35) | 1.36 × 10−19 | 0.42 |
| Secondary signalsd | |||||||||
| rs6763048 | SCN5A | 3 | 38,656,398 | A/G | 0.73 | ++++++++++ | 2.62 (0.38) | 3.75 × 10−12 | 0.74 |
| rs6801957 | SCN10A | 3 | 38,742,319 | T/C | 0.27 | ++++ | 3.36 (0.58) | 9.11 × 10−9 | 0.15 |
Coded allele listed first.
Study-specific direction of β estimates are listed in alphabetical order by study. The + and − symbols represent an increase and decrease, respectively, in the PR interval per copy of the minor allele.
Defined as locus-specific SNP with the lowest P-value.
Defined as significant SNPs after conditional analysis that adjusted for locus-specific primary signal. The conditional analysis for rs6801957 was performed in four cohorts (CHS, HABC, HANDLS and WHI) adjusting for successively less significant SNPs until no genome-wide significant SNPs remained.
Chr, chromosome; se, standard error; p, meta-analysis p-value; phet, Cochran’s Q heterogeneity p-value.
Figure 2.

Regional association plots of six loci associated with PR interval in ten African American cohorts. SNP P-values (represented by circles) at each locus are shown on the −log10(P-value) scale as a function of chromosomal position. Strength of LD is indicated by the color category. Purple diamonds denotes the locus-specific primary signal. Recombination rate is plotted in the background and known genes are shown on the bottom of the plot. A) MEIS1; b) ITGA9; c) SCN5A; d) ARHGAP24; e) CAV1; f) TBX5.
Five of the loci associated with PR were previously identified in populations of European and Asian descent: SCN5A/SCN10A, MEIS1, ARHGAP24, CAV1, and TBX5. Of note, SCN5A/SCN10A, MEIS1, and TBX5 were also reported by the earlier PR GWA study of African Americans.12 The novel locus, ITGA9, was located on chromosome 3, greater than one Mb upstream from the primary SCN5A signal. Several genes resided nearby ITGA9, although only ITGA9 and C3orf35 harbored SNPs in strong to moderate LD with rs267567.
None of the primary or secondary signals reported here were the same as the index SNPs reported in populations of European or Asian ancestry. Although we identified both a primary and secondary SCN5A signal for PR, only one study of European ancestral populations identified SCN5A,10 and this study reported an index SNP (rs11708996) that was monomorphic in HapMap YRI. The SCN10A SNP that we identified (rs6801957) was in low LD with both previously identified SCN10A variants (rs6800541 and rs6795970 (r2 < 0.10, HapMap YRI)), which were reported in European ancestral populations. The MEIS1 index SNP rs3891585 was in moderate LD with the previously described variant rs11897119 (r2 = 0.62, HapMap YRI). Of the two index SNPs reported for ARHGAP24 in populations of European descent, rs7692808 was in high LD (r2 = 0.94, HapMap YRI) and rs7660702 was in low LD (r2 = 0.22, HapMap YRI) with our ARHGAP24 primary signal. Both studies that previously identified CAV1 as a PR-associated locus reported the rs3807989 variant; this SNP was in very high LD with the primary CAV1 SNP presented herein (r2 = 1.0, HapMap YRI). Finally, both GWA studies of PR that identified TBX5 reported the variant rs1895582, which also was in high LD with our TBX5 primary signal, rs1895585 (r2 = 0.84, HapMap YRI).
We identified six loci associated with PR in populations of African descent, yet we were unable to confirm associations at genome-wide significance thresholds for three PR loci that were previously identified in individuals of European descent: NKX2–5, WNT11, and SOX5.8 Although the previously reported chromosome 5 and 11 loci had high minor allele frequencies (MAF) across contributing studies, consistent directions of effect, and little evidence of heterogeneity, neither previously reported index SNP was associated with PR (P > 0.01) (Table 3). Of note, all SNPs residing within a 1 Mb region of these loci had P-values that exceeded 0.0009 (results not shown). Data for the previously reported SOX5 index SNP were only available in six contributing studies and the mean estimated MAF was 0.03. The SOX5 locus also was monomorphic in the HapMap YRI population and all P-values within 1 Mb of this locus exceeded 0.0002 (results not shown).
Table 3.
Associations between PR and three previously reported PR loci10 that were not genome-wide significant in a meta-analysis of 13,415 African-American participants from ten cohort studiesi.
| SNP | Gene | Chr | Position (Build 36) |
Allelesa | Effect Allele Frequency |
Study-specific direction of βb |
β (se) | p | phet |
|---|---|---|---|---|---|---|---|---|---|
| rs251253 | NKX2–5 | 5 | 172,412,942 | T/C | 0.36 | −++++++−++ | 0.77 (0.33) | 1.84 × 10−2 | 0.53 |
| rs4944092 | WNT11 | 11 | 75,587,267 | A/G | 0.57 | ++−+++++−+ | 0.41 (0.32) | 2.05 × 10−1 | 0.18 |
| rs11047543 | SOX5 | 12 | 24,679,606 | A/G | 0.03 | −+???−−−+? | −2.49 (1.25) | 4.57 × 10−2 | 0.12 |
Coded allele listed first.
Study-specific direction of β estimates are listed in alphabetical order of the studies. The + and − symbols represent an increase and decrease, respectively, in the PR interval per copy of the minor allele. A “?” denotes studies that did not contribute to the SNP meta-analysis.
Chr, chromosome; se, standard error; p, meta-analysis p-value; phet, Cochran’s Q heterogeneity p-value.
Discussion
This GWA study and meta-analysis of ten cohorts represents the largest effort in populations of African descent to identify genetic determinants of PR. By building on recent work from the CARe consortium,12 we identify three additional loci associated with PR in African ancestral populations; ARHGAP24, CAV1, and ITGA9. The ITGA9 locus represents a novel finding, having not been identified in any prior GWA studies of PR to date.
ITGA9 is located approximately 1.1 Mb upstream from SCN5A and encodes an alpha integrin, an integral membrane glycoprotein that mediates diverse functions including cell–cell and cell–matrix adhesion, proliferation, and apoptosis.17, 18 ITGA9 also has been associated with hypertension19 and several cancers.20–22 Although, ITGA9 has not been previously implicated in atrioventricular conduction, the extended 3p22–24 region has been shown to harbor variants affecting SCN5A expression. It is therefore possible that ITGA9 marks a distal SCN5A regulatory element.23, 24 Interestingly, pathway analysis suggests a role for ITGA9 in cation binding, hypertrophic cardiomyopathy, and dilated cardiomyopathy.25 Expression QTL studies also have associated variation in ITGA9 with cis expression data from monocytes26 and lymphoblastoid cell lines.27 However, the transferability of associations to cardiac myocyte and conduction tissue warrants further investigation.
In addition to identifying ITGA9 as a potential cis-regulator of SCN5A, we also reported three independent SNPs influencing PR at the 3p21 locus. The 3p21 locus harbors both SCN5A and SCN10A, which encode integral membrane proteins and tetrodotoxin-resistant voltage-gated sodium channel subunits. The NAv1.5 sodium channel alpha-subunit (encoded by SCN5A) is the predominant alpha-subunit expressed in cardiac muscle, and is responsible for the initial upstroke of the action potential in an ECG.28 SCN5A mutations are associated with Brugada syndrome, long-QT syndrome, dilated cardiomyopathy, cardiac conduction disease, idiopathic ventricular fibrillation and atrial fibrillation28 and have been identified in GWA studies of the QT29, 30 and QRS intervals31 in populations of European descent.
The NAv1.8 sodium channel alpha-subunit (encoded by SCN10A) is characterized by a long-duration action potential and preservation of excitability during rapid and sustained stimulation.32 Seven variants at 3p21 have been previously reported,8, 10, 12 and by extending the work of Smith et al.,12 we detected an additional independent signal at genome-wide significance levels. The presence of numerous independent signals at the 3p21 region in African Americans was previously reported by a SCN5A candidate gene study in approximately 3,000 JHS participants, who also contributed to this analysis.33 By including nine additional studies, we validate the previous work by Jeff and colleagues at genome-wide significance levels and identify a neighboring genome-wide significant signal in SCN10A. The ability to identify multiple SCN5A/SCN10A signals may in part be attributable to the greater nucleotide diversity and lower LD in African populations, as 3p21 is characterized by low LD and high recombination.
In addition to SCN5A, we generalized four additional PR loci to populations of African ancestry: ARHGAP24, MEIS1, TBX5, and CAV1, the latter of which was also detected by a GWA study of atrial fibrillation.34 Yet, the importance of NKX2–5, WNT11, and SOX5 in the genetic architecture of PR in African Americans is less clear. Although the “winner’s curse” and inflated genetic effect estimates from initial discovery35 may help explain the inconsistent results, another possibility is that our study was underpowered to detect these loci, especially for the SOX5 locus. In addition, our analysis was conducted in populations that were predominantly female, obese and pre-hypertensive. The degree to which these characteristics influenced the results presented herein remains unclear.
Several limitations of the present study warrant further consideration in order to inform future efforts examining the genetic architecture of PR. The first is study heterogeneity, a common limitation of meta-analyses. In our meta-analysis, studies used common measurement protocols for determining PR and its clinical correlates. In addition, statistical assessments of heterogeneity did not suggest large variation in SNP effects across studies. Another limitation is confounding, either from cryptic population stratification or unmeasured PR risk factors. For example, one potential confounder we were unable to consider was atrial size, given widespread unavailability of echocardiographs. However, we adjusted for BMI, height, and systolic blood pressure, the major contributors to left atrial size. Regarding the potential for bias from population substructure, we adjusted for principal components in study-specific regression models and applied genomic control. These approaches are standard in GWA studies, yet the potential for residual confounding to produce either false-negative or false-positive results remains challenging to determine on a genome-wide level. Finally, we were unable to independently replicate the association with ITGA9 in an independent population given difficulties identifying additional studies of African American participants with ECG measures, extant genotype data, and overlapping analytical timelines. Although results from other ancestral population could provide confirmatory evidence of the association between PR and ITGA9, failure to replicate could simply reflect allelic heterogeneity.
In summary, our results suggest that polymorphisms from six loci on five chromosomes are associated with PR in African Americans, including a novel signal in ITGA9 that may function as a distal SCN5A regulatory element. Our expanded meta-analysis also demonstrates the ability to map novel genes in African Americans and the generalizability of genetic variants associated with PR across global populations. Future work to refine these signals is clearly warranted, including additional examination of the extended chromosome 3p region that harbors SCN5A, SCN10A and ITGA9. GWA studies in other admixed populations, as well as fine-mapping efforts, would be especially useful for further characterization loci identified herein, as well as the identification of new genes influencing atrial arrhythmogenesis.
Materials and Methods
Study populations
A meta-analysis of ten studies was performed to investigate the genetic determinants of PR. Three cohorts were from COGENT including the Health, Aging, and Body Composition Study (Health ABC n=1,054), the Healthy Aging in Neighborhoods of Diversity across the Life Span Study (HANDLS, n=945), and the Women’s Health Initiative (WHI, n=4,149), and four cohorts were available from the CARe consortium, including the ARIC study (n=2,391), the CFS (n=267), the JHS (n=1,962), and MESA (n=1,627). The Baltimore Longitudinal Study of Aging (BLSA, n=155), the Bogalusa Heart Study (BHS, n=191), and the Cardiovascular Health Study (CHS, n=674) were the remaining contributing studies. Additional information on the participating studies is provided in the Supplementary Material. All studies were approved by local ethics committees and all participants provided written informed consent.
PR interval measurement
For each study, certified technicians digitally recorded resting, supine (or semi-recumbent), standard twelve-lead ECGs using comparable procedures for preparing participants, placing electrodes, recording, transmitting, processing and controlling quality (Supplemental Table 4). Participants with the following characteristics were excluded: poor quality ECG, extreme PR (320 ms≤ PR≤80 ms), documented history of atrial fibrillation/flutter, heart failure, myocardial infarction, pacemakers antedating ECG assessment, Wolff-Parkinson-White syndrome, and second/third degree heart block.
Genotype arrays and imputation
Genome-wide SNP genotyping was performed within each cohort using the Affymetrix or Illumina genotyping arrays (Supplemental Table 1). First -degree relatives were excluded in all studies except the family-based CFS and JHS. SNPs were excluded for genotyping call rate thresholds between <95% and <99% and MAF ≤ 1%, the determination of which was study-specific.
Imputation was performed for ~2.5 million autosomal SNPs based on a 1:1 ratio of the HapMap Phase 2 CEU and YRI populations (Supplemental Table 1). SNPs with imputation quality< 0.3 or inconsistent allele designations as per HapMap forward strands were excluded. In addition, SNPs not seen in > 2 studies were excluded from the meta-analyses. After exclusions, 2,845,108 genotyped and imputed SNPs were available.
Statistical analysis
Each study, with the exception of CFS, performed GWA analysis for PR across approximately 2.5 million SNPs based on linear regression under an additive genetic model. The family-based CFS study was analyzed using linear mixed-effects models as implemented in the R GWAF package.36 Specifically, the within pedigree random genetic effects were modeled using a kinship coefficient matrix, with each family having a different covariance pattern. The full N × N kinship variance covariance matrix was generated using the R kinship function within the GWAF software package, according to the algorithm of K. Lange.37 Although the JHS has a limited number of related participants, extensive analyses suggested that results from linear regression or linear mixed effects models were concordant.15 Therefore, JHS results are based on linear regression models unadjusted for family structure.
The association of each SNP with PR was adjusted for age, sex, height, BMI, systolic blood pressure, RR interval, and study site, when appropriate, to maintain consistency with Smith et al.12 All studies included principal components in linear models to adjust for variation in global ancestry (Supplemental Table 1).38 Genotyped data were substituted for imputed data, when available. Individual study results were corrected by their respective genomic inflation factors (λ);39 genomic inflation factors > 1 may indicate sample duplications, unknown or poorly specified familial relationships, a poorly calibrated test statistic, systematic technical bias, or gross population stratification.40
A fixed effects inverse variance meta-analysis was performed to combine beta coefficients and standard errors from study-level regression results for each SNP. Primary signals were defined as the locus-specific SNP with the lowest genome-wide significant P-value (P < 5 × 10−8). Between-study heterogeneity of results was assessed by Cochran’s Q statistic. Meta-analyses were implemented in the software METAL41 and were confirmed by an independent analyst.
A two-stage strategy was used to identify secondary signals. First, LD pruning was performed using PLINK, whereby independent signals were defined as at least two genome-wide significant SNPs in low LD (r2 < 0.20) in the same 1 Mb region. Next, each study performed a conditional analysis by adjusting for the most strongly associated SNP(s) at each locus with at least two bins, restricting to SNPs with P-values < 5.0 × 10−8. SNPs outside 1 Mb of the primary signal were not considered in conditional analyses because no loci exhibited LD patterns that extended beyond 1 Mb, and because conditioning on potential mediators may induce bias, the direction and magnitude of which are difficult to predict.42 Results for secondary signals were presented after conditional adjustment that adjusted for locus-specific primary signals. Additional iterations adjusting for subsequent secondary signals as well as the primary signal were performed in the WHI, HABC, HANDLS, and CHS cohorts (n=5,768, 43% of sample size) until no genome-wide significant associations remained.
As a sensitivity analysis, we assessed the impact of local ancestry by including SNP-specific local ancestry estimates as a covariate in models for genome-wide significant signals. Locus-specific ancestry (i.e. probabilities of whether an individual has 0, 1, or 2 alleles of African ancestry at each locus) was only available for directly genotyped SNPs and was estimated using a Hidden Markov Model and the local haplotype structure to detect transitions in ancestry along the genome.43
Supplementary Material
The PR interval (PR), a potent risk factor for arrhythmia, pacemaker implantation, heart failure, stroke, and all-cause mortality, is influenced by many factors including common and rare genetic variants.Recent genome-wide association (GWA) studies performed in populations of European and Asian descenthave identified several common genetic variants associated with PR, however, limited data exist on loci associated with PR in African Americans. One exception is a PR GWA study by Smith et al. including6,247 participants from four cohorts. Here, we extend this meta-analysis by including an additional 7,168participants fromsix cohorts in order to identify novel loci influencing PR in African Americans. In addition to generalizing four PR loci in ARHGAP24, MEIS1, TBX5, and CAV1 to populations of African ancestry, we identified one novel signal in ITGA9 and two additional independent and genome-wide significant secondary signals in the 3p21 region that harbors SCN5A and SCN10A. Our findings highlight the ability to map novel loci in African Americans, the generalizability of loci associated with PR across populations of African, European and Asian descent, and the new mechanistic insights on biologic processes underlying PR.
Acknowledgments
Funding Sources: The ARIC Study is carried out as a collaborative study supported by National Heart, Lung, and Blood Institute contracts (HHSN268201100005C, HHSN268201100006C, HHSN268201100007C, HHSN268201100008C, HHSN268201100009C, HHSN268201100010C, HHSN268201100011C, and HHSN268201100012C), R01HL087641, R01HL59367 and R01HL086694; National Human Genome Research Institute contract U01HG004402; and NIH contract HHSN268200625226C. Infrastructure was partly supported by Grant Number UL1RR025005, a component of the NIH Roadmap for Medical Research. The BLSA was supported in part by the Intramural Research Program of the NIH/National Institute on Aging. A portion of that support was through a contract with MedStar Research Institute.
The BHS was supported by grants HD-061437 and HD-062783 from the National Institute of Child Health and Human Development, and AG-16592 from the National Institute on Aging. ENS, SSM, and NJS were supported in part by NIH/NCRR Grant Number UL1 RR025774 and Scripps Genomic Medicine.
The CFS was supported by (NIH HL 46380, M01RR00080), Case Western Reserve University. CHS was supported by NHLBI contracts N01-HC-85239, N01-HC-85079 through N01-HC-85086; N01-HC-35129, N01 HC-15103, N01 HC-55222, N01-HC-75150, N01-HC-45133 and NHLBI grants HL080295, HL075366, HL087652, HL085251, HL105756 with additional contribution from NINDS. Additional support was provided through AG-023629, AG-15928, AG-20098, and AG-027058 from the NIA. See also http://www.chs-nhlbi.org/pi.htm. DNA handling and genotyping was supported in part by National Center for Research Resources grant M01-RR00425 to the Cedars-Sinai General Clinical Research Center Genotyping core and National Institute of Diabetes and Digestive and Kidney Diseases grant DK063491 to the Southern California Diabetes Endocrinology Research Center.
The Health ABC study was supported by NIA contracts N01AG62101, N01AG62103, and N01AG62106. The genome-wide association study was funded by NIA grant 1R01AG032098-01A1 to Wake Forest University Health Sciences and genotyping services were provided by the Center for Inherited Disease Research (CIDR). CIDR is fully funded through a federal contract from the NIH to The Johns Hopkins University, contract number HHSN268200782096C. This research was supported in part by the Intramural Research Program of the NIH, National Institute on Aging.
HANDLS was supported by the Intramural Research Program of the NIH, National Institute on Aging and the National Center on Minority Health and Health Disparities (contract # Z01-AG000513 and human subjects protocol # 2009-149). Data analyses for the HANDLS study utilized the high-performance computational capabilities of the Biowulf Linux cluster at the NIH, Bethesda, MD (http://biowulf.nih.gov).
The JHS was supported by NIH contracts N01-HC-95170, N01-HC-95171, and N01-HC-95172 provided by the National Heart, Lung, and Blood Institute and the National Center for Minority Health and Health Disparities.
The MESA was supported by University of Washington (N01-HC-95159), Regents of the University of California (N01-HC-95160), Columbia University (N01-HC-95161), Johns Hopkins University (N01-HC-95162, N01-HC-95168), University of Minnesota (N01-HC-95163), Northwestern University (N01-HC-95164), Wake Forest University (N01-HC-95165), University of Vermont (N01-HC-95166), New England Medical Center (N01-HC-95167), Harbor-UCLA Research and Education Institute (N01-HC-95169), Cedars-Sinai Medical Center (R01-HL-071205), University of Virginia (subcontract to R01-HL-071205).
The WHI was funded by the National Heart, Lung, and Blood Institute, NIH, U.S. Department of Health and Human Services through contracts N01WH22110, 24152, 32100-2, 32105-6, 32108-9, 32111-13, 32115, 32118-32119, 32122, 42107-26, 42129-32, and 44221. Funding for WHI SHARe genotyping was provided by NHLBI Contract N02-HL-64278.
CLA was supported by HL098458 and AMB acknowledges support from CA009330. PTE was supported by grants HL092577, DA027021, HL104156, and HL105780. YL was supported by grants HG006292 and HG006703. Additional support was provided by the National Institute of Environmental Health Sciences (ES017794).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Disclosures: BMP served on a data safety monitoring board (DSMB) for a clinical trial of a device funded by the manufacturer (Zoll-Lifecor). Otherwise, authors have no other conflicts to report.
References
- 1.Cheng S, Keyes MJ, Larson MG, McCabe EL, Newton-Cheh C, Levy D, et al. Long-term outcomes in individuals with prolonged pr interval or first-degree atrioventricular block. JAMA. 2009;301:2571–2577. doi: 10.1001/jama.2009.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lloyd-Jones DM, Wang TJ, Leip EP, Larson MG, Levy D, Vasan RS, et al. Lifetime risk for development of atrial fibrillation: The framingham heart study. Circulation. 2004;110:1042–1046. doi: 10.1161/01.CIR.0000140263.20897.42. [DOI] [PubMed] [Google Scholar]
- 3.Roy D, Talajic M, Dubuc M, Thibault B, Guerra P, Macle L, et al. Atrial fibrillation and congestive heart failure. Curr Opin Cardiol. 2009;24:29–34. doi: 10.1097/hco.0b013e32831c8c58. [DOI] [PubMed] [Google Scholar]
- 4.Havlik RJ, Garrison RJ, Fabsitz R, Feinleib M. Variability of heart rate, p-r, qrs and q-t durations in twins. J Electrocardiol. 1980;13:45–48. doi: 10.1016/s0022-0736(80)80008-2. [DOI] [PubMed] [Google Scholar]
- 5.Smith JG, Lowe JK, Kovvali S, Maller JB, Salit J, Daly MJ, et al. Genome-wide association study of electrocardiographic conduction measures in an isolated founder population: Kosrae. Heart Rhythm. 2009;6:634–641. doi: 10.1016/j.hrthm.2009.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schott JJ, Alshinawi C, Kyndt F, Probst V, Hoorntje TM, Hulsbeek M, et al. Cardiac conduction defects associate with mutations in scn5a. Nat Genet. 1999;23:20–21. doi: 10.1038/12618. [DOI] [PubMed] [Google Scholar]
- 7.Tan HL, Bink-Boelkens MT, Bezzina CR, Viswanathan PC, Beaufort-Krol GC, van Tintelen PJ, et al. A sodium-channel mutation causes isolated cardiac conduction disease. Nature. 2001;409:1043–1047. doi: 10.1038/35059090. [DOI] [PubMed] [Google Scholar]
- 8.Chambers JC, Zhao J, Terracciano CM, Bezzina CR, Zhang W, Kaba R, et al. Genetic variation in scn10a influences cardiac conduction. Nat Genet. 2010;42:149–152. doi: 10.1038/ng.516. [DOI] [PubMed] [Google Scholar]
- 9.Holm H, Gudbjartsson DF, Arnar DO, Thorleifsson G, Thorgeirsson G, Stefansdottir H, et al. Several common variants modulate heart rate, pr interval and qrs duration. Nat Genet. 2010;42:117–122. doi: 10.1038/ng.511. [DOI] [PubMed] [Google Scholar]
- 10.Pfeufer A, van Noord C, Marciante KD, Arking DE, Larson MG, Smith AV, et al. Genome-wide association study of pr interval. Nat Genet. 2010;42:153–159. doi: 10.1038/ng.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Denny JC, Ritchie MD, Crawford DC, Schildcrout JS, Ramirez AH, Pulley JM, et al. Identification of genomic predictors of atrioventricular conduction: Using electronic medical records as a tool for genome science. Circulation. 2010;122:2016–2021. doi: 10.1161/CIRCULATIONAHA.110.948828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith JG, Magnani JW, Palmer C, Meng YA, Soliman EZ, Musani SK, et al. Genome-wide association studies of the pr interval in african americans. PLoS Genet. 2011;7:e1001304. doi: 10.1371/journal.pgen.1001304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Soliman EZ, Prineas RJ, Case LD, Zhang ZM, Goff DC., Jr Ethnic distribution of ecg predictors of atrial fibrillation and its impact on understanding the ethnic distribution of ischemic stroke in the atherosclerosis risk in communities (aric) study. Stroke. 2009;40:1204–1211. doi: 10.1161/STROKEAHA.108.534735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramirez AH, Schildcrout JS, Blakemore DL, Masys DR, Pulley JM, Basford MA, et al. Modulators of normal electrocardiographic intervals identified in a large electronic medical record. Heart Rhythm. 2011;8:271–277. doi: 10.1016/j.hrthm.2010.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reiner AP, Lettre G, Nalls MA, Ganesh SK, Mathias R, Austin MA, et al. Genome-wide association study of white blood cell count in 16,388 african americans: The continental origins and genetic epidemiology network (cogent) PLoS Genet. 2011;7:e1002108. doi: 10.1371/journal.pgen.1002108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Musunuru K, Lettre G, Young T, Farlow DN, Pirruccello JP, Ejebe KG, et al. Candidate gene association resource (care): Design, methods, and proof of concept. Circ Cardiovasc Genet. 2010;3:267–275. doi: 10.1161/CIRCGENETICS.109.882696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo W, Giancotti FG. Integrin signalling during tumour progression. Nat Rev Mol Cell Biol. 2004;5:816–826. doi: 10.1038/nrm1490. [DOI] [PubMed] [Google Scholar]
- 18.Daigo Y, Isomura M, Nishiwaki T, Tamari M, Ishikawa S, Kai M, et al. Characterization of a 1200-kb genomic segment of chromosome 3p22-p21.3. DNA Res. 1999;6:37–44. doi: 10.1093/dnares/6.1.37. [DOI] [PubMed] [Google Scholar]
- 19.Takeuchi F, Isono M, Katsuya T, Yamamoto K, Yokota M, Sugiyama T, et al. Blood pressure and hypertension are associated with 7 loci in the japanese population. Circulation. 2010;121:2302–2309. doi: 10.1161/CIRCULATIONAHA.109.904664. [DOI] [PubMed] [Google Scholar]
- 20.Ghosh A, Ghosh S, Maiti GP, Sabbir MG, Zabarovsky ER, Roy A, et al. Frequent alterations of the candidate genes hmlh1, itga9 and rbsp3 in early dysplastic lesions of head and neck: Clinical and prognostic significance. Cancer Sci. 2010;101:1511–1520. doi: 10.1111/j.1349-7006.2010.01551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gulubova M, Vlaykova T. Immunohistochemical assessment of fibronectin and tenascin and their integrin receptors alpha5beta1 and alpha9beta1 in gastric and colorectal cancers with lymph node and liver metastases. Acta Histochem. 2006;108:25–35. doi: 10.1016/j.acthis.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 22.Ng CC, Yew PY, Puah SM, Krishnan G, Yap LF, Teo SH, et al. A genome-wide association study identifies itga9 conferring risk of nasopharyngeal carcinoma. J Hum Genet. 2009;54:392–397. doi: 10.1038/jhg.2009.49. [DOI] [PubMed] [Google Scholar]
- 23.Weiss R, Barmada MM, Nguyen T, Seibel JS, Cavlovich D, Kornblit CA, et al. Clinical and molecular heterogeneity in the brugada syndrome: A novel gene locus on chromosome 3. Circulation. 2002;105:707–713. doi: 10.1161/hc0602.103618. [DOI] [PubMed] [Google Scholar]
- 24.London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, et al. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (gpd1-l) decreases cardiac na+ current and causes inherited arrhythmias. Circulation. 2007;116:2260–2268. doi: 10.1161/CIRCULATIONAHA.107.703330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, et al. David: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3. [PubMed] [Google Scholar]
- 26.Zeller T, Wild P, Szymczak S, Rotival M, Schillert A, Castagne R, et al. Genetics and beyond--the transcriptome of human monocytes and disease susceptibility. PloS One. 2010;5:e10693. doi: 10.1371/journal.pone.0010693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Montgomery SB, Sammeth M, Gutierrez-Arcelus M, Lach RP, Ingle C, Nisbett J, et al. Transcriptome genetics using second generation sequencing in a caucasian population. Nature. 2010;464:773–777. doi: 10.1038/nature08903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Remme CA, Wilde AA, Bezzina CR. Cardiac sodium channel overlap syndromes: Different faces of scn5a mutations. Trends Cardiovasc Med. 2008;18:78–87. doi: 10.1016/j.tcm.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 29.Newton-Cheh C, Eijgelsheim M, Rice KM, de Bakker PI, Yin X, Estrada K, et al. Common variants at ten loci influence qt interval duration in the qtgen study. Nat Genet. 2009;41:399–406. doi: 10.1038/ng.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pfeufer A, Sanna S, Arking DE, Muller M, Gateva V, Fuchsberger C, et al. Common variants at ten loci modulate the qt interval duration in the qtscd study. Nat Genet. 2009;41:407–414. doi: 10.1038/ng.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sotoodehnia N, Isaacs A, de Bakker PI, Dorr M, Newton-Cheh C, Nolte IM, et al. Common variants in 22 loci are associated with qrs duration and cardiac ventricular conduction. Nat Genet. 2010;42:1068–1076. doi: 10.1038/ng.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Renganathan M, Cummins TR, Waxman SG. Contribution of na(v)1.8 sodium channels to action potential electrogenesis in drg neurons. J Neurophysiol. 2001;86:629–640. doi: 10.1152/jn.2001.86.2.629. [DOI] [PubMed] [Google Scholar]
- 33.Jeff JM, Brown-Gentry K, Buxbaum SG, Sarpong DF, Taylor HA, George AL, Jr, et al. Scn5a variation is associated with electrocardiographic traits in the jackson heart study. Circ Cardiovasc Genet. 2011;4:139–144. doi: 10.1161/CIRCGENETICS.110.958124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ellinor PT, Lunetta KL, Albert CM, Glazer NL, Ritchie MD, Smith AV, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet. 2012;44:670–675. doi: 10.1038/ng.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goring HH, Terwilliger JD, Blangero J. Large upward bias in estimation of locus-specific effects from genomewide scans. Am J Hum Genet. 2001;69:1357–1369. doi: 10.1086/324471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen MH, Yang Q. Gwaf: An r package for genome-wide association analyses with family data. Bioinformatics. 2010;26:580–581. doi: 10.1093/bioinformatics/btp710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lange K. Mathematical and statistical methods for genetic analysis. New York: Springer-Verlag; 2002. [Google Scholar]
- 38.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 40.de Bakker PI, Ferreira MA, Jia X, Neale BM, Raychaudhuri S, Voight BF. Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum Mol Genet. 2008;17:R122–128. doi: 10.1093/hmg/ddn288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Willer CJ, Li Y, Abecasis GR. Metal: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robins JM, Greenland S. Identifiability and exchangeability for direct and indirect effects. Epidemiology. 1992;3:143–155. doi: 10.1097/00001648-199203000-00013. [DOI] [PubMed] [Google Scholar]
- 43.Tang H, Coram M, Wang P, Zhu X, Risch N. Reconstructing genetic ancestry blocks in admixed individuals. Am J Hum Genet. 2006;79:1–12. doi: 10.1086/504302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.