

Abstract
Background
CXC-chemokine receptor 4 (CXCR4) regulates the retention of stem/progenitor cells in the bone marrow (BM), and the CXCR4 antagonist AMD3100 improves recovery from coronary-ligation injury by mobilizing stem/progenitor cells from the BM to the peripheral blood. Thus, we investigated whether AMD3100 also improves recovery from ischemia-reperfusion (IR) injury, which more closely mimics myocardial infarction in patients, because blood flow is only temporarily obstructed.
Methods and Results
Mice were treated with single subcutaneous injections of AMD3100 (5 mg/kg) or saline after IR injury. Three days later, histological measurements of the infarct-area/area-at-risk ratio were smaller in AMD3100-treated mice than in mice administered saline, and echocardiographic measurements of left-ventricular function were greater in the AMD3100-treated mice at week 4. CXCR4+ cells were mobilized for just 1 day in both groups, but the mobilization of sca1+/flk1+ cells endured for 7days in AMD3100-treated mice compared to just 1 day in the saline-treated mice. AMD3100 upregulated BM levels of endothelial nitric oxide synthase (eNOS) and two targets of eNOS signaling, matrix-metalloproteinase 9 and soluble Kit ligand. Furthermore, the loss of BM eNOS expression abolished the benefit of AMD3100 on sca1+/flk1+ cell mobilization without altering the mobilization of CXCR4+ cells, and the cardioprotective effects of AMD3100 were retained in eNOS-knockout mice that had been transplanted with BM from wild-type mice, but not in wild-type mice with eNOS-knockout BM.
Conclusions
AMD3100 prolongs BM progenitor mobilization and improves recovery from IR injury, and these benefits appear to occur through a previously unidentified link between AMD3100 and BM eNOS expression.
Keywords: Drugs, Myocardium, Ischemia, Reperfusion, Nitric oxide synthase
In response to ischemic myocardial injury, stem/progenitor cells are mobilized from the bone marrow (BM) to the peripheral blood (PB) and become incorporated into the injured tissue, where a subset of the mobilized cells including endothelial progenitor cells (EPCs), contribute to cardiac recovery by enhancing vessel growth.1–3 Before mobilization, progenitor cells are sequestered in the BM by interactions between CXC chemokine receptor 4 (CXCR4) and stromal-cell–derived factor 1 (SDF-1).4, 5 Mobilization is triggered when this interaction is disrupted, and SDF-1 expression in the ischemic tissue contributes to the recruitment and incorporation of mobilized EPCs.6 SDF-1 also induces the migration of EPCs in vitro,7 and SDF-1–CXCR4 signaling appears to influence EPC proliferation and survival.8, 9 Thus, the SDF-1/CXCR4 axis is a key regulator of the activity of stem/progenitor including EPCs, particularly the release from BM and the retention/recruitment of progenitors in/to ischemic tissue.
CXCR4 also facilitates cellular entry of the human immunodeficiency virus, which prompted the development of AMD3100, a pharmacological CXCR4 antagonist.10–12 In early pharmacokinetic studies, a single intravenous dose of AMD3100 unexpectedly increased circulating white-blood-cell counts in healthy volunteers,13 and subsequent reports indicate that AMD3100 rapidly mobilizes hematopoietic progenitor cells in both humans and mice by reversibly blocking the SDF-1–CXCR4 interaction.13–16 Previously, we have shown that a single dose of AMD3100 after surgical ligation of the coronary artery17 increases the mobilization of BM progenitor cells (BMPCs), which leads to a greater number of BMPC accumulation in infarcted tissue and to improvements in vascularity and myocardial performance; furthermore, the effect of AMD3100 on BMPC mobilization endured for more than a week, which is somewhat surprising, because the half-life of AMD3100 in serum is just 2–3 hours and, consequently, the acute activity of AMD3100 as a CXCR4 antagonist should dissipate within a day of administration. Here, we investigated whether AMD3100 also improves myocardial recovery after ischemia-reperfusion (IR) injury, which more closely resembles the clinical presentation of acute myocardial infarction, because blood flow is obstructed temporarily, rather than permanently. We also compared the time course and signaling pathways involved in BMPC mobilization to those associated with mobilization of CXCR4+ MNCs.
METHODS
Injury model and treatment
All mice were obtained from The Jackson Laboratories. BM transplantation surgery and IR injury were performed as described previously18–20 and as summarized in the Supplementary Methods. Mice received a single subcutaneous injection of AMD3100 (5 mg/kg, 125 ug in 100 uL; Sigma-Aldrich Co) or an equal volume of saline immediately after surgery was complete. AAR and infarct area were measured as described in the Supplementary Methods; AAR was presented as a percentage of the area of the entire LV, and the infarct area was presented as a percentage of the AAR.
Physiological assessments of LV function
Echocardiographic measurements were performed with a commercially available high-resolution echocardiographic system (VEVO 770™, VisualSonics Inc.). End-systolic and end-diastolic LV areas on the short-axis view were traced at the mid-papillary muscle level according to the instruction of the echocardiographic program, and the calculation of (diastolic LV area - systolic LV area)/diastolic LV area was used for area-fractional shortening (FS%).
PB cell counts
The detailed information is provided in the Supplementary Methods.
Histological and immunofluorescent assessments
The detailed information is provided in the Supplementary Methods.
In vitro assessments with cultured, bone marrow-derived EPCs
The detailed information is provided in the Supplementary Methods.
Luciferase reporter assay
The detailed information is provided in the Supplementary Methods.
Quantitative real-time RT-PCR
The detailed information including primer and probe sequences (Supplementary Table 1) is provided in the Supplementary Methods.
Statistical analysis
All values were expressed as mean±SEM. Comparisons among samples from different mice at single or multiple time points were evaluated by unpaired t-test (bar graph). Comparisons among consecutive samples from identified mouse in a single group were evaluated by one-way ANOVA with Bonferroni post hoc-test (line graph). Comparisons among consecutive samples from identified mouse in multiple groups were evaluated by two-way ANOVA with Bonferroni post hoc-test (line graph, the factors are the groups and time). We applied the Bonferroni-adjustment to both comparisons of the groups within each time point and comparisons of each time point versus baseline within each group only when the data were collected from identical mouse at multiple time points. A two-sided p value of less than 0.05 was considered statistically significant.
RESULTS
The in vivo experiments for histological and echocardiographic analyses were performed separately, and the difference in sample sizes for each group is therefore due to different series of experiments.
AMD3100 treatment reduces infarct size and improves cardiac performance after IR injury
IR injury was induced by surgically occluding the LAD for 60 minutes, and AMD3100 (125 ug in 100 uL) or an equal volume of saline was injected subcutaneously immediately after surgery was complete. Infarct size and the area at risk (AAR) for infarction were evaluated 3 days after IR injury by briefly re-occluding the LAD, perfusing the hearts with microspheres, and then staining sections of heart tissue with TTC (Figure 1A); viable tissue was stained deep red, the infarcted region remained colorless, and the AAR was identified by the absence of microspheres. The size of the AAR was similar in both treatment groups (Figure 1B), but the infarcted regions were significantly smaller in AMD3100-treated mice than in mice administered saline (Figure 1C). AMD3100 treatment was also associated with significantly less apoptosis on day 3 (Figure 1D) and with significantly less fibrosis on day 28 (Figure 1E) after IR injury.
Figure 1.
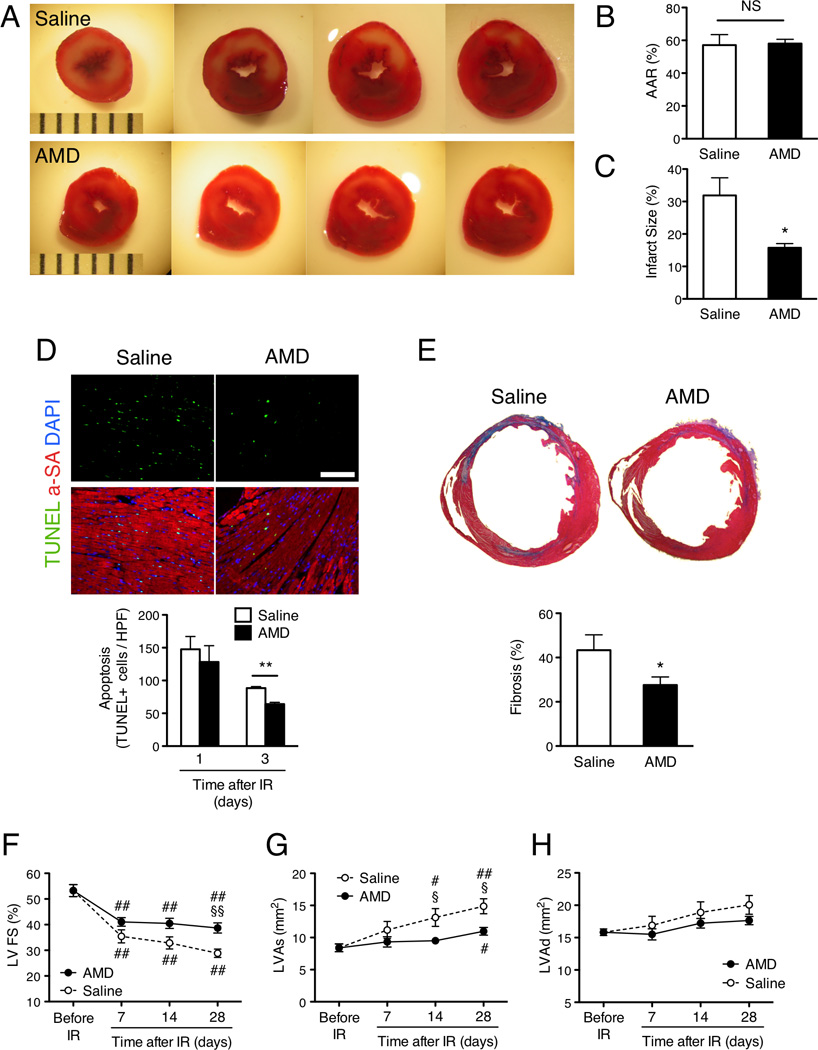
AMD3100 treatment improves cardiac function and reduces infarct size after IR injury. Mice were treated with saline alone or with 100 uL saline containing 125 ug AMD3100 after surgically induced IR injury. (A–C) AAR and infarct size were evaluated 3 days after IR injury via in-vivo microsphere perfusion and TTC-staining. (A) Viable tissue was stained deep red, and the infarcted region is colorless; scale=1 mm increments. (B) The AAR was identified by the absence of microspheres and presented as a percentage of the total left-ventricular (LV) area. (C) Infarct size was normalized to the size of the AAR and presented as a percentage. (D) Apoptosis was evaluated in TUNEL-stained sections of heart tissue from mice sacrificed 1 and 3 days after IR injury; scale bar=100 um. (E) Fibrosis was evaluated in Masson-trichrome-stained heart sections from mice sacrificed 28 days after IR injury, quantified as the ratio of the length of fibrosis (blue) to the LV circumference, and presented as a percentage. (F–H) Echocardiographic assessments of (F) LV fractional shortening (G) (FS), LV systolic area (LVAs), and (H) LV diastolic area (LVAd) were performed before IR injury and 7–28 days afterward; heart rates were maintained at 400–500 beats/minute via isoflurane inhalation. #P<0.05 and ##P<0.01 versus before injection; *P<0.05, **P<0.01, §Bonferroni-adjusted P<0.05 and §§Bonferroni-adjusted P<0.01 versus saline; NS, not significant. Panels B, C: n=3 per treatment group; Panel D: n=3–5 per treatment group; Panel E: n=6–9 per treatment group; Panels F-H: n=10 per treatment group at each time point.
Cardiac function was measured before IR injury and 7, 14, and 28 days afterward via echocardiographic assessments of left-ventricular (LV) fractional shortening (FS), left-ventricular systolic area (LVAs), and left-ventricular diastolic area (LVAd). FS was significantly greater, and LVAs was significantly smaller, in mice administered AMD3100 than in saline-treated mice on day 14 and day 28 after IR injury, while LVAd did not differ significantly between groups at any time point (Figures 1F–H, Supplementary Tables 2 and 3). Collectively, the results from these histological and echocardiographic assessments suggest that a single injection of AMD3100 after IR injury improves cardiac performance by enhancing the preservation and/or recovery of functional myocardial tissue.
AMD3100 preferentially enhances the mobilization of BM cells after IR injury
Because AMD3100 is a CXCR4 antagonist and has been shown to enhance the mobilization of stem/progenitors from BM to PB after permanent ligation of the coronary artery,17 we investigated whether AMD3100 treatment enhanced the mobilization of MNCs, CXCR4+ MNCs, and sca1+/flk1+ MNCs in both uninjured mice and in mice with IR injury. PB levels of the three-types of cells were measured via fluorescence-activated cell sorting (FACS), and FACS measurements of sca1+/flk1+ MNC levels were corroborated via the EPC culture assay. Tie2+ BM progenitor mobilization was also evaluated by monitoring GFP expression in the BM of WT mice transplanted with BM from Tie2-GFP transgenic mice, which express GFP from the endothelial-specific Tie2 promoter.
In the absence of injury, FACS analyses indicated that PB levels of MNCs including sca1+/flk1+ subpopulation tended to increase (Figures 2A and 2C) and CXCR4+ MNCs significantly increased after AMD3100 treatment. (Figures 2B) The time course of mobilization in the three cell types was almost similar: cell counts tended to peak within 3 hours after AMD3100 administration and returned to near-pretreatment levels by hour 24. (Figures 2A–C) After IR injury, AMD3100 treatment did not alter PB MNC levels (Figure 2D), but PB CXCR4+ MNC counts (Figure 2E) and sca1+/flk1+ MNC counts (Figure 2F) were significantly higher 1 and 3 days, respectively, after injury in AMD3100-treated mice than in mice administered saline. The enhanced mobilization of CXCR4+ MNCs diminished by day 3, whereas PB sca1+/flk1+ MNC counts remained significantly higher in the AMD3100-treated mice than in the saline-treatment group through day 7. When evaluated via culture assay, PB EPC levels (i.e., the number of cells stained positively for both lectin and acetylated low-density lipoprotein [acLDL]) were significantly higher in the AMD3100-treated mice than in the saline-treated mice on day 3 and day 7 after IR injury (Supplementary Figure 1), and in mice with Tie2-GFP BM, GFP-expressing cells were significantly less common in BM from the AMD3100-treatment group than in BM from saline-treated animals on day 5 after IR injury (Figure 2G). Thus, AMD3100 appears to rapidly, but briefly, enhance the mobilization of CXCR4+ MNCs after IR injury, which is consistent with the role of AMD3100 as a CXCR4 antagonist. However, the effect of AMD3100 on sca1+/flk1+ MNC mobilization is delayed, more durable, and, consequently, likely mediated by a different mechanism.
Figure 2.

AMD3100 enhances the mobilization of circulating CXCR4+ MNCs and Sca1/Flk1 positive cells after IR injury. (A–C) Peripheral-blood (PB) levels of (A) MNCs, (B) CXCR4+ MNCs, and (C) sca1+/flk1+ cells were determined in uninjured mice before injection of AMD3100 (125 ug in100 uL saline) and from 1–24 hours afterward. (D–F) PB levels of (D) MNCs, (E) CXCR4+ MNCs, and (F) sca1+/flk1+ cells were determined in mice before IR injury and treatment with AMD3100 or saline and from 1–28 days afterward. MNC levels were measured with a HemaVet hematology system, and the levels of CXCR4+ MNCs and sca1+/flk1+ cells were measured via FACS analyses of MNCs labeled with fluorescent CXCR4 antibodies (CXCR4+ MNCs) or double-labeled with fluorescent Sca1 antibodies and Flk1 antibodies. (G) IR injury was surgically induced in WT mice that had been transplanted with BM from mice with Tie2-regulated GFP expression. Mice were treated with AMD3100 or saline after injury, and the number of GFP+ BM cells was determined 5 days later; scale bar=100 um. #Bonferroni-adjusted P<0.05 and ##Bonferroni-adjusted P<0.01 versus before injection/IR; §Bonferroni-adjusted P<0.05, §§Bonferroni-adjusted P<0.01 and **P<0.01 versus saline. Panels A–C: n=3, Panels D–F: n=3–5 per treatment group at each time point, Panel G: n=8 per treatment group. SEM are too small to be visible graphically for Panel A, Hour 1; Panel B, Hour 1 and Hour 24; Panel E, Before IR, Day 7 (saline), and Day 28; and Panel F, Day 7 (saline) and Day 28.
AMD3100 increases the contribution of BM-derived progenitors to vascular growth after IR injury
To determine whether the enhanced BMPC mobilization observed in mice treated with AMD3100 after IR injury was accompanied by improved vascularity in the AAR, the functional vasculature of injured mice was stained via in-vivo perfusion with BS-1 lectin before sacrifice. Experiments were performed in WT mice transplanted with BM from eGFP-expressing mice to enable identification of BM-derived cells in the vasculature. Compared to observations in saline-treated mice, the AAR of AMD3100-treated mice contained significantly more GFP+ cells on day 3 after injury (Figures 3A and 3B) and significantly more GFP+ cells, GFP-lectin double-positive cells (Figures 3C and 3D), and lectin+ vessel density (Figures 3C and 3E) on day 28. AMD3100 treatment also kept elevated SDF-1 expression levels in AAR through day 3 to day 7 after IR injury in mice with WT BM (Supplementary Figure 2), which likely contributed to the enhanced incorporation of BM-derived cells, perhaps, including EPCs.
Figure 3.

AMD3100 increases capillary density and the number of BM-derived endothelial cells in the myocardium after IR injury. AMD3100 or saline was subcutaneously injected after IR injury in WT mice that had been transplanted with BM from GFP-expressing mice. (A, B) BM-derived (i.e., GFP-expressing) cells (green) were identified in the AAR and quantified on day 1, day 3, and day 28 after injury; scale bar=100 um. (C–E) On day 28 after IR injury, mice were perfused with BS1-lectin before sacrifice, and sections from the AAR were stained with fluorescent anti-lectin antibodies. (C) BM-derived endothelial cells were quantified as the number of cells positive for both GFP expression and lectin staining; scale bar=100 um. (D) BM-derived cells (green, GFP fluorescence) and functional vascular structures (red, lectin fluorescence) were identified in the AAR, and (E) capillary density was quantified as the number of lectin+ vascular structures. *P<0.05 and **P<0.01 versus saline. Panel B: n=3–6 per treatment group at each time point; Panels D, E: n=3–4 per treatment group.
AMD3100 increases BM eNOS expression and the number of eNOS-expressing BM-derived cells in the AAR after IR injury
Endothelial nitric oxide synthase (eNOS) is a key regulator of endothelial-cell growth and migration, vascular remodeling, and angiogenesis21–23 and has recently been shown to have an important role in the activity of stem and progenitor cells. We investigated whether the enhanced functional recovery and BMPC mobilization associated with AMD3100 administration after IR injury is accompanied by increases in eNOS activity.
From day 1 through day 7 after IR injury, eNOS-expressing cells were significantly more common in the BM of AMD3100-treated mice than in the BM of mice administered saline (Figure 4A). AMD3100 treatment was also associated with higher BM protein levels of matrix metalloproteinase 9 (MMP-9) and soluble Kit ligand (sKitL),24 two downstream components of the eNOS pathway, from day 1 and day 3, respectively, through day 7 (Figures 4B and 3C) and with higher PB levels of nitrate and nitrite (i.e., the final metabolites of nitric oxide) on day 3 (Figure 4D). In mice transplanted with BM from transgenic GFP-expressing mice, the number of cells in the AAR that expressed eNOS, GFP, or both eNOS and GFP was significantly higher in AMD3100-treated mice than in saline-treated mice on day 3 after injury (Figures 4E–H). Thus, AMD3100 administration after IR injury appears to increase eNOS activity in both the BM and the ischemic region.
Figure 4.
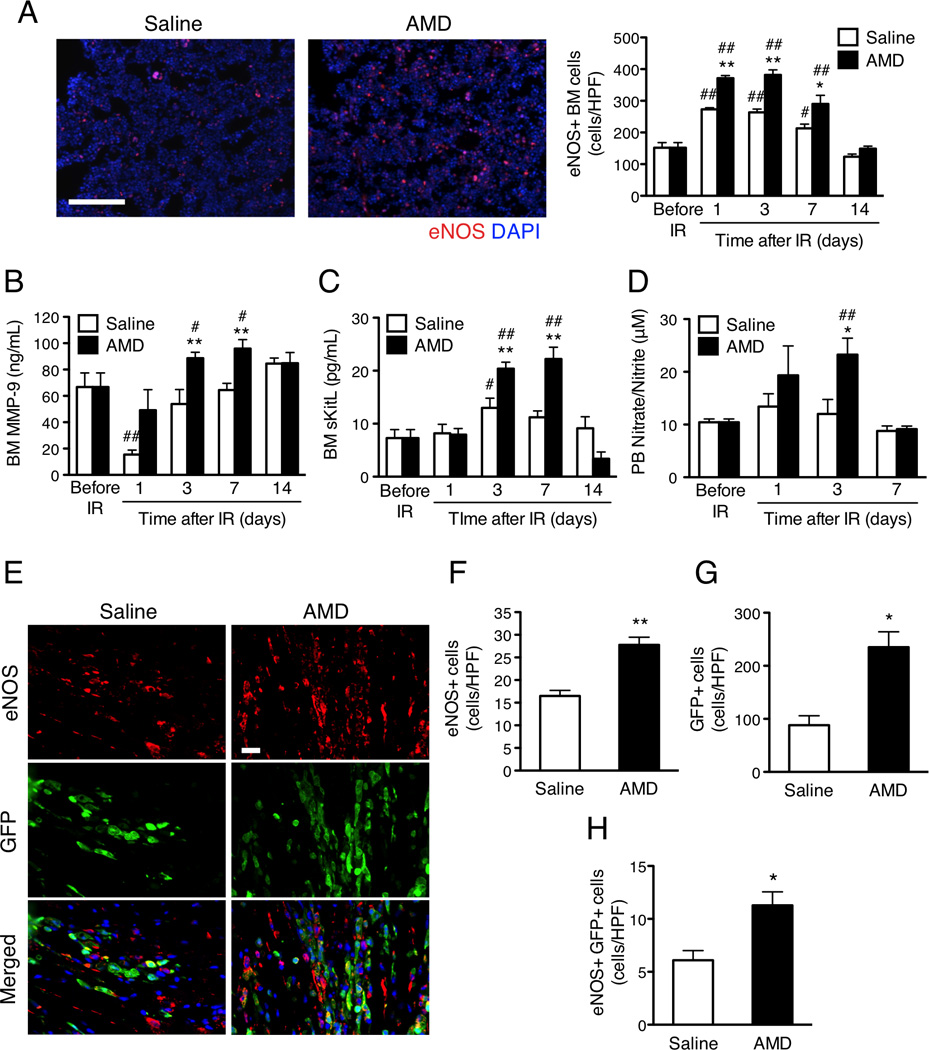
AMD3100 increases BM eNOS expression and the number of eNOS-expressing BM-derived cells in the AAR after IR injury. AMD3100 or saline was subcutaneously injected after IR injury in (A–D) WT mice and (E–H) WT mice that had been transplanted with BM from GFP-expressing mice. (A–C) BM was harvested from mice sacrificed before IR injury and from 1–14 days afterward. (A) BM cells were labeled with fluorescent anti-eNOS antibodies (red), and eNOS+ cells were quantified; scale bar=100 um. (B, C) BM plasma levels of (B) MMP-9 and (C) sKitL protein were determined via ELISA. (D) PB levels of nitrate and nitrite were determined via colorimetric assay before IR injury and 1–14 days afterward. (E) eNOS (red) and GFP (green) expression was evaluated in sections from the AAR of mice sacrificed 3 days after IR injury; scale bar=20 um. Cells positive for the expression of (F) eNOS, (G) GFP, or (H) both eNOS and GFP were quantified. #P<0.05 and ##P<0.01 versus before injection; *P<0.05 and **P<0.01 versus saline. Panels A–D: n=4–8 per treatment group at each time point; Panels F–H: n=3–5 per treatment group.
The direct influence of AMD3100 on eNOS activity was investigated by determining whether AMD3100 treatment altered eNOS mRNA expression and nitrate/nitrite production in cultured BMPCs or luciferase activity in murine endothelial cells transfected with a gene coding for luciferase expression from the eNOS promoter. AMD3100 treatment was associated with higher levels of both eNOS expression and nitrate/nitrite production in BMPCs (Supplementary Figures 3A and 3B), and AMD3100 dose-dependently increased luciferase activity in transfected endothelial cells (Supplementary Figure 3C). Collectively, these observations suggest that the benefits associated with AMD3100 administration are accompanied by increases in eNOS activity.
The benefit of AMD3100 treatment after IR injury is dependent on eNOS expression in the BM but not in the ischemic region
To determine whether eNOS expression contributes to the benefits associated with AMD3100 administration after IR injury, and if so, whether that contribution comes from BM cells or from cells already present in the ischemic tissue, the influence of AMD3100 on myocardial recovery was evaluated in eNOS-knockout mice that had been transplanted with BM from WT mice (eNOS-KO/WTBM) and in WT mice transplanted with BM from eNOS-KO mice (WT/eNOS-KOBM). An identical set of assessments was performed in WT mice transplanted with WT BM (WT/WTBM).
In eNOS-KO/WTBM mice, left-ventricular FS on day 14 and day 28 after IR injury was significantly greater with AMD3100 treatment than with saline treatment, but the functional benefit of AMD3100 treatment was not observed in WT/eNOS-KOBM mice at any time point (Figures 5A and 5B, Supplementary Table 2). Similarly, AMD3100 treatment in eNOS-KO/WTBM mice, but not in WT/eNOS-KOBM mice, was associated with greater numbers of eNOS+ BM cells, elevated BM MMP-9 and sKitL protein expression, higher PB sca1+/flk1+ MNC counts, less cardiac apoptosis, and smaller infarcts on day 3 after injury (Figures 5D–I), and with less cardiac fibrosis and greater capillary density on day 28 (Figures 5J and 5K). The results associated with AMD3100 treatment in WT/WTBM mice matched those observed in eNOS-KO/WTBM mice (Figures 5C–K), and the only treatment-related effect observed in all three chimeric mouse lines was the enhanced mobilization of CXCR4+ MNCs, which occurred on day 1 after injury and diminished by day 3 (Figure 5L). Thus, the benefits associated with AMD3100 administration after IR injury require eNOS expression in the BM, but not in the ischemic region, and eNOS appears to have a role in the mobilization of sca1+/flk1+ MNCs, but not CXCR4+ MNCs.
Figure 5.
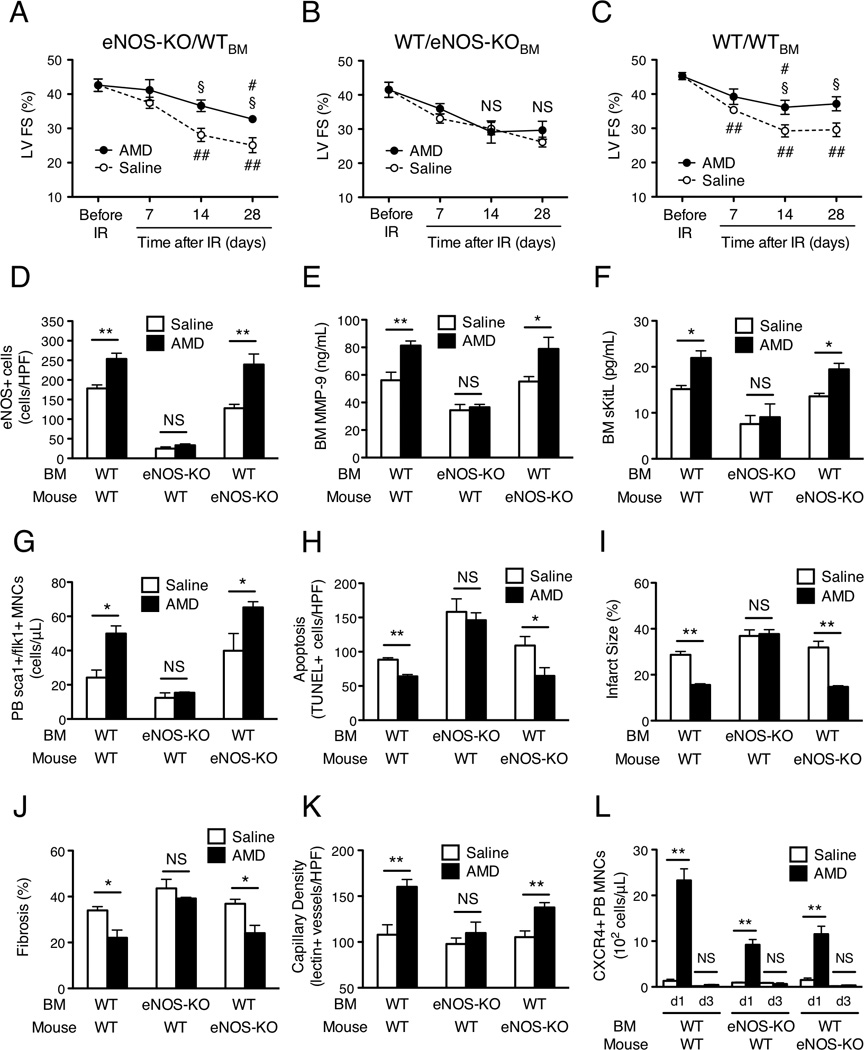
The benefit of AMD3100 treatment after IR injury is dependent on eNOS expression in the bone marrow but not in the ischemic tissue. AMD3100 or saline was subcutaneously injected after IR injury in eNOS-knockout mice that had been transplanted with BM from WT mice (eNOS-KO/WTBM), in WT mice transplanted with BM from eNOS-KO mice (WT/eNOS-KOBM), and in WT mice transplanted with WT BM (WT/WTBM). (A–C) Echocardiographic assessments of LV fractional shortening (FS) were performed before IR injury and 7–28 days afterward. (D–F) BM was harvested 3 days after IR injury. (D) BM cells were labeled with fluorescent anti-eNOS antibodies, and positively stained cells were quantified. (E, F) BM plasma levels of (E) MMP-9 and (F) sKitL protein were determined via ELISA. (G) Three days after IR injury, MNCs were isolated from the PB, labeled with fluorescent anti-Sca1 and anti-Flk1 antibodies, and the number of MNCs positive for both Sca1 and Flk1 expression were determined via FACS. (H) Sections from the AAR of mice sacrificed on day 3 after IR injury were TUNEL stained, and apoptosis was quantified as the number of positively stained cells. (I) Infarct size and AAR were assessed via in-vivo microsphere perfusion and TTC staining in mice sacrificed on day 3 after IR injury; the ratio of the area of the infarct to the AAR was presented as a percentage. (J) Fibrosis on day 28 after IR injury was assessed in Masson-trichrome–stained heart sections, quantified as the ratio of the length of fibrosis to the LV circumference, and presented as a percentage. (K) Capillary density was assessed in mice that had been perfused with BS1-lectin before sacrifice on day 28 after IR injury and quantified as the number of lectin+ vascular structures. (L) MNCs were harvested from the PB on day 1 (d1) and day 3 (d3) after IR injury, labeled with fluorescent anti-CXCR4 antibodies, and the number of CXCR4+ MNCs was determined via FACS. ## Bonferroni-adjusted P<0.01 versus before injection; §Bonferroni-adjusted P<0.05, *P<0.05 and **P<0.01 versus saline; NS, not significant. Panels A–C: n=4–8 per treatment group at each time point; Panel D: n=3–4 per treatment group; Panel E: n=4–8 per treatment group; Panel F: n=5–9 per treatment group; Panels G–J: n=3–6 per treatment group; Panel K: n=3 per treatment group; Panel L: n=3–6 per treatment group at each time point. SEM is too small to be visible graphically for Panel C, Day 7 (saline)
The expression of eNOS by PB EPCs contributes to myocardial recovery but is not required for EPC incorporation
Because BM eNOS expression is required for both the beneficial effects of AMD3100 treatment after IR injury and for the mobilization of progenitors from BM to PB, we investigated whether eNOS expression in circulating BMPCs contributes to myocardial recovery. BMPCs were isolated from WT mice and eNOS-KO mice, cultured for 4 days, and then intravenously injected into WT mice 24 hours after IR injury; a third group of mice was injected with saline. Fourteen and 28 days after IR injury, LV FS was significantly greater in mice administered WT BMPCs than in mice administered saline, but the difference between treatment with eNOS-KO BMPCs and saline administration did not reach statistical significance (Figure 6A, Supplementary Table 2).
Figure 6.

The expression of eNOS by circulating bone marrow progenitor cells (BMPCs) contributes to myocardial recovery. (A) BMPCs from WT mice, BMPCs from eNOS-KO mice, or saline were intravenously injected into WT mice 24 hours after IR injury. Echocardiographic assessments of LV fractional shortening (FS) were performed before IR injury and 7–28 days afterward. (B–D) DiI-labeled WT BMPCs, DiI-labeled eNOS-KO BMPCs, or saline was intravenously injected into WT mice 24 hours after IR injury and treatment with subcutaneous injections of AMD3100 or saline; mice were sacrificed 2 days later (i.e., 3 days after IR injury). §Bonferroni-adjusted P<0.05 versus saline; #P<0.05 and ##P<0.01 versus before injection. (B) Incorporation of the injected cells was evaluated by quantifying the number of DiI-positive cells in the AAR; scale bar=100 um. (C) Sections from the AAR were TUNEL-stained (green), stained with fluorescent anti-alpha-sarcomeric actin antibodies (red), and counterstained with DAPI (blue); scale bar=100 um. Apoptosis was quantified as the number of TUNEL+ cells. (D) Infarct size and AAR were assessed via in-vivo microsphere perfusion and TTC staining; viable tissue appears deep red and the infarcted region is colorless. The ratio of the area of the infarct to the AAR was presented as a percentage. *P<0.05 and **P<0.01; #P<0.05 and ##P<0.01 versus the same intravenous injection (i.e., saline, WT BMPCs, or eNOS-KO BMPCs) in animals treated with subcutaneous injections of saline; NS, not significant. Panel A: n=4–5 per treatment group at each time point; Panel B: n=3–5 per group; Panel C: n=3–7 per group; Panel D: n=4–7 per group.
To determine whether the expression of eNOS by circulating BMPCs contributes to myocardial recovery by increasing the incorporation, and whether AMD3100 enhances the incorporation of circulating BMPCs, WT and eNOS-KO EPCs were DiI-labeled and intravenously injected into WT mice 24 hours after IR injury and treatment with AMD3100 or saline; mice were sacrificed 3 days after IR injury for histological analyses. Neither the type of cell injected (i.e., eNOS-KO or WT) nor the treatment administered (i.e., AMD3100 or saline) significantly influenced the incorporation of injected cells (Figure 6B). Nevertheless, apoptotic cells were significantly less common, and infarct sizes were significantly smaller, in mice administered WT BMPCs than in mice administered eNOS-KO BMPCs, regardless of treatment group, and measurements in mice administered eNOS-KO BMPCs did not differ significantly from those in saline-treated mice (Figures 6C and 6D).
Collectively, these observations suggest that the expression of eNOS by circulating BMPCs does not have a role in their recruitment and incorporation, but does contribute to cardiac protection. Therefore, the greater number of BM-derived endothelial cells observed in the AAR of AMD3100-treated mice (Figure 3C) appears to evolve primarily through enhanced BMPC mobilization, which subsequently increases the number of the progenitors available in the circulation, rather than by directly influencing the recruitment and incorporation of circulating cells.
DISCUSSION
In the present study, we have shown that AMD3100, a CXCR4 antagonist, had a beneficial effect on cardiac IR injury that closely mimics coronary intervention in AMI patients. AMD3100 enhanced the mobilization of BM-derived progenitor cells, including EPCs, which were incorporated into AAR and exerted cardioprotective effects by anti-apoptosis and anti-inflammation in acute phase and also revascularizing effects in remote phase, resulting in minimization of scar size and preservation of cardiac functions.
AMD3100 has been approved by the US Food and Drug Administration for use as a stem cell mobilizing agent25; however, few reports have evaluated the time course and subpopulations of cells mobilized by AMD3100 administration after IR injury. Our results indicate that rapid accumulation of circulating MNCs specifically inflammatory cells into AAR account for the decreased number of MNCs in PB on day 1 after IR injury. On the other hand, IR injury could be a trigger to mobilize progenitors (i.e. CXCR4+ or sca-1/flk1+ MNCs including EPCs) from BM resulting in the increased number of progenitors in PB. AMD3100 increased the mobilization of CXCR4+ cells on day 1 after IR injury by disrupting SDF-1–CXCR4 binding in BM, but this enhancement dissipated before the expression of SDF-1, a ligand of CXCR4, was upregulated in ischemic myocardium on day 3 following IR injury. The delay in SDF-1 upregulation may explain why the rapid mobilization of CXCR4+ cells did not result in dramatic increase of BM-derived cell recruitment to the AAR on day 1 after IR injury. The AMD3100-induced delayed enhancement of sca1/flk1+ MNC mobilization coincided with elevated BM levels of eNOS and two eNOS-targeted proteins, MMP-9 and sKitL that have been linked to progenitor-cell mobilization26,27 and the enhanced mobilization of progenitors required BM eNOS expression.
eNOS is known to protect cardiomyocytes against apoptosis,28, 29 and AMD3100 treatment led to increases in the number of eNOS-expressing cells, to declines in the number of apoptotic cells, and to reduction of infarct sizes in AAR on day 3 after IR injury. Similarly, systemically injected eNOS expressing WT BMPCs, but not eNOS-KO BMPCs, were associated with less apoptosis and smaller infarct sizes in both AMD3100-treated and saline-treated mice on day 3 after injury, even though the number of recruited cell in both cell types in AAR was similar. Additionally, in IR injured mice without AMD3100 treatment, eNOS expression in BM was upregulated by myocardial transient ischemia alone from day 1 through day 7, therefore, AMD3100 was assumed to give an extra elevation of eNOS production in BM cells over ischemic insult.
In the mechanistic aspect, it was reported that eNOS expressing cells enhanced VEGF protein production,30 and also, as known broadly, VEGF upregulates eNOS expression, suggesting that initial upregulation of eNOS expression sustained up to day 7 after IR injury with AMD3100 treatment via an autocrine mechanism involving VEGF-eNOS signaling pathway. We previously showed that VEGF played a crucial role to mobilize progenitors from BM into PB in a mouse coronary-ligation model. However, in the present study, we confirmed a direct effect of AMD3100 on eNOS upregulation by reporter assay (Supplemental Figure 3C). Also, eNOS mRNA expression was upregulated more than 2-fold in the AMD3100-treated BMPCs compared to control BM PCs, while VEGF mRNA expression limited to less than 2-fold increase in the AMD3100-treated group (Supplementary Figure 4). Thus, AMD3100 could directly enhance eNOS production in the recruited cultured BMPCs in ischemic myocardium, suggesting that AMD3100 might exhibit cardioprotective effect via the recruited “BM PC”-derived eNOS production, at least in part, in the WT and eNOS-KO BMPC injection experiment. In addition, based on the evidences that eNOS promoted angiogenesis and reduced apoptosis by inhibition of TGF-β1 signaling,31 imported BMPC-derived eNOS may also contribute to enhance capillary density after IR injury via a similar mechanism. Thus, the cardioprotective effects of AMD3100-induced eNOS activity are a crucial component of the response to IR injury, but these effects appear to evolve primarily from BM-derived progenitors, rather than from cells already present in AAR.
In terms of contribution of the mobilized BM-derived progenitors by AMD3100 to ischemic myocardium, though there is no direct evidence that circulating CXCR4+ cells incorporated into AAR in acute phase after IR, the peak number of mobilized CXCR4+ cells was striking (10 times more than that of mobilized sca1/flk1+ cells) and CXCR4+ cells may therefore play a role in AAR. Referring to the reports in which CXCR4 expressing cells were shown to enhance incorporation into ischemic area and improve cardiac function after myocardial infarction32, 33 and the fact that a plasma half-life of AMD3100 is very short (3.5 hours) in circulation, mobilized CXCR4+ cells might not be interfered with its recruitment to SDF-1 releasing sites of ischemia at day 1–3 after IR injury. Even though eNOS KO- or WT-cultured BMPC infusion study did not show significant difference of “cultured BMPC” recruitment to sites of ischemia, endogenous progenitors namely CXCR4+ cells and sca1/flk1+ cells mobilized by AMD3100 might recruit to ischemic myocardium exhibiting cardioprotective effects, because SDF-1 and VEGF are released from ischemic myocardium and recruit circulating progenitors expressing receptors of CXCR432 and flk1.34
In conclusion, the findings presented here indicate that AMD3100 improves the recovery of cardiac function after IR injury, and that the beneficial effect of AMD3100 on ischemic heart with IR injury requires eNOS expression in BM but not in myocardium. AMD3100 sustains the mobilization of sca1/flk1+ MNCs rather than CXCR4+ MNCs resulting in the increased number of recruited BM-derived eNOS-expressing cells, and contributes to limit infarct size reducing cardiac apoptosis and increasing vascularity in ischemic myocardium. Collectively, single treatment with AMD3100 may give rise to a novel supportive therapy in PTCA and stenting for acute coronary syndrome via a cardioprotection-/pro-angiogenesis-dependent mechanism.
Study limitation
In this study, we have not definitively identified the mobilized BM-derived cells by AMD3100 including sca1+/flk1+ MNCs and CXCR4+ cells providing direct evidences for the incorporation into vascular structure, however, we and others have previously demonstrated that not the majority but a certain extent of these BM-derived cells were exactly incorporated into neovasculature in ischemic tissue.32, 33, 35–37 Also, since we have focused on the therapeutic effect of AMD3100 with its eNOS-dependent BMPC mobilization in peripheral blood and the recruitment to sites of IR injury, we have not tried to identify what type of cells, so-called EPCs, defined by multiple cell surface markers are incorporated into neovasculature. The precise definition of human/mouse EPCs still remains unclear, but it would be substantially difficult.
Supplementary Material
ACKNOWLEDGMENTS
We thank Kari Krueger for administrative assistance and W. Kevin Meisner, PhD, ELS for editorial support. The work presented in this manuscript was performed at the Feinberg Cardiovascular Research Institute, Northwestern University Feinberg School of Medicine, Chicago, IL.
FUNDING SOURCES: This work was supported in part by the US National Institutes of Health (grants: HL053354-14, HL057516-12, HL080137-05, HL095874-04, HL093439, and HL113541).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
CONFLICT OF INTEREST DISCLOSURES: Dr. Losordo is an employee of Baxter Healthcare.
References
- 1.Murasawa S, Kawamoto A, Horii M, Nakamori S, Asahara T. Niche-dependent translineage commitment of endothelial progenitor cells, not cell fusion in general, into myocardial lineage cells. Arterioscler Thromb Vasc Biol. 2005;25:1388–1394. doi: 10.1161/01.ATV.0000168409.69960.e9. [DOI] [PubMed] [Google Scholar]
- 2.Shintani S, Murohara T, Ikeda H, Ueno T, Honma T, Katoh A, Sasaki K, Shimada T, Oike Y, Imaizumi T. Mobilization of endothelial progenitor cells in patients with acute myocardial infarction. Circulation. 2001;103:2776–2779. doi: 10.1161/hc2301.092122. [DOI] [PubMed] [Google Scholar]
- 3.Brenner W, Aicher A, Eckey T, Massoudi S, Zuhayra M, Koehl U, Heeschen C, Kampen WU, Zeiher AM, Dimmeler S, Henze E. 111in-labeled cd34+ hematopoietic progenitor cells in a rat myocardial infarction model. J Nucl Med. 2004;45:512–518. [PubMed] [Google Scholar]
- 4.Mohle R, Bautz F, Rafii S, Moore MA, Brugger W, Kanz L. The chemokine receptor cxcr-4 is expressed on cd34+ hematopoietic progenitors and leukemic cells and mediates transendothelial migration induced by stromal cell-derived factor-1. Blood. 1998;91:4523–4530. [PubMed] [Google Scholar]
- 5.Peled A, Grabovsky V, Habler L, Sandbank J, Arenzana-Seisdedos F, Petit I, Ben-Hur H, Lapidot T, Alon R. The chemokine sdf-1 stimulates integrin-mediated arrest of cd34(+) cells on vascular endothelium under shear flow. J Clin Invest. 1999;104:1199–1211. doi: 10.1172/JCI7615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yamaguchi J, Kusano KF, Masuo O, Kawamoto A, Silver M, Murasawa S, Bosch-Marce M, Masuda H, Losordo DW, Isner JM, Asahara T. Stromal cell-derived factor-1 effects on ex vivo expanded endothelial progenitor cell recruitment for ischemic neovascularization. Circulation. 2003;107:1322–1328. doi: 10.1161/01.cir.0000055313.77510.22. [DOI] [PubMed] [Google Scholar]
- 7.Aiuti A, Webb IJ, Bleul C, Springer T, Gutierrez-Ramos JC. The chemokine sdf-1 is a chemoattractant for human cd34+ hematopoietic progenitor cells and provides a new mechanism to explain the mobilization of cd34+ progenitors to peripheral blood. J Exp Med. 1997;185:111–120. doi: 10.1084/jem.185.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang JF, Liu ZY, Groopman JE. The alpha-chemokine receptor cxcr4 is expressed on the megakaryocytic lineage from progenitor to platelets and modulates migration and adhesion. Blood. 1998;92:756–764. [PubMed] [Google Scholar]
- 9.Lataillade JJ, Clay D, Dupuy C, Rigal S, Jasmin C, Bourin P, Le Bousse-Kerdiles MC. Chemokine sdf-1 enhances circulating cd34(+) cell proliferation in synergy with cytokines: Possible role in progenitor survival. Blood. 2000;95:756–768. [PubMed] [Google Scholar]
- 10.Este JA, Cabrera C, De Clercq E, Struyf S, Van Damme J, Bridger G, Skerlj RT, Abrams MJ, Henson G, Gutierrez A, Clotet B, Schols D. Activity of different bicyclam derivatives against human immunodeficiency virus depends on their interaction with the cxcr4 chemokine receptor. Mol Pharmacol. 1999;55:67–73. doi: 10.1124/mol.55.1.67. [DOI] [PubMed] [Google Scholar]
- 11.Gerlach LO, Skerlj RT, Bridger GJ, Schwartz TW. Molecular interactions of cyclam and bicyclam non-peptide antagonists with the cxcr4 chemokine receptor. J Biol Chem. 2001;276:14153–14160. doi: 10.1074/jbc.M010429200. [DOI] [PubMed] [Google Scholar]
- 12.Hatse S, Princen K, Bridger G, De Clercq E, Schols D. Chemokine receptor inhibition by amd3100 is strictly confined to cxcr4. FEBS Lett. 2002;527:255–262. doi: 10.1016/s0014-5793(02)03143-5. [DOI] [PubMed] [Google Scholar]
- 13.Hendrix CW, Flexner C, MacFarland RT, Giandomenico C, Fuchs EJ, Redpath E, Bridger G, Henson GW. Pharmacokinetics and safety of amd-3100, a novel antagonist of the cxcr-4 chemokine receptor, in human volunteers. Antimicrob Agents Chemother. 2000;44:1667–1673. doi: 10.1128/aac.44.6.1667-1673.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA, Liles WC, Li X, Graham-Evans B, Campbell TB, Calandra G, Bridger G, Dale DC, Srour EF. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with amd3100, a cxcr4 antagonist. J Exp Med. 2005;201:1307–1318. doi: 10.1084/jem.20041385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liles WC, Broxmeyer HE, Rodger E, Wood B, Hubel K, Cooper S, Hangoc G, Bridger GJ, Henson GW, Calandra G, Dale DC. Mobilization of hematopoietic progenitor cells in healthy volunteers by amd3100, a cxcr4 antagonist. Blood. 2003;102:2728–2730. doi: 10.1182/blood-2003-02-0663. [DOI] [PubMed] [Google Scholar]
- 16.Devine SM, Flomenberg N, Vesole DH, Liesveld J, Weisdorf D, Badel K, Calandra G, DiPersio JF. Rapid mobilization of cd34+ cells following administration of the cxcr4 antagonist amd3100 to patients with multiple myeloma and non-hodgkin's lymphoma. J Clin Oncol. 2004;22:1095–1102. doi: 10.1200/JCO.2004.07.131. [DOI] [PubMed] [Google Scholar]
- 17.Jujo K, Hamada H, Iwakura A, Thorne T, Sekiguchi H, Clarke T, Ito A, Misener S, Tanaka T, Klyachko E, Kobayashi K, Tongers J, Roncalli J, Tsurumi Y, Hagiwara N, Losordo DW. Cxcr4 blockade augments bone marrow progenitor cell recruitment to the neovasculature and reduces mortality after myocardial infarction. Proc Natl Acad Sci U S A. 2010;107:11008–11013. doi: 10.1073/pnas.0914248107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Asahara T, Masuda H, Takahashi T, Kalka C, Pastore C, Silver M, Kearne M, Magner M, Isner JM. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res. 1999;85:221–228. doi: 10.1161/01.res.85.3.221. [DOI] [PubMed] [Google Scholar]
- 19.Asahara T, Takahashi T, Masuda H, Kalka C, Chen D, Iwaguro H, Inai Y, Silver M, Isner JM. Vegf contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. Embo J. 1999;18:3964–3972. doi: 10.1093/emboj/18.14.3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ii M, Nishimura H, Iwakura A, Wecker A, Eaton E, Asahara T, Losordo DW. Endothelial progenitor cells are rapidly recruited to myocardium and mediate protective effect of ischemic preconditioning via "imported" nitric oxide synthase activity. Circulation. 2005;111:1114–1120. doi: 10.1161/01.CIR.0000157144.24888.7E. [DOI] [PubMed] [Google Scholar]
- 21.Pipili-Synetos E, Sakkoula E, Maragoudakis ME. Nitric oxide is involved in the regulation of angiogenesis. Br J Pharmacol. 1993;108:855–857. doi: 10.1111/j.1476-5381.1993.tb13476.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ziche M, Morbidelli L, Masini E, Granger HJ, Gepetti P, Ledda F. Nitric oxide promotes DNA synthesis and cyclic gmp formation in endothelial cells from post capillary venules. Biochem Biophys Res Commun. 1993;192:1198–1203. doi: 10.1006/bbrc.1993.1543. [DOI] [PubMed] [Google Scholar]
- 23.Ziche M, Morbidelli L, Masini E, Amerini S, Granger HJ, Maggi CA, Geppetti P, Ledda F. Nitric oxide mediates angiogenesis in vivo and endothelial cell growth and migration in vitro promoted by substance p. J Clin Invest. 1994;94:2036–2044. doi: 10.1172/JCI117557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-Ihling K, Zeiher AM, Dimmeler S. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med. 2003;9:1370–1376. doi: 10.1038/nm948. [DOI] [PubMed] [Google Scholar]
- 25.Steinberg M, Silva M. Plerixafor: A chemokine receptor-4 antagonist for mobilization of hematopoietic stem cells for transplantation after high-dose chemotherapy for non-hodgkin's lymphoma or multiple myeloma. Clin Ther. 2010;32:821–843. doi: 10.1016/j.clinthera.2010.05.007. [DOI] [PubMed] [Google Scholar]
- 26.Aicher A, Heeschen C, Mildner-Rihm C, Urbich C, Ihling C, Technau-Ihling K, Zeiher AM, Dimmeler S. Essential role of endothelial nitric oxide synthase for mobilization of stem and progenitor cells. Nat Med. 2003;9:1370–1376. doi: 10.1038/nm948. [DOI] [PubMed] [Google Scholar]
- 27.Heissig B, Hattori K, Dias S, Friedrich M, Ferris B, Hackett NR, Crystal RG, Besmer P, Lyden D, Moore MA, Werb Z, Rafii S. Recruitment of stem and progenitor cells from the bone marrow niche requires mmp-9 mediated release of kit-ligand. Cell. 2002;109:625–637. doi: 10.1016/s0092-8674(02)00754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones SP, Girod WG, Palazzo AJ, Granger DN, Grisham MB, Jourd'Heuil D, Huang PL, Lefer DJ. Myocardial ischemia-reperfusion injury is exacerbated in absence of endothelial cell nitric oxide synthase. Am J Physiol. 1999;276:H1567–H1573. doi: 10.1152/ajpheart.1999.276.5.H1567. [DOI] [PubMed] [Google Scholar]
- 29.Razavi HM, Hamilton JA, Feng Q. Modulation of apoptosis by nitric oxide: Implications in myocardial ischemia and heart failure. Pharmacol Ther. 2005;106:147–162. doi: 10.1016/j.pharmthera.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 30.Mees B, Recalde A, Loinard C, Tempel D, Godinho M, Vilar J, van Haperen R, Levy B, de Crom R, Silvestre JS. Endothelial nitric oxide synthase overexpression restores the efficiency of bone marrow mononuclear cell-based therapy. Am J Pathol. 178:55–60. doi: 10.1016/j.ajpath.2010.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen LL, Yin H, Huang J. Inhibition of tgf-beta1 signaling by enos gene transfer improves ventricular remodeling after myocardial infarction through angiogenesis and reduction of apoptosis. Cardiovasc Pathol. 2007;16:221–230. doi: 10.1016/j.carpath.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 32.Zhang D, Fan GC, Zhou X, Zhao T, Pasha Z, Xu M, Zhu Y, Ashraf M, Wang Y. Over-expression of cxcr4 on mesenchymal stem cells augments myoangiogenesis in the infarcted myocardium. J Mol Cell Cardiol. 2008;44:281–292. doi: 10.1016/j.yjmcc.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morimoto H, Takahashi M, Shiba Y, Izawa A, Ise H, Hongo M, Hatake K, Motoyoshi K, Ikeda U. Bone marrow-derived cxcr4+ cells mobilized by macrophage colony-stimulating factor participate in the reduction of infarct area and improvement of cardiac remodeling after myocardial infarction in mice. Am J Pathol. 2007;171:755–766. doi: 10.2353/ajpath.2007.061276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li J, Brown LF, Hibberd MG, Grossman JD, Morgan JP, Simons M. Vegf, flk-1, and flt-1 expression in a rat myocardial infarction model of angiogenesis. Am J Physiol. 1996;270:H1803–H1811. doi: 10.1152/ajpheart.1996.270.5.H1803. [DOI] [PubMed] [Google Scholar]
- 35.Cho HJ, Lee N, Lee JY, Choi YJ, Ii M, Wecker A, Jeong JO, Curry C, Qin G, Yoon YS. Role of host tissues for sustained humoral effects after endothelial progenitor cell transplantation into the ischemic heart. J Exp Med. 2007;204:3257–3269. doi: 10.1084/jem.20070166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawamoto A, Gwon HC, Iwaguro H, Yamaguchi JI, Uchida S, Masuda H, Silver M, Ma H, Kearney M, Isner JM, Asahara T. Therapeutic potential of ex vivo expanded endothelial progenitor cells for myocardial ischemia. Circulation. 2001;103:634–637. doi: 10.1161/01.cir.103.5.634. [DOI] [PubMed] [Google Scholar]
- 37.Chavakis E, Aicher A, Heeschen C, Sasaki K, Kaiser R, El Makhfi N, Urbich C, Peters T, Scharffetter-Kochanek K, Zeiher AM, Chavakis T, Dimmeler S. Role of beta2-integrins for homing and neovascularization capacity of endothelial progenitor cells. J Exp Med. 2005;201:63–72. doi: 10.1084/jem.20041402. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.