

Abstract
We previously demonstrated that intra-peritoneal delivery of adeno-associated virus serotype 8 (AAV8) stably transduces the pancreas, including the β-cells in the endogenous islets. We also demonstrated the ability to deliver and express genes specifically in β-cells for at least 6 months using a murine insulin promoter (mIP) in a double-stranded, self-complementary AAV vector (dsAAV8-mIP). Here we evaluated the effects of dsAAV8-mIP mediated delivery of interleukin 4 (mIL-4) to endogenous β-cells in NOD mice. In 4 week old NOD mice, the extent of gene transfer and expression in endogenous β-cells following i.p. delivery of dsAAV8-mIP-eGFP was comparable to normal BALB/c mice. Furthermore, following i.p. delivery of dsAAV8-mIP-IL4, expression of mIL-4 was detected in islets isolated and cultured from the treated mice. AAV8-mIP mediated gene expression of mIL-4 to endogenous β-cells of 4 and 8 week old NOD mice prevented the onset of hyperglycemia in NOD mice and reduced the severity of insulitis. Moreover, expression of mIL-4 also maintained the level of CD4+CD25+FoxP3+ cells and adoptive co-transfer of splenocytes from diabetes-free IL-4 vector recipients and splenocytes from wild type diabetic NOD mice prevented the onset diabetes. Taken together, these results demonstrate that local expression of mIL-4 in islets prevents islet destruction and blocks autoimmunity, in part, through regulation of T cell function. These results also demonstrate the utility of using dsAAV8-mIP gene transfer to endogenous NOD β-cells to examine the role of specific gene products in preventing or exacerbating the onset of type 1 diabetes.
INTRODUCTION
Autoimmune type 1 diabetes (T1D) is an organ specific disease, characterized by infiltration of auto reactive T cells into the pancreas, leading to destruction of pancreatic islet β-cells. About 5%–10% of diabetes cases in North America are type 1, and the disease is most prominent among young children, with about 40 cases per 10,000 children and peak age of onset at 11–12 years of age. Insulin replacement can ameliorate the symptoms of the disease, but it has no effect on the autoimmune process, eventually leading to complications such as nephropathy, micro-vascular disease, blindness, and atherosclerosis. Preventing the progression of T1D in its early stages therefore requires controlling the autoimmune component of the disease. A murine model for human type 1 diabetes is the non-obese diabetic (NOD) mouse that, depending upon housing conditions and type of food, spontaneously develops insulitis around 4 weeks of age followed by the onset of hyperglycemia between 12 and 18 weeks of age.
T1D is mediated by T cells because it can be prevented by treatment with anti-T–cell antibodies [1, 2] and can be transferred to pre-diabetic animals with T cells from diabetic mice [3, 4]. T1D develops, in part, from a deficiency in the function of regulatory T cells (T-reg) that fail to control disease pathogenesis [5]. TH1 cells appear responsible for mediating autoimmunity in NOD mice, whereas endogenous TH2 and regulatory T cells can protect mice against diabetes development [3, 6]. The inflammatory process in early diabetes is initiated and propagated by the effect of TH1-secreted cytokines such as interferon (IFN-γ) and suppressed by TH2-secreted anti-inflammatory cytokines such as IL-4 and IL-10. Numerous studies suggest that the intrinsic TH1/TH2 balance in NOD mice is tilted toward TH1 and manipulations that correct this balance result in protection from destructive insulitis and diabetes [7, 8].
Delivery of TH2 cytokines prevents the onset of diabetes in NOD mice [3, 6], low dose streptozotocin-treated mice [9], and BioBreeding rats [10]. In particular, IL-4 and IL-10 have been delivered therapeutically as recombinant proteins[3], by gene transfer of expression plasmids[6, 11–13], by transplantation of genetically modified dendritic cells [7, 14] or by gene transfer using recombinant adeno-associated virus vectors[10]. Additionally, transgenic NOD mice expressing IL-4 specifically in β-cells has been used to demonstrate that pancreas specific expression of these TH2 cytokines was sufficient to block the development of hyperglycemia[15, 16]These results all suggest that the TH2 cytokines IL-10 and, in particular, IL-4, may be therapeutic in preventing or reversing T1D.
Systemic administration of anti-inflammatory mediators such as recombinant cytokines requires repeated administration due to the short half-life of these molecules and can result in non-specific side effects. Sustained, systemic expression of cytokines has been achieved by gene transfer to different tissues such as muscle, but there is a concern that the systemic cytokine levels may have adverse effects on normal immune function. Thus approaches to deliver anti-inflammatory or anti-apoptotic agents to endogenous islets have been investigated. Adenoviral vectors are effective for transduction of both human and rodent islets in culture, resulting in gene transfer of the β-cells within the intact islet without interfering with β-cell function [17, 18]. Adenoviral vectors also are able to transduce of endogenous islets in vivo, albeit at a low efficiency. The disadvantage of adenoviral vectors is that gene expression is transient, due in part to the inherent immunogenicity of first generation adenoviral vectors. Adeno-associated virus (AAV) based vectors can infect both dividing and non-dividing cells resulting in long-term efficient transgene expression in animal models with limited immunogenicity [19]. The use of self-complementary (double stranded) AAV vectors partially overcomes the need for high MOI of ssAAV (single stranded AAV), conferring rapid and efficient transduction. Also, the efficiency of AAV infection has been improved through the use of certain serotypes of AAV that have different tropisms in vivo. We previously have shown the efficient transduction of both rodent and human islets with dsAAV vectors packaged in AAV2, 6 and 8 capsids in culture [20]. Moreover, we have demonstrated that intravenous, intraperitoneal and intraductal injection of AAV6 and, in particular, AAV8 results in efficient transduction of endogenous murine islets[21]. Moreover, we have demonstrated that the use of a murine insulin promoter (mIP) with the AAV vector results in β-cell specific transgene expression for more than six months without any apparent side effect[21].
In the present study, we have investigated the therapeutic effects of dsAAV8 gene transfer of murine IL-4 into endogenous β-cells of 4 and 8 week old NOD mice. Expression of IL-4, but not IL-10 or IκB, in β-cells reduced insulitis maintained the number of CD4+CD25+ FoxP3+ cells and prevented the onset of hyperglycemia in NOD mice. Adoptive transfer of splenocytes from the non-diabetic dsAAV8-mIP-IL-4 mice to NODscid mice was able to block the diabetes induced by splenocytes co-adoptively transferred form non-diabetic dsAAV-mIP-eGFP mice. Taken together, these results demonstrate that local expression of IL-4 in islets prevents islet destruction by blocking autoimmunity through regulation of diabetogenic T cells, in part through maintenance of CD4+CD25+ FoxP3+ cells. These results also demonstrate the utility of using the dsAAV8-mIP-mediated gene transfer to endogenous NOD β-cells to examine the role of specific agents conferring or preventing T1D.
RESULTS
Intra-peritoneal delivery of dsAAV8 transduces pancreatic islets and specifically β-cells
We demonstrated previously that intra-peritoneal (i.p.), systemic (i.v.) and intra-ductal (i.d.) administration of dsAAV8-CMV-GFP results in significant transduction of both exocrine and endocrine cells of the pancreas along with other organs in both C57BL/6 and BALB/c mice[21]. The use of the murine preproinsulin promoter (mIP) shown to be β cell specific[22], in the dsAAV vector limited eGFP expression to the pancreatic β-cells[21]. The ability to express genes specifically in endogenous β-cells allows for the examination of the ability of specific gene products to regulate progression of diabetes in NOD mice when expressed in endogenous β-cells at certain times prior to onset of hyperglycemia.
Initially, we investigated the ability of dsAAV8 carrying the mIP-eGFP cassette to transduce pancreatic β-cells in young (4 week old) female NOD mice compared to normal BALB/C mice. As shown in figure 1, dsAAV8-mIP-eGFP transduction and expression of eGFP in pancreatic isolated islets of young female NOD mice (Fig. 1c) was as efficient as in normal BALB/C mice (Fig. 1b) and was specifically expressed in β-cells (Figs. 1d–f). Moreover, the GFP expression was absent in exocrine pancreatic tissue, liver, and spleen (Figs. 1g–i), and no eGFP expression was seen in vehicle control (Fig. 1a), similar to our previous results[21].
Figure 1. Expression of eGFP in pancreatic islets transduced with dsAAV8 in vivo.

4X10^11 v.g. of dsAAV8 expressing eGFP under mIP was injected i.p., in to BALB/c and NOD mice, subsequently, two weeks later the islets were isolated as described in methods. Expression of eGFP in isolated islets was visualized by two-photon laser scanning confocal microscopy. eGFP expression was similar in both BALB/c and NOD islets receiving dsAAV8 (b-c) whereas, the mice receiving vehicle control showed no eGFP in islets (a). Further, the eGFP expression was specifically present in β-cells as shown by double immunofluorescence staining of GFP (d) insulin (e), or merged image of both (f). Moreover, the GFP expression was completely absent in exocrine pancreatic tissue, liver, and spleen (Figs. 1g–i). The photos are representative of two independent experiments performed in duplicate. Magnification a–c and g–i, 100X; d–f 1000X.
Expression of murine IL-4 (mIL-4) in islets following transduction with dsAAV8-mIP-IL-4 in culture
To determine if local expression of IL-4 in the endogenous β-cells in NOD mice could block diabetes progression, we generated a dsAAV vector carrying the mIL-4 cDNA under the regulation of the mIP. The ability of the dsAAV8-mIP-IL-4 to express IL-4 following infection of β-cells was tested initially by infecting a group of 100 IEQ, isolated from BALB/C mice, with 10,000 v.g of AAV-mIP-IL-4 per islet cell in vitro in triplicate in a 12 well plate. At four and six days post-infection, the level of IL-4 secreted into the media was determined by ELISA. As shown in figure 2a, the levels of IL-4 secreted by the β-cells increased with time whereas only negligible amounts of IL-4 were detected in islets transduced with AAV expressing GFP and mock (control) infected islets. Similarly, significant amounts of IL-4 was detected in islet homogenates infected with dsAAV expressing IL-4 as compared to mock (control) and GFP transduced islets (Fig 2b).
Figure 2. mIL-4 secretion in media from dsAAV8mIP-eGFP and mIL-4 transduced islets.
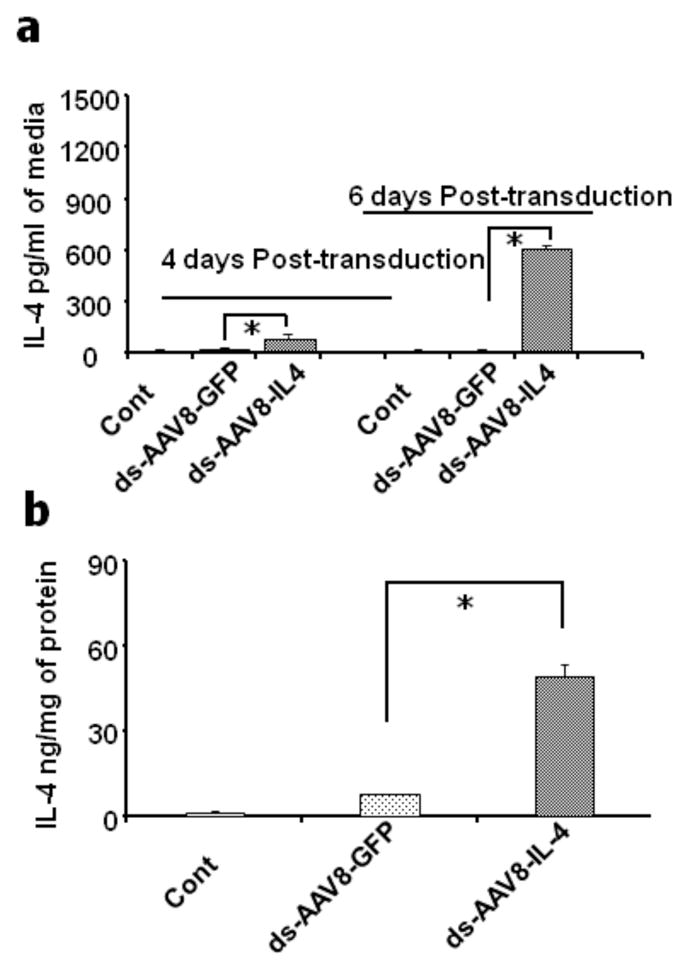
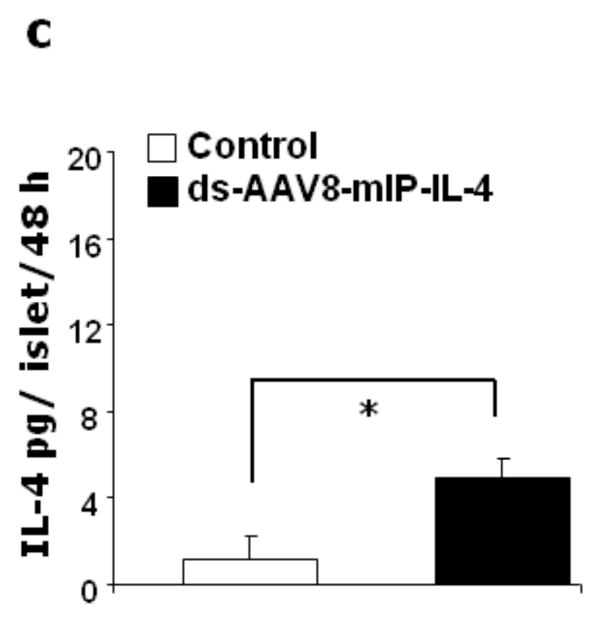
A group of 100 isolated islets were transduced with dsAAV8 expressing either GFP, or mIL-4 under mIP and subsequently cultured in RPMI for 6 days and the mIL-4 secreted into media was assayed by ELISA. The mIL-4 release in eGFP and AAV8-mIL-4 transduced islets on day 4 and 6 of cultured media (a) and in the islet lysate on day 6 (b) respectively post-transduction and culture. Secretion of mIL-4 from dsAAV8mIP-eGFP and mIL-4 in vivo transduced NOD islets. 4X1011 v.g. of dsAAV8-mIP expressing either eGFP or mIL-4 was injected i.p. and the islets subsequently isolated as described in methods. A group of 100 isolated islets were cultured in RPMI for 48 hr and the levels of mIL-4 in the media were determined as above. The mIL-4 release in eGFP (control) and mIL-4 (2c) transduced islets in 48 h culture media. The data represent the mean ± SEM of two independent experiments in triplicate (* p<0.001).
Subsequently, to demonstrate expression of IL-4 following transduction of endogenous islets, 4 week old female NOD mice were injected i.p. with dsAAV8-mIP-IL-4. The islets (100) from 5 mice were isolated two weeks post-injection and cultured in a 12 well plate. As shown in figure 2c, significant expression of IL-4 (Fig. 2c) was detected in NOD islets infected with dsAAV8-mIP-IL-4, as compared to eGFP (control). Although it is possible that some of the IL-4 expression could be from endogenous cells, the majority of IL-4 expression detected is exogenous IL-4 from the AAV-transduced β-cells. Also, it is important to note that no expression of IL-4 was detected in the serum of mice for each of the three groups (data not shown).
Prevention of onset of diabetes in NOD mice treated with dsAAV-mIP-IL-4
Non-obese diabetic (NOD) mice spontaneously develop insulitis around 4 weeks of age followed by the onset of hyperglycemia between 12 and 18 weeks of age in 80% to 90% of females. It has been reported that the inflammatory process in early diabetes is initiated and propagated by the effect of TH1-secreted cytokines such as interferon-γ (IFN-γ), and suppressed by TH2-secreted anti-inflammatory cytokines such as IL-4 and IL-10[3, 6, 11–13]. To determine the possible therapeutic effects of IL-4 expression from endogenous β-cells on the course of T1D in NOD mice, 4 week-old female NOD mice at the time of onset of insulitis were injected intra-peritoneally with 4 X 1011 v.g. of the recombinant AAV vector expressing mIL-4. Blood glucose was monitored weekly until the experiment was terminated at 40 weeks of age. As shown in Figure 3a, dsAAV mediated intra-pancreatic β-cell expression of IL-4 significantly reduced the frequency of onset of hyperglycemia compared to GFP and vehicle control mice. However, in the GFP control mice, the onset of disease was only slightly delayed as compared to saline controls (Fig. 3a). In contrast, treatment with dsAAV vectors expressing IL-10 and IκB super-repressor appeared to have no therapeutic effect on onset of disease (Fig 3c&d).
Figure 3. Murine IL-4 expression in pancreatic islet β-cells of female NOD mice reduced the onset of hyperglycemia.

Four week old female NOD mice received dsAAV8 expressing either vehicle (n=35), GFP (n=24), mIL-4 (n=29), mIL-10 (n=20), mIκB (n=13) super-repressor and 8 week old NOD mice received ds-AAV-mIL-4 (n=10). The onset of diabetes was monitored as described in methods. A significant number of 4 week old (a) and eight week old (b) mice treated with dsAAV8-mIP-IL-4 were normal as compared to eGFP and vehicle control recipient mice. Whereas, there was no difference in the incidence of diabetes in mIL-10 and mIκB treated mice as compared to controls. A p value of less than 0.05 by long rank test analysis was used to indicate statistically significant percentage normo-glycemic (* p<0.001, # non-significant).
To determine the possible therapeutic effects of IL-4 expression in NOD mice with progressive expansion of insulitis, 8 week old female NOD mice were injected intra-peritoneally with 4 X 1011 v.g. of dsAAV-mIP-IL-4 and monitored for blood glucose. As shown in figure 3b, the β-cell specific expression of mIL-4 even in 8 week old mice significantly reduced the onset of hyperglycemia as compared to saline and eGFP controls.
Evaluation of insulitis by histology
The extent of insulitis in the 8 and 21 week euglycemic mice was examined by histological analysis of pancreatic sections. In 8 week old mice, there was mild to severe mononuclear cell infiltration in and around islets of the saline and GFP treated mice (Figs. 4a–b) whereas the dsAAV8-IL-4 treated mice showed limited mononuclear cell infiltration in significantly fewer numbers of islets (Fig. 4c). In the 21 week old NOD mice there was 50 to 100% of mono-nuclear cell infiltration in the islets of saline and eGFP control mice (Figs. 4d–e). In contrast, expression of mIL-4 in the β-cells limited insulitits to the periphery of islet (peri-insulitis; Fig. 4f).
Figure 4. Histochemical analysis of infiltrating leukocytes in twenty week normo-glycemic mice.

Photomicrographs of H&E for infiltrating leukocytes in female 8 and 21 week normoglycemic female NOD mice showed mild to severe mononuclear cell infiltration in and around islets of saline and GFP treated mice (Figs. 4a–b; e–f), whereas the dsAAV8-mIP-IL-4 treated mice showed limited peri-islet mononuclear cell infiltration in significantly fewer numbers of islets (Figs. 4c, f). Figure 4g shows the insulitis score determined as mentioned in methods. Magnification 100X.
Overall, the insulitis score at 21 weeks of age of dsAAV-mIP-IL-4 recipient mice (1.365 ± 1.205) was lower than that of mice received dsAAV-mIP-eGFP (2.009 ± 1.438; Fig. 4g). The percent of total intact islets and with mild peri-insulitis in dsAAV-mIP-IL-4 recipient mice was 73%, approx 37% higher than that of eGFP recipient mice (36%; Fig. 4g). Most of the insulitis (33%) in AAV-mIP-IL-4 recipient mice occurred in the range of moderate insulitis (insulitis score 1); whereas 53% of the islets measured in AAV-eGFP recipient mice had severe insulitis (Fig. 4g).
Expression of IL-4 in the pancreatic β-cells modulates CD4+CD25+FoxP3+ regulatory T-cells
To examine the mechanism of suppression of disease onset mediated by IL-4 expression in the β-cells, we initially analyzed whether there was a change in the T-reg cell population in the spleen. As shown in figure 5, mIL-4 expression in pancreatic β-cells for four weeks resulted in normal reported levels of CD4+CD25+FoxP3+ T-cells (3.3±0.29) in the splenocytes, whereas there was significant (p=0.04)_decrease in the CD4+CD25+FoxP3+ T-cell population in the AAV-GFP (1.83±0.38) and saline control (1.6±0.21) treated mice (figure 5).
Figure 5. CD4+CD25+FoxP3+ T cells from spleens of 8 week old female NOD mice treated with dsAAV8-mIP-IL-4.

Four week old NOD mice received dsAAV8-mIP expressing either mIL-4 or eGFP. Four weeks (Fig. 5) post-injection, the spleens were collected and CD4+CD25+FoxP3+ T cells analyzed by FACS. Splenocytes stained for CD4+CD25+ and isotype control for FoxP3 (top panel); and splenocytes stained for CD4+CD25+FoxP3+ (bottom panel). The bar diagram showing percentage of CD4+CD25+Foxp3+ cells in splenocytes of control, AAV-GFP and AAV-IL4 injected mice. The values are Mean±SD of three independent samples. (*p<0.04)
Splenocytes from mice treated with dsAAV8-mIP-IL-4 are unable to adoptively transfer diabetes
To address the possibility that regulatory T cell were being modulated by IL-4 in conferring protection from diabetes, adoptive transfer experiments were performed. Splenocytes from normo-glycemic 21 week old NOD mice administered with dsAAV expressing mIL-4 were injected into NODscid mice. As shown in figure 6c and 6d, the total splenocytes from NOD mice treated with the dsAAV8-mIP-IL-4 were unable to confer disease whereas the splenocytes from dsAAV-mIP-GFP treated, but euglycemic, mice rapidly transferred disease (Fig. 6b). The control, saline injected NOD.scid also were euglycemic (6a).
Figure 6. Adoptive transfer of diabetes in NODscid.

Splenocytes isolated from normal 21 week dsAAV-mIP-GFP or normal 21 and 40 week mIL-4 transduced mice were injected intra-peritoneally in to NODscid mice and blood glucose levels were monitored weekly. The NODscid mice which received splenocytes from GFP treated mice became diabetic (Fig. 6b), whereas the splenocytes from 22 and 40 week mIL-4 treated mice didn’t transfer the disease to NODscid (Figs. 6c–d). The NODscid mice receiving saline only also did not develop disease (Fig. 6a). To determine the suppressive effects of mIL-4 treated NOD mice splenocytes, co-adoptive transfer was performed as described in methods. Three of the 4 mice which received both splenocytes from mIL-4 and eGFP treated mice did not develop diabetes (Fig. 6e).
To determine whether T cells from splenocytes of 21 week old normal NOD mice treated with AAV-mIL-4 would inhibit transfer of disease conferred by diabetogenic T cells from eGFP treated mice, an adoptive co-transfer experiment was performed. An equal number of splenocytes from the AAV-mIL-4 treated mice were co-administered with splenocytes from the AAV-eGFP treated mice. As shown in Figure 6e, three of the four mice were protected from development of diabetes in NOD.scid mice. These results suggest that local expression of mIL-4 from the islets modulates regulatory T cell subsets. However, there may be additional mechanism through which localized expression of IL-4 in β-cells suppresses onset of hyperglycemia.
DISCUSSION
We previously demonstrated dsAAV8-based vectors can confer long term, stable gene transfer and expression in pancreatic β-cells when delivered intra-peritoneally, intravenously and intra-ductally into wild type C57Bl/6 and BALB/c mice[21]. In the present study, we demonstrate the efficient gene transfer to endogenous β-cells in NOD mice using a dsAAV8-mIP-eGFP vector injected intra-peritoneally, similar to our previous results in BALB/c mice[21]. The ability to genetically modify endogenous β-cells in a stable manner allows for the analysis of specific gene products in regulating the progression of T1D in animal models. The advantage of gene transfer and expression of transgene only in endogenous β-cells using mIP as compared to transgenic mice is that gene delivery can be facilitated at different stages of insulitis to examine therapeutic efficacy. Also, it is significantly easier to use AAV gene transfer to deliver transgenes to islets than generating and breeding transgenic mice. Thus, we initiated experiments to examine the therapeutic efficacy of specific immunosuppressive gene products in preventing diabetes in NOD mice following delivery to endogenous β-cells. Here we have demonstrated that dsAAV mediated delivery of the mIL-4 gene to the endogenous β-cells of 4 and 8 week old NOD mice reduced insulitis and prevented onset of hyperglycemia compared to controls.
It has been shown that TH1 cells are responsible for autoimmunity in NOD mice and that endogenous TH2 cells can protect mice against diabetes development [7, 8, 15, 23]. The inflammatory process in early diabetes is reported to be initiated and propagated, in part, by TH1 cytokines and suppressed by TH2 anti-inflammatory cytokines. For example, systemic administration of IL-4 limits insulitis and T1D by reversing CD4 T cell hypo responsiveness and potentiating TH2 cell function [8, 24, 25]. Systemic administration of recombinant IL-4 in to young NOD mice or expression of IL-4 within the islets of transgenic NOD mice reduced the incidence of diabetes [23–25]. The protection was attributed to a reversal in CD4 T cell hypo responsiveness and the capacity to produce IL-4 [23, 25]. It also has been reported that IL-4 expressed in the islets of transgenic mice does not prevent the generation of pathogenic islet responses, but instead induces islet Ag-specific Th2 T cells that block the action of diabetogenic T cells in the pancreas.
In examining the mechanism of regulation in NOD mice, we observed that no disease developed in NODscid receiving splenocytes from 21 and 40 week old NOD mice treated with dsAAV-IL-4, as well as in co-adoptive transfer where splenocytes from 21 week old NOD mice treated with dsAAV-IL-4 were mixed with diabetogenic splenocytes. This is in contrast to the NODscid mice that developed diabetes after adoptive transfer of splenocytes from the disease-free AAV-mIP-eGFP mice. These results demonstrate the presence of regulatory splenocytes, presumably T cells, in the AAV-IL4 treated mice. Consistent with this result is the observation that mIL-4 expression in pancreatic β-cells resulted in normal levels of CD4+CD25+FoxP3+ T-cells in the spleen. Thus, it appears as if β-cell specific expression of IL-4 inhibits disease-causing lymphocytes, in part, by maintaining the number of regulatory T cells.
Regulatory T cells arise in the thymus as a consequence of positive selection and play an active role in the maintenance of immune homeostasis. Recent studies have shown there is a reduction in the number of CD4+CD25+ regulatory T cells in NOD mice as disease progresses [26, 27]. The regulatory activity is confined to CD62L− and/or CD25-expressing CD4+ T cell subsets [28, 29]. In addition, CD4+CD25+ T cells effectively protected NODscid mice in co-transfer experiments with diabetogenic CD28−/− cells [27]. Our results demonstrate that CD4+CD25+ regulatory T cells are affected in mice after 4 weeks of IL-4 expression in β-cells, whereas a significantly smaller T-reg population was found in saline and GFP control mice. Moreover, the T-regs decreased more prominently with age in control mice as compared to IL-4 recipient mice (data not shown).
Immunization with self-antigens or antigenic epitopes derived from these β-cell products can prevent diabetes onset in NOD mice and various models of induced diabetes by generation of auto reactive regulatory T cells[30–35]. The precise role of individual effector molecules produced by regulatory T cells appears to be a function of the experimental model system employed, nature of the immunizing antigen and the presence of immunosuppressive agents such as TGF-β, IL-10, IL-4, and CTLA-4[30–35]. We speculate that the dose and route of administration of IL-4 also effects the regulation of the immune response, leading to the prevention of diabetes.
We have demonstrated that gene transfer of IκB super repressor and mIL-10 by AAV to endogenous NOD β-cells was unable to delay onset of hyperglycemia. These results with AAV gene transfer of IL-10 and IκB demonstrate the specificity of localized mIL-4 expression in blocking the onset of hyperglycemia. It is important to note that ds-AAV-mIP-eGFP gene transfer to β-cells slightly delayed the onset of the disease as compared to the saline control. It is possible that the marginal, non-significant delay in onset of diabetes in AAV8-mIP eGFP control treated NOD could be due to either the effect of eGFP expression or AAV infection.
In conclusion, in this report, we have demonstrated the ability of dsAAV8 to transduce endogenous β-cells when delivered i.p. in NOD mice. Moreover, we have demonstrated the ability to prevent disease onset by local expression of mIL-4 from the islets. Thus the delivery to islets of anti-inflammatory cytokines, cytoprotective antioxidants, and anti-inflammatory enzymes, along with anti-apoptotic molecules or growth promoting factors by dsAAV may prevent the onset of type 1 diabetes and may offer a promising form of immunotherapy.
MATERIAL AND METHODS
Experimental animals
Four week old female NOD/LTJ mice were purchased from Jackson Laboratories (Bar Harbor, Maine) and housed in the specific pathogen free animal facility of University of Pittsburgh. All the studies were performed in compliance with the US Department of Agriculture and National Institutes of Health regulations. All animal experiments were conducted and monitored under protocols reviewed and approved by the Institutional Animal Care and Use Committee.
Construction of AAV vector
AAV vectors, dsAAV-CMV-GFP were generated by triple plasmid transfection of 293 cells as described [36–38]. The AAV vector plasmid dsAAV-mIP-GFP was made by replacing the CMV promoter of dsAAV-CMV-GFP with a 1.13-kb mouse preproinsulin gene II promoter. mIP was obtained by PCR from plasmid Ad.Ins-C-GFP, as published previously [21], which includes full-length promoter, intron 1, noncoding sequence of exon 1 and exon 2 of mouse preproinsulin gene II [21, 23, 39, 40]. The pseudotyped AAV packaging plasmid contained the AAV8 serotype capsid gene coupled with the AAV2 rep gene [36, 41]. The AAV viruses were purified twice with CsCl gradient ultracentrifugation [38] and the titers of vector-genome (v.g.) particles determined by a standard dot-blot assay [38]. The double stranded AAV vector expressing murine IL-4 (mIL-4) and IL-10 (mIL-10) was generated by replacing the GFP expression cassette with mIL-4 and mIL-10 which was obtained by PCR from plasmid pAdlox-mIL-4 and IL-10 [42, 43]. Murine IκB super-repressor was a kind gift from Denis Guttridge (Ohio State University). To test the functional efficacy of the these AAV viruses, a group of 100 isolated islets were transduced with 10,000 v.g. per islet cell and incubated at 37°C for 48 h and media was replaced with fresh media and subsequently the mIL-4 levels were determined after 4 and 6 days of transduction and culture in media and islet lysates at the end of 6 days of culture (IL-10 and IκB super-repressor data don’t shown).
In vivo pancreatic islet AAV transduction, islet isolation and culture
Saline or AAV8-mIP-GFP or AAV8-mIP-IL-4 were injected at a dose of 4 X 1011 viral genomes (v.g.) per mouse intra-peritoneally into 4 week old female NOD/LTJ mice. Two weeks later islets were isolated from the mouse pancreata by intraductal collagenase digestion (Type IX, 1.75 mg/ml, Sigma Chemical Co., St. Louis, MO) as described [20, 44]. The isolated islets were further purified by Ficoll density gradient centrifugation and were hand picked under stereomicroscope. Purity of the islets was determined by dithizone staining and was >95% in all isolations. Islets of approximately 150 μM in diameter determined by standard algorithm were expressed as islet equivalent (IEQ), and a group of 100 IEQ was handpicked, incubated for 48 h in RPMI-1640 supplemented with 20 mM L-glutamine, 100 μg/ml streptomycin, 100 U/ml penicillin and 10% heat-inactivated fetal bovine serum in a humidified 5% CO2 incubator at 37°C. The level of secreted mIL-4 was determined by standard ELISA. The GFP expression was analyzed by two-photon confocal microscope [20, 44].
Intra-peritoneal injection of AAV-mIP-IL-4, IL-10, IκB super-repressor or eGFP in female NOD mice
4 X 1011 vg of AAV8-mIP-GFP or mIL-4 vector genomes were injected in 1 ml of saline intra-peritoneally into 4 and 8 week old female NOD/LTJ mice. Similarly. 4 week old female NOD received i.p., either AAV8-mIL-10 or AAV8-IκB super-repressor. The controls received 1 ml of vehicle buffer (saline). The animals were monitored by blood glucose readings from the tail vein and only mice with blood glucose over 300 mg/dl (Ascensia Breeze, Bayer) on at least two different days were considered as hyperglycemic.
Flow cytometric analysis of T-reg cells in mIL-4 treated NOD mice
Groups of four week old female NOD/LTJ mice were injected intraperitoneally with buffer or 4X10x11 v.g. of AAV expressing either GFP or mIL-4. Eight weeks following i.p. delivery of dsAAV expressing eGFP or mIL-4 mice were sacrificed and spleens were harvested. Splenocytes were obtained and then immunostained for surface expression of CD4 and CD25, using appropriate concentrations of FITC-conjugated anti-mouse CD4 and APC-conjugated anti-mouse CD25 antibodies as described by manufacturers protocol (eBioscience, San Diego, CA). The staining was performed at 4° C for 30 min, with antibodies suspended in staining buffer. After washing to remove unbound antibodies, intracellular detection of FoxP3 was performed by incubating a cross-reactive, directly conjugated murine FoxP3 antibody at 4 °C for 30 min (PE conjugated, anti-mouse/rat FoxP3, clone FJK-16s and the permeabilization and fix/perm buffers that accompany the antibody (FoxP3 Staining Set, eBioscience). A PE conjugated rat IgG2a antibody was used as the isotype control. Following two additional washes in permeabilization buffer, the cells were resuspended in FACS buffer and kept at 4 °C prior to analysis. The percentage of T-reg cells were calculated based on the percentage of CD4+CD25+FoxP3+cells within the overall cell population.
Adoptive transfer of splenocytes
Groups of six normo-glycemic female NOD/LTJ mice, 21 and 40 (mIL-4 recipient) weeks old, previously treated by intraperitoneal injection of AAV expressing either GFP or mIL-4 were used as donors. Spleens were harvested and equal numbers of splenocytes (20 × 106/mouse, six donors per six recipients) were infused by i.p. route into recipient NODscid mice. To study the suppressive effects of disease transferring T cells, co-adoptive transfer experiments were performed. Ten million splenocytes from dsAAV-IL-4 and eGFP treated mice were injected i.p. in NOD.scid mice. It has been reported in literature that ten million splenocytes injected systemically are sufficient to transfer disease in NOD.Scid mice [45]. All the animals were monitored by blood glucose readings from the tail vein and only mice with blood glucose over 300 mg/dl (Ascensia Breeze, Bayer) on at least two different days were considered as hyperglycemic.
Histochemistry
Eight week and 21 week euglycemic NOD mice sacrificed, the pancreata were collected for immunohistochemistry. The 5 μm sections were deparaffinized, histological analysis of infiltrating mononuclear cells was performed by routine hematoxilin and eosin staining and subsequently examined by light microscopy for scoring of insulitis. The mice were scored by investigators who were blinded to the treatment groups, and for each mouse between 20 and 40 islets were examined. To accomplish an accurate statistical analysis, each islet was instead scored as having or not having intra-islet insulitis according to following classification scheme: 0=no lymphocytic infiltration; 1=peri-insulitis; 2=intrainsulitis affecting <1/3 of the islet area; 3=insulitis comprising 1/3 to 2/3 of the islet; 4=insulitis comprising >2/3 of the islet. The histology score index was calculated by dividing the sum of all individual islet scores by the total number of islets evaluated [46].
Statistical analysis
Statistics were performed using the Stata 8.2 (STATA Corp., College Station, Texas) software package and data collected were expressed as mean ± SEM and percentage of non-diabetic mice was analyzed by Kaplan-Meier survival analysis. A p value of less than 0.05 by ANOVA and long rank test analysis was used to indicate statistically significant differences.
Acknowledgments
This work was supported in part by grants from the Juvenile Diabetes Research Foundation (4-1999-845 and 7-2005-1154) and the American Diabetes Association (1-04-ISLET-23). We would like to thank Nick Giannoukakis, Suzanne Bertera, Nicole Bianco for critical reading of manuscript and Maliha Zahid for statistical analysis.
References
- 1.Chatenoud L, Thervet E, Primo J, Bach JF. Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:123–127. doi: 10.1073/pnas.91.1.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ogawa N, Minamimura K, Kodaka T, Maki T. Short administration of polyclonal anti-T cell antibody (ALS) in NOD mice with extensive insulitis prevents subsequent development of autoimmune diabetes. Journal of Autoimmunity. 2006;26:225–231. doi: 10.1016/j.jaut.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 3.Cameron MJ, et al. IL-4 prevents insulitis and insulin-dependent diabetes mellitus in nonobese diabetic mice by potentiation of regulatory T helper-2 cell function. J Immunol. 1997;159:4686–4692. [PubMed] [Google Scholar]
- 4.Maki T, Ichikawa T, Blanco R, Porter J. Long-term abrogation of autoimmune diabetes in nonobese diabetic mice by immunotherapy with anti-lymphocyte serum. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:3434–3438. doi: 10.1073/pnas.89.8.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miyazaki A, et al. Predominance of T lymphocytes in pancreatic islets and spleen of pre-diabetic non-obese diabetic (NOD) mice: a longitudinal study. Clinical & Experimental Immunology. 1985;60:622–630. [PMC free article] [PubMed] [Google Scholar]
- 6.Lee M, Koh JJ, Han SO, Ko KS, Ki SW. Prevention of autoimmune insulitis by delivery of interleukin-4 plasmid using a soluble and biodegradable polymeric carrier. Pharmaceutical research. 2002;19:246–249. doi: 10.1023/a:1014478515005. [DOI] [PubMed] [Google Scholar]
- 7.Pennline KJ, Roque-Gaffney E, Monahan M. Recombinant human IL-10 prevents the onset of diabetes in the nonobese diabetic mouse. Clin Immunol Immunopathol. 1994;71:169–175. doi: 10.1006/clin.1994.1068. [DOI] [PubMed] [Google Scholar]
- 8.Rapoport MJ, et al. Interleukin 4 reverses T cell proliferative unresponsiveness and prevents the onset of diabetes in nonobese diabetic mice. Journal of Experimental Medicine. 1993;178:87–99. doi: 10.1084/jem.178.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wood SC, Rao TD, Frey AB. Multidose streptozotocin induction of diabetes in BALB/cBy mice induces a T cell proliferation defect in thymocytes which is reversible by interleukin-4. Cellular immunology. 1999;192:1–12. doi: 10.1006/cimm.1998.1413. [DOI] [PubMed] [Google Scholar]
- 10.Zipris D, Karnieli E. A single treatment with IL-4 via retrovirally transduced lymphocytes partially protects against diabetes in BioBreeding (BB) rats. Jop. 2002;3:76–82. [PubMed] [Google Scholar]
- 11.Hayashi T, Yasutomi Y, Hasegawa K, Sasaki Y, Onodera T. Interleukin-4-expressing plasmid DNA inhibits reovirus type-2-triggered autoimmune insulitis in DBA/1 J suckling mice. International journal of experimental pathology. 2003;84:101–106. doi: 10.1046/j.1365-2613.2003.00341.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ko KS, Lee M, Koh JJ, Kim SW. Combined administration of plasmids encoding IL-4 and IL-10 prevents the development of autoimmune diabetes in nonobese diabetic mice. Mol Ther. 2001;4:313–316. doi: 10.1006/mthe.2001.0459. [DOI] [PubMed] [Google Scholar]
- 13.Wolfe T, et al. Endogenous expression levels of autoantigens influence success or failure of DNA immunizations to prevent type 1 diabetes: addition of IL-4 increases safety. European journal of immunology. 2002;32:113–121. doi: 10.1002/1521-4141(200201)32:1<113::AID-IMMU113>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 14.Feili-Hariri M, et al. Dendritic cells transduced to express interleukin-4 prevent diabetes in nonobese diabetic mice with advanced insulitis. Human gene therapy. 2003;14:13–23. doi: 10.1089/10430340360464679. [DOI] [PubMed] [Google Scholar]
- 15.Gallichan WS, Balasa B, Davies JD, Sarvetnick N. Pancreatic IL-4 expression results in islet-reactive Th2 cells that inhibit diabetogenic lymphocytes in the nonobese diabetic mouse. Journal of Immunology. 1999;163:1696–1703. [PubMed] [Google Scholar]
- 16.Mueller R, Krahl T, Sarvetnick N. Pancreatic expression of interleukin-4 abrogates insulitis and autoimmune diabetes in nonobese diabetic (NOD) mice. Journal of Experimental Medicine. 1996;184:1093–1099. doi: 10.1084/jem.184.3.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Csete ME, Afra R, Mullen Y, Drazan KE, Benhamou PY, Shaked A. Adenoviral-mediated gene transfer to pancreatic islets does not alter islet function. Transplant Proc. 1994;26:756–757. [PubMed] [Google Scholar]
- 18.Csete ME, et al. Efficient gene transfer to pancreatic islets mediated by adenoviral vectors. Transplantation. 1995;59:263–268. [PubMed] [Google Scholar]
- 19.Flotte T, et al. Efficient ex vivo transduction of pancreatic islet cells with recombinant adeno-associated virus vectors. Diabetes. 2001;50:515–520. doi: 10.2337/diabetes.50.3.515. [DOI] [PubMed] [Google Scholar]
- 20.Rehman KK, et al. Efficient gene delivery to human and rodent islets with double-stranded (ds) AAV-based vectors. Gene Therapy. 2005;12:1313–1323. doi: 10.1038/sj.gt.3302530. [DOI] [PubMed] [Google Scholar]
- 21.Wang Z, et al. Widespread and stable pancreatic gene transfer by adeno-associated virus vectors via different routes. Diabetes. 2006;55:875–884. doi: 10.2337/diabetes.55.04.06.db05-0927. [DOI] [PubMed] [Google Scholar]
- 22.Watkins S, Geng X, Li L, Papworth G, Robbins PD, Drain P. Imaging secretory vesicles by fluorescent protein insertion in propeptide rather than mature secreted peptide. Traffic. 2002;3:461–471. doi: 10.1034/j.1600-0854.2002.30703.x. [DOI] [PubMed] [Google Scholar]
- 23.Debray-Sachs M, et al. Prevention of diabetes in NOD mice treated with antibody to murine IFN gamma. J Autoimmun. 1991;4:237–248. doi: 10.1016/0896-8411(91)90021-4. [DOI] [PubMed] [Google Scholar]
- 24.Inobe JI, Chen Y, Weiner HL. In vivo administration of IL-4 induces TGF-beta-producing cells and protects animals from experimental autoimmune encephalomyelitis. Ann N Y Acad Sci. 1996;778:390–392. doi: 10.1111/j.1749-6632.1996.tb21153.x. [DOI] [PubMed] [Google Scholar]
- 25.Tominaga Y, et al. Administration of IL-4 prevents autoimmune diabetes but enhances pancreatic insulitis in NOD mice. Clin Immunol Immunopathol. 1998;86:209–218. doi: 10.1006/clin.1997.4471. [DOI] [PubMed] [Google Scholar]
- 26.Green EA, Choi Y, Flavell RA. Pancreatic lymph node-derived CD4(+)CD25(+) Treg cells: highly potent regulators of diabetes that require TRANCE-RANK signals. Immunity. 2002;16:183–191. doi: 10.1016/s1074-7613(02)00279-0. [DOI] [PubMed] [Google Scholar]
- 27.Salomon B, et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 28.Herbelin A, Gombert JM, Lepault F, Bach JF, Chatenoud L. Mature mainstream TCR alpha beta+CD4+ thymocytes expressing L-selectin mediate “active tolerance” in the nonobese diabetic mouse. J Immunol. 1998;161:2620–2628. [PubMed] [Google Scholar]
- 29.Lepault F, Gagnerault MC. Characterization of peripheral regulatory CD4+ T cells that prevent diabetes onset in nonobese diabetic mice. J Immunol. 2000;164:240–247. doi: 10.4049/jimmunol.164.1.240. [DOI] [PubMed] [Google Scholar]
- 30.Belghith M, Bluestone JA, Barriot S, Megret J, Bach JF, Chatenoud L. TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med. 2003;9:1202–1208. doi: 10.1038/nm924. [DOI] [PubMed] [Google Scholar]
- 31.Bergerot I, et al. Insulin B-chain reactive CD4+ regulatory T-cells induced by oral insulin treatment protect from type 1 diabetes by blocking the cytokine secretion and pancreatic infiltration of diabetogenic effector T-cells. Diabetes. 1999;48:1720–1729. doi: 10.2337/diabetes.48.9.1720. [DOI] [PubMed] [Google Scholar]
- 32.Maron R, Melican NS, Weiner HL. Regulatory Th2-type T cell lines against insulin and GAD peptides derived from orally- and nasally-treated NOD mice suppress diabetes. J Autoimmun. 1999;12:251–258. doi: 10.1006/jaut.1999.0278. [DOI] [PubMed] [Google Scholar]
- 33.Maron R, Palanivel V, Weiner HL, Harn DA. Oral administration of schistosome egg antigens and insulin B-chain generates and enhances Th2-type responses in NOD mice. Clin Immunol Immunopathol. 1998;87:85–92. doi: 10.1006/clin.1997.4506. [DOI] [PubMed] [Google Scholar]
- 34.Tian J, et al. Modulating autoimmune responses to GAD inhibits disease progression and prolongs islet graft survival in diabetes-prone mice. Nat Med. 1996;2:1348–1353. doi: 10.1038/nm1296-1348. [DOI] [PubMed] [Google Scholar]
- 35.von Herrath MG, Dyrberg T, Oldstone MB. Oral insulin treatment suppresses virus-induced antigen-specific destruction of beta cells and prevents autoimmune diabetes in transgenic mice. J Clin Invest. 1996;98:1324–1331. doi: 10.1172/JCI118919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rabinowitz JE, et al. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. Journal of Virology. 2002;76:791–801. doi: 10.1128/JVI.76.2.791-801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Z, Ma HI, Li J, Sun L, Zhang J, Xiao X. Rapid and highly efficient transduction by double-stranded adeno-associated virus vectors in vitro and in vivo. Gene Therapy. 2003;10:2105–2111. doi: 10.1038/sj.gt.3302133. [DOI] [PubMed] [Google Scholar]
- 38.Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenovirus. Journal of Virology. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Geng X, Li L, Watkins S, Robbins PD, Drain P. The insulin secretory granule is the major site of K(ATP) channels of the endocrine pancreas. Diabetes. 2003;52:767–776. doi: 10.2337/diabetes.52.3.767. [DOI] [PubMed] [Google Scholar]
- 40.Wentworth BM, Schaefer IM, Villa-Komaroff L, Chirgwin JM. Characterization of the two nonallelic genes encoding mouse preproinsulin. Journal of Molecular Evolution. 1986;23:305–312. doi: 10.1007/BF02100639. [DOI] [PubMed] [Google Scholar]
- 41.Rutledge EA, Halbert CL, Russell DW. Infectious clones and vectors derived from adeno-associated virus (AAV) serotypes other than AAV type 2. Journal of Virology. 1998;72:309–319. doi: 10.1128/jvi.72.1.309-319.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim SH, Kim S, Evans CH, Ghivizzani SC, Oligino T, Robbins PD. Effective treatment of established murine collagen-induced arthritis by systemic administration of dendritic cells genetically modified to express IL-4. Journal of Immunology. 2001;166:3499–3505. doi: 10.4049/jimmunol.166.5.3499. [DOI] [PubMed] [Google Scholar]
- 43.Kim SH, et al. Exosomes derived from IL-10-treated dendritic cells can suppress inflammation and collagen-induced arthritis. J Immunol. 2005;174:6440–6448. doi: 10.4049/jimmunol.174.10.6440. [DOI] [PubMed] [Google Scholar]
- 44.Rehman KK, et al. Protection of islets by in situ peptide-mediated transduction of the Ikappa B kinase inhibitor Nemo-binding domain peptide. J Biol Chem. 2003;278:9862–9868. doi: 10.1074/jbc.M207700200. [DOI] [PubMed] [Google Scholar]
- 45.Fuchtenbusch M, Larger E, Thebault K, Boitard C. Transfer of diabetes from prediabetic NOD mice to NOD-SCID/SCID mice: association with pancreatic insulin content. Horm Metab Res. 2005;37:63–67. doi: 10.1055/s-2005-861155. [DOI] [PubMed] [Google Scholar]
- 46.Rietz C, Screpanti V, Brenden N, Bohme J, Fernandez C. Overexpression of bcl-2 in T cells affects insulitis in the nonobese diabetic mouse. Scand J Immunol. 2003;57:342–349. doi: 10.1046/j.1365-3083.2003.01244.x. [DOI] [PubMed] [Google Scholar]