

Abstract
A variety of physiological functions, not only restricted to the nervous system, are discussed for the cellular prion protein (PrPC). A prominent, non-physiological property of PrPC is the conversion into its pathogenic isoform (PrPSc) during fatal, transmissible, and neurodegenerative prion diseases. The prion protein is subject to posttranslational proteolytic processing and these cleavage events have been shown i) to regulate its physiological functions, ii) to produce biologically active fragments, and iii) to potentially influence the course of prion disease. Here, we give an overview on the proteolytic processing under physiological and pathological conditions and critically review what is currently known about the three main cleavage events of the prion protein, namely α-cleavage, β-cleavage, and ectodomain shedding. The biological relevance of resulting fragments as well as controversies regarding candidate proteases, with special emphasis on members of the A-disintegrin-and-metalloproteinase (ADAM) family, will be discussed. In addition, we make suggestions aimed at facilitating clarity and progress in this important research field. The better understanding of this issue will not only answer basic questions in prion biology but will likely impact research on other neurodegenerative diseases as well.
Keywords: Prion protein, proteolytic processing, α-cleavage, β-cleavage, ectodomain shedding, ADAM10, ADAM17
Introduction
The mature cellular prion protein (PrPC), coded by the PRNP gene, is a membrane-anchored protein of 208 amino acids and a molecular weight of approximately 35 kDa (in humans and mice) with two variably occupied N-glycosylation sites. The flexible N-terminal part harbors an octameric repeat region (OR), a neurotoxic domain (ND, discussed later), and a hydrophobic core (HC) (Figure 1). The C-terminal part of the protein, on the other hand, is more structured and comprises three alpha-helices, two beta-strands, loop domains, up to two N-glycans, a disulfide bond, and the glycosylphosphatidylinositol (GPI)-anchor for attachment to the outer leaflet of membranes [1,2]. Due to its GPI-anchor the protein is mainly located within cholesterol and sphingolipid-rich microdomains, termed lipid rafts [3,4].
Figure 1.
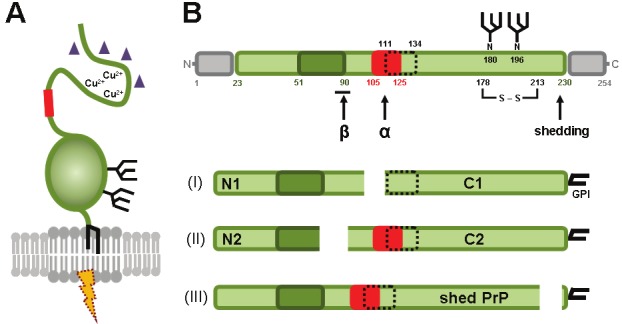
Schematic representation of the prion protein. (A) The prion protein is located in lipid rafts and attached to the outer leaflet of the cellular membrane via a GPI-anchor. The flexible N-terminal part of the protein - among other features - harbors a neurotoxic domain (red box) and is able to bind copper ions and oligomeric amyloid β (purple triangles). The C-terminal part of PrPC has a globular structure and comprises up to two N-glycan side chains. Involvement of PrPC in protective or toxic signalling (dotted thunderbolt) requires accessory molecules (not shown) to bypass the lipid bilayer. (B) Linear representation of the primary sequence of murine PrPC showing important protein domains. After removal of the N-terminal signal sequence (aa 1-22; grey box) by signal peptidases in the ER and the C-terminal signal sequence for the attachment of the GPI-anchor (aa 231-254; grey box), the mature prion protein comprises an octameric repeat region (aa 51-90; dark green), a neurotoxic domain (aa 105-125; red box), a hydrophobic core (aa 111-134; dotted box), a disulfide bridge (between aa 178 and 213), and two variably occupied N-glycosylation sites (aa 180 and 196). The three most important cleavage events are indicated by arrows. (I) α-cleavage gives rise to a soluble N1 fragment of 11 kDa and a membrane-bound C1 fragment of 18 kDa. Of note, this cleavage destroys the neurotoxic domain. (II) β-cleavage at the end of the octameric repeat region produces N2 (9 kDa) and C2 (20 kDa) fragments. (III) Ectodomain shedding close to the GPI-anchor results in the release of nearly full-length PrP from the membrane. References are given in the text.
PrPC is discussed to fulfil several physiological functions [5-7]. These range from involvement in neuro-, synapto-, and neuritogenesis as well as differentiation [8-10], cell adhesion [11,12], neuroprotection [13,14], and copper-homeostasis [15], to receptor properties and participation in cellular signalling pathways. In signalling, PrPC can either have a central role [16-19] or act as a regulatory cofactor [20]. In both situations, accessory molecules are required since PrPC does not span the plasma membrane and is thus unable to transduce signals into the cytosol.
A non-physiological property of PrPC is its conversion into the pathogenic isoform (PrPSc; Sc for scrapie, a prion disease of sheep) giving rise to prion diseases or transmissible spongiform encephalopathies (TSE). Prion diseases are fatal neurodegenerative conditions of sporadic or genetic aetiology or may be acquired by exposure to infectious prions [21]. They affect - in different subtypes and peculiarities - humans [22] and other mammalian species [23]. PrPSc, produced by a template-driven conformational change of PrPC, is partially resistant to proteinase K (PK) digestion, has amyloidogenic properties, tends to aggregate, and is thought to be the main, if not the sole, component of the transmissible agent termed “prion” [24]. However, recent data indicates that neurotoxicity is not necessarily linked to transmissibility and the nature of the neurotoxic agent in prion diseases remains to be defined [25]. Loss of physiological functions of PrPC is also assumed to contribute to the neurodegeneration seen in prion disease [26].
In addition to its pivotal role in prion disease [27,28], PrPC might play a role in other neurodegenerative conditions such as Alzheimer`s disease (AD) (reviewed in [2]). For instance, recent studies indicate an influence of PrPC on the neurotoxicity of oligomeric species of amyloid β (Aβ) [20,29], a peptide causally involved in disease initiation and progression of AD, and of other β-sheet-rich conformers produced in neurodegenerative diseases [19]. If and how binding of oligomeric Aβ to PrPC really leads to neurotoxic effects in AD is currently under discussion [29-33]. Furthermore, PrPC negatively regulates the activity of beta-site APP-cleaving enzyme 1 (BACE1) thereby reducing the amyloidogenic processing of the amyloid precursor protein (APP) to Aβ [34,35].
PrPC is subject to proteolytic processing (Figure 1B). Accumulating data indicate the importance of these processing steps which seem to regulate the multiple functions of PrPC under physiological conditions and produce biologically active fragments. In addition, processing of the prion protein is thought to influence not only the course of prion diseases but also other neurodegenerative diseases. Here, we review the three most important cleavage events, i.e. the α-cleavage, β-cleavage, and ectodomain shedding, with regard to their biological relevance, role in disease, and responsible proteases. Regulating these cleavages might be of therapeutic interest. We will not discuss artificial proteolytic processing of the prion protein such as treatment with PK or thermolysin which is routinely performed for research and diagnostic purposes [36-39].
Alpha-cleavage of PrPC
Description
The mature prion protein is composed of two structurally divergent parts, each of them making up roughly one half of the full-length protein: the less-structured N-terminal part with its flexible N-terminus and the compact and globular C-terminal domain (Figure 1). Since, in general, structure determines protein function, it was reasonable to expect that a cleavage in the middle of PrPC, separating these two unequal parts, would produce two fragments with diverse functions distinct from the function of full length PrPC.
Indeed, as initially reported for chicken PrP and later extended for mammalian PrPC, a released soluble fragment of approximately 11kD (termed N1) and a N-terminally truncated membrane-attached counterpart of approximately 18 kD (termed C1, which after deglycosylation runs at approximately 16 kD [40,41]) were found in transfected cell lines and, more importantly, under physiological conditions in brain homogenates and cerebrospinal fluid [42-44]. The corresponding cleavage site was termed α (discriminating it from another upstream β-cleavage site [see below]) and was identified by sequencing and epitope mapping at positions K110/H111 or H111/M112 (human sequence) located directly upstream of the hydrophobic core domain [44-46]. In fact, α-cleavage is the main proteolytic processing event yielding C1 fragments that accumulate at the plasma membrane and make up 5-50% of total PrPC levels depending on the cell type and brain region [43,44,47]. Several cellular compartments have been suggested for this cleavage event. Since Brefeldin A and lysosomal inhibitors [43] as well as cholesterol depletion [48] block production of N1 or C1 fragments, α-cleavage was thought to occur in acidic endosomal compartments or in detergent resistant microdomains of the plasma membrane respectively. In contrast to this, a more recent study using different constructs of mammalian PrP indicates that α-cleavage does not require lipid raft localization and seems to take place while PrPC is traversing the late secretory pathway [49]. This finding is also supported by a cell-culture based study investigating processing of bovine PrPC [50].
Identifying the protease
Controversy remains regarding the identity of the protease(s) responsible for α-cleavage. Initial reports suggested an involvement of lysosomal serine proteases [43]. However, the use of chicken PrP and potential occurrence of PrP fragments resulting from lysosomal degradation impede interpretation of these data. The serine protease plasmin is yet another enzyme that has been suggested for this cleavage in vitro, yet in vivo data could not confirm this [51]. Inhibitor-based in vitro experiments analyzing the α-cleavage of fluorescently labelled PrPC suggested involvement of calpain-like activity, but again in vivo proof is still lacking [52].
Most of the current work on the identification of the responsible protease and probably best experimental support so far focuses on the A-disintegrin-and-metalloproteinase (ADAM) family of proteases. Many of these membrane-bound and zinc-dependent proteases of the adamalysin subfamily perform proteolytic ectodomain shedding of a broad range of substrates in various physiological and pathological processes [53,54]. Mainly two members of this group, ADAM10 and ADAM17 (TNFα-converting enzyme, TACE), have been suggested to be responsible for α-cleavage of PrPC. At first, it was shown that production of N1 and C1 fragments could be increased in HEK cells upon phorbolester-driven activation of protein kinase C (PKC) [46]. This observation was systematically followed by inhibitor studies that suggested ADAM10 and ADAM17 as PrPC-cleaving enzymes with ADAM10 being responsible for the constitutive α-cleavage while processing via ADAM17 was stimulus-dependent [55]. ADAM9, another member of the ADAM family, was suggested to contribute indirectly to the production of N1/C1, namely by shedding of ADAM10 from the membrane [56]. These results could be confirmed by overexpression of ADAM10 in cell culture [57] and by the finding that inhibitors of metalloproteases were able to block α-cleavage [58]. A role of ADAM10 in the constitutive α-cleavage was then further supported by a study investigating the content of PrPC fragments in human cerebral cortex. Despite inter-individual variations, this study found a positive correlation between C1 amounts and levels of active ADAM10 [59].
However, there is also strong controversy. An initial opposing report challenging the role of ADAM10 in this cleavage came from a study where neither ectopic expression nor siRNA depletion of ADAM10 in HEK cells affected C1 levels [60]. Another study was likewise unable to detect enhanced amounts of specific cleavage products of PrPC in transgenic mice that overexpressed ADAM10 in neurons [61]. Recently, ADAM8 and not ADAM10 was identified as the main contributor to α-cleavage in skeletal muscle, although it should be noted, that expression of ADAM10 in muscle is only 2% of its level in brain and thus the situation in the CNS might differ [62]. These authors also described a self-regulatory loop with PrPC being able to upregulate ADAM8 thereby modifying its own cleavage and the production of C1/N1 fragments. Finally, in vivo data showed unaltered levels of C1 and N1 fragments in mice deficient in ADAM10 in neuronal precursor cells [41]. Thus, involvement of this protease in α-cleavage remains controversial although it is certain that interspecies and inter-tissue differences exist and a certain degree of redundancy [54] complicates these investigations as it is possible that additional proteases might take over ADAM10-functions in the absence of ADAM10.
As mentioned before, ADAM17 is another candidate protease for the α-cleavage of PrPC. Not only its contribution to cleavage but also the mechanism of regulation has been investigated in detail (reviewed in [63]). In brief, studies on cell lines and primary neurons showed that stimulation of muscarinic receptor subtypes M1 and M3 by cholinergic agonists induces a cascade of events involving the activation of certain isoforms of protein kinase C as well as extracellular regulated kinase-1 (ERK-1). The latter leads to phosphorylation of ADAM17 thereby upregulating its activity, which then culminates in increased α-cleavage of PrPC [64,65]. Furthermore, ERK-1 not only controls proteolysis of PrPC but also its expression levels via promoter transactivation in a regulatory cascade involving the transcription factor AP-1 [66]. A similar transcriptional control of PrPC has previously been shown for the amyloid intracellular domain (AICD) that is produced by γ-secretase mediated cleavage of APP and acts on PrPC transcription via p53 upregulation [67]. However, neither the regulation of PrPC by AICD nor the involvement of ADAM17 in PrPC endoproteolysis could be confirmed by follow-up studies using cell culture models or transgenic mice [60,68].
While the primary sequence around the cleavage site was reported by one group to be of significant importance for the generation of N1 and C1 [69], studies of others indicate that the protease is surprisingly tolerant towards different kinds of modifications within the PrPC sequence but might depend on the highly conserved HC domain as well as on PrPC membrane-anchoring [70,71]. Of note, sequence differences at the cleavage site (H111/M112 in humans compared to H110/V111 in mice) may account for interspecies differences regarding the α-cleavage with ADAM17 showing preference for murine PrPC [69,72]. However, since there is uncertainty on the true identity of the protease responsible for α-cleavage, the term “α-PrPase” seems justified [70]. This also avoids confusion with the “α-secretase” that performs the non-amyloidogenic processing of APP and has recently been identified to be ADAM10 [73-76].
N1-fragment
Despite the enigma concerning the nature of the α-PrPase, several recent findings highlight the physiological importance of α-cleavage of PrPC. Firstly, several functions of PrPC have been attributed to the N-terminal part of the protein and binding of a variety of ligands was shown to take place to different motifs of this part (reviewed in [77]). Therefore, α-cleavage can be seen as a way to negatively regulate these functions. Secondly, produced N1 and C1 fragments have intrinsic functions. For soluble N1, a role in intercellular communication and neuroprotective functions were suggested [78] (Figure 2). Moreover, N1 production was shown to interfere with the neurotoxicity of Aβ oligomers, the proposed neurotoxic species in AD. Recently, two stretches (residues 23-27 and 95-110) within the N-terminus of PrPC have been reported to build a high-affinity platform for the binding of Aβ oligomers [29,79]. Thus, in addition to the neuroprotective signalling, this effect might in part be achieved by binding of soluble N1 to amyloid β-oligomers thereby blocking neurotoxic signalling pathways [80] (Figure 2). Interestingly, this blocking and neuroprotective function of N1 might not be limited solely to Aβ oligomers but could be a more general mechanism of competing with toxic β-sheet-rich conformers found in several neurodegenerative diseases [19,81]. By releasing the N-terminus of PrPC, α-cleavage might have a dual protective function: producing the neuroprotective catabolite N1 as well as inhibiting neurotoxic signalling which is thought to require full-length PrPC not only in prion disease [28,82] but also in other neurodegenerative conditions [19,29,30,83]. In line with this, expression of N-terminally truncated or deleted constructs that are unable to undergo this cleavage lead to toxicity in transgenic mice [84,85].
Figure 2.
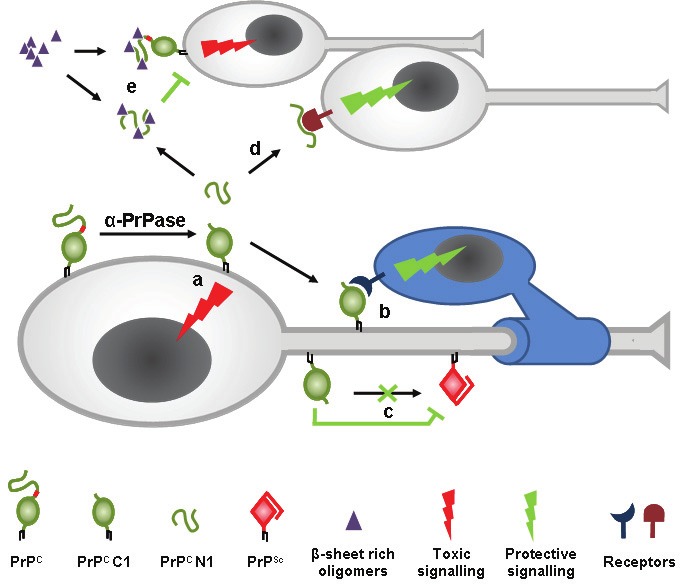
Model of the α-cleavage of PrPC. The α-PrPase produces membrane-attached C1 and soluble N1 fragments. C1 plays a dual role by initiating apoptotic signalling (a) and its involvement in trophic signalling onto Schwann cells to maintain the myelin sheath (b). Due to the loss of the neurotoxic domain, C1 is unable to misfold into the pathogenic isoform PrPSc and, in addition, might be a dominant negative inhibitor of the conversion process (c). The released N1 fragment is involved in neuroprotective signalling (d). While β-sheet-rich oligomers are thought to bind to the N-terminus of PrPC to induce toxic signalling, α-cleavage not only prevents this interaction but produces N1 that might bind these oligomers and block their toxicity (e). References are given in the text.
C1-fragment
The physiological function of the membrane-attached C1 fragment is controversial (Figure 2). On the one hand, cell culture based studies suggest that C1, similar to full-length PrPC [86,87], is able to initiate a p53-dependent apoptotic cascade resulting in increased caspase-3 activation [88]. In contrast to full-length PrPC, this effect of C1 does not depend on clathrin-mediated endocytosis and seems to involve a different pathway leading to p53 activation [89].
On the other hand, a protective function of the C1 fragment has recently been shown [40]. More than a decade ago, it was already found that transgenic mice expressing N-terminal deletion mutants of PrPC show diverse signs of neurotoxicity and demyelination, which, although milder, could also be observed in PRNP0/0 mice [84,90,91]. Interestingly, all of these deletions included the α-cleavage site and could be rescued by coexpression of PrPC. It was postulated that axonal C1 expression is linked to the maintenance of the myelin sheath in the peripheral nervous system [40]. Since expression of C1 at the axonal surface is able to rescue myelination, C1 acts in trans on adjacent Schwann cells to initiate protective signalling that helps to maintain the myelin sheath. It might be hypothesized, that these signals should be rather longstanding than transient. In line with this assumption, C1 was shown to have a much longer half-life than PrPC and to accumulate on the cellular surface [43]. In fact, the study of Bremer et al. found that sciatic nerves contain even more C1 than full-length PrPC [40]. Unlike PrPC, C1 is unable to undergo copper-stimulated [92-94] and clathrin-mediated endocytosis, which has been shown by several groups to depend on the N-terminus [95-97] and its interaction with LRP1 [98,99].
This characteristic of C1 might account for its involvement in longstanding signalling events that might not be limited to the role mentioned above.
In contrast to the findings of Bremer et al., another group reported that N1 rather than C1 might be responsible for the myelinotrophic effects and that C1 is not neuroprotective [100]. However, the α-cleavage seems to be fundamental in this regard.
Since α-cleavage takes place directly N-terminal of the HC, the resulting C1 fragment with its exposed stretch of hydrophobic amino acids is reminiscent of some viral fusion proteins (e.g. those of paramyxoviruses) which after proteolytic activation also harbor a free stretch of approximately 20 hydrophobic amino acids, termed the fusion peptide. This fusion peptide inserts into the host-cell membrane to initiate fusion [101,102]. Although data in this regard is lacking, we hypothesize that the N-terminus of C1 might likewise be able to insert into membranes, for instance of neighbouring cells. Given the suggested role of PrPC in cell adhesion [11,12], this aspect might deserve further investigation.
Alpha-cleavage protects PrPC from misfolding
The α-cleavage site is located within the neurotoxic domain (ND) of PrPC (amino acids 106-126 in human and 105-125 in murine sequence) which constitutes a structural prerequisite for the conformational conversion to PrPSc and is thought to be a main contributor to the amyloidogenic properties of PrPSc [21,44,103]. Interestingly, a synthetic peptide spanning this short region tends to form fibrils, is neurotoxic, and can induce proliferation of astrocytes [104,105]. In addition to this neurotoxic domain, the N-terminus of PrPC might play a substantial role in prion pathogenesis as indicated by a recent report [106]. Thus, α-cleavage releases N1 and cleaves PrPC within the ND, thereby preventing misfolding of residual C1 (Figure 2). Supporting the importance of an intact ND for the misfolding, one study found that all PK-resistant PrPSc cores studied in brains from sCJD or GSS patients exhibited an intact α-cleavage site [58]. A palindromic sequence (112-AGAAAAGA-119) is located directly downstream of the cleavage site and was shown to be of importance for the physical interaction of PrPC with PrPSc during the conformational conversion [107]. Although this primary sequence would remain intact, α-cleavage might alter the secondary structure of this region and thus block this interaction.
On the other hand, after conversion of PrPC to PrPSc, α-cleavage was found to be impaired. This fact might be explained by a sterical hindrance preventing the protease from getting access to the cleavage site. Recent reports using different experimental setups and prion strains show that increasing the level of C1 relative to full-length PrPC has protective effects in prion disease [47,108]. In the latter study, transgenic mice only expressing C1 in the absence of full-length PrPC were not susceptible to prion infection, did not accumulate PrPSc, and did not show any signs of neurodegeneration. Moreover, coexpression of PrPC and C1 led to reduced PrPSc production and prolonged incubation times after prion inoculation compared to wildtype mice. In view of the unphysiological overexpression of C1 in this study, it remains questionable whether C1 really is a dominant-negative inhibitor of the conversion process [47] (Figure 2) or if the observed effect is just due to the dilution of convertible full-length PrPC. Nevertheless, these data clearly demonstrate the protective effect of the α-cleavage.
Beta-cleavage of the prion protein
An additional but under physiological conditions less prominent cleavage of PrPC takes place upstream of the α-cleavage site at the end of the octameric repeat region (Q90 in the murine sequence). Cleavage at this β-site produces soluble N2 and membrane-bound C2 fragments of ~9 kDa and ~20 kDa respectively. The latter fragment was found to be the main cleavage product of PrP in prion infected neuroblastoma cells [109] and in brains of CJD patients [44,58] indicating a pathophysiological relevance. Since C2 shares several characteristics (i.e. its insolubility in nondenaturating detergents and the electrophoretic mobility [110,111]) it was regarded as the “in vivo homologue” of the protease-resistant core of PrPSc (referred to as PrP (27-30)) that is experimentally derived after treatment with PK [44,112]. In fact, PK cleavage sites within PrPSc are located at the end of the octameric repeat region [84]. Initially it was thought that the C2 fragment resulted from an incomplete degradation of PrPSc by lysosomal proteases [109]. Later, using pharmacological and genetic approaches, it was found that C2 was produced by calpains, and Ca2+-regulated cysteine proteases. Inhibition of lysosomal proteases, caspases, and the proteasome instead had no effect on C2 levels [113].
Similar C2 fragments were also found to be drastically increased in cultured cells that were stressed with reactive oxygen species (ROS) and cleavage at the β-site was dependent on copper ions [45,58,114]. Of note, PrPC was attributed to be involved in protection against oxidative stress [115] and, in turn, oxidative stress was thought to contribute to the pathologic events during prion disease [116-119]. Thus, β-cleavage might be one step in the mechanism by which PrPC fulfils its cellular protection against oxidative stress [120]. Furthermore, it was shown that this cleavage i) can be directly performed by ROS [114], ii) is dependent on specific Trp residues within the octameric repeat region [120,121], iii) takes place at the cellular surface, and - in contrast to what was published before [113] - iv) seems not to depend on calpains [120].
Thus, cleavage at the β-site at the end of the octameric repeat region can be achieved by two distinct mechanisms: proteolytic processing in prion diseases and cleavage under conditions of oxidative stress (performed by ROS) [45]. To avoid confusion, it would be reasonable to consider a new nomenclature separating these two events. However, in contrast to the C1/N1 fragments derived from α-cleavage, there is no experimental data in favor of a physiological function of the resulting C2/N2 fragments [78,88].
Shedding and anchorless PrP
A third physiological cleavage of PrPC occurs in close proximity to its GPI-anchor and results in the release of the almost full-length protein from the plasma membrane. This protease-mediated cleavage at the very C-terminus is termed shedding [122]. This is distinct from experimentally performed cleavage of the GPI-anchor by phospholipases which was fundamental for the identification of the GPI-anchorage of PrPC [42,123]. However, since phospholipases are cytoplasmic enzymes they are unlikely to encounter PrPC under physiological conditions. In this regard it was surprising when Harris et al. studied the processing of chicken PrP in neuroblastoma cells and found that parts of the released full-length protein and C1 fragments still contained some anchor structure [42]. This finding might be explained by the existence of phospholipases in the cell culture serum as observed by others [124].
In cell culture, a small but significant fraction of total PrPC is slowly but constitutively shed into the media and shed PrPC lacks any parts of the GPI-modification indicating cleavage by a protease [122]. A shed and soluble form of PrPC was also found in human CSF [125] and blood [124,126,127] indicating a physiological relevance. Thus, shedding of PrPC not only occurs in neurons but also in lymphoid cells [124]. Although PrPC was shown to be less sensitive to phospholipase cleavage compared to other GPI-anchored proteins, it has a much shorter half-life at the cell surface [128], a notion that further supported involvement of a “sheddase”. Proteolytic shedding was also confirmed for bovine PrPC in two cell culture models [50].
Identification of the PrP sheddase
Even though the mechanism releasing PrPC from the surface was apparent, it took some time until candidate proteases for this event were suggested. In vitro studies using inhibitors and stimulators of zinc metalloproteases suggested that shedding of PrPC requires a zinc metalloproteases and that this event is remarkably reminiscent of the α-secretase-mediated cleavage of APP [122,129]. Finally, cell culture experiments identified ADAM10 as the sheddase of PrPC [60]. This study confirmed the previously suggested cleavage site by mass spectrometry at position Gly228/Arg229 [130] and found that ADAM9 has an indirect influence on the shedding by regulating ADAM10 activity, a finding that was also observed by others [56,131,132]. In addition, ADAM17 does not seem to be involved in PrP shedding [60].
A somewhat opposing report showed that overexpression of a novel sorting nexin, SNX33, interfered with the constitutive endocytosis of PrPC thereby leading to elevated amounts of surface PrPC that was counterbalanced by increased release. This release into the culture medium however appeared to be independent of ADAM10 and was rather performed by phospholipase cleavage of the GPI-anchor [57]. In vivo data supporting this SNX33-regulated and ADAM10-independent release of PrPC is lacking to date. Proteolytic processing of PrPC and the role of ADAM10 was also investigated using mice that moderately overexpressed the protease [61]. In this study, the authors did not find increased production of C1/N1 fragments or shed PrP. Instead they found reduced PrPC mRNA levels and suggested that ADAM10 overexpression transcriptionally downregulates PrPC expression.
In contrast, in vivo data from our group confirmed ADAM10 as the main functionally relevant sheddase of PrPC [41]. First, mice with a knockout of ADAM10 in neural precursor cells had increased amounts of PrPC (Figure 3A) while mRNA levels remained unaffected compared to controls. Second, shedding was absent in primary neurons derived from these mice (Figure 3B). Third, genetic reintroduction of ADAM10 into neural stem cells of these knockout mice restored shedding of PrPC after neuronal differentiation. It remains to be elucidated if i) ADAM10 is able to shed PrPC in trans as shown for Ephrin, another substrate of ADAM10 [133,134], ii) ADAM10 from other cell types contributes to the release of PrPC from neuronal membranes, and iii) the protease also plays a role in the sorting of PrPC to specific regions prior to cleavage, as shown for the processing of its substrate CD23 [135,136]. Additionally, it has recently been shown that ADAM10 activity towards other substrates, i.e. APP, is tightly regulated on different levels including modulation of its expression, its proteolysis, and its trafficking [131,132,137,138]. It deserves further investigations if shedding of PrPC is likewise regulated.
Figure 3.
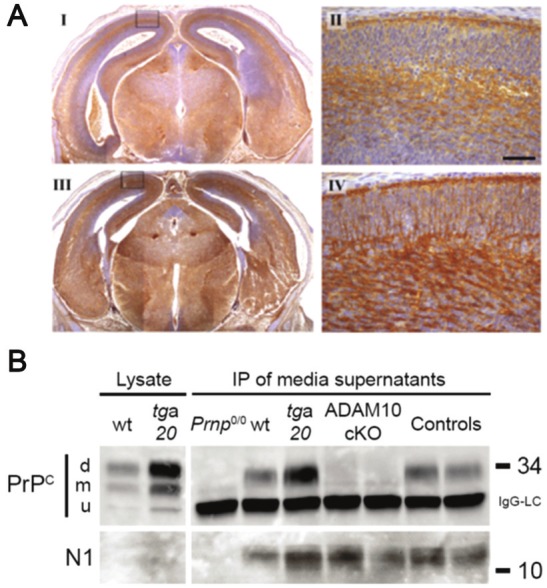
Lack of ADAM10 results in elevated amounts of PrPC and loss of PrP shedding. (A) Increased immunoreactivity for PrPC (mouse monoclonal antibody POM1 was used) in brains of embryonic mice with an ADAM10 knockout in neural precursor cells (III and IV) compared to littermate controls (I and II). II and IV represent magnifications of cortex area (see boxes in I and III respectively). Scale bar is 50 μm. (B) Immunoprecipitation (IP) of released PrP fragments in media supernatants of primary neurons derived from embryonic PrPC knockout (Prnp0/0), wildtype (wt), and PrPC-overexpressing (tga20) mice as well as from ADAM10 conditional knockout (ADAM10 cKO) and littermate controls reveals a loss of shedding when ADAM10 is lacking while production of the N1 fragment is not affected (d, m, u = di-, mono-, unglycosylated forms of PrPC; IgG-LC = light chain of capture antibody POM2). (B) is taken from [41] originally published by BioMed Central.
Is there a physiological relevance for shedding PrPC?
Like α-cleavage and clathrin-mediated endocytosis, shedding can be seen as a mechanism to regulate PrPC levels at the plasma membrane. This is of outstanding importance regarding the multiple functions discussed for PrPC, especially its receptor properties and its involvement in toxic signalling cascades. In fact, primary neurons of mice lacking ADAM10 accumulated PrPC in compartments of the early secretory pathway rather than tolerating increased levels at the cellular surface [41]. Surprisingly, ADAM10-knockout mice also showed reduced activity of BACE1 [74]. This unexpected finding could be explained by the inhibitory effect of PrPC towards BACE1 in the secretory pathway [34,35].
In addition, shed PrP itself may have intrinsic functions differing from membrane-attached PrPC [41,42,124]. For instance, it might act as a soluble trophic factor onto neighbouring or distant cells. Furthermore, as discussed for the N1 fragment, shed PrP could potentially bind β-sheet-rich oligomers thereby blocking their deleterious effects and directing them to phagocytosis and degradation (Figure 4).
Figure 4.

Ectodomain shedding and the role of anchorless PrP. ADAM10-mediated shedding releases nearly full-length PrP (and C1 fragments) from the plasma membrane (a) thereby impairing toxic and protective signaling via PrPC. By reducing the substrate for prion conversion, shedding might have an protective role in prion disease (b). On the other hand, shed PrP was shown to be convertible to PrPSc (c) and shedding might release PrPSc molecules from the membrane (d). Anchorless PrPSc might facilitate the spread of infectivity and induce neurotoxicity and misfolding of PrPC in other cells (e). References are given in the text. It might be speculated that shed PrP acts as a trophic factor and is involved in neuroprotective signalling (f). In addition and as shown for the N1 fragment, shed PrP might likewise be able to bind β-sheet-rich oligomers thereby blocking their toxic effects (g) and potentially guiding them towards phagocytosis and degradation (h).
It has been shown that Aβ oligomers damage neurons by the production of ROS in a process that involves activation of the N-methyl-D-aspartate receptor (NMDAR) [139]. This neurotoxic interaction is influenced by copper ions and PrPC with PrPC limiting excessive NMDAR activity by toxic oligomers [20]. An inhibitory effect of PrPC towards NMDAR has previously been shown by others [140]. Interestingly, neurons react to Aβ oligomer treatment by increasing the amount of PrPC at the plasma membrane [141]. Of note, a recent report indicates that activation of NMDAR leads to upregulation of the PrP sheddase ADAM10 in a Wnt/MAPK-dependent cascade [142]. We hypothesize that these initially unrelated findings might combine to a regulatory feedback loop in the control of Aβ oligomer-mediated neurotoxicity. In this scenario, increased surface expression and shedding of PrPC might be a mechanism to block the effects of toxic oligomers.
PrP-shedding and prion disease
The role of shedding in the course of prion disease is not fully understood to date. Two opposing scenarios are conceivable (Figure 4): On the one hand, shedding of PrPC may be protective against prion disease since shedding releases PrPC from the surface and reduces the substrate for conversion [57,143]. In fact, early experiments with prion-infected cell culture models showed that artificially induced release of surface PrPC by treatment with phospholipase C or with the drug filipin, which both mimic ectodomain shedding, interfered with the formation of PrPSc [143-146]. However, it has to be taken into account that this forced release is much more effective than the physiological shedding. In line with this notion, Taylor et al. did not find any alterations in the generation of PrPSc by inhibition or activation of ADAM10-mediated shedding [60]. On the other hand, shedding of PrPC may favor prion disease. In this scenario, shedding of misfolded prions - as released factors - could facilitate prion-spread throughout the nervous system. Accordingly, artificial removal of the GPI-anchor from PrPSc in brain-homogenates from prion-infected mice by cathepsin D did not inhibit further PrPSc formation and infectivity [147]. In contrast to phospholipase C, which was shown to be unable to release PrPSc without prior denaturation of the substrate [147,148], ADAM10 can shed PrPSc in infected neuroblastoma cells [60]. In addition, anchorless mutant versions of PrPC can be converted to PrPSc in cell-culture and cell-free systems [149,150].
Besides all these hints from in vitro studies, the role of shedding and anchorless PrP are only poorly understood in vivo. Endres et al. found prolonged incubation times in prion-infected mice that overexpressed ADAM10 [61]. Although these authors attributed this to transcriptional downregulation of PrPC by ADAM10, which could not be confirmed in another study [41], this finding underlines the first scenario, with ADAM10-mediated shedding of PrPC having a protective effect during prion disease. In contrast, the second rather deleterious scenario is supported by results showing that transgenically expressed anchorless, secreted PrP leads to widespread formation of PrPSc when mice are prion infected [151]. Although some defects in learning and memory were later found in these mice [152], lack of clinical signs of prion disease and a normal life-span were surprising but could later be explained by a low expression of the anchorless PrP (~50% of PrPC expression in wildtype mice). Indeed, a follow-up study by the same group using mice with a two-fold expression of anchorless PrP found a new type of prion disease upon infection, characterized by a high degree of infectivity and the appearance of dense PK-resistant plaques, whereas grey matter spongiosis was reduced compared to nontransgenic control mice [153]. Another group increased the expression of anchorless PrP in mice 1.7-fold compared to wildtype PrPC expression and found that these animals spontaneously developed neurological signs reminiscent of Gerstmann-Sträussler-Scheinker (GSS) disease that was accompanied by the generation of anchorless, transmissible bona fide prions [106]. In fact, some mutations in the PRNP gene resulting in anchorless forms of the proteins were found in patients to cause GSS [154,155].
Although none of these studies directly investigated ADAM10-mediated shedding, the anchorless version that was used is highly reminiscent of physiologically shed PrPC. Thus, these findings support the second scenario with shed PrP being able to misfold and contribute to PrPSc propagation and infectivity. However, since incubation time to terminal prion disease in “anchorless PrP” mice was prolonged, neurodegeneration seems to depend on membrane-anchoring of PrPC [153], a finding that again indicates the involvement of different entities in neurotoxicity and prion propagation [25].
Outlook
Here we summarized the current knowledge on the proteolytic processing of the prion protein. Cleavage of this protein is of outmost importance for the regulation of the functions of PrPC and the generation of fragments with autonomous functions such as neuroprotective signalling or myelin maintenance. Furthermore, it has become obvious that at least two cleavage events, i.e. α-cleavage and ectodomain shedding, very likely have the capacity to influence neurodegenerative diseases. Since candidate proteases would provide an attractive goal for therapeutic treatment of these devastating conditions, more research aiming at a deeper understanding of the mechanisms, relevance, and key players of prion protein processing is required. For instance, the therapeutic potential of ADAM inhibition and activation has long been in the focus of researchers in the context of various diseases (as reviewed by [156]). This knowledge should be adopted to and intensified for the field of prion diseases and other neurodegenerative disorders as well.
Acknowledgements
We thank Kerstin Rehm (Department of Medical Microbiology, Virology and Hygiene; University Medical Center Hamburg-Eppendorf) for proofreading of the manuscript. HCA is supported by the Leibniz graduate school “Model systems for infectious diseases”. This work is supported by grants of the Deutsche Forschungsgemeinschaft (especially FOR885 and GRK1459), the Landesexzellenzinitiative of Hamburg (SDI-LEXI), and the Bundesministerium für Bildung und Forschung to ERA-Net Neuron: ADTest.
References
- 1.Riek R, Hornemann S, Wider G, Glockshuber R, Wüthrich K. NMR characterization of the full-length recombinant murine prion protein, mPrP(23-231) FEBS Lett. 1997;413:282–288. doi: 10.1016/s0014-5793(97)00920-4. [DOI] [PubMed] [Google Scholar]
- 2.Biasini E, Turnbaugh JA, Unterberger U, Harris DA. Prion protein at the crossroads of physiology and disease. Trends Neurosci. 2012;35:92–103. doi: 10.1016/j.tins.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brugger B, Graham C, Leibrecht I, Mombelli E, Jen A, Wieland F, Morris R. The membrane domains occupied by glycosylphosphatidylinositol-anchored prion protein and Thy-1 differ in lipid composition. J Biol Chem. 2004;279:7530–7536. doi: 10.1074/jbc.M310207200. [DOI] [PubMed] [Google Scholar]
- 4.Taylor DR, Hooper NM. The prion protein and lipid rafts. Mol Membr Biol. 2006;23:89–99. doi: 10.1080/09687860500449994. [DOI] [PubMed] [Google Scholar]
- 5.Linden R, Martins VR, Prado MA, Cammarota M, Izquierdo I, Brentani RR. Physiology of the prion protein. Physiol Rev. 2008;88:673–728. doi: 10.1152/physrev.00007.2007. [DOI] [PubMed] [Google Scholar]
- 6.Aguzzi A, Polymenidou M. Mammalian prion biology: one century of evolving concepts. Cell. 2004;116:313–327. doi: 10.1016/s0092-8674(03)01031-6. [DOI] [PubMed] [Google Scholar]
- 7.Aguzzi A, Baumann F, Bremer J. The prion's elusive reason for being. Annu Rev Neurosci. 2008;31:439–477. doi: 10.1146/annurev.neuro.31.060407.125620. [DOI] [PubMed] [Google Scholar]
- 8.Graner E, Mercadante AF, Zanata SM, Forlenza OV, Cabral AL, Veiga SS, Juliano MA, Roesler R, Walz R, Minetti A, Izquierdo I, Martins VR, Brentani RR. Cellular prion protein binds laminin and mediates neuritogenesis. Brain Res Mol Brain Res. 2000;76:85–92. doi: 10.1016/s0169-328x(99)00334-4. [DOI] [PubMed] [Google Scholar]
- 9.Steele AD, Emsley JG, Ozdinler PH, Lindquist S, Macklis JD. Prion protein (PrPc) positively regulates neural precursor proliferation during developmental and adult mammalian neurogenesis. Proc Natl Acad Sci U S A. 2006;103:3416–3421. doi: 10.1073/pnas.0511290103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hajj GN, Lopes MH, Mercadante AF, Veiga SS, da Silveira RB, Santos TG, Ribeiro KC, Juliano MA, Jacchieri SG, Zanata SM, Martins VR. Cellular prion protein interaction with vitronectin supports axonal growth and is compensated by integrins. J Cell Sci. 2007;120:1915–1926. doi: 10.1242/jcs.03459. [DOI] [PubMed] [Google Scholar]
- 11.Mange A, Milhavet O, Umlauf D, Harris D, Lehmann S. PrP-dependent cell adhesion in N2a neuroblastoma cells. FEBS Lett. 2002;514:159–162. doi: 10.1016/s0014-5793(02)02338-4. [DOI] [PubMed] [Google Scholar]
- 12.Malaga-Trillo E, Solis GP, Schrock Y, Geiss C, Luncz L, Thomanetz V, Stuermer CA. Regulation of embryonic cell adhesion by the prion protein. PLoS Biol. 2009;7:e55. doi: 10.1371/journal.pbio.1000055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiarini LB, Freitas AR, Zanata SM, Brentani RR, Martins VR, Linden R. Cellular prion protein transduces neuroprotective signals. EMBO J. 2002;21:3317–3326. doi: 10.1093/emboj/cdf324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rambold AS, Muller V, Ron U, Ben-Tal N, Winklhofer KF, Tatzelt J. Stress-protective signalling of prion protein is corrupted by scrapie prions. EMBO J. 2008;27:1974–1984. doi: 10.1038/emboj.2008.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brown DR. Prion protein expression modulates neuronal copper content. J Neurochem. 2003;87:377–385. doi: 10.1046/j.1471-4159.2003.02046.x. [DOI] [PubMed] [Google Scholar]
- 16.Mouillet-Richard S, Ermonval M, Chebassier C, Laplanche JL, Lehmann S, Launay JM, Kellermann O. Signal transduction through prion protein. Science. 2000;289:1925–1928. doi: 10.1126/science.289.5486.1925. [DOI] [PubMed] [Google Scholar]
- 17.Mouillet-Richard S, Schneider B, Pradines E, Pietri M, Ermonval M, Grassi J, Richards JG, Mutel V, Launay JM, Kellermann O. Cellular prion protein signaling in serotonergic neuronal cells. Ann N Y Acad Sci. 2007;1096:106–119. doi: 10.1196/annals.1397.076. [DOI] [PubMed] [Google Scholar]
- 18.Solforosi L, Criado JR, McGavern DB, Wirz S, Sanchez-Alavez M, Sugama S, DeGiorgio LA, Volpe BT, Wiseman E, Abalos G, Masliah E, Gilden D, Oldstone MB, Conti B, Williamson RA. Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science. 2004;303:1514–1516. doi: 10.1126/science.1094273. [DOI] [PubMed] [Google Scholar]
- 19.Resenberger UK, Harmeier A, Woerner AC, Goodman JL, Muller V, Krishnan R, Vabulas RM, Kretzschmar HA, Lindquist S, Hartl FU, Multhaup G, Winklhofer KF, Tatzelt J. The cellular prion protein mediates neurotoxic signalling of beta-sheet-rich conformers independent of prion replication. EMBO J. 2011;30:2057–2070. doi: 10.1038/emboj.2011.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.You H, Tsutsui S, Hameed S, Kannanayakal TJ, Chen L, Xia P, Engbers JD, Lipton SA, Stys PK, Zamponi GW. Abeta neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc Natl Acad Sci U S A. 2012;109:1737–1742. doi: 10.1073/pnas.1110789109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prusiner SB. The prion diseases. Brain Pathol. 1998;8:499–513. doi: 10.1111/j.1750-3639.1998.tb00171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Imran M, Mahmood S. An overview of human prion diseases. Virol J. 2011;8:559. doi: 10.1186/1743-422X-8-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Imran M, Mahmood S. An overview of animal prion diseases. Virol J. 2011;8:493. doi: 10.1186/1743-422X-8-493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prusiner SB. Prions. Proc Natl Acad Sci U S A. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sandberg MK, Al-Doujaily H, Sharps B, Clarke AR, Collinge J. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature. 2011;470:540–542. doi: 10.1038/nature09768. [DOI] [PubMed] [Google Scholar]
- 26.Aguzzi A, Calella AM. Prions: protein aggregation and infectious diseases. Physiol Rev. 2009;89:1105–1152. doi: 10.1152/physrev.00006.2009. [DOI] [PubMed] [Google Scholar]
- 27.Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73:1339–1347. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- 28.Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature. 1996;379:339–343. doi: 10.1038/379339a0. [DOI] [PubMed] [Google Scholar]
- 29.Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature. 2009;457:1128–1132. doi: 10.1038/nature07761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Lauren J, Gimbel ZA, Strittmatter SM. Memory impairment in transgenic Alzheimer mice requires cellular prion protein. J Neurosci. 2010;30:6367–6374. doi: 10.1523/JNEUROSCI.0395-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Balducci C, Beeg M, Stravalaci M, Bastone A, Sclip A, Biasini E, Tapella L, Colombo L, Manzoni C, Borsello T, Chiesa R, Gobbi M, Salmona M, Forloni G. Synthetic amyloid-beta oligomers impair long-term memory independently of cellular prion protein. Proc Natl Acad Sci U S A. 2010;107:2295–2300. doi: 10.1073/pnas.0911829107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Calella AM, Farinelli M, Nuvolone M, Mirante O, Moos R, Falsig J, Mansuy IM, Aguzzi A. Prion protein and Abeta-related synaptic toxicity impairment. EMBO Mol Med. 2010;2:306–314. doi: 10.1002/emmm.201000082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kessels HW, Nguyen LN, Nabavi S, Malinow R. The prion protein as a receptor for amyloid-beta. Nature. 2010;466:E3–4. doi: 10.1038/nature09217. discussion E4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parkin ET, Watt NT, Hussain I, Eckman EA, Eckman CB, Manson JC, Baybutt HN, Turner AJ, Hooper NM. Cellular prion protein regulates beta-secretase cleavage of the Alzheimer's amyloid precursor protein. Proc Natl Acad Sci U S A. 2007;104:11062–11067. doi: 10.1073/pnas.0609621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Griffiths HH, Whitehouse IJ, Baybutt H, Brown D, Kellett KA, Jackson CD, Turner AJ, Piccardo P, Manson JC, Hooper NM. Prion protein interacts with BACE1 protein and differentially regulates its activity toward wild type and Swedish mutant amyloid precursor protein. J Biol Chem. 2011;286:33489–33500. doi: 10.1074/jbc.M111.278556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Notari S, Capellari S, Giese A, Westner I, Baruzzi A, Ghetti B, Gambetti P, Kretzschmar HA, Parchi P. Effects of different experimental conditions on the PrPSc core generated by protease digestion: implications for strain typing and molecular classification of CJD. J Biol Chem. 2004;279:16797–16804. doi: 10.1074/jbc.M313220200. [DOI] [PubMed] [Google Scholar]
- 37.Owen JP, Maddison BC, Whitelam GC, Gough KC. Use of thermolysin in the diagnosis of prion diseases. Mol Biotechnol. 2007;35:161–170. doi: 10.1007/BF02686111. [DOI] [PubMed] [Google Scholar]
- 38.Cronier S, Gros N, Tattum MH, Jackson GS, Clarke AR, Collinge J, Wadsworth JD. Detection and characterization of proteinase K-sensitive disease-related prion protein with thermolysin. Biochem J. 2008;416:297–305. doi: 10.1042/BJ20081235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nicot S, Baron TG. Strain-specific proteolytic processing of the prion protein in prion diseases of ruminants transmitted in ovine transgenic mice. J Gen Virol. 2010;91:570–574. doi: 10.1099/vir.0.014464-0. [DOI] [PubMed] [Google Scholar]
- 40.Bremer J, Baumann F, Tiberi C, Wessig C, Fischer H, Schwarz P, Steele AD, Toyka KV, Nave KA, Weis J, Aguzzi A. Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci. 2010;13:310–318. doi: 10.1038/nn.2483. [DOI] [PubMed] [Google Scholar]
- 41.Altmeppen HC, Prox J, Puig B, Kluth MA, Bernreuther C, Thurm D, Jorissen E, Petrowitz B, Bartsch U, De Strooper B, Saftig P, Glatzel M. Lack of a-disintegrin-and-metalloproteinase ADAM10 leads to intracellular accumulation and loss of shedding of the cellular prion protein in vivo. Mol Neurodegener. 2011;6:36. doi: 10.1186/1750-1326-6-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harris DA, Huber MT, van Dijken P, Shyng SL, Chait BT, Wang R. Processing of a cellular prion protein: identification of N- and C-terminal cleavage sites. Biochemistry. 1993;32:1009–1016. doi: 10.1021/bi00055a003. [DOI] [PubMed] [Google Scholar]
- 43.Shyng SL, Huber MT, Harris DA. A prion protein cycles between the cell surface and an endocytic compartment in cultured neuroblastoma cells. J Biol Chem. 1993;268:15922–15928. [PubMed] [Google Scholar]
- 44.Chen SG, Teplow DB, Parchi P, Teller JK, Gambetti P, Autilio-Gambetti L. Truncated forms of the human prion protein in normal brain and in prion diseases. J Biol Chem. 1995;270:19173–19180. doi: 10.1074/jbc.270.32.19173. [DOI] [PubMed] [Google Scholar]
- 45.Mange A, Beranger F, Peoc'h K, Onodera T, Frobert Y, Lehmann S. Alpha- and beta-cleavages of the amino-terminus of the cellular prion protein. Biol Cell. 2004;96:125–132. doi: 10.1016/j.biolcel.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 46.Vincent B, Paitel E, Frobert Y, Lehmann S, Grassi J, Checler F. Phorbol ester-regulated cleavage of normal prion protein in HEK293 human cells and murine neurons. J Biol Chem. 2000;275:35612–35616. doi: 10.1074/jbc.M004628200. [DOI] [PubMed] [Google Scholar]
- 47.Westergard L, Turnbaugh JA, Harris DA. A naturally occurring C-terminal fragment of the prion protein (PrP) delays disease and acts as a dominant-negative inhibitor of PrPSc formation. J Biol Chem. 2011;286:44234–44242. doi: 10.1074/jbc.M111.286195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Taraboulos A, Scott M, Semenov A, Avrahami D, Laszlo L, Prusiner SB. Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J Cell Biol. 1995;129:121–132. doi: 10.1083/jcb.129.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Walmsley AR, Watt NT, Taylor DR, Perera WS, Hooper NM. alpha-cleavage of the prion protein occurs in a late compartment of the secretory pathway and is independent of lipid rafts. Mol Cell Neurosci. 2009;40:242–248. doi: 10.1016/j.mcn.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 50.Zhao H, Klingeborn M, Simonsson M, Linne T. Proteolytic cleavage and shedding of the bovine prion protein in two cell culture systems. Virus Res. 2006;115:43–55. doi: 10.1016/j.virusres.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 51.Barnewitz K, Maringer M, Mitteregger G, Giese A, Bertsch U, Kretzschmar HA. Unaltered prion protein cleavage in plasminogen-deficient mice. Neuroreport. 2006;17:527–530. doi: 10.1097/01.wnr.0000209003.55728.ac. [DOI] [PubMed] [Google Scholar]
- 52.Hachiya N, Komata Y, Harguem S, Nishijima K, Kaneko K. Possible involvement of calpainlike activity in normal processing of cellular prion protein. Neurosci Lett. 2011;490:150–155. doi: 10.1016/j.neulet.2010.12.046. [DOI] [PubMed] [Google Scholar]
- 53.Reiss K, Saftig P. The "a disintegrin and metalloprotease" (ADAM) family of sheddases: physiological and cellular functions. Semin Cell Dev Biol. 2009;20:126–137. doi: 10.1016/j.semcdb.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 54.Klein T, Bischoff R. Active metalloproteases of the A Disintegrin and Metalloprotease (ADAM) family: biological function and structure. J Proteome Res. 2011;10:17–33. doi: 10.1021/pr100556z. [DOI] [PubMed] [Google Scholar]
- 55.Vincent B, Paitel E, Saftig P, Frobert Y, Hartmann D, De Strooper B, Grassi J, Lopez-Perez E, Checler F. The disintegrins ADAM10 and TACE contribute to the constitutive and phorbol ester-regulated normal cleavage of the cellular prion protein. J Biol Chem. 2001;276:37743–37746. doi: 10.1074/jbc.M105677200. [DOI] [PubMed] [Google Scholar]
- 56.Cisse MA, Sunyach C, Lefranc-Jullien S, Postina R, Vincent B, Checler F. The disintegrin ADAM9 indirectly contributes to the physiological processing of cellular prion by modulating ADAM10 activity. J Biol Chem. 2005;280:40624–40631. doi: 10.1074/jbc.M506069200. [DOI] [PubMed] [Google Scholar]
- 57.Heiseke A, Schobel S, Lichtenthaler SF, Vorberg I, Groschup MH, Kretzschmar H, Schatzl HM, Nunziante M. The novel sorting nexin SNX33 interferes with cellular PrP formation by modulation of PrP shedding. Traffic. 2008;9:1116–1129. doi: 10.1111/j.1600-0854.2008.00750.x. [DOI] [PubMed] [Google Scholar]
- 58.Jimenez-Huete A, Lievens PM, Vidal R, Piccardo P, Ghetti B, Tagliavini F, Frangione B, Prelli F. Endogenous proteolytic cleavage of normal and disease-associated isoforms of the human prion protein in neural and non-neural tissues. Am J Pathol. 1998;153:1561–1572. doi: 10.1016/S0002-9440(10)65744-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Laffont-Proust I, Faucheux BA, Hassig R, Sazdovitch V, Simon S, Grassi J, Hauw JJ, Moya KL, Haik S. The N-terminal cleavage of cellular prion protein in the human brain. FEBS Lett. 2005;579:6333–6337. doi: 10.1016/j.febslet.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 60.Taylor DR, Parkin ET, Cocklin SL, Ault JR, Ashcroft AE, Turner AJ, Hooper NM. Role of ADAMs in the ectodomain shedding and conformational conversion of the prion protein. J Biol Chem. 2009;284:22590–22600. doi: 10.1074/jbc.M109.032599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Endres K, Mitteregger G, Kojro E, Kretzschmar H, Fahrenholz F. Influence of ADAM10 on prion protein processing and scrapie infectiosity in vivo. Neurobiol Dis. 2009;36:233–241. doi: 10.1016/j.nbd.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 62.Liang J, Wang W, Sorensen D, Medina S, Ilchenko S, Kiselar J, Surewicz WK, Booth SA, Kong Q. Cellular prion protein regulates its own alpha-cleavage through ADAM8 in skeletal muscle. J Biol Chem. 2012 doi: 10.1074/jbc.M112.360891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Checler F. Two-steps control of cellular prion physiology by the Extracellular Regulated Kinase-1 (ERK1) Prion. 2012;6:23–25. doi: 10.4161/pri.6.1.18004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alfa Cisse M, Sunyach C, Slack BE, Fisher A, Vincent B, Checler F. M1 and M3 muscarinic receptors control physiological processing of cellular prion by modulating ADAM17 phosphorylation and activity. J Neurosci. 2007;27:4083–4092. doi: 10.1523/JNEUROSCI.5293-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alfa Cisse M, Louis K, Braun U, Mari B, Leitges M, Slack BE, Fisher A, Auberger P, Checler F, Vincent B. Isoform-specific contribution of protein kinase C to prion processing. Mol Cell Neurosci. 2008;39:400–410. doi: 10.1016/j.mcn.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 66.Cisse M, Duplan E, Guillot-Sestier MV, Rumigny J, Bauer C, Pages G, Orzechowski HD, Slack BE, Checler F, Vincent B. The extracellular regulated kinase-1 (ERK1) controls regulated alpha-secretase-mediated processing, promoter transactivation, and mRNA levels of the cellular prion protein. J Biol Chem. 2011;286:29192–29206. doi: 10.1074/jbc.M110.208249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vincent B, Sunyach C, Orzechowski HD, St George-Hyslop P, Checler F. p53-Dependent transcriptional control of cellular prion by presenilins. J Neurosci. 2009;29:6752–6760. doi: 10.1523/JNEUROSCI.0789-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lewis V, Whitehouse IJ, Baybutt H, Manson JC, Collins SJ, Hooper NM. Cellular prion protein expression is not regulated by the Alzheimer's amyloid precursor protein intracellular domain. PLoS One. 2012;7:e31754. doi: 10.1371/journal.pone.0031754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Haigh CL, Lewis VA, Vella LJ, Masters CL, Hill AF, Lawson VA, Collins SJ. PrPC-related signal transduction is influenced by copper, membrane integrity and the alpha cleavage site. Cell Res. 2009;19:1062–1078. doi: 10.1038/cr.2009.86. [DOI] [PubMed] [Google Scholar]
- 70.Oliveira-Martins JB, Yusa S, Calella AM, Bridel C, Baumann F, Dametto P, Aguzzi A. Unexpected tolerance of alpha-cleavage of the prion protein to sequence variations. PLoS One. 2010;5:e9107. doi: 10.1371/journal.pone.0009107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lutz J, Brabeck C, Niemann HH, Kloz U, Korth C, Lingappa VR, Burkle A. Microdeletions within the hydrophobic core region of cellular prion protein alter its topology and metabolism. Biochem Biophys Res Commun. 2010;393:439–444. doi: 10.1016/j.bbrc.2010.02.015. [DOI] [PubMed] [Google Scholar]
- 72.Mohan MJ, Seaton T, Mitchell J, Howe A, Blackburn K, Burkhart W, Moyer M, Patel I, Waitt GM, Becherer JD, Moss ML, Milla ME. The tumor necrosis factor-alpha converting enzyme (TACE): a unique metalloproteinase with highly defined substrate selectivity. Biochemistry. 2002;41:9462–9469. doi: 10.1021/bi0260132. [DOI] [PubMed] [Google Scholar]
- 73.Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F. Constitutive and regulated alpha-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci U S A. 1999;96:3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jorissen E, Prox J, Bernreuther C, Weber S, Schwanbeck R, Serneels L, Snellinx A, Craessaerts K, Thathiah A, Tesseur I, Bartsch U, Weskamp G, Blobel CP, Glatzel M, De Strooper B, Saftig P. The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J Neurosci. 2010;30:4833–4844. doi: 10.1523/JNEUROSCI.5221-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kuhn PH, Wang H, Dislich B, Colombo A, Zeitschel U, Ellwart JW, Kremmer E, Rossner S, Lichtenthaler SF. ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010;29:3020–3032. doi: 10.1038/emboj.2010.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Epis R, Marcello E, Gardoni F, Vastagh C, Malinverno M, Balducci C, Colombo A, Borroni B, Vara H, Dell'Agli M, Cattabeni F, Giustetto M, Borsello T, Forloni G, Padovani A, Di Luca M. Blocking ADAM10 synaptic trafficking generates a model of sporadic Alzheimer's disease. Brain. 2010;133:3323–3335. doi: 10.1093/brain/awq217. [DOI] [PubMed] [Google Scholar]
- 77.Beland M, Roucou X. The prion protein unstructured N-terminal region is a broad-spectrum molecular sensor with diverse and contrasting potential functions. J Neurochem. 2012;120:853–868. doi: 10.1111/j.1471-4159.2011.07613.x. [DOI] [PubMed] [Google Scholar]
- 78.Guillot-Sestier MV, Sunyach C, Druon C, Scarzello S, Checler F. The alpha-secretase-derived N-terminal product of cellular prion, N1, displays neuroprotective function in vitro and in vivo. J Biol Chem. 2009;284:35973–35986. doi: 10.1074/jbc.M109.051086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen S, Yadav SP, Surewicz WK. Interaction between human prion protein and amyloid-beta (Abeta) oligomers: role OF N-terminal residues. J Biol Chem. 2010;285:26377–26383. doi: 10.1074/jbc.M110.145516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guillot-Sestier MV, Sunyach C, Ferreira ST, Marzolo MP, Bauer C, Thevenet A, Checler F. alpha-Secretase-derived fragment of cellular prion, N1, protects against monomeric and oligomeric amyloid beta (Abeta)-associated cell death. J Biol Chem. 2012;287:5021–5032. doi: 10.1074/jbc.M111.323626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Resenberger UK, Winklhofer KF, Tatzelt J. Cellular Prion Protein Mediates Toxic Signaling of Amyloid Beta. Neurodegener Dis. 2011 doi: 10.1159/000332596. [DOI] [PubMed] [Google Scholar]
- 82.Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302:871–874. doi: 10.1126/science.1090187. [DOI] [PubMed] [Google Scholar]
- 83.Chung E, Ji Y, Sun Y, Kascsak RJ, Kascsak RB, Mehta PD, Strittmatter SM, Wisniewski T. Anti-PrPC monoclonal antibody infusion as a novel treatment for cognitive deficits in an Alzheimer's disease model mouse. BMC Neurosci. 2010;11:130. doi: 10.1186/1471-2202-11-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shmerling D, Hegyi I, Fischer M, Blattler T, Brandner S, Gotz J, Rulicke T, Flechsig E, Cozzio A, von Mering C, Hangartner C, Aguzzi A, Weissmann C. Expression of amino-terminally truncated PrP in the mouse leading to ataxia and specific cerebellar lesions. Cell. 1998;93:203–214. doi: 10.1016/s0092-8674(00)81572-x. [DOI] [PubMed] [Google Scholar]
- 85.Li A, Barmada SJ, Roth KA, Harris DA. N-terminally deleted forms of the prion protein activate both Bax-dependent and Bax-independent neurotoxic pathways. J Neurosci. 2007;27:852–859. doi: 10.1523/JNEUROSCI.4244-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Paitel E, Fahraeus R, Checler F. Cellular prion protein sensitizes neurons to apoptotic stimuli through Mdm2-regulated and p53-dependent caspase 3-like activation. J Biol Chem. 2003;278:10061–10066. doi: 10.1074/jbc.M211580200. [DOI] [PubMed] [Google Scholar]
- 87.Paitel E, Sunyach C, Alves da Costa C, Bourdon JC, Vincent B, Checler F. Primary cultured neurons devoid of cellular prion display lower responsiveness to staurosporine through the control of p53 at both transcriptional and post-transcriptional levels. J Biol Chem. 2004;279:612–618. doi: 10.1074/jbc.M310453200. [DOI] [PubMed] [Google Scholar]
- 88.Sunyach C, Cisse MA, da Costa CA, Vincent B, Checler F. The C-terminal products of cellular prion protein processing, C1 and C2, exert distinct influence on p53-dependent staurosporine-induced caspase-3 activation. J Biol Chem. 2007;282:1956–1963. doi: 10.1074/jbc.M609663200. [DOI] [PubMed] [Google Scholar]
- 89.Sunyach C, Checler F. Combined pharmacological, mutational and cell biology approaches indicate that p53-dependent caspase 3 activation triggered by cellular prion is dependent on its endocytosis. J Neurochem. 2005;92:1399–1407. doi: 10.1111/j.1471-4159.2004.02989.x. [DOI] [PubMed] [Google Scholar]
- 90.Radovanovic I, Braun N, Giger OT, Mertz K, Miele G, Prinz M, Navarro B, Aguzzi A. Truncated prion protein and Doppel are myelinotoxic in the absence of oligodendrocytic PrPC. J Neurosci. 2005;25:4879–4888. doi: 10.1523/JNEUROSCI.0328-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nishida N, Tremblay P, Sugimoto T, Shigematsu K, Shirabe S, Petromilli C, Erpel SP, Nakaoke R, Atarashi R, Houtani T, Torchia M, Sakaguchi S, DeArmond SJ, Prusiner SB, Katamine S. A mouse prion protein transgene rescues mice deficient for the prion protein gene from purkinje cell degeneration and demyelination. Lab Invest. 1999;79:689–697. [PubMed] [Google Scholar]
- 92.Pauly PC, Harris DA. Copper stimulates endocytosis of the prion protein. J Biol Chem. 1998;273:33107–33110. doi: 10.1074/jbc.273.50.33107. [DOI] [PubMed] [Google Scholar]
- 93.Perera WS, Hooper NM. Ablation of the metal ion-induced endocytosis of the prion protein by disease-associated mutation of the octarepeat region. Curr Biol. 2001;11:519–523. doi: 10.1016/s0960-9822(01)00147-6. [DOI] [PubMed] [Google Scholar]
- 94.Haigh CL, Edwards K, Brown DR. Copper binding is the governing determinant of prion protein turnover. Mol Cell Neurosci. 2005;30:186–196. doi: 10.1016/j.mcn.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 95.Nunziante M, Gilch S, Schatzl HM. Essential role of the prion protein N terminus in subcellular trafficking and half-life of cellular prion protein. J Biol Chem. 2003;278:3726–3734. doi: 10.1074/jbc.M206313200. [DOI] [PubMed] [Google Scholar]
- 96.Sunyach C, Jen A, Deng J, Fitzgerald KT, Frobert Y, Grassi J, McCaffrey MW, Morris R. The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. EMBO J. 2003;22:3591–3601. doi: 10.1093/emboj/cdg344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Taylor DR, Watt NT, Perera WS, Hooper NM. Assigning functions to distinct regions of the N-terminus of the prion protein that are involved in its copper-stimulated, clathrin-dependent endocytosis. J Cell Sci. 2005;118:5141–5153. doi: 10.1242/jcs.02627. [DOI] [PubMed] [Google Scholar]
- 98.Taylor DR, Hooper NM. The low-density lipoprotein receptor-related protein 1 (LRP1) mediates the endocytosis of the cellular prion protein. Biochem J. 2007;402:17–23. doi: 10.1042/BJ20061736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Parkyn CJ, Vermeulen EG, Mootoosamy RC, Sunyach C, Jacobsen C, Oxvig C, Moestrup S, Liu Q, Bu G, Jen A, Morris RJ. LRP1 controls biosynthetic and endocytic trafficking of neuronal prion protein. J Cell Sci. 2008;121:773–783. doi: 10.1242/jcs.021816. [DOI] [PubMed] [Google Scholar]
- 100.Turnbaugh JA, Westergard L, Unterberger U, Biasini E, Harris DA. The N-terminal, polybasic region is critical for prion protein neuroprotective activity. PLoS One. 2011;6:e25675. doi: 10.1371/journal.pone.0025675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Diederich S, Maisner A. Molecular characteristics of the Nipah virus glycoproteins. Ann N Y Acad Sci. 2007;1102:39–50. doi: 10.1196/annals.1408.003. [DOI] [PubMed] [Google Scholar]
- 102.Diederich S, Sauerhering L, Weis M, Altmeppen H, Schaschke N, Reinheckel T, Erbar S, Maisner A. Activation of the Nipah virus fusion protein in MDCK cells is mediated by cathepsin B within the endosome-recycling compartment. J Virol. 2012;86:3736–3745. doi: 10.1128/JVI.06628-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gasset M, Baldwin MA, Lloyd DH, Gabriel JM, Holtzman DM, Cohen F, Fletterick R, Prusiner SB. Predicted alpha-helical regions of the prion protein when synthesized as peptides form amyloid. Proc Natl Acad Sci U S A. 1992;89:10940–10944. doi: 10.1073/pnas.89.22.10940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Forloni G, Angeretti N, Chiesa R, Monzani E, Salmona M, Bugiani O, Tagliavini F. Neurotoxicity of a prion protein fragment. Nature. 1993;362:543–546. doi: 10.1038/362543a0. [DOI] [PubMed] [Google Scholar]
- 105.Tagliavini F, Prelli F, Verga L, Giaccone G, Sarma R, Gorevic P, Ghetti B, Passerini F, Ghibaudi E, Forloni G, et al. Synthetic peptides homologous to prion protein residues 106-147 form amyloid-like fibrils in vitro. Proc Natl Acad Sci U S A. 1993;90:9678–9682. doi: 10.1073/pnas.90.20.9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Stohr J, Watts JC, Legname G, Oehler A, Lemus A, Nguyen HO, Sussman J, Wille H, DeArmond SJ, Prusiner SB, Giles K. Spontaneous generation of anchorless prions in transgenic mice. Proc Natl Acad Sci U S A. 2011;108:21223–21228. doi: 10.1073/pnas.1117827108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Norstrom EM, Mastrianni JA. The AGAAAAGA palindrome in PrP is required to generate a productive PrPSc-PrPC complex that leads to prion propagation. J Biol Chem. 2005;280:27236–27243. doi: 10.1074/jbc.M413441200. [DOI] [PubMed] [Google Scholar]
- 108.Lewis V, Hill AF, Haigh CL, Klug GM, Masters CL, Lawson VA, Collins SJ. Increased proportions of C1 truncated prion protein protect against cellular M1000 prion infection. J Neuropathol Exp Neurol. 2009;68:1125–1135. doi: 10.1097/NEN.0b013e3181b96981. [DOI] [PubMed] [Google Scholar]
- 109.Caughey B, Raymond GJ, Ernst D, Race RE. N-terminal truncation of the scrapie-associated form of PrP by lysosomal protease(s): implications regarding the site of conversion of PrP to the protease-resistant state. J Virol. 1991;65:6597–6603. doi: 10.1128/jvi.65.12.6597-6603.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bolton DC, Bendheim PE, Marmorstein AD, Potempska A. Isolation and structural studies of the intact scrapie agent protein. Arch Biochem Biophys. 1987;258:579–590. doi: 10.1016/0003-9861(87)90380-8. [DOI] [PubMed] [Google Scholar]
- 111.Stahl N, Baldwin MA, Teplow DB, Hood L, Gibson BW, Burlingame AL, Prusiner SB. Structural studies of the scrapie prion protein using mass spectrometry and amino acid sequencing. Biochemistry. 1993;32:1991–2002. doi: 10.1021/bi00059a016. [DOI] [PubMed] [Google Scholar]
- 112.Oesch B, Westaway D, Walchli M, McKinley MP, Kent SB, Aebersold R, Barry RA, Tempst P, Teplow DB, Hood LE, et al. A cellular gene encodes scrapie PrP 27-30 protein. Cell. 1985;40:735–746. doi: 10.1016/0092-8674(85)90333-2. [DOI] [PubMed] [Google Scholar]
- 113.Yadavalli R, Guttmann RP, Seward T, Centers AP, Williamson RA, Telling GC. Calpain-dependent endoproteolytic cleavage of PrPSc modulates scrapie prion propagation. J Biol Chem. 2004;279:21948–21956. doi: 10.1074/jbc.M400793200. [DOI] [PubMed] [Google Scholar]
- 114.McMahon HE, Mange A, Nishida N, Creminon C, Casanova D, Lehmann S. Cleavage of the amino terminus of the prion protein by reactive oxygen species. J Biol Chem. 2001;276:2286–2291. doi: 10.1074/jbc.M007243200. [DOI] [PubMed] [Google Scholar]
- 115.Milhavet O, Lehmann S. Oxidative stress and the prion protein in transmissible spongiform encephalopathies. Brain Res Brain Res Rev. 2002;38:328–339. doi: 10.1016/s0165-0173(01)00150-3. [DOI] [PubMed] [Google Scholar]
- 116.Lee DW, Sohn HO, Lim HB, Lee YG, Kim YS, Carp RI, Wisniewski HM. Alteration of free radical metabolism in the brain of mice infected with scrapie agent. Free Radic Res. 1999;30:499–507. doi: 10.1080/10715769900300541. [DOI] [PubMed] [Google Scholar]
- 117.Guentchev M, Voigtlander T, Haberler C, Groschup MH, Budka H. Evidence for oxidative stress in experimental prion disease. Neurobiol Dis. 2000;7:270–273. doi: 10.1006/nbdi.2000.0290. [DOI] [PubMed] [Google Scholar]
- 118.Choi SI, Ju WK, Choi EK, Kim J, Lea HZ, Carp RI, Wisniewski HM, Kim YS. Mitochondrial dysfunction induced by oxidative stress in the brains of hamsters infected with the 263 K scrapie agent. Acta Neuropathol. 1998;96:279–286. doi: 10.1007/s004010050895. [DOI] [PubMed] [Google Scholar]
- 119.Milhavet O, McMahon HE, Rachidi W, Nishida N, Katamine S, Mange A, Arlotto M, Casanova D, Riondel J, Favier A, Lehmann S. Prion infection impairs the cellular response to oxidative stress. Proc Natl Acad Sci U S A. 2000;97:13937–13942. doi: 10.1073/pnas.250289197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Watt NT, Taylor DR, Gillott A, Thomas DA, Perera WS, Hooper NM. Reactive oxygen species-mediated beta-cleavage of the prion protein in the cellular response to oxidative stress. J Biol Chem. 2005;280:35914–35921. doi: 10.1074/jbc.M507327200. [DOI] [PubMed] [Google Scholar]
- 121.Pushie MJ, Vogel HJ. Modeling by assembly and molecular dynamics simulations of the low Cu2+ occupancy form of the mammalian prion protein octarepeat region: gaining insight into Cu2+-mediated beta-cleavage. Biophys J. 2008;95:5084–5091. doi: 10.1529/biophysj.108.139568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Borchelt DR, Rogers M, Stahl N, Telling G, Prusiner SB. Release of the cellular prion protein from cultured cells after loss of its glycoinositol phospholipid anchor. Glycobiology. 1993;3:319–329. doi: 10.1093/glycob/3.4.319. [DOI] [PubMed] [Google Scholar]
- 123.Stahl N, Borchelt DR, Hsiao K, Prusiner SB. Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell. 1987;51:229–240. doi: 10.1016/0092-8674(87)90150-4. [DOI] [PubMed] [Google Scholar]
- 124.Parizek P, Roeckl C, Weber J, Flechsig E, Aguzzi A, Raeber AJ. Similar turnover and shedding of the cellular prion protein in primary lymphoid and neuronal cells. J Biol Chem. 2001;276:44627–44632. doi: 10.1074/jbc.M107458200. [DOI] [PubMed] [Google Scholar]
- 125.Tagliavini F, Prelli F, Porro M, Salmona M, Bugiani O, Frangione B. A soluble form of prion protein in human cerebrospinal fluid: implications for prion-related encephalopathies. Biochem Biophys Res Commun. 1992;184:1398–1404. doi: 10.1016/s0006-291x(05)80038-5. [DOI] [PubMed] [Google Scholar]
- 126.Perini F, Vidal R, Ghetti B, Tagliavini F, Frangione B, Prelli F. PrP27-30 is a normal soluble prion protein fragment released by human platelets. Biochem Biophys Res Commun. 1996;223:572–577. doi: 10.1006/bbrc.1996.0936. [DOI] [PubMed] [Google Scholar]
- 127.MacGregor I, Hope J, Barnard G, Kirby L, Drummond O, Pepper D, Hornsey V, Barclay R, Bessos H, Turner M, Prowse C. Application of a time-resolved fluoroimmunoassay for the analysis of normal prion protein in human blood and its components. Vox Sang. 1999;77:88–96. doi: 10.1159/000031082. [DOI] [PubMed] [Google Scholar]
- 128.Li R, Liu T, Yoshihiro F, Tary-Lehmann M, Obrenovich M, Kuekrek H, Kang SC, Pan T, Wong BS, Medof ME, Sy MS. On the same cell type GPI-anchored normal cellular prion and DAF protein exhibit different biological properties. Biochem Biophys Res Commun. 2003;303:446–451. doi: 10.1016/s0006-291x(03)00354-1. [DOI] [PubMed] [Google Scholar]
- 129.Parkin ET, Watt NT, Turner AJ, Hooper NM. Dual mechanisms for shedding of the cellular prion protein. J Biol Chem. 2004;279:11170–11178. doi: 10.1074/jbc.M312105200. [DOI] [PubMed] [Google Scholar]
- 130.Stahl N, Baldwin MA, Burlingame AL, Prusiner SB. Identification of glycoinositol phospholipid linked and truncated forms of the scrapie prion protein. Biochemistry. 1990;29:8879–8884. doi: 10.1021/bi00490a001. [DOI] [PubMed] [Google Scholar]
- 131.Tousseyn T, Thathiah A, Jorissen E, Raemaekers T, Konietzko U, Reiss K, Maes E, Snellinx A, Serneels L, Nyabi O, Annaert W, Saftig P, Hartmann D, De Strooper B. ADAM10, the rate-limiting protease of regulated intramembrane proteolysis of Notch and other proteins, is processed by ADAMS-9, ADAMS-15, and the gamma-secretase. J Biol Chem. 2009;284:11738–11747. doi: 10.1074/jbc.M805894200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Moss ML, Powell G, Miller MA, Edwards L, Qi B, Sang QX, De Strooper B, Tesseur I, Lichtenthaler SF, Taverna M, Zhong JL, Dingwall C, Ferdous T, Schlomann U, Zhou P, Griffith LG, Lauffenburger DA, Petrovich R, Bartsch JW. ADAM9 inhibition increases membrane activity of ADAM10 and controls alpha-secretase processing of amyloid precursor protein. J Biol Chem. 2011;286:40443–40451. doi: 10.1074/jbc.M111.280495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Janes PW, Saha N, Barton WA, Kolev MV, Wimmer-Kleikamp SH, Nievergall E, Blobel CP, Himanen JP, Lackmann M, Nikolov DB. Adam meets Eph: an ADAM substrate recognition module acts as a molecular switch for ephrin cleavage in trans. Cell. 2005;123:291–304. doi: 10.1016/j.cell.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 134.Janes PW, Wimmer-Kleikamp SH, Frangakis AS, Treble K, Griesshaber B, Sabet O, Grabenbauer M, Ting AY, Saftig P, Bastiaens PI, Lackmann M. Cytoplasmic relaxation of active Eph controls ephrin shedding by ADAM10. PLoS Biol. 2009;7:e1000215. doi: 10.1371/journal.pbio.1000215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Weskamp G, Ford JW, Sturgill J, Martin S, Docherty AJ, Swendeman S, Broadway N, Hartmann D, Saftig P, Umland S, Sehara-Fujisawa A, Black RA, Ludwig A, Becherer JD, Conrad DH, Blobel CP. ADAM10 is a principal 'sheddase' of the low-affinity immunoglobulin E receptor CD23. Nat Immunol. 2006;7:1293–1298. doi: 10.1038/ni1399. [DOI] [PubMed] [Google Scholar]
- 136.Mathews JA, Gibb DR, Chen BH, Scherle P, Conrad DH. CD23 Sheddase A disintegrin and metalloproteinase 10 (ADAM10) is also required for CD23 sorting into B cell-derived exosomes. J Biol Chem. 2010;285:37531–37541. doi: 10.1074/jbc.M110.141556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Donmez G, Wang D, Cohen DE, Guarente L. SIRT1 suppresses beta-amyloid production by activating the alpha-secretase gene ADAM10. Cell. 2010;142:320–332. doi: 10.1016/j.cell.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 138.Prox J, Willenbrock M, Weber S, Lehmann T, Schmidt-Arras D, Schwanbeck R, Saftig P, Schwake M. Tetraspanin15 regulates cellular trafficking and activity of the ectodomain sheddase ADAM10. Cell Mol Life Sci. 2012 doi: 10.1007/s00018-012-0960-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.De Felice FG, Velasco PT, Lambert MP, Viola K, Fernandez SJ, Ferreira ST, Klein WL. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J Biol Chem. 2007;282:11590–11601. doi: 10.1074/jbc.M607483200. [DOI] [PubMed] [Google Scholar]
- 140.Khosravani H, Zhang Y, Tsutsui S, Hameed S, Altier C, Hamid J, Chen L, Villemaire M, Ali Z, Jirik FR, Zamponi GW. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J Cell Biol. 2008;181:551–565. doi: 10.1083/jcb.200711002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Caetano FA, Beraldo FH, Hajj GN, Guimaraes AL, Jurgensen S, Wasilewska-Sampaio AP, Hirata PH, Souza I, Machado CF, Wong DY, De Felice FG, Ferreira ST, Prado VF, Rylett RJ, Martins VR, Prado MA. Amyloid-beta oligomers increase the localization of prion protein at the cell surface. J Neurochem. 2011;117:538–553. doi: 10.1111/j.1471-4159.2011.07225.x. [DOI] [PubMed] [Google Scholar]
- 142.Wan XZ, Li B, Li YC, Yang XL, Zhang W, Zhong L, Tang SJ. Activation of NMDA Receptors Upregulates A Disintegrin and Metalloproteinase 10 via a Wnt/MAPK Signaling Pathway. J Neurosci. 2012;32:3910–3916. doi: 10.1523/JNEUROSCI.3916-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Marella M, Lehmann S, Grassi J, Chabry J. Filipin prevents pathological prion protein accumulation by reducing endocytosis and inducing cellular PrP release. J Biol Chem. 2002;277:25457–25464. doi: 10.1074/jbc.M203248200. [DOI] [PubMed] [Google Scholar]
- 144.Caughey B, Raymond GJ. The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J Biol Chem. 1991;266:18217–18223. [PubMed] [Google Scholar]
- 145.Borchelt DR, Taraboulos A, Prusiner SB. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J Biol Chem. 1992;267:16188–16199. [PubMed] [Google Scholar]
- 146.Enari M, Flechsig E, Weissmann C. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc Natl Acad Sci U S A. 2001;98:9295–9299. doi: 10.1073/pnas.151242598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lewis PA, Properzi F, Prodromidou K, Clarke AR, Collinge J, Jackson GS. Removal of the glycosylphosphatidylinositol anchor from PrP(Sc) by cathepsin D does not reduce prion infectivity. Biochem J. 2006;395:443–448. doi: 10.1042/BJ20051677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Stahl N, Borchelt DR, Prusiner SB. Differential release of cellular and scrapie prion proteins from cellular membranes by phosphatidylinositol-specific phospholipase C. Biochemistry. 1990;29:5405–5412. doi: 10.1021/bi00474a028. [DOI] [PubMed] [Google Scholar]
- 149.Rogers M, Yehiely F, Scott M, Prusiner SB. Conversion of truncated and elongated prion proteins into the scrapie isoform in cultured cells. Proc Natl Acad Sci U S A. 1993;90:3182–3186. doi: 10.1073/pnas.90.8.3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kocisko DA, Come JH, Priola SA, Chesebro B, Raymond GJ, Lansbury PT, Caughey B. Cell-free formation of protease-resistant prion protein. Nature. 1994;370:471–474. doi: 10.1038/370471a0. [DOI] [PubMed] [Google Scholar]
- 151.Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, Caughey B, Masliah E, Oldstone M. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005;308:1435–1439. doi: 10.1126/science.1110837. [DOI] [PubMed] [Google Scholar]
- 152.Trifilo MJ, Sanchez-Alavez M, Solforosi L, Bernard-Trifilo J, Kunz S, McGavern D, Oldstone MB. Scrapie-induced defects in learning and memory of transgenic mice expressing anchorless prion protein are associated with alterations in the gamma aminobutyric acidergic pathway. J Virol. 2008;82:9890–9899. doi: 10.1128/JVI.00486-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chesebro B, Race B, Meade-White K, Lacasse R, Race R, Klingeborn M, Striebel J, Dorward D, McGovern G, Jeffrey M. Fatal transmissible amyloid encephalopathy: a new type of prion disease associated with lack of prion protein membrane anchoring. PLoS Pathog. 2010;6:e1000800. doi: 10.1371/journal.ppat.1000800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ghetti B, Tagliavini F, Takao M, Bugiani O, Piccardo P. Hereditary prion protein amyloidoses. Clin Lab Med. 2003;23:65–85. viii. doi: 10.1016/s0272-2712(02)00064-1. [DOI] [PubMed] [Google Scholar]
- 155.Jansen C, Parchi P, Capellari S, Vermeij AJ, Corrado P, Baas F, Strammiello R, van Gool WA, van Swieten JC, Rozemuller AJ. Prion protein amyloidosis with divergent phenotype associated with two novel nonsense mutations in PRNP. Acta Neuropathol. 2010;119:189–197. doi: 10.1007/s00401-009-0609-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Saftig P, Reiss K. The "A Disintegrin And Metalloproteases" ADAM10 and ADAM17: novel drug targets with therapeutic potential? Eur J Cell Biol. 2011;90:527–535. doi: 10.1016/j.ejcb.2010.11.005. [DOI] [PubMed] [Google Scholar]