

Abstract
The amyloid β precursor protein (APP) is a single-pass transmembrane glycoprotein that is ubiquitously expressed in many cell types, including neurons. Amyloidogenic processing of APP by β- and γ-secretases leads to the production of amyloid-β (Aβ) peptides that can oligomerize and aggregate into amyloid plaques, a characteristic hallmark of Alzheimer’s disease (AD) brains. Multiple reports suggest that dimerization of APP may play a role in Aβ production; however, it is not yet clear whether APP dimers increase or decrease Aβ and the mechanism is not fully understood. To better understand the relationship between APP dimerization and production of Aβ, a high throughput screen for small molecule modulators of APP dimerization was conducted using APP-Firefly luciferase enzyme complementation to detect APP dimerization. Selected modulators identified from a compound library of 77,440 compounds were tested for their effects on Aβ generation. Two molecules that inhibited APP dimerization produced a reduction in Aβ levels as measured by ELISA. The inhibitors did not change sAPPα or γ-CTF levels, but lowered sAPPβ levels, suggesting that blocking the dimerization is preventing the cleavage by β-secretase in the amyloidogenic processing of APP. To our knowledge, this is the first High Throughput Screen (HTS) effort to identify small molecule modulators of APP dimerization. Inhibition of APP dimerization has previously been suggested as a therapeutic target in AD. The findings reported here further support that modulation of APP dimerization may be a viable means of reducing the production of Aβ.
Keywords: Alzheimer disease, amyloid-β precursor protein, protein dimerization, amyloid beta-peptides, high-throughput screening, firefly luciferase complementation
Introduction
The amyloid-β precursor protein (APP) is known to play a role in the pathogenesis of Alzheimer’s disease (AD) as its proteolysis results in the generation of amyloid-β peptides (Aβ) and mutations in APP cause autosomal dominant early onset AD [1]. Aβ can oligomerize and aggregate into extracellular deposits of amyloid plaques, a classic pathologic hallmark of AD associated with perturbed neuronal functions, disrupted cellular networks, cell death and tissue degeneration [2]. The resulting lesions can have a significant impact and dire clinical consequences on cognitive functions in patients. Unfortunately, current therapies for AD remain largely symptomatic and disease-modifying drugs have yet to be discovered.
Production of Aβ from APP occurs by an amyloidogenic-processing pathway. First, APP is cleaved by β-secretase (also known as the β site APP cleaving enzyme (BACE1), or memapsin-2 [3,4]) at the extracellular domain to release a secreted ectodomain fragment, sAPPβ, and a membrane-bound C-terminal fragment, CTFβ or C99. Then, CTFβ is cleaved by γ-secretase (a large multi-subunit complex comprised of presenilin (PS), nicastrin (Nct), anterior pharynx defective (Aph-1) and the presenilin enhancer (Pen-2) [5]) via a process called regulated intramembrane proteolysis (RIP). This results in the production of Aβ and an intracellular domain fragment, AICD (APP intracellular domain), which can translocate to the nucleus and has been implicated in the regulation of gene expression [6]. Depending on the location of the γ-secretase cleavage, Aβ peptides of 39 to 42 amino acids are made [7,8]. In AD, Aβ40 and Aβ42 are of most concern, with Aβ42 being the most toxic to neurons and synapses. Alternatively, under non-amyloidogenic condition, APP is first cleaved by α-secretase (members include the A disintegrin and metalloproteinase proteins, ADAM9, ADAM10 and ADAM17/tumor necrosis factor a-converting enzyme (TACE) [9-11]) to release a truncated ectodomain fragment, sAPPα. The remaining C-terminal fragment, CTFα or C83, is then cleaved by γ-secretase to release a p3 peptide and AICD. Most APP molecules undergo this non-amyloidogenic processing pathway.
APP is a single-pass transmembrane glycoprotein with a large extracellular N-terminal domain and a short intracellular C-terminal domain. Its structure resembles a cell-surface receptor [12] that can bind ligands and participate in protein-protein interactions. Like many receptor proteins that dimerize upon ligand activation, APP can also form homodimers [12,13], as well as heterodimers with Notch for cellular signaling [14-16] and with its homologs the amyloid precursor like proteins 1 and 2 (APLP1 and 2) for cellular adhesion and cell-cell communication [17,18]. The precise function of APP and the significance of its dimerization have yet to be determined. However, many biological activities have been linked to the protein, including that of platelet aggregation [19], metal homeostasis [20-22], gene transcription and cellular metabolism [6,23], and various neuronal processes such as cellular growth, differentiation, migration, aborization, axonal transport, memory formation, and neuroprotection [24-31].
APP has a few notable dimerization sites. First, the E1 growth factor like domain (GFLD) has 3 cysteine bridges where the bridge between Cys98 and Cys105 forms an important beta hairpin loop for dimerization [32]. Secondly, the residues His147, His151, Tyr168, and Met170 at the E1 copper-binding domain (CuBD) form a tetrahedral structure that can coordinate metals such as copper and zinc that can regulate APP dimerization [33,34]. The E2 collagen-binding site (CBD) can bind ligands such as Type I collagen and heparin for cellular adhesion and participate in both cis- and trans- APP dimerization [35,36]. Additionally, binding of Aβ to the juxtamembrane region facilitates APP multimerization, which may be crucial for cell death signaling [37]. Finally, 3 consecutive GxxxG motifs at the transmembrane domain (Aβ residues 25-37) [8], and the GXXXA sequence that comes directly after [38], are the most studied sites for APP dimerization.
Recent studies suggest that APP dimerization may influence Aβ production. For example, replacement of a lysine residue by cysteine at the juxtamembrane region of APP (K624C mutant) resulted in constitutive APP dimerization via a disulphide bond, and a concomitant increase in Aβ levels [13]. Likewise, mutations that disrupted the GxxxG transmembrane dimerization motif of APP resulted in a corresponding decrease of toxic Aβ42 under a ToxR system. In particular, a G33I mutant abolished both dimerization and Aβ production [8]. However, in a follow-up study on GxxxG mutations, it was found that increased APP dimerization at the C-terminal domain by these mutants lead to a decrease in Aβ [39]. Also, when examining familial APP mutants that cause early onset AD, the mutations destabilized APP transmembrane dimerization and increased Aβ42/40 ratio in a way that negatively correlated with the mean age of onset of AD symptoms [38]. Furthermore, when using an FKBP/rapamycin system to examine APP dimerization and its effects on Aβ generation in the absence of mutations, it was found that induction of dimerization decreased Aβ production [40]. Regardless of whether APP dimerization increases or decreases Aβ, multiple groups report that it does have an effect on Aβ levels demonstrating the importance of APP dimerization in AD pathogenesis and that further evaluation of this process is warranted.
To better understand the relationship between APP dimerization and Aβ production, we developed an assay to identify small molecule APP dimerization inhibitors and subsequently tested their effects on Aβ generation. A high throughput screen (HTS) was conducted using the method of Firefly luciferase enzyme fragment complementation (FLuc EFC). This technique has successfully been used by others to study epidermal growth factor receptor dimerization [41], chemokine receptor dimerization CXCR4 and 7 [42], rapamycin induced FRB/FKBP association [43], phospho-dependent association of Cdc25C/14-3-3ε in cell cycle division [43], IFN-γ induced nuclear translocation of STAT1/STAT1 complexes [43], cellular marconuclear delivery vehicles, [44] as well as protein-protein interactions in plants [45]. Applications of the method also allow visualization of luminescence in live cells and animals [43,44]. Here, we applied the same principle with the goal of detecting APP-APP interactions. Two inactive deletion mutants of the FLuc enzyme were separately fused to APP. When APP is in monomeric form, the FLuc fragments remain inactive. However, upon APP-APP dimerization, the two inactive FLuc fragments are brought in close proximity to form a fully functional enzyme. Quantification of APP-APP interactions is then made possible by measuring chemiluminescence following the addition of luciferin. The FLuc EFC is an effective way to monitor APP-APP interactions that could be used for a better understanding of APP processing and Aβ production.
Materials and methods
APP-firefly luciferase constructs
Full-length APP Firefly luciferase (APP FLuc FL) construct was prepared using a pGL3-basic vector (Promega) as template. An AgeI site was inserted at the start of the luciferase gene using sense (5'-GGC ATT CCG GTA CTG TAC CGG TAG CCA CCA TGG AAG AC-3') and anti-sense (5'-GTC TTC CAT GGT GGC TAC CGG TAC AGT ACC GGA ATG CC-3') primers, followed by excision of the luciferase insert using the restriction enzymes AgeI and XbaI (New England Biolabs). In parallel, an APP YC pcDNA1 plasmid [14] was also digested using the same restriction enzymes to release the APP pcDNA1 vector. The resulting products were then ligated with a XbaI/NotI sense (5’-GGC CGA GCT CTG TTA-3’) and anti-sense (5’-CTA GTA ACA GAG CTC-3’) adapter.
For the construction of the APP firefly luciferase N-terminal fragment (APP FLucN) from the APP FLuc FL, a stop codon was added after amino acid 475 using the sense (5'-GGT CTT CCC GAC TAA GAC GCC GGT GAA CTT CCC-3') and antisense (5'-GGG AAG TTC ACC GGC GTC TTA GTC GGG AAG ACC-3') primers.
The APP firefly luciferase C-terminal fragment (APP FLucC) was constructed by adding an AgeI site before amino acid 265 using sense (5'-GAT ATG TGG ATT TCG AGT ACC GGT AAT GTA TAG ATT TGA AG-3') and anti-sense (5'-CT TCA AAT CTA TAC ATT ACC GGT ACT CGA AAT CCA CAT ATC-3') primers. Following that, the DNA encoding for amino acids 1-264 of luciferase was excised using AgeI, and the remaining plasmid was self-ligated.
To make APP Firefly luciferase plasmids suitable for stable cell lines, the plasmids in pcDNA1 vector were cloned into the pcDNA3.1 mammalian expression vector with the geneticin (G418) antibiotic resistance gene for eukaryotic cells. The APP luciferase insert was excised from the pcDNA1 vector using HindII and XbaI. Likewise, the Klotho pcDNA3.1 plasmid [46] was also digested with the same enzymes to release the Klotho insert. The resulting enzyme digested products were then ligated to create APP FLucN and APP FLucC plasmids in pcDNA3.1. All plasmid sequences were confirmed by DNA sequencing at Tufts University.
Cell maintenance and transfection
HEK293 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (CellGro) supplemented with 10% fetal bovine serum (FBS) (Atlantic Biologicals) and 1% penicillin-streptomycin (CellGro) at 37°C and 5% CO2. The day before transfection, cells were split using 0.25% Trypsin-EDTA into 6 well plates at a density of 1x105 cells per well or into 96 well plates at a density of 8,000 cells per well. DNA were transfected into cells using Nanofect (Qiagen). Forty-eight hours post transfection, cells were lysed in lysis buffer (100mM potassium phosphate, pH7.8; 0.2% Triton X-100; Complete mini protease cocktail inhibitor tablet (Roche)). Lysates were stored at -80°C until used.
APP-firefly luciferase stably transfected cells
HEK293 cells stably over-expressing APP-Firefly luciferase complementing fragments (APP FLuc EFC) were made by the limiting dilution method. One μg each of the APP FLucN and APP FLucC plasmids were co-transfected into cells in 10cm plates using FugeneHD (Roche). For selection, 800mg/mL geneticin (Cellgro) was placed in culture medium. Single cells were grown into colonies, and positive clones were assessed for optimal protein expression by western blotting and luciferase activity by luminescence assay.
Sodium dodecyl sulphate - polyacrylamide (SDS-PAGE) electrophoresis and western blotting
Protein concentration in cell lysates was measured by the Pierce Bicinchoninic Acid (BCA) Assay in accordance with manufacturer’s protocol. Ten μg of total protein was denatured in Laemmli buffer and separated on 8% Tris-Glycine gels at 150V for 60min. Proteins were then transferred onto 0.4mm Immobilon Hybridization nitrocellulose filter membranes (Millipore). The primary and secondary antibodies used are described below. The Supersignal West Pico Chemiluminescent Substrate (Thermo scientific) was added to the membrane for 5min and the blots were then developed on Phoenix Research Products F-BX810 Blue X-Ray Films.
Antibodies
Primary antibodies for western blots were mouse monoclonal 6E10 (Covance, 1:1000) against amino acids 1-17 of Aβ but also recognizing full length APP and sAPPα; rabbit polyclonal APP C8 antibody recognizing amino acids 676-695 of APP for detecting C-terminal fragments (γ-CTFs), a kind gift from Dr. D. Selkoe (1:1000) [47]; mouse monoclonal 192wt (Elan Pharmaceuticals, 1:1000) recognizing amino acids 590-596 of APP for detecting sAPPb; and mouse monoclonal beta-tubulin (Invitrogen, 1:10,000). Secondary antibodies used for western blots were peroxidase labeled goat anti-mouse IgG (H+L) and peroxidase labeled goat anti-rabbit IgG (H+L) (KPL) both at 1:5000.
Aβ enzyme-linked immunosorbent (ELISA) assay
HEK293 cells stably transfected with wtAPP, a kind gift from Dr. D. Selkoe, were plated onto 6-well plates at a density of 4x105 cells per well. The next day, 600mL of fresh medium was incubated with cells with 1μM of compounds overnight. Media were then collected and centrifuged at 16,873g for 5min at 4°C to discard cell debris. Cell lysates were also prepared, and both medium and lysates were stored at -80°C until used. ELISAs were carried out using the human Aβ40 and Aβ42 ELISA kits (Invitrogen) in accordance with manufacturer’s protocol with samples diluted 1:2 in diluent buffer.
Firefly luciferase and dual luciferase reporter assay
The luciferase assay used to assess the APP FLuc EFC system was carried out using the Promega Bright-Glo Luciferase Assay System in accordance with manufacturer’s protocol. One hundred μL of cell lysate was added to 96 well white microtiter plates (Thermo Electron Corporation), followed by the addition of 100 μL substrate, and read immediately in the Promega GloMax-Multi microplate reader. The luciferase assay used to assess whether the compounds inhibited the luciferase signal was performed using the Promega Dual-Luciferase Reporter Assay. Cells in 96-well plates were transfected with the SV40 luciferase and TK Renilla plasmids (Promega). The next day, 1μM of compound was added to cells. After 24 hours, luminescence was measured following substrate addition, and the Firefly luciferase signal was normalized to the Renilla luciferase signal.
Compound library
The compound library consists of 77,440 small molecules that include Food and Drug Administration (FDA) approved drugs, purified natural products, synthetic compound collections from Peakdale (High Peak, UK), Maybridge PIc (Cornwall, UK), Cerep (Paris, France), Bionet Research Ltd. (Cornwall UK), Prestwick (Ilkirsch, France), Specs and Biospecs (CP Rijswijk, Netherlands), ENAMINE (Kiev, Ukraine), I.F. Lab LTD (Burlington, Ontario Canada), Chemical Diversity Labs LTD (San Diego, CA), ChemBridge (San Diego, CA), and various academic institutions around the world.
High throughput screening (HTS)
Stably transfected cells were maintained in 2-CellSTACK chambers (Corning) in culture medium (10% FBS, 1% Penicillin/Strptomycin, 800mg/mL geneticin, in phenol red DMEM) at 37°C and 5% CO2. The day before screening, cells were dissociated using cell dissociation buffer (Sigma) and split into 384-well Perkin Elmer white CulturPlateTM at 8,000 cells per well using the Thermo Electron Corporation Multidrop 384, in plating medium (10% FBS and 1% P/S in non-phenol red DMEM). Compound and DMSO control plates were diluted in media and transferred to cells using the Beckman Coulter Biomek® NX Laboratory Automation Workstation for a final compound concentration of ~1μM in the assay and incubated with cells overnight. Luciferase detection was performed using the Perkin Elmer BriteLite Plus Ultra-High Sensitivity Luminescence Reporter Gene Assay System. Substrate was added to cells using the Biomek® FX Laboratory Automation Workstation and incubated for 5min in the dark at room temperature to allow luminescence to reach peak development. Finally, luminescence was measured using the Perkin Elmer EnVisionTM multilabel plate reader. Any compound that lowered luciferase signal ≥ 50% than the average DMSO control was considered as potential hit. Potential hits were reviewed by medicinal chemistry to confirm that they do not affect luciferase activities.
12-Point dose response assays
Selected compounds were re-ordered and dissolved in DMSO to produce 50μM stock solutions and then serially diluted to produce twelve different concentrations starting at 50μM final in the assay. Compounds and DMSO control were first diluted into 96-well plates and then quadmapped into 384-well plates and assayed as described above.
Cell toxicity and cell proliferation assays
To assess the effect of compounds on cell toxicity, the Promega CellTiter-Glo Luminescent Cell Viability Assay was used in accordance with manufacturer’s protocol in parallel with the primary screening assay. Cells were plated into 384-well plates and allowed to settle overnight. The next day, 10 μL substrate was added to cells for 10min at room temperature, and read on the plate reader. To assess the effect of compounds on cell proliferation, the Promega CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (MTS) was used. Cells were plated in 96 well plates and compounds were added to cells the next day. The following day, absorbance was measured following substrate addition.
Data analysis
The Graphpad Prism 5 software was used for data analysis. Statistical differences were evaluated using the non-parametric Kruskal-Wallis test, followed by a post-hoc Dunn’s multiple comparison test. For the 12-point dose response experiments, data were plotted using a non-linear log (inhibitor) vs. response - variable slope (four parameters) curve fit to calculate the concentration at 50% Inhibition (IC50) values. Protein expression in Western blots was analyzed by densitometry using ImageJ software.
Results
Development of APP-firefly luciferase enzyme fragment complementation (APP Fluc EFC) system
The APP FLuc EFC system optimized for HTS is illustrated in Figure 1. APP tagged to full-length luciferase enzyme is represented as APP Fluc FL. There is a 13 amino acid linker region (STVPRARDPPVAT) between APP and the luciferase fragment. The full-length luciferase enzyme is then split into two fragments, one corresponding to its N- terminal end with amino acids 1-475, and one corresponding to its C-terminal end with amino acids 265-550. These complementing fragments have been shown to produce the most optimal luciferase signal [44]. When APP is in monomeric form, there is no luciferase activity as the luciferase enzyme is fragmented. However, when APP dimerizes, the luciferase fragments reconstitute into a fully functional active enzyme. Addition of a luciferase substrate produces luminescence, which allows for the quantification of APP dimerization.
Figure 1.
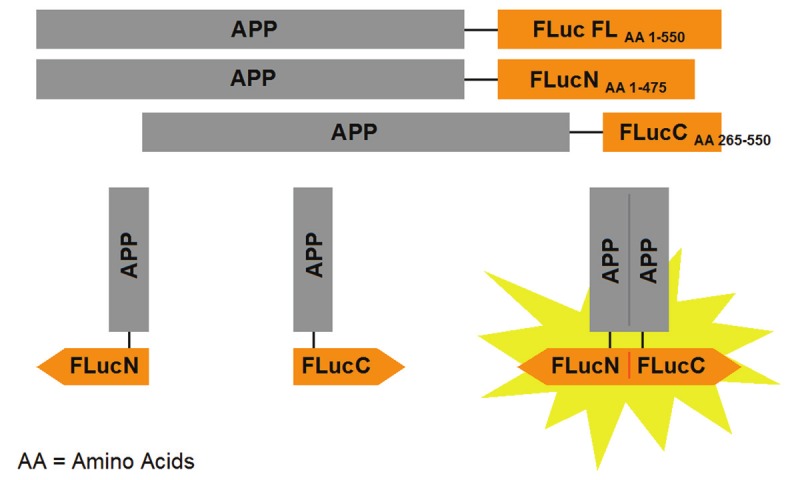
Schematic diagram of the APP-Firefly luciferase enzyme fragment complementation (APP FLuc EFC) system. Deletion mutants of the overlapping Firefly luciferase fragments were generated, where FLucN corresponds to the N-terminal fragment of luciferase with amino acids 1-475, and FLucC corresponds to the C-terminal fragment of luciferase with amino acids 265-550. APP tagged to full-length Firefly luciferase (APP FLuc FL) was constructed as a positive control for luminescence-inducing activity.
Protein expression of APP firefly luciferase constructs
Protein expression of the cloned APP Firefly luciferase (APP FLuc) constructs in transfected HEK293 cells was analyzed by SDS-PAGE followed by western blotting. Figure 2B shows a representative blot of the constructs. When cells were transfected with the pcDNA3.1 empty vector, no protein bands were detected. When cells were transfected with the APP FLuc FL construct, there was a protein band at ~172kDa, which corresponds to the 110kDa APP, along with the 62 kDa luciferase enzyme. When cells were transfected with the APP FLucN fragment, a smaller protein band at ~162kDa was seen, as the N-terminal fragment consists of only 475 amino acids of the full-length luciferase. When cells were transfected with the APP FLucC fragment, an even smaller protein band at ~142 kDa was seen, as the C-terminal fragment consists of only 285 amino acids of the full-length luciferase. Finally, when cells were transfected with both the APP FLucN and the APP FLucC fragments, protein bands corresponding to each respective constructs were seen. Notice that in all samples, an additional protein band for the immature, non glycosylated, form of APP was also seen. Accordingly, the results show that the APP FLuc constructs can be expressed appropriately in HEK293 cells.
Figure 2.
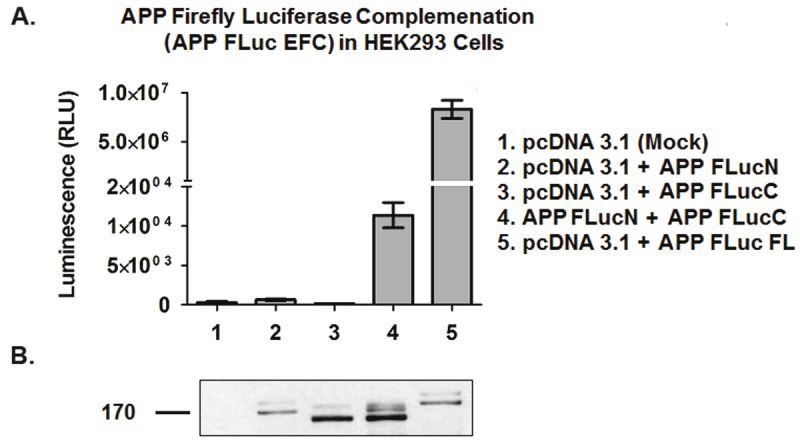
Validation of APP-Firefly luciferase system. A. Quantification of APP dimerization by measurement of luminescence. No luminescence is detected from HEK293 cells transfected with pcDNA1, or cells transfected with the APP FLucN or APP FLucC fragments alone. Luminescence is detected only in cells transfected with the APP FLuc FL control or cells cotransfected with both the APP FLucN and APP FLucC fragments (APP FLuc EFC). APP FLuc EFC activity is correlated to APP dimerization. Results shown are mean ± standard error, n = 3. B. Protein expression of APP Firefly luciferase constructs. HEK293 cell lysates were ran on 8% Tris-Glycine gel. Protein samples in each lane correspond to the luminescence samples in Figure 2A. 6E10 antibody was used at 1:1000 dilution.
Quantification of APP dimerization by measurement of firefly luciferase activity
To test for the functionality of the APP FLuc system, luminescence of transfected cell lysates was measured in a luciferase assay (Figure 2A). Only background luminescence was observed for cells transfected with pcDNA3.1, and for cells transfected with the APP FLucN or the APP FlucC fragments alone. On the other hand, luminescence significantly higher than background can be seen from cells transfected with APP FLuc FL or co-transfected with both APP FLucN and APP FLucC (APP FLuc EFC). Notice that the complemented luciferase activity is lower than the full-length luciferase control. This is not surprising given that only a portion of the APP dimers would result in the correct complementation. Nevertheless, the luminescence from the APP FLuc EFC cell lysates corresponds to APP forming dimers where luciferase fragments were brought close enough to complement each other to form an active luciferase enzyme. Thus, the APP FLuc EFC assay can readily be used to quantify the amount of APP dimerization in cell systems. After validating the use of the APP FLuc EFC system, a stable cell line was generated for the APP luciferase constructs for HTS.
High throughput screening results
For the HTS, cells stably transfected with the APP FLuc EFC fragments were treated with different compounds to assess changes in luciferase activity. A total of 77,440 compounds were screened; of those, 113 compounds lowered luciferase signal 50% or more. This amounts to a final hit rate of 0.15%. Figure 3A shows the results of ~10,000 representative compounds that were tested. Data points that fall lower than the mean represent potential APP dimerization inhibitors. Figure 3B shows the percent inhibition for each compound from Figure 3A. Any compound that inhibited greater than 50% inhibition line was considered a potential hit.
Figure 3.
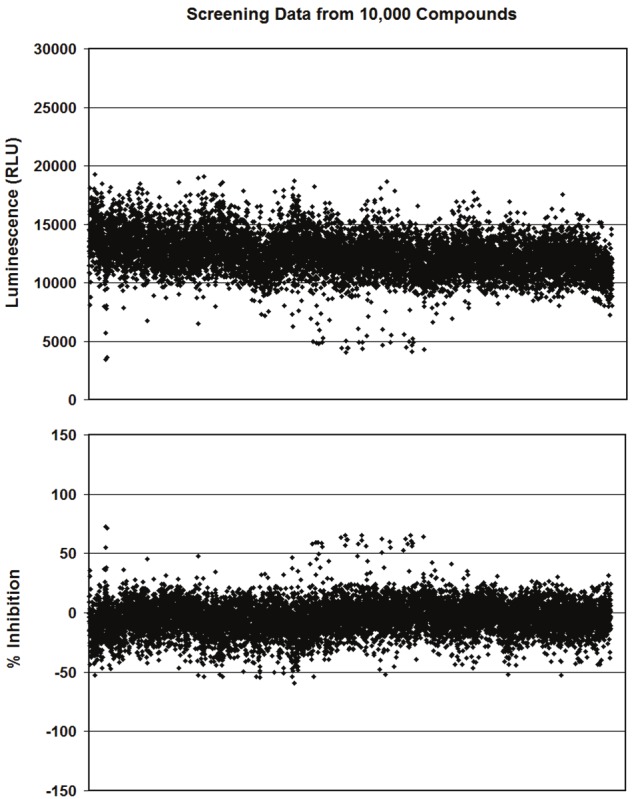
Representative results from HTS. Effects of ~10,000 compounds on stably transfected APP FLuc EFC cells are shown, which represent ~1/7 of the total compounds screened. Each data point represents a single compound. A. Luminescence results. The mean luciferase signal is about 12,000 RLU. Data points that fall lower than the mean represent potential APP dimerization inhibitors. B. Percent (%) inhibition as calculated for each compound as compared to DMSO control. % inhibition is calculated as [(Average DMSO signal - Compound Signal)/ Average DMSO Signal] x 100. Only data points that fall over the 50% or higher cutoff point are considered as potential hits.
12-Point dose response and toxicity assays
12-point dose response experiments were conducted using the APP FLuc assay system to confirm the effects of compound treatment on APP dimerization and evaluate potency. Since the luciferase signal in this assay could be reduced if the compounds were toxic to the cells, cell viability was tested in parallel to rule out toxic compounds. Of the 113 candidate compounds identified in the initial HTS, only 14 of the retested compounds displayed dose-response effects and were non-cytotoxic. Of these 14 compounds, two compounds, LDN-0128964 (compound X) and LDN-0004724 (compound Y), were selected for further studies based on potency and lack of toxicity (Figure 4). Both compounds had IC50 values in the low nM range.
Figure 4.
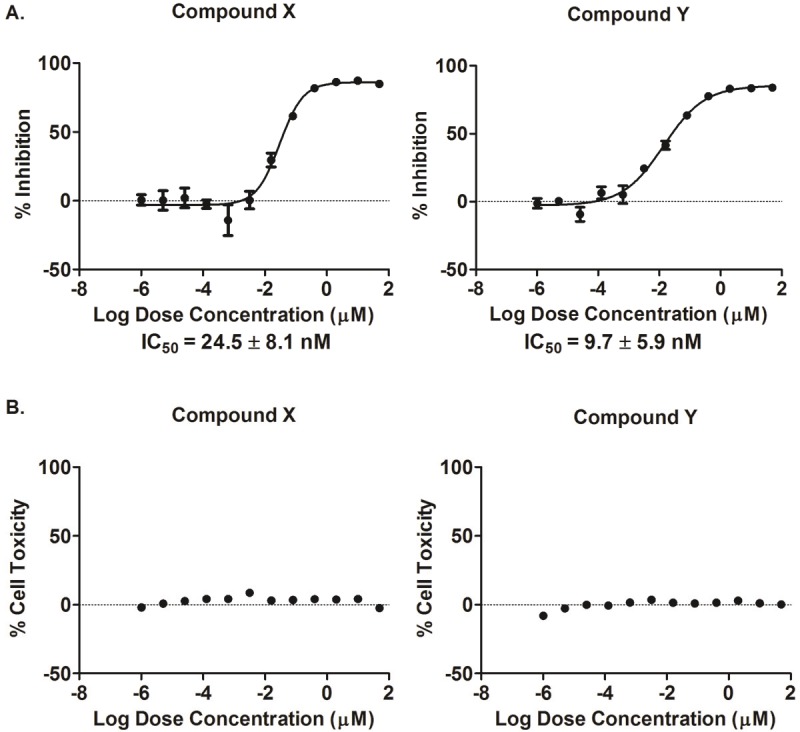
12-Point dose response and toxicity curves. Representative 12-point dose response and toxicity curves for compounds X and Y are shown. A. The dose-response curves demonstrate that increasing compound concentration increased inhibition of assay signal, which can be correlated to inhibition of APP dimerization. B. In the accompanying toxicity curves, the 12 compound doses are shown to be non-toxic to cells. IC50 values are mean ± standard deviation, n = 2, in quadruplicates.
Effect of compounds on luciferase activities
The effect of compounds X and Y on luciferase activity was analyzed to ensure that the decrease in luminescence by the compounds is due to inhibiting APP dimerization, and not by affecting luciferase activity. Cells that were transiently transfected with luciferase plasmids were used. Figure 5A shows the effects of the compounds on luciferase activity as fold change compared to DMSO control. Neither compound X nor Y affect luciferase activity and can be categorized as novel small molecule APP dimerization inhibitors. A corresponding cell proliferation assay in Figure 5B was used to demonstrate that any differences seen in luciferase signal is not due to a change in cell number.
Figure 5.
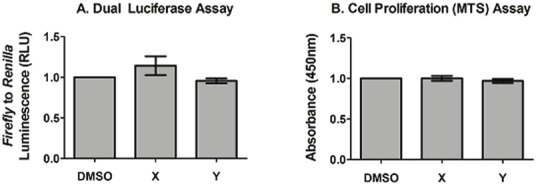
Effect of compounds on luciferase signal. A. Dual Luciferase assay. Compounds X and Y did not significantly affect luciferase signal in transfected HEK293 cells as compared to DMSO. B. Cell proliferation assay. Compounds X and Y did not affect cell number in transfected HEK293 cells as measured by MTS viability assay . Results are fold changes over DMSO control, mean ± standard error, n = 4.
Effect of compounds on Aβ production by ELISA
After validating that the selected compounds do inhibit APP dimerization, their effects on Aβ production was investigated. Cells stably transfected with untagged WT APP cells were used. After correcting for APP protein expression by Western blots, cells treated with compound X did not had a significant effect on Aβ production as compared to cells that were treated with the DMSO vehicle control, while compound Y significantly reduced both Aβ40 or Aβ42 levels (Figure 6A and B). The Aβ42 to Aβ40 ratios remained unchanged for cells treated with either of the compounds as compared to DMSO control (Figure 6C). These results are valid for the 1 μM concentration at which the compounds were tested, where both compounds X and Y maximally inhibited dimerization at ~70-80%.
Figure 6.
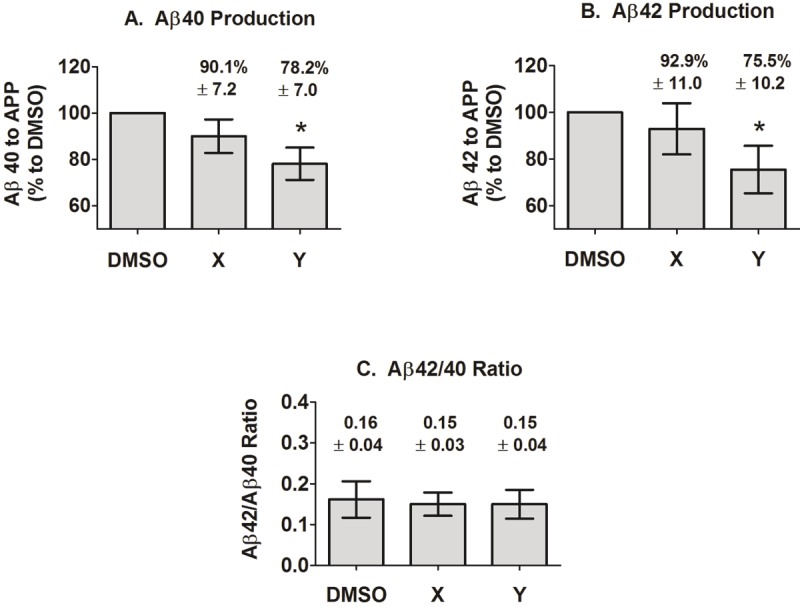
Effects of compounds on Aβ40 and 42 production. Aβ was measured from the media of stably transfected WT APP cells treated with DMSO or with 1 μM of compound X or Y by ELISA. Aβ levels are then normalized to APP protein expression via densitometry of western blots. A. Compound X did not significantly affect Aβ40 level as compared to DMSO vehicle control, but there is a trend towards a decrease. Compound Y significantly reduced Aβ40 with p<0.05. B. Similarly, compound X did not significantly affect Aβ42 as compared to DMSO vehicle control, but there is a trend towards a decrease. Compound Y significantly reduced Aβ42 with p<0.05. C. Both compounds X and Y did not affect Aβ42 to Aβ40 ratio. Results are mean ± standard error, n = 7.
Effect of compounds on APP processing
The effects of compounds X and Y on APP protein expression and APP processing were also examined in stably transfected WT APP cells. Figure 7 is a representative Western blot of treated cells and the expression level of total APP; the level of sAPPα, which indicates α-secretase processing; the level of sAPPβ, which indicates β-secretase processing, and the levels of the C-terminal fragments (γ-CTFs), C99 and C83, which indicate γ-secretase processing following β or α secretase, respectively. The results indicate that the expression of APP and most of its processed products were unaltered in the presence of the compounds as compared to DMSO control; however, there is a significant reduction in sAPPβ release in the presence of compound Y. As there is no change in the C99 level, this may suggest that compound Y may lower Aβ40 and Aβ42 production by altering β-secretase activity.
Figure 7.
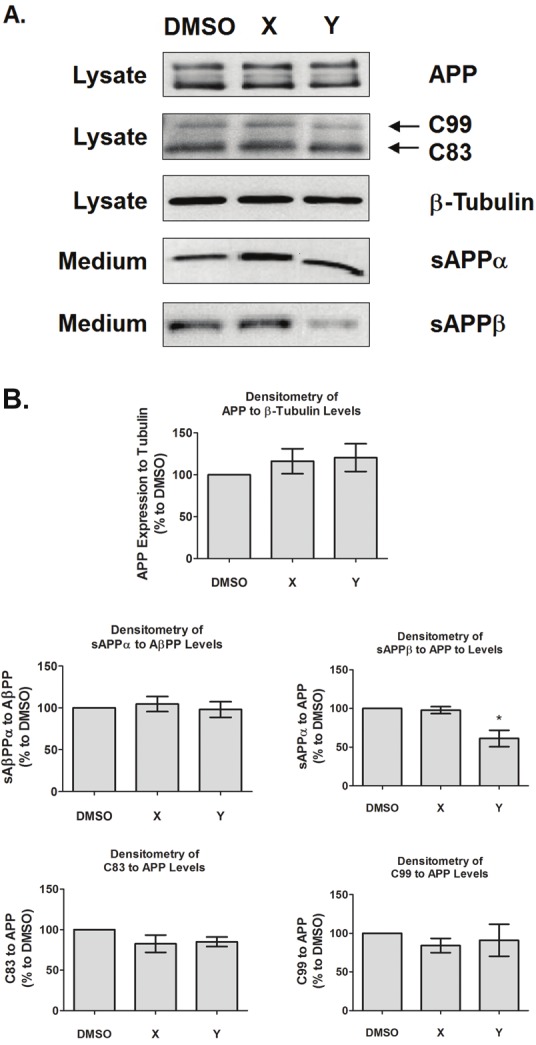
Effect of compounds on APP processing. A. Cell lysates and media from stably transfected WT APP cells treated with DMSO or with 1 μM of compound X or Y were collected and ran on 8% SDS-PAGE Tris-Glycine gel followed by western blot. The representative blots indicate the effects of compounds X and Y on APP protein expression and the APP processing products sAPPα, sAPPβ, and the γ-CTFs, C99 ad C83. B. Upon quantification, densitometric plots indicate that compound X does not affect APP expression and its processing products as compared to DMSO control. However, compound Y significantly reduced the sAPPβ at p<0.05. Results are mean ± standard error, n = 7.
Discussion
APP dimerization has been reported by several groups to affect Aβ levels. However, it is unclear whether dimerization increases or decreases Aβ [8,13,32,38-40]. The methods used to determine the effects of APP dimerization on Aβ generation also vary widely, possibly accounting for some of the inconsistencies. These approaches include introducing mutations to cross-link monomers into dimers (K624C APP) [13], introducing mutations to disrupt dimerization sites (GxxxG mutants) [8,38,39], using complementing peptides to the APP growth factor-like domain (GFLD) loop domain [32] and the GxxxG motif [48] to block dimer formation, analyzing familial APP mutants in micelles, phospholipid bilayers [38] and in cellular systems (So et al., manuscript in preparation), and inducing dimerization with FKBP, an rapamycin analog [40]. In this study, we developed an additional technique to monitor APP dimerization. An advantage of our APP FLuc system is that it eliminates the issue of introducing mutations that can affect γ-secretase processing. At the same time, it measures dimer formation in cells more naturally as compared to inducing dimer formation with exogenous drugs [40]. The luciferase fragments are also small in size, which minimizes interference with the normal biological activities of APP. Moreover, the complemented APP proteins are fully reversible into monomers [43]. A similar method using β-galactosidase EFC has previously been described by our group [49], and another method using Renilla luciferase has been described by others to monitor APP and γ-secretase interactions [50]. One of the most important features of luciferase complementation is that it can be miniaturized and adapted to a high-throughput format for drug screening. Using this method, we screened a large compound library to identify APP dimerization modulators with the hope to better understand the relationship between APP dimerization and Aβ production.
Two potent inhibitors,LDN-0128964 (Compound X) and LDN-0004724 (Compound Y), were identified by HTS. To our knowledge, this is the first report of small molecules that can inhibit APP dimerization. The IC50 values of the compounds are at a nanomolar range, which signifies their potencies. However, whether these compounds prevent de novo APP dimer formation and/or break existing dimers apart is unknown. Further evaluation is necessary to fully understand the mechanisms of the compounds’ actions. We also hypothesized that modulating dimerization would affect Aβ production. Whether dimerization increases or decreases Aβ, our results would provide more information as to the relationship between these two processes. To our surprise, compound X had no significant effect on Aβ production, APP protein expression, and APP processing; however, there is a small trend towards decrease in both Aβ40 and 42. Note that the effects of the compound was tested only at 1 μM, which according to the 12-point dose curves, is at maximum inhibition of ~70-80%. It is possible that at different concentrations, a larger effect may be seen. It is also possible that significant inhibition may be seen if the compound structure were to be optimized with medicinal chemistry. On the other hand, compound Y significantly decreased both Aβ40 and 42 and sAPPβ levels. Thus, our results add further support to the current literature that suggests inhibition of APP dimerization inhibits Aβ production. In the APP dimerization assay we used APPF Luc EFC and X and Y exhibited a 70-80% inhibition of dimerization, while only compound Y significantly reduced Aβ levels but only by 25% when tested by ELISA of Aβ secreted from HEK293 cells stably overexpressing untagged APP. The two systems employed are very different and that could explain this discrepancy. Nevertheless, a reduction of only 25% in Aβ has been shown recently to be enough to rapidly revert cognitive dysfunction in an AD animal model treated with bexarotene, an oral RXR agonist [51].
Until now, published data have shown promise in terms of utilizing APP dimerization as a therapeutic target. For example, APPsα is known to be neuroprotective for neurons. By introducing sAPPα to serum-starved neuroblastoma cells, there is an increased amount of disrupted APP dimers on the cell surface, along with an increase in survival of neurons. This implies that the neuroprotective effects of sAPPα may be the result of disrupting APP dimers [31]. Also, the G33 residue (Aβ numbering) in the APP GxxxG transmembrane domain is the key residue responsible for the dimerization of APP [39] and oligomerization and toxicity of Aβ [52]. When designing peptides that complemented the GxxxG motif in Aβ40 and 42, the tendency of Aβ to form fibrils was reduced, and pre-formed mature fibrils also depolymerized. These peptides were also able to reduce the toxicity of Aβ42 and increase the survival of cultured rat cortical neurons [48]. In addition, γ-secretase modulators (GSMs) such as some NSAIDS are known to inhibit APP processing, lower Aβ production, and inhibit Aβ aggregation [53]. It was once thought that these GSMs bind to γ-secretase and prevent its ability to bind to and cleave APP γ-CTFs into Aβ, but when observing photo-labeled GSMs fluoroprobes, it was found that they actually bind to APP and Aβ, and not γ-secretase [52]. Complementary results of surface plasmon resonance (SPR) and nuclear magnetic resonance (NMR) analysis showed that the Aβ lowering NSAIDs bind to the APP-TMS GxxxG dimerization motif. In fact, sulindac sulfide and its derivatives destabilized APP TMS dimer in a concentration dependent manner, which correlated with the binding strength and Aβ lowering activity [54]. Furthermore, introducing mutations at the GxxxG motif in familial APP mutants decreased Aβ levels and rescued the effects of familial AD. These results suggest that the APP transmembrane dimerization GxxxG motif is not only important in WT APP, but also in familial APP mutants [55]. Given these data, it seems that dimeric APP is a promising drug target that could be relevant to both sporadic and early onset AD. Our data further support the notion that inhibiting APP dimerization with small molecules can be beneficial by reducing Aβ. Future plans of this study include working with medicinal chemistry to modify the chemical structures and synthesize analogues of the lead compounds to enhance their ability to further lower Aβ levels. It would also be interesting to examine whether the APP dimerization activators identified in this screen would increase Aβ production. In this study, about 80 compounds from the screening library were identified as activators that increased the luciferase signal 50% or more when compared to DMSO controls. These compounds could prove to be an additional set of tool molecules to investigate the role of APP dimerization in AD. In summary, we have shown here that modulation of APP dimerization is a promising therapeutic target for AD; however, we also believe that more research in regards to the biological significance of APP dimers and their impact on Aβ production is needed before we can fully exploit this target for AD drug discovery. These new tools are an important step towards this goal.
Acknowledgements
We would like to thank Dr. Dennis Selkoe, Harvard Medical School, for his gifts of the APP C8 antibody and the HEK293 cell line stably overexpressing wt APP. We also thank Dr. Howard Cabral, Boston University School of Medicine, for expert help with statistical analyses. This work was supported by NIH U24 NS04933901 to MAG and the Alzheimer’s Association IIRG-10-1744475 award to CRA.
References
- 1.Selkoe D, Mandelkow E, Holtzman D. Deciphering Alzheimer disease. Cold Spring Harbor perspectives in medicine. 2012;2:a011460. doi: 10.1101/cshperspect.a011460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, Citron M. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- 4.Lin X, Koelsch G, Wu S, Downs D, Dashti A, Tang J. Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein. Proc Natl Acad Sci U S A. 2000;97:1456–1460. doi: 10.1073/pnas.97.4.1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Strooper B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron. 2003;38:9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- 6.Cao X, Sudhof TC. Dissection of amyloid-beta precursor protein-dependent transcriptional transactivation. J Biol Chem. 2004;279:24601–24611. doi: 10.1074/jbc.M402248200. [DOI] [PubMed] [Google Scholar]
- 7.Takami M, Nagashima Y, Sano Y, Ishihara S, Morishima-Kawashima M, Funamoto S, Ihara Y. gamma-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J Neurosci. 2009;29:13042–13052. doi: 10.1523/JNEUROSCI.2362-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Munter LM, Voigt P, Harmeier A, Kaden D, Gottschalk KE, Weise C, Pipkorn R, Schaefer M, Langosch D, Multhaup G. GxxxG motifs within the amyloid precursor protein transmembrane sequence are critical for the etiology of Abeta42. Embo J. 2007;26:1702–1712. doi: 10.1038/sj.emboj.7601616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Asai M, Hattori C, Szabo B, Sasagawa N, Maruyama K, Tanuma S, Ishiura S. Putative function of ADAM9, ADAM10, and ADAM17 as APP alpha-secretase. Biochem Biophys Res Commun. 2003;301:231–235. doi: 10.1016/s0006-291x(02)02999-6. [DOI] [PubMed] [Google Scholar]
- 10.Buxbaum JD, Liu KN, Luo Y, Slack JL, Stocking KL, Peschon JJ, Johnson RS, Castner BJ, Cerretti DP, Black RA. Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem. 1998;273:27765–27767. doi: 10.1074/jbc.273.43.27765. [DOI] [PubMed] [Google Scholar]
- 11.Slack BE, Ma LK, Seah CC. Constitutive shedding of the amyloid precursor protein ectodomain is up-regulated by tumour necrosis factor-alpha converting enzyme. Biochem J. 2001;357:787–794. doi: 10.1042/0264-6021:3570787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 13.Scheuermann S, Hambsch B, Hesse L, Stumm J, Schmidt C, Beher D, Bayer TA, Beyreuther K, Multhaup G. Homodimerization of amyloid precursor protein and its implication in the amyloidogenic pathway of Alzheimer's disease. J Biol Chem. 2001;276:33923–33929. doi: 10.1074/jbc.M105410200. [DOI] [PubMed] [Google Scholar]
- 14.Chen CD, Oh SY, Hinman JD, Abraham CR. Visualization of APP dimerization and APP-Notch2 heterodimerization in living cells using bimolecular fluorescence complementation. J Neurochem. 2006;97:30–43. doi: 10.1111/j.1471-4159.2006.03705.x. [DOI] [PubMed] [Google Scholar]
- 15.Oh SY, Ellenstein A, Chen CD, Hinman JD, Berg EA, Costello CE, Yamin R, Neve RL, Abraham CR. Amyloid precursor protein interacts with notch receptors. J Neurosci Res. 2005;82:32–42. doi: 10.1002/jnr.20625. [DOI] [PubMed] [Google Scholar]
- 16.Oh SY, Chen CD, Abraham CR. Cell-type dependent modulation of Notch signaling by the amyloid precursor protein. J Neurochem. 2010;113:262–274. doi: 10.1111/j.1471-4159.2010.06603.x. [DOI] [PubMed] [Google Scholar]
- 17.Soba P, Eggert S, Wagner K, Zentgraf H, Siehl K, Kreger S, Lower A, Langer A, Merdes G, Paro R, Masters CL, Muller U, Kins S, Beyreuther K. Homo- and heterodimerization of APP family members promotes intercellular adhesion. Embo J. 2005;24:3624–3634. doi: 10.1038/sj.emboj.7600824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaden D, Voigt P, Munter LM, Bobowski KD, Schaefer M, Multhaup G. Subcellular localization and dimerization of APLP1 are strikingly different from APP and APLP2. J Cell Sci. 2009;122:368–377. doi: 10.1242/jcs.034058. [DOI] [PubMed] [Google Scholar]
- 19.Bush AI, Martins RN, Rumble B, Moir R, Fuller S, Milward E, Currie J, Ames D, Weidemann A, Fischer P, et al. The amyloid precursor protein of Alzheimer's disease is released by human platelets. J Biol Chem. 1990;265:15977–15983. [PubMed] [Google Scholar]
- 20.Bellingham SA, Ciccotosto GD, Needham BE, Fodero LR, White AR, Masters CL, Cappai R, Camakaris J. Gene knockout of amyloid precursor protein and amyloid precursor-like protein-2 increases cellular copper levels in primary mouse cortical neurons and embryonic fibroblasts. J Neurochem. 2004;91:423–428. doi: 10.1111/j.1471-4159.2004.02731.x. [DOI] [PubMed] [Google Scholar]
- 21.Duce JA, Tsatsanis A, Cater MA, James SA, Robb E, Wikhe K, Leong SL, Perez K, Johanssen T, Greenough MA, Cho HH, Galatis D, Moir RD, Masters CL, McLean C, Tanzi RE, Cappai R, Barnham KJ, Ciccotosto GD, Rogers JT, Bush AI. Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer's disease. Cell. 2010;142:857–867. doi: 10.1016/j.cell.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mattson MP, Cheng B, Culwell AR, Esch FS, Lieberburg I, Rydel RE. Evidence for excito-protective and intraneuronal calcium-regulating roles for secreted forms of the beta-amyloid precursor protein. Neuron. 1993;10:243–254. doi: 10.1016/0896-6273(93)90315-i. [DOI] [PubMed] [Google Scholar]
- 23.von Rotz RC, Kohli BM, Bosset J, Meier M, Suzuki T, Nitsch RM, Konietzko U. The APP intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. J Cell Sci. 2004;117:4435–4448. doi: 10.1242/jcs.01323. [DOI] [PubMed] [Google Scholar]
- 24.Klar A, Baldassare M, Jessell TM. F-spondin: a gene expressed at high levels in the floor plate encodes a secreted protein that promotes neural cell adhesion and neurite extension. Cell. 1992;69:95–110. doi: 10.1016/0092-8674(92)90121-r. [DOI] [PubMed] [Google Scholar]
- 25.Kwak YD, Brannen CL, Qu T, Kim HM, Dong X, Soba P, Majumdar A, Kaplan A, Beyreuther K, Sugaya K. Amyloid precursor protein regulates differentiation of human neural stem cells. Stem Cells Dev. 2006;15:381–389. doi: 10.1089/scd.2006.15.381. [DOI] [PubMed] [Google Scholar]
- 26.Young-Pearse TL, Bai J, Chang R, Zheng JB, LoTurco JJ, Selkoe DJ. A critical function for beta-amyloid precursor protein in neuronal migration revealed by in utero RNA interference. J Neurosci. 2007;27:14459–14469. doi: 10.1523/JNEUROSCI.4701-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Qiu WQ, Ferreira A, Miller C, Koo EH, Selkoe DJ. Cell-surface beta-amyloid precursor protein stimulates neurite outgrowth of hippocampal neurons in an isoform-dependent manner. J Neurosci. 1995;15:2157–2167. doi: 10.1523/JNEUROSCI.15-03-02157.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leyssen M, Ayaz D, Hebert SS, Reeve S, De Strooper B, Hassan BA. Amyloid precursor protein promotes post-developmental neurite arborization in the Drosophila brain. EMBO J. 2005;24:2944–2955. doi: 10.1038/sj.emboj.7600757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kamal A, Almenar-Queralt A, LeBlanc JF, Roberts EA, Goldstein LS. Kinesin-mediated axonal transport of a membrane compartment containing beta-secretase and presenilin-1 requires APP. Nature. 2001;414:643–648. doi: 10.1038/414643a. [DOI] [PubMed] [Google Scholar]
- 30.Meziane H, Dodart JC, Mathis C, Little S, Clemens J, Paul SM, Ungerer A. Memory-enhancing effects of secreted forms of the beta-amyloid precursor protein in normal and amnestic mice. Proc Natl Acad Sci U S A. 1998;95:12683–12688. doi: 10.1073/pnas.95.21.12683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gralle M, Botelho MG, Wouters FS. Neuroprotective secreted amyloid precursor protein acts by disrupting amyloid precursor protein dimers. J Biol Chem. 2009;284:15016–15025. doi: 10.1074/jbc.M808755200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kaden D, Munter LM, Joshi M, Treiber C, Weise C, Bethge T, Voigt P, Schaefer M, Beyermann M, Reif B, Multhaup G. Homophilic interactions of the amyloid precursor protein (APP) ectodomain are regulated by the loop region and affect beta-secretase cleavage of APP. J Biol Chem. 2008;283:7271–7279. doi: 10.1074/jbc.M708046200. [DOI] [PubMed] [Google Scholar]
- 33.Kong GK, Miles LA, Crespi GA, Morton CJ, Ng HL, Barnham KJ, McKinstry WJ, Cappai R, Parker MW. Copper binding to the Alzheimer's disease amyloid precursor protein. Eur Biophys J. 2008;37:269–279. doi: 10.1007/s00249-007-0234-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bush AI, Multhaup G, Moir RD, Williamson TG, Small DH, Rumble B, Pollwein P, Beyreuther K, Masters CL. A novel zinc(II) binding site modulates the function of the beta A4 amyloid protein precursor of Alzheimer's disease. J Biol Chem. 1993;268:16109–16112. [PubMed] [Google Scholar]
- 35.Beher D, Hesse L, Masters CL, Multhaup G. Regulation of amyloid protein precursor (APP) binding to collagen and mapping of the binding sites on APP and collagen type I. J Biol Chem. 1996;271:1613–1620. doi: 10.1074/jbc.271.3.1613. [DOI] [PubMed] [Google Scholar]
- 36.Wang Y, Ha Y. The X-ray structure of an antiparallel dimer of the human amyloid precursor protein E2 domain. Mol Cell. 2004;15:343–353. doi: 10.1016/j.molcel.2004.06.037. [DOI] [PubMed] [Google Scholar]
- 37.Shaked GM, Kummer MP, Lu DC, Galvan V, Bredesen DE, Koo EH. Abeta induces cell death by direct interaction with its cognate extracellular domain on APP (APP 597-624) FASEB J. 2006;20:1254–1256. doi: 10.1096/fj.05-5032fje. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gorman PM, Kim S, Guo M, Melnyk RA, McLaurin J, Fraser PE, Bowie JU, Chakrabartty A. Dimerization of the transmembrane domain of amyloid precursor proteins and familial Alzheimer's disease mutants. BMC Neurosci. 2008;9:17. doi: 10.1186/1471-2202-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kienlen-Campard P, Tasiaux B, Van Hees J, Li M, Huysseune S, Sato T, Fei JZ, Aimoto S, Courtoy PJ, Smith SO, Constantinescu SN, Octave JN. Amyloidogenic processing but not amyloid precursor protein (APP) intracellular C-terminal domain production requires a precisely oriented APP dimer assembled by transmembrane GXXXG motifs. J Biol Chem. 2008;283:7733–7744. doi: 10.1074/jbc.M707142200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eggert S, Midthune B, Cottrell B, Koo EH. Induced dimerization of the amyloid precursor protein leads to decreased amyloid-beta protein production. J Biol Chem. 2009;284:28943–28952. doi: 10.1074/jbc.M109.038646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang KS, Ilagan MX, Piwnica-Worms D, Pike LJ. Luciferase fragment complementation imaging of conformational changes in the epidermal growth factor receptor. J Biol Chem. 2009;284:7474–7482. doi: 10.1074/jbc.M808041200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luker KE, Gupta M, Luker GD. Imaging chemokine receptor dimerization with firefly luciferase complementation. FASEB J. 2009;23:823–834. doi: 10.1096/fj.08-116749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Luker KE, Smith MC, Luker GD, Gammon ST, Piwnica-Worms H, Piwnica-Worms D. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc Natl Acad Sci U S A. 2004;101:12288–12293. doi: 10.1073/pnas.0404041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paulmurugan R, Gambhir SS. Firefly luciferase enzyme fragment complementation for imaging in cells and living animals. Anal Chem. 2005;77:1295–1302. doi: 10.1021/ac0484777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen H, Zou Y, Shang Y, Lin H, Wang Y, Cai R, Tang X, Zhou JM. Firefly luciferase complementation imaging assay for protein-protein interactions in plants. Plant Physiol. 2008;146:368–376. doi: 10.1104/pp.107.111740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen CD, Podvin S, Gillespie E, Leeman SE, Abraham CR. Insulin stimulates the cleavage and release of the extracellular domain of Klotho by ADAM10 and ADAM17. Proc Natl Acad Sci U S A. 2007;104:19796–19801. doi: 10.1073/pnas.0709805104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Selkoe DJ, Podlisny MB, Joachim CL, Vickers EA, Lee G, Fritz LC, Oltersdorf T. Beta-amyloid precursor protein of Alzheimer disease occurs as 110- to 135-kilodalton membrane-associated proteins in neural and nonneural tissues. Proc Natl Acad Sci U S A. 1988;85:7341–7345. doi: 10.1073/pnas.85.19.7341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sato T, Kienlen-Campard P, Ahmed M, Liu W, Li H, Elliott JI, Aimoto S, Constantinescu SN, Octave JN, Smith SO. Inhibitors of amyloid toxicity based on beta-sheet packing of Abeta40 and Abeta42. Biochemistry. 2006;45:5503–5516. doi: 10.1021/bi052485f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.So PP, Chen CD, Abraham CR. Detection of Amyloid-beta Precursor Protein Homo-Interactions Using Beta-Galactosidase Enzyme Fragment Complementation. J Alzheimers Dis. 2011 doi: 10.3233/JAD-2011-110323. [DOI] [PubMed] [Google Scholar]
- 50.Asada-Utsugi M, Uemura K, Noda Y, Kuzuya A, Maesako M, Ando K, Kubota M, Watanabe K, Takahashi M, Kihara T, Shimohama S, Takahashi R, Berezovska O, Kinoshita A. N-cadherin enhances APP dimerization at the extracellular domain and modulates Abeta production. J Neurochem. 2011 doi: 10.1111/j.1471-4159.2011.07364.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, Casali BT, Restivo JL, Goebel WD, James MJ, Brunden KR, Wilson DA, Landreth GE. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–1506. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harmeier A, Wozny C, Rost BR, Munter LM, Hua H, Georgiev O, Beyermann M, Hildebrand PW, Weise C, Schaffner W, Schmitz D, Multhaup G. Role of amyloid-beta glycine 33 in oligomerization, toxicity, and neuronal plasticity. J Neurosci. 2009;29:7582–7590. doi: 10.1523/JNEUROSCI.1336-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Page RM, Gutsmiedl A, Fukumori A, Winkler E, Haass C, Steiner H. Beta-amyloid precursor protein mutants respond to gamma-secretase modulators. J Biol Chem. 2010;285:17798–17810. doi: 10.1074/jbc.M110.103283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Richter L, Munter LM, Ness J, Hildebrand PW, Dasari M, Unterreitmeier S, Bulic B, Beyermann M, Gust R, Reif B, Weggen S, Langosch D, Multhaup G. Amyloid beta 42 peptide (Abeta42)-lowering compounds directly bind to Abeta and interfere with amyloid precursor protein (APP) transmembrane dimerization. Proc Natl Acad Sci U S A. 2010;107:14597–14602. doi: 10.1073/pnas.1003026107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Munter LM, Botev A, Richter L, Hildebrand PW, Althoff V, Weise C, Kaden D, Multhaup G. Aberrant amyloid precursor protein (APP) processing in hereditary forms of Alzheimer disease caused by APP familial Alzheimer disease mutations can be rescued by mutations in the APP GxxxG motif. J Biol Chem. 2010;285:21636–21643. doi: 10.1074/jbc.M109.088005. [DOI] [PMC free article] [PubMed] [Google Scholar]