

Abstract
Amyotrophic lateral sclerosis (ALS) is a fatal neuromuscular disease for which effective therapeutic interventions and an understanding of underlying disease mechanism are lacking. A variety of biochemical pathways are believed to contribute to the pathophysiology of ALS that are common to both sporadic and familial forms of the disease. Evidence from both human and animal studies indicates that expression of retinoid signaling genes is altered in ALS and may contribute to motor neuron loss. Our goals were to examine the expression and distribution of proteins of the retinoid signaling pathway in spinal cord samples from patients with sporadic and familial ALS and to evaluate the role of these proteins in motor neuron cell survival. In sporadic ALS, the cytoplasmic binding protein that facilitates nuclear translocation of retinoic acid, cellular retinoic acid binding protein-II (CRABP-II), was localized to the nucleus and retinoic acid receptor β (RARβ) was significantly increased in motor neuron nuclei when compared to either familial ALS patients or non-neurologic disease controls. Motor neurons with increased nuclear RARβ were negative for markers of apoptosis. Pre-treatment of primary motor neuron-enriched cultures with a pan-RAR or RARβ-specific agonist decreased motor neuron cell death associated with oxidative injury/stress while a RARβ-specific antagonist enhanced cell death. Our data suggest retinoid signaling is altered in ALS and increased nuclear RARβ occurs in motor neurons of sporadic ALS patients. Activation of RARβ protects motor neurons from oxidative-induced cell death.
Keywords: Retinoid signaling, retinoic acid receptors, nuclear receptor, amyotrophic lateral sclerosis, motor neurons, oxidative stress-induced cell death, motor neuron
Introduction
Amyotrophic lateral sclerosis (ALS), the most common adult onset motor neuron disease, is a progressive, fatal neurodegenerative disorder which is poorly understood and for which effective therapeutic interventions are lacking. The prevalence of ALS is approximately 4-6 cases per 100,000, the average age at disease onset is approximately 55 years and the majority of cases (over 90%) are idiopathic in nature. For familial forms of ALS, the list of associated genes continues to grow [reviewed in [1]; however, no mechanistic differences between sporadic and familial forms have been identified to date.
The clinical manifestations of ALS include both upper motor neuron (enhanced and pathological reflexes and spasticity) and lower motor neuron (focal and multifocal muscle weakness and atrophy, fasciculations and cramps) involvement [2]. A variety of processes (i.e., oxidative stress, mitochondrial dysfunction, abnormal axonal transport and protein aggregation) are believed to play a role in motor neuron degeneration although it remains unclear how each contributes to the pathogenesis of ALS.
Retinoic acid (RA), a member of the retinoid family of lipids and mediator of vitamin A activity, is an essential morphogen in vertebrate development and is critical for neural development/specification and neural plasticity and regeneration [3,4]. Studies also suggest a role for this molecule in adulthood with evidence that retinoid signaling can promote regeneration of the adult rodent nervous system [5-7] or impact the process of neurodegeneration [8,9]. In addition, RA has been shown to modulate the antioxidant defense system and superoxide dismutase-1 (SOD1) gene expression [10,11] further implicating a role for RA in ALS.
Prior studies have demonstrated that genes for retinoid pathway proteins and genes regulated by retinoid signaling are differentially expressed in post-mortem tissues from ALS patients [12-16]. Moreover, in transgenic animal models of familial ALS (FALS), spinal cord gene expression profiling has revealed altered expression of genes of the retinoid signaling pathway [17]. A study in the mutant G93A SOD1 transgenic rat model of ALS indicates that changes in the retinoid receptor expression profile occur in the spinal cord of early pre-symptomatic and terminal stage disease both at the RNA and protein levels [18]. Furthermore, Corcoran and colleagues observed astrocytosis, accumulation of neurofilaments and motor neuron loss in the lumbar spinal cord of vitamin A deficient animals [19]. As systemic vitamin A deficiency in patients with ALS has not been observed [20], it remains unclear how this signaling pathway plays a role in this disease.
Within the cell, the level of free versus bound retinol and RA are regulated by cellular retinol-binding protein (CRBPI) and cellular retinoic acid-binding proteins (CRABP-I and II), respectively [21]. CRBPI binds retinol in the cytoplasm and promotes its metabolism to retinaldehyde [22] and esterification to the retinyl ester for storage [23]. CRABP-I has been suggested to promote the catabolism of RA [24] whereas CRABP-II is thought to mediate the transport of RA to the nucleus for association with its receptors [25]. RA mediates its effects on gene transcription through the retinoic acid receptors (RARs) and the retinoid X receptors (RXRs) [26,27]. Each receptor has three isotypes (α , β and γ) with multiple isoforms of each subtype generated by alternative splicing and differential promoter usage [26]. RARs are activated by either all-trans retinoic acid (ATRA) or 9-cis-retinoic acid (9-cis-RA) and mediate gene expression by forming heterodimers with RXRs whereas RXRs are activated only by 9-cis-RA and modulate gene expression either as homodimers or by forming heterodimers with RARs or a variety of orphan receptors [28-30]. In the absence of ligand, RA nuclear receptors function as transcriptional repressors [4].
The goal of this study was to investigate the role of proteins involved in the RA signaling pathway in ALS. Our overall hypothesis was that retinoid signaling is altered in ALS and thereby contributes to motor neuron cell death. We analyzed post-mortem lumbar spinal cord tissue from individuals with sporadic ALS (SALS), FALS, and non-neurologic disease controls to determine the levels and subcellular localization of proteins involved in this pathway. We observed increased nuclear localization for both CRABP-II and RARβ in motor neurons of patients with SALS. In primary motor neuron-enriched cultures, pan-RAR or RARβ-specific agonists were neuroprotective indicating that signaling through RARβ can promote motor neuron cell survival. These data indicate a role for retinoid signaling in ALS and support further investigation of this signaling pathway as a therapeutic opportunity in ALS.
Materials and methods
Subjects
Lumbar spinal cord tissue was obtained from the ALS Tissue Bank at the University of Pittsburgh Medical Center (RB, Director). Approval was obtained from the University of Pittsburgh Institutional Review Board. In selecting cases for this study, efforts were made to control for age, gender and post-mortem interval with cases with available frozen spinal cord tissue whenever possible. SALS subjects (n = 20) included in this study were clinically diagnosed using the revised El Escorial criteria. FALS patients (n = 4) had a family history of ALS. Two of the SALS and one of the FALS cases harbor C9ORF72 repeat expansions (Table 1), as determined using the repeat-primed polymerase chain reaction (PCR) method previously described [31]. The genetic cause of the remaining FALS cases remains undefined. Control subjects (n = 9) lacked clinical or neuropathological evidence of neurologic disease. The age range for all individuals was 40-95 years old. The average age at death was 60.25 ± 12.66 years for SALS (range, 40 to 82 years) and was not significantly different from FALS (52.75 ± 8.02 years; range, 45 to 63 years; p = 0.27) or control subjects (64.78 ± 15.60 years; range, 51 to 95 years; p = 0.41). The average post-mortem interval time for SALS subjects was 5.50 ± 2.12 hours (range, 2 to 11 hours) and not significantly different from FALS (6.50 ± 5.07 hours; range 3 to 14 hours; p = 0.51) or control subjects (7.89 ± 5.71 hours; range 2 to 20 hours; p = 0.11).
Table 1.
Cases utilized for this study
| Age | Gender | PMI (hours) | Percentage of Motor Neurons with Nuclear CRABP-II Localization | Percentage of Motor Neurons with Increased RARβ Nuclear Localization | |
|---|---|---|---|---|---|
| SALS1 | 45 | M | 11 | 40% (4 of 10) | 55% (6 of 11) |
| SALS2 | 51 | M | 7 | 50% (4 of 8) | 60% (6 of 10) |
| SALS3 | 45 | M | 7 | 82% (9 of 11) | 64% (7 of 11) |
| SALS4 | 62 | M | 7 | 70% (7 of 10) | 14% (1 of 7) |
| SALS5 | 73 | M | 6 | 82% (9 of 11) | 55% (6 of 11) |
| SALS6 | 80 | F | 4 | 64% (9 of 14) | 88% (22 of 25) |
| SALS7 | 82 | M | 5 | 64% (9 of 14) | 73% (22 of 30) |
| SALS8 | 72 | F | 9 | 50% (4 of 8) | 70% (7 of 10) |
| SALS9 | 56 | M | 3 | 63% (5 of 8) | 62% (8 of 13) |
| SALS10 | 63 | F | 5 | 67% (6 of 9) | 18% (2 of 11) |
| SALS11 | 59 | M | 4 | 67% (8 of 12) | 33% (5 of 15) |
| SALS12* | 43 | M | 6 | 70% (7 of 10) | 86% (6 of 7) |
| SALS13* | 67 | M | 4 | 41% (7 of 17) | 13% (2 of 16) |
| SALS14 | 51 | M | 4 | 78% (7 of 9) | 58% (7 of 12) |
| SALS15 | 40 | M | 6 | 38% (3 of 8) | 44% (4 of 9) |
| SALS16 | 77 | F | 4 | 89% (8 of 9) | 50% (5 of 10) |
| SALS17 | 69 | M | 5 | 80% (8 of 10) | 56% (5 of 9) |
| SALS18 | 60 | F | 2 | 70% (7 of 10) | 27% (3 of 11) |
| SALS19 | 50 | F | 7 | 88% (7 of 8) | 73% (8 of 11) |
| SALS20 | 60 | F | 4 | 78% (7 of 9) | 50% (5 of 10) |
| FALS1 | 45 | M | 3 | 17% (2 of 12) | 8% (1 of 12) |
| FALS2 | 48 | F | 5 | 17% (2 of 12) | 20% (2 of 10) |
| FALS3 | 55 | M | 14 | 56% (5 of 9) | 20% (2 of 10) |
| FALS4* | 63 | F | 4 | 50% (4 of 8) | 20% (2 of 10) |
| CON1 | 54 | M | 6 | 26% (6 of 23) | 13% (2 of 15) |
| CON2 | 51 | F | 5 | 38% (3 of 8) | 18% (2 of 11) |
| CON3 | 76 | M | 13 | 31% (4 of 13) | 29% (9 of 31) |
| CON4 | 57 | M | 2 | 25% (3 of 12) | 4% (2 of 47) |
| CON5 | 58 | F | 5 | 73% (8 of 11) | 25% (6 of 24) |
| CON6 | 82 | F | 5 | 11% (1 of 9) | 19% (4 of 21) |
| CON7 | 57 | F | 11 | ND | 21% (7 of 34) |
| CON8 | 95 | M | 20 | 50% (5 of 10) | 0% (0 of 7) |
| CON9 | 53 | F | 4 | 20% (2 of 10) | 6% (1 of 16) |
The age and post-mortem interval (PMI) in hours (average SALS 5.50 hours; average FALS 6.5 hours; average control 7.89 hours) are indicated for the SALS (60.25 years), FALS (52.75 years) and control (64.78 years) cases. 17 of 20 SALS cases exhibited increased nuclear localization of RARβ as compared to controls and FALS.
signifies presence of the C9ORF72 repeat expansion within the subject.
ND = not determined
Antibodies
Monoclonal antibodies were used to detect CRBPI (G4E4; Santa Cruz Biotechnology, Santa Cruz, CA), CRABP-I (C-1; Novus Biologicals, Littleton, CO), RARα (763; Chemicon International, Temecula, CA) and RARγ (1371; Chemicon International). Polyclonal antibodies were used to detect CRABP-II (K-13; Santa Cruz Biotechnology) and RARβ (Chemicon International). For immunohistochemistry, these antibodies were used at dilutions of 1:50 (RARα and RARβ), 1:100 (CRABP-I and RARγ), 1:250 (CRBPI) and 1:2500 (CRABP-II). Western blots were probed with the antibodies listed above at 1:500 (CRABP-II) or alternate polyclonal antibodies to detect RARα (C-20; Santa Cruz Biotechnology) and RARβ (C-19; Santa Cruz Biotechnology) used at a dilution of 1:500.
Tissue homogenates and protein extraction
Lumbar spinal cord tissue homogenates were prepared from a subset of patients (9 SALS; 4 FALS; 5 control) for co-immunoprecipitation studies. The age range and post-mortem interval for this patient subgroup did not differ from that of all subjects nor across the subgroups.
Spinal cord frozen tissue samples from control and both sporadic and familial ALS cases were homogenized and analyzed as previously described [32]. For total cell lysates, spinal cord tissue samples were homogenized using an Omni Tissue Homogenizer (Omni International, Marietta, GA) set at 15,000 rpm for 45 seconds in lysis buffer containing 25 mmol/L HEPES (pH 7.4), 50 mmol/L NaCl, protease inhibitor cocktail II (Sigma, St. Louis, MO) and 1% Triton X-100. The homogenized product was spun at 14,000 rpm at 4°C and the supernatant saved as the total cell lysate. Nuclear and post-nuclear extracts were prepared as described previously [33]. Following homogenization with a Potter-Elvehjem grinder (Omni International) in buffer containing 10 mmol/L Tris (pH 8.0), 10 mmol/L MgCl2, 15 mmol/L NaCl, 0.5 mmol/L phenylmethyl sulfonyl fluoride (PMSF), 2 μg/mL pepstatin A and 1 μg/mL leupeptin, nuclei were collected via low-speed centrifugation at 800g for 5 minutes. The resulting supernatant was saved as the post-nuclear extract and the nuclei further extracted with high-salt buffer (20% glycerol, 20 mmol/L HEPES (pH 7.9), 0.42 mol/L NaCl, 0.1% Nonidet P-40, 0.5 mmol/L PMSF, 2 μg/mL pepstatin A and 1 μg/mL leupeptin) on ice for 10 minutes. Remaining insoluble material was removed via centrifugation at 14,000 rpm for 5 minutes. The resulting supernatant fraction was collected as the nuclear-enriched fraction. Protein concentrations were determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Rockford, IL) per the manufacturer’s instructions.
Immunohistochemistry and TUNEL staining
Paraffin-embedded tissue sections (8 μM) were deparaffinized and a hydrogen peroxide/methanol mixture used to block endogenous peroxidase activity. Sections were steamed for 20 minutes in Antigen Retrieval Citra (pH 6.0) (BioGenex, San Ramon, CA) or Target Retrieval Solution (pH 9.0) (Dako North America, Carpinteria, CA). After washing, blocking was performed with Protein Blocking Agent (Thermo Fisher Scientific) or Power Block Universal Blocking Reagent (BioGenex, San Ramon, CA). Primary antibodies were diluted in phosphate-buffered saline (PBS) or Super Block (ScyTek Laboratories, Logan, Utah) and incubated for 1 hour at room temperature in a humidified chamber. After washing, sections were incubated with the appropriate biotinylated secondary antibody (1:200 dilution; Vector Laboratories, Burlingame, CA) for 30 minutes at room temperature in a humidified chamber. The signal was amplified using Vectastain ABC Reagent (Vector Laboratories) according to the manufacturer’s protocol. NovaRED (Vector Laboratories) served as the chromagen and all sections were counterstained with hematoxylin.
For terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick end-labeling (TUNEL), sections (4 μM) were deparaffinized, treated with proteinase K and endogenous peroxidase activity blocked with a hydrogen peroxide/methanol mixture. TdT enzyme was then added for 25 minutes at 37°C. Anti-digoxigenin conjugate (rhodamin) was added for 30 minutes at room temperature. After washing, sections were incubated in 3,3'-diaminobenzidine (DAB) solution and counterstained with hematoxylin.
Motor neurons located within the ventral horn of the spinal cord were counted for each case by two independent investigators. The number of motor neurons with nuclear CRABP-II or RARβ was reported as a percentage of the total number of motor neurons counted for each case (Table 1) and by grouping (i.e., control, SALS or FALS). Only motor neurons for which the nucleus could be observed were counted. One slide was counted for each case although consistent staining patterns were observed over multiple slides. SALS cases (n = 6) with demonstrated nuclear RARβ were utilized to evaluate the co-localization of nuclear RARβ and TUNEL staining.
Protein-protein interactions
Co-immunoprecipitation (co-IP) of nuclear-enriched fractions was performed using the MultiMACS Protein A/G Kit and μMACS Protein A/G MicroBeads (Miltenyi Biotech, Inc., Auburn, CA). Lysates were incubated with primary antibody for two hours at 4°C with gentle rocking. Protein A/G MicroBeads were then added to the immune complex. After washing, extracts were added and the magnetically labeled protein retained on the column. The eluted immunoprecipitates were analyzed by immunoblot as described below.
Immunoblot
Immunoprecipitated proteins from nuclear-enriched fractions were fractionated on 12% gels by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were transferred to a polyvinylidene difluoride (PVDF) membrane (PerkinElmer, Boston, MA) and blocked with 5% non-fat milk/Tris-buffered saline with Tween-20 (TBST). Blots were probed with primary antibody overnight at 4°C in 5% milk/TBST and then washed with TBST. Horseradish peroxidase-conjugated secondary antibodies specific for each primary antibody were added for 2-3 hours at room temperature after which blots were washed with TBST. The final reaction products were visualized using enhanced chemiluminescence (PerkinElmer) and the density of bands measured using NIH ImageJ software (National Institutes of Health).
Motor neuron-enriched primary cultures
Motor neurons were isolated from the spinal cords of Sasco Sprague-Dawley rat embryos at gestation day 14 (E14) as previously described [34-36]. Briefly, following removal of the spinal cords, tissue was trypsinized and the dissociated cells spun over a metrizamide density gradient. Cells above the metrizamide layer (larger, dense motor neurons) were collected and motor neurons plated in 24-well culture trays coated with poly-D lysine (PDL) and then human placental laminin (HPL) at a density of approximately 12,000 cells/well in neurobasal base media (Invitrogen, Carlsbad, CA) supplemented with B27 (variations +/- RA) (Invitrogen), BDNF, CNTF and GDNF (Peprotech, Rocky Hill, NJ), and as described [34,35]. Cultures were incubated at 37°C at 5% CO2 and half of the culture supernatant exchanged with fresh medium after 24 hours and every 2 to 3 days thereafter. For RNA extractions, cultures were treated with the Qiagen RNeasy Kit (Valencia, CA) per the manufacturer’s instructions. RNA (1.0 μg) was reverse-transcribed to complementary DNA (cDNA) using SuperScript III reverse transcriptase (Invitrogen) prior to analysis by real-time PCR. Products were resolved on 1.5% agarose gels and visualized with ethidium bromide.
RA pathway modulators
All-trans retinoic acid (ATRA) was obtained from Sigma (St. Louis, MO). Adapalene (RARβ and RARγ agonist) and LE-135 (RARβ antagonist) were obtained from Tocris Bioscience (Ellisville, MO). Stock solutions were prepared in dimethyl sulfoxide (DMSO) such that a constant concentration of 0.1% DMSO was present for all treatment groups. Motor neuron-enriched cultures were pre-treated with these agents for approximately 1 hour prior to the addition of hydrogen peroxide (H2O2).
Cell viability/live cell imaging
After 24 hours in culture, motor neurons were treated with the RA pathway modulators (ATRA final concentrations of 1.0, 10 or 100 μM; adapalene final concentrations of 0.25, 2.5 or 25 μM; LE-135 final concentrations of 1.0, 10 or 100 μM) in combination with 400 μM H2O2. Propidium iodide (PI) was added at a final concentration of 1 μL/500 μL medium. Cells were kept in 24-well plates in a previously described time-lapsed microscopic imaging system (Automated Cell, Pittsburgh, PA) where bright-field and fluorescent images were acquired every 10 minutes [37]. These images were analyzed using NIH Image J software and the number of cells in the bright-field and PI-positive cells quantified at 0, 4, 6, 12, 18, 24, 36, 48, 60 and 72 hour time points. The percentage of cell death at these time points was then determined for each treatment. For a typical experiment, each treatment was added to three wells and three separate fields averaging 10-20 cells each were counted per well. The percentage of cell death was determined for each field by binning at the indicated time points, and each experiment repeated at least twice.
Statistical methods
Comparisons between any two groups of data were accomplished using the unpaired t test at the 95% confidence interval. Multi-group comparisons (SALS vs. FALS vs. controls) were performed using ANOVA followed by the Tukey post-test. Comparisons between treatment groups were accomplished using two-way ANOVA followed by Bonferroni post-tests. A p value ≤ 0.05 was considered statistically significant.
Results
Subcellular distribution of CRABP-II is altered in SALS spinal cord motor neurons
We first examined the localization of cytoplasmic retinol and retinoic acid (RA) binding proteins in the lumbar spinal cord of ALS and control subjects using commercially available antibodies. CRBPI, the cytoplasmic protein that mediates metabolism and storage, was not detected in spinal cord motor neurons of SALS, FALS or control subjects (Figure 1A, A-C). CRABP-I was noted in all three subject groups and the immunostaining pattern and intensity were not significantly different across subject groups (Figure 1A, D-F). However, CRABP-II, the protein responsible for mediating nuclear translocation and facilitating nuclear receptor interactions, exhibited enhanced nuclear and perinuclear immunoreactivity in spinal cord motor neurons of SALS cases (Figure 1A, H). CRABP-II was observed in a more diffuse, cytoplasmic pattern in both FALS and control subjects (Figure 1A, G and I). When morphologically distinct motor neurons from multiple lumbar spinal cord sections of SALS, FALS and control cases were counted, the percentage of motor neurons with nuclear staining for CRABP-II was 66% (135 of 205) for SALS cases, 32% (13 of 41) for FALS cases and 33% (32 of 96) for control cases. These differences were statistically significant between and across subject groups (Figure 1B). The percentage of motor neurons with punctate nuclear CRABP-II for each individual subject is shown in Table 1.
Figure 1.
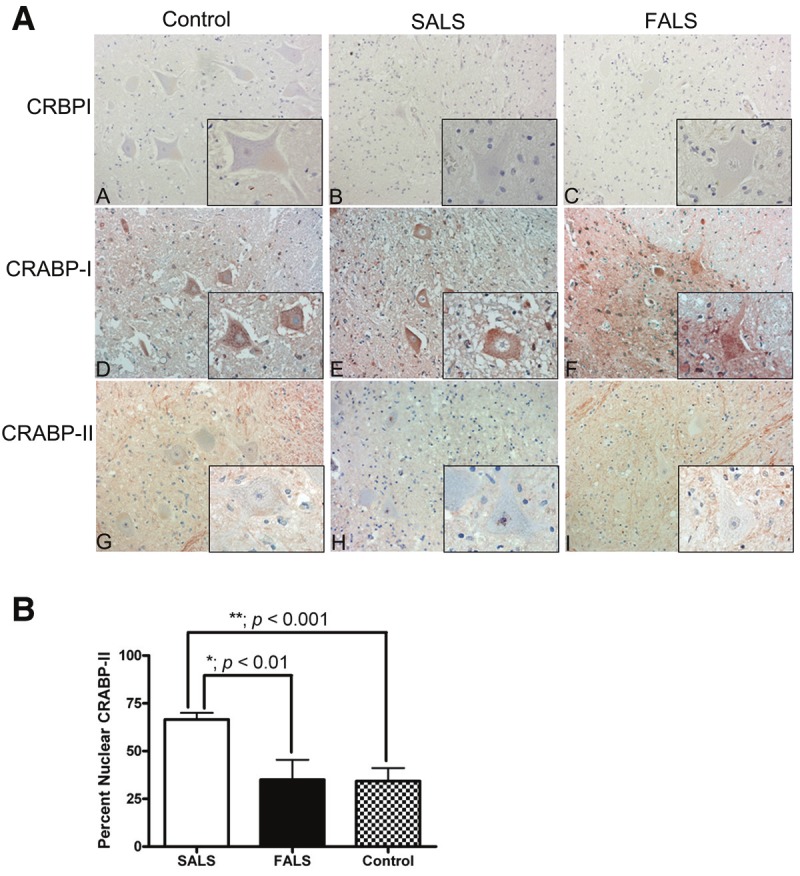
Immunohistochemical analysis of cytoplasmic binding proteins of the retinoic acid signaling pathway in human spinal cord tissue. A: Lumbar spinal cord sections were immunostained for CRBPI (A-C), CRABP-I (D-F) and CRABP-II (G-I) in red and counterstained with hematoxylin. Insets represent a high power magnification of each panel. Original magnifications: 200X (A to I); 400X (insets in A to I). B: The localization of CRABP-II was assessed for morphologically distinct motor neurons from each patient group. The percent of motor neurons with nuclear CRABP-II was significantly greater in patients with SALS (white bars) as compared to FALS (black bars; p < 0.01) or controls (stippled bars; p < 0.001). There was no significant difference between FALS patients and controls (p > 0.05).
RARβ immunoreactivity is significantly increased in motor neuron nuclei of SALS patients and does not correlate with TUNEL staining
We next examined the levels and localization of the three retinoic acid receptor (RAR) isotypes (RARα, RARβ and RARγ) in lumbar spinal cord motor neurons as these receptors are activated by the predominant RA isoform, all-trans RA. RARα was observed in motor neurons of all ALS cases as well as controls and was localized predominantly to the cytoplasm (Figure 2A, A-C). The level of this protein did not differ significantly between motor neurons of the three patient groups but was noted in distal processes of motor neurons of SALS and, to a lesser extent, control subjects. For RARβ, there was a significant increase in the immunostaining of motor neuron nuclei in SALS subjects (Figure 2A, E). In FALS and non-neurologic disease controls, RARβ exhibited a predominantly cytoplasmic distribution in lumbar spinal cord motor neurons (Figure 2A, D and F). The percentage of motor neurons with nuclear RARβ immunostaining was 55% (137 of 249) for SALS cases, 17% (7 of 42) for FALS cases and 16% (33 of 206) for control cases (Figure 2B). This difference in nuclear RARβ was statistically significant when motor neurons from SALS were compared to FALS patients (p < 0.01) and controls (p < 0.001) (Figure 2B). The percentage of motor neurons with increased nuclear RARβ per subject is shown in Table 1. We did not detect any correlation of CRABPII or RARβ distribution to the presence of C9ORF72 repeat expansions that were seen in three patients (Table 1). RARγ was not detected in spinal cord motor neurons of SALS, FALS or control subjects (Figure 2A, G-I).
Figure 2.
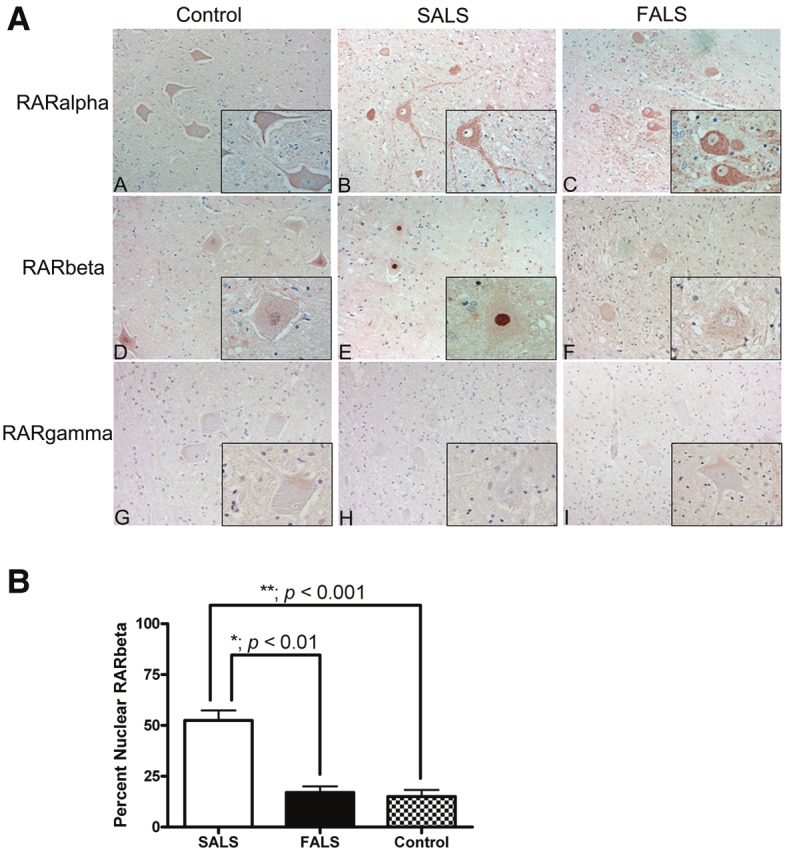
Immunohistochemical analysis of retinoic acid receptor (RAR) proteins in human spinal cord tissue. A: Lumbar spinal cord sections were immunostained for RARα (A-C), RARβ (D-F) and RARγ (G-I) in red and counterstained with hematoxylin. Insets represent a high power magnification of each panel. Original magnifications: 200X (A to I); 400X (insets in A to I). B: Nuclear RARβ immunoreactivity was determined by counting morphologically distinct motor neurons from each patient group. The percent of motor neurons with increased nuclear RARβ was significantly greater in patients with SALS (white bars) when compared to FALS (black bars; p < 0.01) or controls (stippled bars; p < 0.001). There was no significant difference between FALS patients and controls (p > 0.05).
RARβ has been implicated in molecular mechanisms regulating cell death in multiple cell types [38-40]. Conversely, this RAR isotype has been shown to mediate both neurotrophic and neuritogenic effects of retinoic acid [41]. To determine whether this response was apoptotic or pro-survival, we used immunohistochemistry to evaluate the co-localization of TUNEL with RARβ in motor neurons. Serial sections of lumbar spinal cord from six SALS patients were immunostained for RARβ and TUNEL, and the same motor neurons evaluated from both sections as denoted by tissue landmarks. We did not observe TUNEL staining in motor neurons with RARβ nuclear localization (Figure 3A) but did observe TUNEL-positive nuclei in motor neurons lacking nuclear RARβ (representative image in Figure 3B). Overall, 79% (26 of 33) of motor neurons with nuclear RARβ immunostaining were TUNEL negative across multiple SALS subjects.
Figure 3.
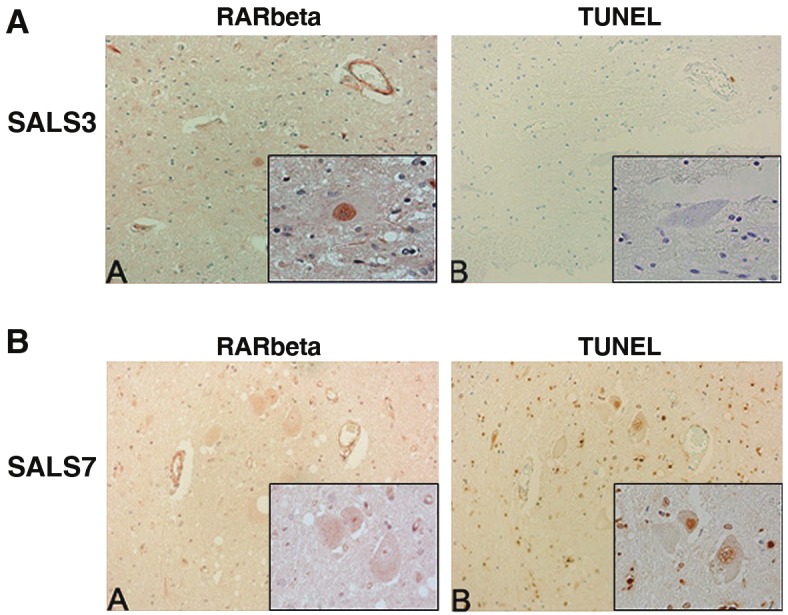
Immunohistochemical analysis of RARβ and TUNEL in human spinal cord. Serial lumbar spinal cord sections from SALS cases were immunostained for RARβ and TUNEL and representative cases are shown. Identical motor neurons were imaged as determined by anatomical hallmarks and location within the tissue. A: Motor neurons with intense nuclear RARβ immunostaining (A) lacked TUNEL staining (B). B: Motor neurons within the spinal cord without nuclear RARβ immunostaining (A) were TUNEL positive (B). Insets represent a high power magnification of each panel. Original magnifications: 200X; 400X (insets).
Protein interactions between CRABP-II and the nuclear receptors do not differ between ALS and control groups
We next examined protein-protein interactions between CRABP-II and either RARα or RARβ to determine the functional consequences of CRABP-II localization to the nucleus. We immunoprecipitated CRABP-II from nuclear-enriched fractions and determined whether the nuclear receptors co-immunoprecipitated with CRABP-II. We observed an interaction between CRABP-II and both RARα (Figure 4A) and RARβ (Figure 4B) although there was no significant difference between ALS and control groups (Figure 4C; p = 0.9042 for RARα in ALS versus controls; p = 0.8881 for RARβ in ALS versus controls).
Figure 4.
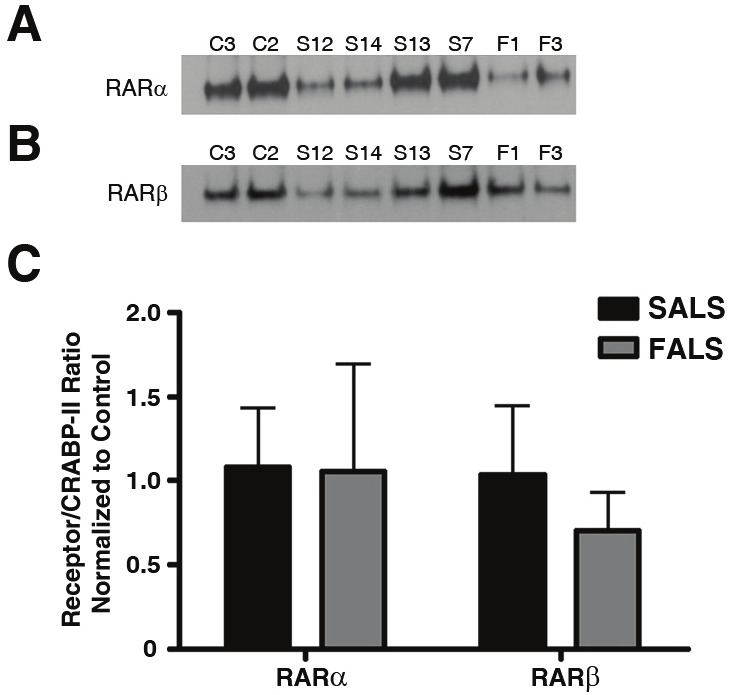
Protein-protein interactions in nuclear-enriched fractions from human lumbar spinal cord. Protein extracts from ALS and controls were prepared as described in the Materials and Methods. Protein (500 μg) from each extract was immunoprecipitated with CRABP-II and resulting blots were probed with antibodies specific to RARα (A) or RARβ (B). A: Immunoblots of nuclear-enriched fractions from lumbar spinal cord co-immunoprecipitated with CRABP-II and probed for RARα. B: Immunoblots of nuclear-enriched fractions from lumbar spinal cord coimmunoprecipitated with CRABP-II and probed for RARβ. C: Graph representing the results from densitometry quantification. RAR Receptor levels were normalized to that of CRABP-II for each sample. Black bars represent control cases (n = 2) and checkered bars are ALS cases (n = 6). Statistical analyses were performed using an unpaired t-test with a 95% confidence interval. The p values for RARα and RARβ were 0.9042 and 0.8881, respectively. Letters and numbers correspond to patients in Table 1.
Motor neuron-enriched cell cultures and retinoic acid treatment
To further explore the functional role of RAR signaling in motor neurons, we utilized a primary motor neuron-enriched culture system. The procedure for purifying spinal motor neurons was modified from that of Camu and Henderson [42,43] and utilized a density gradient to separate low-density cells from the spinal cord cell suspension. PCR was used to determine which of the RARs were expressed in this in vitro system. Using the primers listed in Table 2, we detected expression of RARα and RARβ but not RARγ (Figure 5).
Table 2.
Primer sets for PCR analysis
| Forward Primer | Reverse Primer | |
|---|---|---|
| RARα | 5’-GGCTACCACTATGGGGTTAGCGC-3’ | 5’-GGTTCCGGGTCACCTTGTTGAT-3’ |
| RARβ | 5’-AGTCATCGGGCTACCACTATGGC-3’ | 5’-AGCACTTCTGCAGGCGGCA-3’ |
| RARγ | 5’-CATTTGAGATGCTGAGCCCCA-3’ | 5’-CATACAAAGCACGGCTTATAGACCC-3’ |
| Cyclophilin | 5’-CCCCACCGTGTTCTTCGACA-3’ | 5’-CCCAGTGCTCAGAGCACGAAA-3’ |
Figure 5.
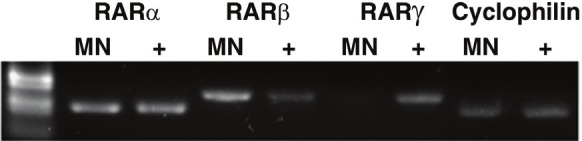
In vitro expression of the RARs. Expression of the three RAR isotypes was evaluated in the motor neuron-enriched cell culture system. Polymerase chain reaction (PCR) analysis of RNA isolated from primary motor neuron cultures after 24 hours in culture. RARα and RARβ but not RARγ were expressed. “MN” denotes RNA from primary motor neurons; “+” denotes RNA isolated from JM2 cells used as a positive control. Cyclophilin was used as a PCR positive control for each cell type.
As vitamin A deprivation has been characterized by increased reactive oxygen species (ROS) formation and oxidative stress has been implicated in ALS pathogenesis [reviewed in [44]], H2O2 was used as a model of oxidative stress/injury to determine the effect of stimulating the RARs on motor neuron cell survival. After 24 hours in culture, cells were treated with increasing concentrations of H2O2 to establish a dose-response curve (data not shown), and the concentration used for subsequent experiments (400 μM H2O2) resulted in approximately 50% cell death after 24 hours.
Eliminating RA from the media reduced cell viability at the 24 hour time point and thereafter (Figure 6A) as assessed by live cell imaging/propidium iodide staining. The presence of RA increased motor neuron survival in the presence of H2O2 as significant protection was observed as early as 6 hours post-treatment (Figure 6B).
Figure 6.
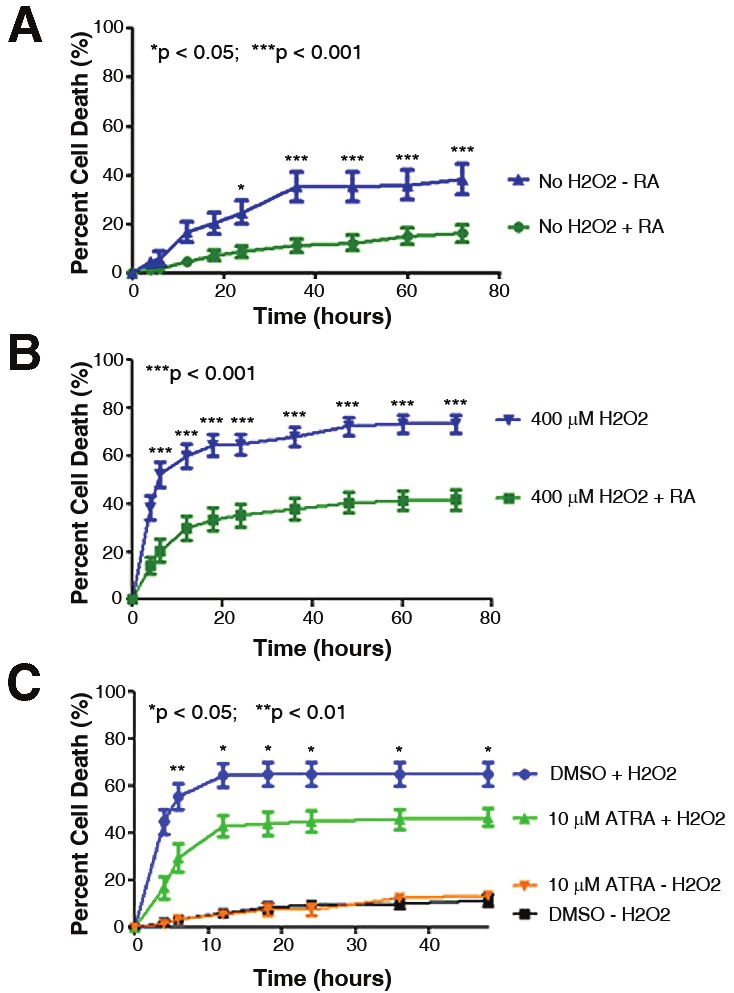
The presence of RA modulates cell death. The percentage of motor neuron cell death was evaluated using live cell imaging and propidium iodide staining over time. A: The absence of RA in the B27 media supplement (blue curve) resulted in significantly increased cell death in the absence of any insult. B: When treated with H2O2 to model oxidative stress/injury, the absence of RA in the B27 media supplement (blue curve) resulted in significantly increased percentages of cell death beginning at the 6.0 hour time point. C: Motor neuron-enriched cultures were pre-treated with ATRA for 1.0 hour prior to addition of H2O2 to induce cell death. In the presence of H2O2, ATRA significantly decreased the percentage of cell death as compared to the control treatment (DMSO + H2O2). Control treatments without H2O2 (ATRA or DMSO alone) were included for comparison (orange and black curves). Error bars represent the mean percentage of cell death ± standard error of the mean from at least two separate experiments. *p < 0.05; **p < 0.01; ***p < 0.001.
Stimulating RAR-mediated signaling delays oxidative-induced cell death
The ability of ATRA, a pan-RAR agonist, to protect motor neurons from H2O2-induced cell death was evaluated. When cells were pretreated with 10 μM ATRA, the percentage of cell death was significantly decreased as compared to control-treated cells (Figure 6C). The localization of RARα and RARβ during ATRA treatment was also assessed, although no difference in RARα or RARβ protein localization was observed in the presence of ATRA (data not shown).
Specific agonists and antagonists of RARβ were then used to determine if modulating this specific nuclear receptor could recapitulate the protective effects observed with ATRA. When pretreated for 1.0 hour with adapalene (RARβ agonist) prior to the addition of H2O2, the percentage of motor neuron cell death was significantly decreased as compared to the control treatment (Figure 7A). However, when motor neuron-enriched cultures were pre-treated for 1.0 hour with a RARβ-selective antagonist (LE-135), the percentage of cell death was significantly increased over control treatment (Figure 7B).
Figure 7.
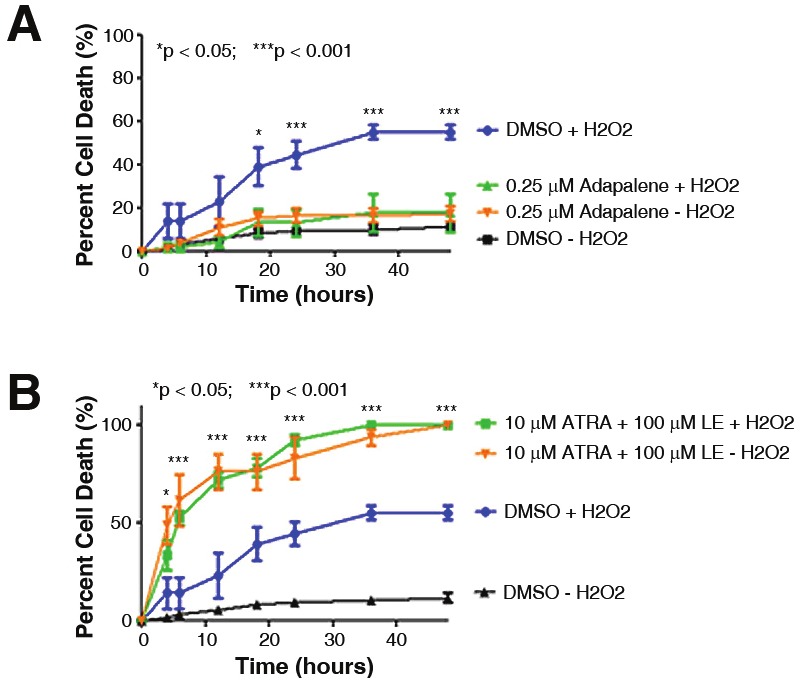
RARβ activity regulates H2O2 induced cell death. Motor neuron cultures were pre-treated for 1.0 hour with agents that specifically target RARβ prior to the addition of toxin. A: In the presence of adapalene, a RARβ agonist, the percentage of cell death induced by H2O2 was significantly reduced when compared to the control treatment of DMSO plus H2O2 (compare blue and green curves). Controls of DMSO alone or adapalene alone measured baseline level of cell death over time (black and orange curves). B: Pre-treatment of cells with a combination of ATRA and LE-135, a selective RARβ antagonist, eliminated the protective effects of ATRA and exacerbated the toxicity in the presence or absence of H2O2 (green and orange curves). Control treatments of DMSO alone (black curve) or DMSO plus H2O2 (blue curve) are shown. Error bars represent the mean percentage of cell death ± standard error of the mean from at least two separate experiments. *p < 0.05; ***p < 0.001.
Discussion
The retinoid signaling pathway is critical for neuronal development with increasing evidence that it has equally important functions in adulthood. Our prior studies identified altered post-translational modifications of transthyretin in the CSF of ALS patients [45,46], suggesting that regulated delivery of retinol throughout the CNS may be abnormal in ALS. Although gene expression studies suggest that the RA signaling pathway and genes regulated by this pathway are altered in ALS, the role of this pathway in ALS remains unclear. The goal of the current study was to examine cytoplasmic binding proteins and nuclear receptors of the retinoid signaling pathway in lumbar spinal cord motor neurons of control and ALS patients. We also utilized a primary motor neuron-enriched cell culture system to demonstrate that RARβ-mediated signaling events modulate oxidative stress-induced cell death.
Surprisingly, we noted differences between SALS and FALS. More specifically, we observed increased nuclear immunoreactivity for both CRABP-II and RARβ in motor neurons of SALS patients. In SALS cases with increased CRABP-II in motor neuron nuclei, 85% (17 of 20) also had increased nuclear RARβ in motor neurons. The lack of RARβ nuclear immunostaining in FALS suggests differences in underlying disease mechanism between familial and sporadic forms of the disease. However, further studies are necessary to validate these initial findings.
We did not observe alterations between ALS and controls with respect to CRABP-II and nuclear receptor interaction via coimmunoprecipitation. Due to low cellular CRABP-II levels and multiple RAR binding partners, the reverse co-immunoprecipitation experiment was not successful. We suggest that downstream interactions between RA (ligand), RARβ (receptor) and nuclear response element (RARE) may explain the altered subcellular distribution we observed in motor neurons of SALS patients rather than upstream protein-protein interactions.
Although RA signaling has been shown to mediate apoptosis in a variety of cells [38,47-49], this pathway has also been shown to inhibit apoptosis in specific cell types [50-52]. We propose that redistribution of RARβ to motor neuron nuclei represents a regenerative, protective response as demonstrated previously [41]. Our inability to co-localize nuclear RARβ with TUNEL or activated caspase-3 (data not shown) suggests that RA signaling through RARβ is a non-apoptotic response. In addition, our in vitro data indicates that RARβ mediates the neuroprotective properties of RA to oxidative damage. RARβ-mediated pro-survival and regenerative responses have been reported previously in models of spinal cord and axonal injury [5,7,53,54].
The intracellular distribution of the RARs is an indicator of the transcriptional activity of RA [55,56]. This localization is believed to be determined, in part, by the presence of ligand. Previous reports suggest that over 95% of RARβ translocated to the nucleus in the presence of RA [57]. Although both ligand binding and receptor heterodimerization with RXRs are required for transcriptional activation, only heterodimerization is necessary to ensure binding to retinoic acid response elements. Based on the tissue-specific expression of the RXRs [58], we would expect RXRβ (expressed in most tissues) and possibly RXRγ (expressed mostly in muscle and brain) to heterodimerize with RARβ in the spinal cord. However we did not observe any change in the localization of RXRβ in spinal cord motor neurons between control and ALS subjects (data not shown). While the overall effect of RARβ on gene expression during ALS remains to be determined, several genes known to be modulated by RA have been implicated in motor neuron disease including various cytokines, growth factors, matrix metalloproteases (MMPs), and the amyloid precursor protein (APP) [59-65] (summarized in Figure 8).
Figure 8.
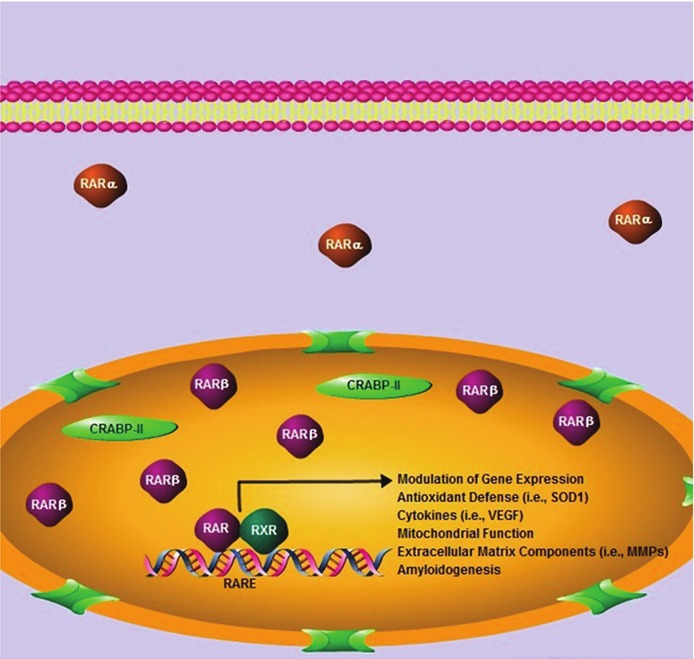
Schematic representation of retinoid signaling in SALS. Increased nuclear localization of RARβ occurs in SALS. Nuclear CRABP-II and cytoplasmic RARα localization were also observed. Signaling through RARβ in vitro was neuroprotective in the presence of oxidative stress. Processes shown to be mediated by the RARs are provided as potential downstream targets mediating this protection. Pharmacologic agents that stimulate RARβ may be promising therapeutic targets that could delay or prevent motor neuron cell death in ALS.
Our findings contradict prior studies investigating the role of retinoid signaling proteins, RARs and RXRs in animal models of chronic spinal cord degeneration. For example, Jokic and colleagues observed diffuse RARα and RARβ immunostaining in lumbar spinal cord motor neurons at pre-symptomatic stages of a rat model of ALS, which declined in end-stage disease [18]. A similar decline in RARα immunoreactivity was reported in rats fed a vitamin A-deficient diet [19]. Although we are unable to perform a longitudinal study in human tissue, we did not observe a difference between ALS and control subjects with respect to the level or subcellular localization of RARα. We believe this is due, at least in part, to species and mechanistic differences between the transgenic SOD1 animal models of ALS and the human disease.
We acknowledge that protein lysates generated from spinal cord tissue include multiple cell types that contribute to our immunoblot and coimmunoprecipitation data. However, these cells impact motor neuron survival [66] and are able to respond to RA signaling [67]. In fact, cells within the white matter were immunopositive for RARα and RARβ suggesting that they are responding to the disease process. Further studies using a combination of laser capture microdissection and quantitative real-time polymerase chain reaction (qPCR) will examine protein and mRNA levels on an individual cell basis.
Our cell culture experiments support a direct role for RARβ in motor neuron survival and neuroprotection. The fact that motor neuron viability was decreased in the absence of RA (Figure 6A) was surprising considering the multitude of growth factors and supplements included in the culture media. However, this suggests that RA modulated signaling is critically important for motor neuron survival and this is further supported by the neuroprotective properties of RA in the presence of oxidative damage. Moreover, RARβ specific agonists mediate the neuroprotective effects in this model system, indicating a particular nuclear receptor in the retinoid signaling pathway for motor neuron survival. Inhibition of RARβ exacerbated motor neuron cell death, suggesting that up- or down-regulation of specific downstream RARβ targets may prevent this response.
As noted above, ATRA and adapalene were neuroprotective even following toxic insult, suggesting that these agents could be useful therapeutically. Although the post-insult neuroprotective effects were modest and occurred at early time points, it may be that stimulating RARβ may be most effective when initiated early in the disease process, and could possibly protect motor neurons that have not yet been affected by the disease. Adapalene is an FDA-approved agent for the topical treatment of acne with few and relatively minor side effects [68]. An adapalene delivery system has been used for the treatment of cervical intraepithelial neoplasia without side effects [69] although an ALS-relevant therapy may require an analog that could cross the blood-brain barrier for optimal long-term treatment.
In conclusion, we have determined that the retinoid signaling pathway exhibits distinct alterations in SALS but not FALS patients. In particular, altered localization of two key components of the retinoid signaling pathway (CRABP-II and RARβ) were observed in spinal cord motor neurons of patients with SALS. Increased nuclear RARβ in lumbar spinal cord motor neurons may increase the ability of these cells to survive in the spinal cord microenvironment during ALS as it did not co-localize with markers of apoptosis. Furthermore, our in vitro data indicates RARβ signaling mediates neuroprotection of motor neurons under oxidative stress. Our findings support a role for retinoid signaling in the pathophysiology of ALS and suggest new potential therapeutic targets for ALS.
Acknowledgments
The authors wish to thank Dr. Johnny Huard, Dr. Lauren Drowley and Jonathan Proto for help with live cell imaging experiments. We also wish to thank Dr. Aaron Bell for assistance with primer design and Dr. Richard Bodnar for his suggestions and constructive criticism. Sources of funding for this work include NIH grant NS061867 (RB), NIH T32 EB001026 Predoctoral Training Grant (CK) and NIH T32 05 TL1 RR024155-02 (CK). R.B. is a co-founder of Knopp Biosciences, a private company developing drug therapies for ALS.
References
- 1.Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol. 2011;7:603–615. doi: 10.1038/nrneurol.2011.150. [DOI] [PubMed] [Google Scholar]
- 2.Cudkowicz M, Qureshi M, Shefner J. Measures and markers in amyotrophic lateral sclerosis. NeuroRx. 2004;1:273–283. doi: 10.1602/neurorx.1.2.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wichterle H, Lieberam I, Porter JA, Jessell TM. Directed differentiation of embryonic stem cells into motor neurons. Cell. 2002;110:385–397. doi: 10.1016/s0092-8674(02)00835-8. [DOI] [PubMed] [Google Scholar]
- 4.Mey J, McCaffery P. Retinoic acid signaling in the nervous system of adult vertebrates. Neuroscientist. 2004;10:409–421. doi: 10.1177/1073858404263520. [DOI] [PubMed] [Google Scholar]
- 5.Wong LF, Yip PK, Battaglia A, Grist J, Corcoran J, Maden M, Azzouz M, Kingsman SM, Kingsman AJ, Mazarakis ND, McMahon SB. Retinoic acid receptor beta2 promotes functional regeneration of sensory axons in the spinal cord. Nat Neurosci. 2006;9:243–250. doi: 10.1038/nn1622. [DOI] [PubMed] [Google Scholar]
- 6.Harvey BK, Shen H, Chen GJ, Yoshida Y, Wang Y. Midkine and retinoic acid reduce cerebral infarction induced by middle cerebral artery ligation in rats. Neurosci Lett. 2004;369:138–141. doi: 10.1016/j.neulet.2004.07.086. [DOI] [PubMed] [Google Scholar]
- 7.Corcoran J, So PL, Barber RD, Vincent KJ, Mazarakis ND, Mitrophanous KA, Kingsman SM, Maden M. Retinoic acid receptor beta2 and neurite outgrowth in the adult mouse spinal cord in vitro. J Cell Sci. 2002;115:3779–3786. doi: 10.1242/jcs.00046. [DOI] [PubMed] [Google Scholar]
- 8.Corcoran JP, So PL, Maden M. Disruption of the retinoid signalling pathway causes a deposition of amyloid beta in the adult rat brain. Eur J Neurosci. 2004;20:896–902. doi: 10.1111/j.1460-9568.2004.03563.x. [DOI] [PubMed] [Google Scholar]
- 9.Craft NE, Haitema TB, Garnett KM, Fitch KA, Dorey CK. Carotenoid, tocopherol, and retinol concentrations in elderly human brain. J Nutr Health Aging. 2004;8:156–162. [PubMed] [Google Scholar]
- 10.Ahlemeyer B, Bauerbach E, Plath M, Steuber M, Heers C, Tegtmeier F, Krieglstein J. Retinoic acid reduces apoptosis and oxidative stress by preservation of SOD protein level. Free Radic Biol Med. 2001;30:1067–1077. doi: 10.1016/s0891-5849(01)00495-6. [DOI] [PubMed] [Google Scholar]
- 11.Yoo HY, Chang MS, Rho HM. Induction of the rat Cu/Zn superoxide dismutase gene through the peroxisome proliferator-responsive element by arachidonic acid. Gene. 1999;234:87–91. doi: 10.1016/s0378-1119(99)00176-6. [DOI] [PubMed] [Google Scholar]
- 12.Malaspina A, Kaushik N, de Belleroche J. A 14-3-3 mRNA is up-regulated in amyotrophic lateral sclerosis spinal cord. J Neurochem. 2000;75:2511–2520. doi: 10.1046/j.1471-4159.2000.0752511.x. [DOI] [PubMed] [Google Scholar]
- 13.Malaspina A, Kaushik N, de Belleroche J. Differential expression of 14 genes in amyotrophic lateral sclerosis spinal cord detected using gridded cDNA arrays. J Neurochem. 2001;77:132–145. doi: 10.1046/j.1471-4159.2001.t01-1-00231.x. [DOI] [PubMed] [Google Scholar]
- 14.Malaspina A, de Belleroche J. Spinal cord molecular profiling provides a better understanding of amyotrophic lateral sclerosis pathogenesis. Brain Res Brain Res Rev. 2004;45:213–229. doi: 10.1016/j.brainresrev.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 15.Jiang YM, Yamamoto M, Kobayashi Y, Yoshihara T, Liang Y, Terao S, Takeuchi H, Ishigaki S, Katsuno M, Adachi H, Niwa J, Tanaka F, Doyu M, Yoshida M, Hashizume Y, Sobue G. Gene expression profile of spinal motor neurons in sporadic amyotrophic lateral sclerosis. Ann Neurol. 2005;57:236–251. doi: 10.1002/ana.20379. [DOI] [PubMed] [Google Scholar]
- 16.Dangond F, Hwang D, Camelo S, Pasinelli P, Frosch MP, Stephanopoulos G, Stephanopoulos G, Brown RH Jr, Gullans SR. Molecular signature of late-stage human ALS revealed by expression profiling of postmortem spinal cord gray matter. Physiol Genomics. 2004;16:229–239. doi: 10.1152/physiolgenomics.00087.2001. [DOI] [PubMed] [Google Scholar]
- 17.Yoshihara T, Ishigaki S, Yamamoto M, Liang Y, Niwa J, Takeuchi H, Doyu M, Sobue G. Differential expression of inflammation- and apoptosis-related genes in spinal cords of a mutant SOD1 transgenic mouse model of familial amyotrophic lateral sclerosis. J Neurochem. 2002;80:158–167. doi: 10.1046/j.0022-3042.2001.00683.x. [DOI] [PubMed] [Google Scholar]
- 18.Jokic N, Ling YY, Ward RE, Michael-Titus AT, Priestley JV, Malaspina A. Retinoid receptors in chronic degeneration of the spinal cord: observations in a rat model of amyotrophic lateral sclerosis. J Neurochem. 2007;103:1821–1833. doi: 10.1111/j.1471-4159.2007.04893.x. [DOI] [PubMed] [Google Scholar]
- 19.Corcoran J, So PL, Maden M. Absence of retinoids can induce motoneuron disease in the adult rat and a retinoid defect is present in motoneuron disease patients. J Cell Sci. 2002;115:4735–4741. doi: 10.1242/jcs.00169. [DOI] [PubMed] [Google Scholar]
- 20.Malaspina A, Turkheimer F. A review of the functional role and of the expression profile of retinoid signaling and of nuclear receptors in human spinal cord. Brain Res Bull. 2007;71:437–446. doi: 10.1016/j.brainresbull.2006.10.032. [DOI] [PubMed] [Google Scholar]
- 21.Siegenthaler G. Extra-and intracellular transport of retinoids: a reappraisal. Horm Res. 1996;45:122–127. doi: 10.1159/000184774. [DOI] [PubMed] [Google Scholar]
- 22.Napoli JL. Biosynthesis and metabolism of retinoic acid: roles of CRBP and CRABP in retinoic acid: roles of CRBP and CRABP in retinoic acid homeostasis. J Nutr. 1993;123:362–366. doi: 10.1093/jn/123.suppl_2.362. [DOI] [PubMed] [Google Scholar]
- 23.Ghyselinck NB, Bavik C, Sapin V, Mark M, Bonnier D, Hindelang C, Dierich A, Nilsson CB, Hakansson H, Sauvant P, Azais-Braesco V, Frasson M, Picaud S, Chambon P. Cellular retinol-binding protein I is essential for vitamin A homeostasis. Embo J. 1999;18:4903–4914. doi: 10.1093/emboj/18.18.4903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Boylan JF, Gudas LJ. The level of CRABP-I expression influences the amounts and types of all-trans-retinoic acid metabolites in F9 teratocarcinoma stem cells. J Biol Chem. 1992;267:21486–21491. [PubMed] [Google Scholar]
- 25.Delva L, Bastie JN, Rochette-Egly C, Kraiba R, Balitrand N, Despouy G, Chambon P, Chomienne C. Physical and functional interactions between cellular retinoic acid binding protein II and the retinoic acid-dependent nuclear complex. Mol Cell Biol. 1999;19:7158–7167. doi: 10.1128/mcb.19.10.7158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leid M, Kastner P, Chambon P. Multiplicity generates diversity in the retinoic acid signalling pathways. Trends Biochem Sci. 1992;17:427–433. doi: 10.1016/0968-0004(92)90014-z. [DOI] [PubMed] [Google Scholar]
- 27.Jonk LJ, de Jonge ME, Vervaart JM, Wissink S, Kruijer W. Isolation and developmental expression of retinoic-acid-induced genes. Dev Biol. 1994;161:604–614. doi: 10.1006/dbio.1994.1056. [DOI] [PubMed] [Google Scholar]
- 28.Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–850. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 29.Kastner P, Chambon P, Leid M. Role of nuclear retinoic acid receptors in the regulation of gene expression. In: Blomhoff R, editor. Vitamin A in Health and Disease. New York: Marcel Dekker Inc.; 1994. pp. 189–238. [Google Scholar]
- 30.Kliewer SA, Umesono K, Evans RM, Mangelsdorf DJ. The retinoid X receptors: modulators of multiple hormonal signalling pathways. In: Blomhoff R, editor. Vitamin A in Health and Disease. New York: Marcel Dekker Inc.; 1994. pp. 239–255. [Google Scholar]
- 31.Renton AE, Majounie E, Waite A, Simon-Sanchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L, Kalimo H, Paetau A, Abramzon Y, Remes AM, Kaganovich A, Scholz SW, Duckworth J, Ding J, Harmer DW, Hernandez DG, Johnson JO, Mok K, Ryten M, Trabzuni D, Guerreiro RJ, Orrell RW, Neal J, Murray A, Pearson J, Jansen IE, Sondervan D, Seelaar H, Blake D, Young K, Halliwell N, Callister JB, Toulson G, Richardson A, Gerhard A, Snowden J, Mann D, Neary D, Nalls MA, Peuralinna T, Jansson L, Isoviita V-M, Kaivorinee A-L, Holtta-Vuori M, Ikonen E, Sulkava R, Benatar M, Wuu J, Chio A, Restagno G, Borghero G, Sabatelli M, Consortium TI, Heckerman D, Rogaeva E, Zinman L, Rothstein JD, Sendtner M, Drepper C, Eichler EE, Alkan C, Abdullaey Z, Pack SD, Dutra A, Pak E, Hardy J, Singleton A, Williams NM, Heutink P, Pickering-Brown S, Morris HR, Tienari PJ, Traynor BJ. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ranganathan S, Bowser R. Alterations in G (1) to S phase cell-cycle regulators during amyotrophic lateral sclerosis. Am J Pathol. 2003;162:823–835. doi: 10.1016/S0002-9440(10)63879-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jordan-Sciutto KL, Dragich JM, Bowser R. DNA binding activity of the fetal Alz-50 clone 1 (FAC1) protein is enhanced by phosphorylation. Biochem Biophys Res Commun. 1999;260:785–789. doi: 10.1006/bbrc.1999.0986. [DOI] [PubMed] [Google Scholar]
- 34.Hanson MG Jr, Shen S, Wiemelt AP, McMorris FA, Barres BA. Cyclic AMP elevation is sufficient to promote the survival of spinal motor neurons in vitro. J Neurosci. 1998;18:7361–7371. doi: 10.1523/JNEUROSCI.18-18-07361.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ullian EM, Harris BT, Wu A, Chan JR, Barres BA. Schwann cells and astrocytes induce synapse formation by spinal motor neurons in culture. Mol Cell Neurosci. 2004;25:241–251. doi: 10.1016/j.mcn.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 36.Stommel EW, van Hoff RM, Graber DJ, Bercury KK, Langford GM, Harris BT. Tumor necrosis factor-alpha induces changes in mitochondrial cellular distribution in motor neurons. Neuroscience. 2007;146:1013–1019. doi: 10.1016/j.neuroscience.2007.02.036. [DOI] [PubMed] [Google Scholar]
- 37.Deasy BM, Lu A, Tebbets JC, Feduska JM, Schugar RC, Pollett JB, Sun B, Urish KL, Gharaibeh BM, Cao B, Rubin RT, Huard J. A role for cell sex in stem cell-mediated skeletal muscle regeneration: female cells have higher muscle regeneration efficiency. J Cell Biol. 2007;177:73–86. doi: 10.1083/jcb.200612094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kada N, Suzuki T, Aizawa K, Matsumura T, Ishibashi N, Suzuki N, Takeda N, Munemasa Y, Sawaki D, Ishikawa T, Nagai R. Acyclic retinoid inhibits neointima formation through retinoic acid receptor beta-induced apoptosis. Arterioscler Thromb Vasc Biol. 2007;27:1535–1541. doi: 10.1161/ATVBAHA.106.134114. [DOI] [PubMed] [Google Scholar]
- 39.Chang Q, Chen Z, You J, McNutt MA, Zhang T, Han Z, Zhang X, Gong E, Gu J. All-transretinoic acid induces cell growth arrest in a human medulloblastoma cell line. J Neurooncol. 2007;84:263–267. doi: 10.1007/s11060-007-9380-9. [DOI] [PubMed] [Google Scholar]
- 40.Morosetti R, Servidei T, Mirabella M, Rutella S, Mangiola A, Maira G, Mastrangelo R, Koeffler HP. The PPARgamma ligands PGJ2 and rosiglitazone show a differential ability to inhibit proliferation and to induce apoptosis and differentiation of human glioblastoma cell lines. Int J Oncol. 2004;25:493–502. [PubMed] [Google Scholar]
- 41.Mey J. New therapeutic target for CNS injury? The role of retinoic acid signaling after nerve lesions. J Neurobiol. 2006;66:757–779. doi: 10.1002/neu.20238. [DOI] [PubMed] [Google Scholar]
- 42.Camu W, Henderson CE. Purification of embryonic rat motoneurons by panning on a monoclonal antibody to the low-affinity NGF receptor. J Neurosci Methods. 1992;44:59–70. doi: 10.1016/0165-0270(92)90114-s. [DOI] [PubMed] [Google Scholar]
- 43.Camu W, Henderson CE. Rapid purification of embryonic rat motoneurons: an in vitro model for studying MND/ALS pathogenesis. J Neurol Sci. 1994;124(Suppl):73–74. doi: 10.1016/0022-510x(94)90185-6. [DOI] [PubMed] [Google Scholar]
- 44.Barber SC, Shaw PJ. Oxidative stress in ALS: key role in motor neuron injury and therapeutic target. Free Radic Biol Med. 2010;48:629–641. doi: 10.1016/j.freeradbiomed.2009.11.018. [DOI] [PubMed] [Google Scholar]
- 45.Ranganathan S, Williams E, Ganchev P, Gopalakrishnan V, Lacomis D, Urbinelli L, Newhall K, Cudkowicz ME, Brown RH Jr, Bowser R. Proteomic profiling of cerebrospinal fluid identifies biomarkers for amyotrophic lateral sclerosis. J Neurochem. 2005;95:1461–1471. doi: 10.1111/j.1471-4159.2005.03478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ryberg H, An J, Darko S, Lustgarten JL, Jaffa M, Gopalakrishnan V, Lacomis D, Cudkowicz M, Bowser R. Discovery and verification of amyotrophic lateral sclerosis biomarkers by proteomics. Muscle Nerve. 2010;42:104–111. doi: 10.1002/mus.21683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mailhos C, Howard MK, Latchman DS. A common pathway mediates retinoic acid and PMA-dependent programmed cell death (apoptosis) of neuronal cells. Brain Res. 1994;664:7–12. doi: 10.1016/0006-8993(94)90339-5. [DOI] [PubMed] [Google Scholar]
- 48.Okazawa H, Shimizu J, Kamei M, Imafuku I, Hamada H, Kanazawa I. Bcl-2 inhibits retinoic acid-induced apoptosis during the neural differentiation of embryonal stem cells. J Cell Biol. 1996;132:955–968. doi: 10.1083/jcb.132.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tong L, Werrbach-Perez K, Perez-Polo JR. Retinoic acid induces apoptosis in PC12 cells independent of neurotrophic factors. J Neurochem. 1997;68:1424–1434. doi: 10.1046/j.1471-4159.1997.68041424.x. [DOI] [PubMed] [Google Scholar]
- 50.Ahlemeyer B, Krieglstein J. Retinoic acid reduces staurosporine-induced apoptotic damage in chick embryonic neurons by suppressing reactive oxygen species production. Neurosci Lett. 1998;246:93–96. doi: 10.1016/s0304-3940(98)00242-0. [DOI] [PubMed] [Google Scholar]
- 51.Moreno-Manzano V, Ishikawa Y, Lucio-Cazana J, Kitamura M. Suppression of apoptosis by all-trans-retinoic acid. Dual intervention in the c-Jun n-terminal kinase-AP-1 pathway. J Biol Chem. 1999;274:20251–20258. doi: 10.1074/jbc.274.29.20251. [DOI] [PubMed] [Google Scholar]
- 52.Ahlemeyer B, Krieglstein J. Inhibition of glutathione depletion by retinoic acid and tocopherol protects cultured neurons from staurosporine-induced oxidative stress and apoptosis. Neurochem Int. 2000;36:1–5. doi: 10.1016/s0197-0186(99)00101-1. [DOI] [PubMed] [Google Scholar]
- 53.Corcoran J, Maden M. Nerve growth factor acts via retinoic acid synthesis to stimulate neurite outgrowth. Nat Neurosci. 1999;2:307–308. doi: 10.1038/7214. [DOI] [PubMed] [Google Scholar]
- 54.Corcoran J, Shroot B, Pizzey J, Maden M. The role of retinoic acid receptors in neurite outgrowth from different populations of embryonic mouse dorsal root ganglia. J Cell Sci. 2000;113(Pt 14):2567–2574. doi: 10.1242/jcs.113.14.2567. [DOI] [PubMed] [Google Scholar]
- 55.Fukunaka K, Saito T, Wataba K, Ashihara K, Ito E, Kudo R. Changes in expression and subcellular localization of nuclear retinoic acid receptors in human endometrial epithelium during the menstrual cycle. Mol Hum Reprod. 2001;7:437–446. doi: 10.1093/molehr/7.5.437. [DOI] [PubMed] [Google Scholar]
- 56.Braun KW, Tribley WA, Griswold MD, Kim KH. Follicle-stimulating hormone inhibits all-trans-retinoic acid-induced retinoic acid receptor alpha nuclear localization and transcriptional activation in mouse Sertoli cell lines. J Biol Chem. 2000;275:4145–4151. doi: 10.1074/jbc.275.6.4145. [DOI] [PubMed] [Google Scholar]
- 57.Maruvada P, Baumann CT, Hager GL, Yen PM. Dynamic shuttling and intranuclear mobility of nuclear hormone receptors. J Biol Chem. 2003;278:12425–12432. doi: 10.1074/jbc.M202752200. [DOI] [PubMed] [Google Scholar]
- 58.Pavan B, Dalpiaz A, Biondi C, Nieddu M, De Luca A, Prasad PD, Paganetto G, Favaloro B. An RPE cell line as a useful in vitro model for studying retinoic acid receptor beta: expression and affinity. Biosci Rep. 2008;28:327–334. doi: 10.1042/BSR20080103. [DOI] [PubMed] [Google Scholar]
- 59.Balmer JE, Blomhoff R. Gene expression regulation by retinoic acid. J Lipid Res. 2002;43:1773–1808. doi: 10.1194/jlr.r100015-jlr200. [DOI] [PubMed] [Google Scholar]
- 60.Calingasan NY, Chen J, Kiaei M, Beal MF. Beta-amyloid 42 accumulation in the lumbar spinal cord motor neurons of amyotrophic lateral sclerosis patients. Neurobiol Dis. 2005;19:340–347. doi: 10.1016/j.nbd.2005.01.012. [DOI] [PubMed] [Google Scholar]
- 61.Koistinen H, Prinjha R, Soden P, Harper A, Banner SJ, Pradat PF, Loeffler JP, Dingwall C. Elevated levels of amyloid precursor protein in muscle of patients with amyotrophic lateral sclerosis and a mouse model of the disease. Muscle Nerve. 2006;34:444–450. doi: 10.1002/mus.20612. [DOI] [PubMed] [Google Scholar]
- 62.Hamilton RL, Bowser R. Alzheimer disease pathology in amyotrophic lateral sclerosis. Acta Neuropathol. 2004;107:515–522. doi: 10.1007/s00401-004-0843-1. [DOI] [PubMed] [Google Scholar]
- 63.Beuche W, Yushchenko M, Mader M, Maliszewska M, Felgenhauer K, Weber F. Matrix metalloproteinase-9 is elevated in serum of patients with amyotrophic lateral sclerosis. Neuroreport. 2000;11:3419–3422. doi: 10.1097/00001756-200011090-00003. [DOI] [PubMed] [Google Scholar]
- 64.Demestre M, Parkin-Smith G, Petzold A, Pullen AH. The pro and the active form of matrix metalloproteinase-9 is increased in serum of patients with amyotrophic lateral sclerosis. J Neuroimmunol. 2005;159:146–154. doi: 10.1016/j.jneuroim.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 65.Lorenzl S, Albers DS, LeWitt PA, Chirichigno JW, Hilgenberg SL, Cudkowicz ME, Beal MF. Tissue inhibitors of matrix metalloproteinases are elevated in cerebrospinal fluid of neurodegenerative diseases. J Neurol Sci. 2003;207:71–76. doi: 10.1016/s0022-510x(02)00398-2. [DOI] [PubMed] [Google Scholar]
- 66.He BP, Wen W, Strong MJ. Activated microglia (BV-2) facilitation of TNF-alpha-mediated motor neuron death in vitro. J Neuroimmunol. 2002;128:31–38. doi: 10.1016/s0165-5728(02)00141-8. [DOI] [PubMed] [Google Scholar]
- 67.Schrage K, Koopmans G, Joosten EA, Mey J. Macrophages and neurons are targets of retinoic acid signaling after spinal cord contusion injury. Eur J Neurosci. 2006;23:285–295. doi: 10.1111/j.1460-9568.2005.04534.x. [DOI] [PubMed] [Google Scholar]
- 68.Czernielewski J, Michel S, Bouclier M, Baker M, Hensby JC. Adapalene biochemistry and the evolution of a new topical retinoid for treatment of acne. J Eur Acad Dermatol Venereol. 2001;15(Suppl 3):5–12. doi: 10.1046/j.0926-9959.2001.00006.x. [DOI] [PubMed] [Google Scholar]
- 69.DiSilvestro PA, DiSilvestro JM, Lernhardt W, Pfahl M, Mannel RS. Treatment of cervical intraepithelial neoplasia levels 2 and 3 with adapalene, a retinoid-related molecule. J Low Genit Tract Dis. 2001;5:33–37. doi: 10.1046/j.1526-0976.2001.51007.x. [DOI] [PubMed] [Google Scholar]