

Abstract
The most rigorous scenario for testing a candidate rheumatoid arthritis therapeutic would be to use clinically relevant biomarkers and readouts to monitor disease development in an animal model that has a mechanism of disease that reflects the human condition. Treatment should begin when the full spectrum of arthritic processes, including bone damage, is present. We have tried to take this approach to evaluate a novel EP4 receptor antagonist (ER-886046) for its anti-arthritic potential. This work aimed not only to test a potential drug, but to also demonstrate a strategy for performing a more clinically relevant evaluation of future candidate arthritis treatments. A variety of biomarkers including: radiographic evaluation, clinical scoring, histology analysis, F4/80 macrophage immunohistochemistry, luminol bioluminescent imaging and 99mTc-MDP-SPECT imaging were evaluated as disease readouts in the mouse anti-collagen antibody induced arthritis model (CAIA). CAIA mice were treated either prophylactically or therapeutically with ER-886046 and the compound’s efficacy was probed using the various biomarkers and compared to the reference drugs prednisolone and celecoxib. The various biomarkers effectively measured different aspects of arthritis pathology and consistently demonstrated the efficacy of ER-886046. The compound was found to be effective even when dosed therapeutically after bone damaging processes had initiated. The results presented herein demonstrate how biomarkers and a clinically relevant experimental design can be used to evaluate a candidate therapeutic. Utilization of clinically relevant biomarkers may provide a means for more translatable pre-clinical testing of candidate therapeutics and may provide information on their mechanism of action.
Keywords: Arthritis, EP4 receptor, PGE2, translational medicine, drug development, biomarkers
Introduction
A variety of methods can be used in humans and mice for monitoring rheumatoid arthritis (RA) and its various pathological manifestations and it is important to choose the most appropriate ones for determining drug efficacy. Detecting bone damage can best be accomplished using imaging technologies such as X-ray and 99mTc-MDP-SPECT and both of these methods are used clinically. Radiographically detectable bone damage is the benchmark for assessing RA and prevention of RA-associated bone erosions and joint destruction is the gold standard by which treatments are evaluated [1]. Optical bioluminescent imaging is another technology that can be used in preclinical studies to detect inflammation and a recent report has suggested these methods can discriminate between DMARD and non-DMARD treatments [2]. A measurement used in clinical trials for assessing arthritis is CD68 immunohistochemistry (IHC) of joint biopsies. Infiltration of CD68 positive macrophages has been reported to give an accurate indication of drug efficacy in clinical trials [3]. IHC analysis for F4/80 macrophages in mouse paws is an analogous measurement that can be performed in preclinical studies and is a surrogate for CD68 staining in humans. Histological analysis of joint damage is another technique for assessing arthritic damage in humans and mice and can provide further insight into disease pathogenesis. Combined, these techniques can provide a great deal of information about a compound’s anti-arthritic activity and its mechanism of action and by employing them in preclinical studies a candidate therapeutic may be accurately assessed for its potential clinical efficacy. However, these highly relevant techniques are not always used in preclinical drug evaluation studies and thus there is an opportunity to make preclinical drug evaluation studies more translatable and predictive.
The production of PGE2 downstream of the enzyme cyclooxygenase (COX) has been associated with inflammation as PGE2 can activate multiple inflammatory cells. Much of the inflammatory activity of PGE2 is through activation of the EP4 receptor. Activation of EP4 has been demonstrated to stimulate T cell activity [4] as well as innate immune cells such as DCs [5,6]. Furthermore, preclinical studies have suggested that activation of the EP4 receptor may play a role in several diseases including multiple sclerosis, dermatitis, colitis, and arthritis [7] and an EP4 polymorphism has been associated with an increased risk for multiple sclerosis [8] and Crohn’s disease [9,10]. Using EP4 antagonists and knockout mice it has been shown that blocking activation of the EP4 receptor can reduce arthritis development in models of arthritis such as mouse collagen-induced arthritis (CIA) [5,11], rat adjuvant arthritis [12-14], mouse G6PI-induced arthritis [5] and collagen antibody-induced arthritis (CAIA) [15]. Thus, targeting EP4 with an antagonist may be a strategy to provide medical benefit for several different indications, particularly RA. We have endeavored to produce an EP4 antagonist with drug-like properties that may be used for several indications, but have focused here on evaluating its potential as a rheumatoid arthritis (RA) treatment.
The goals for the work presented in this report were to first determine if clinically relevant biomarkers could be employed in a way that could increase the translatability of preclinical drug evaluation studies and second to test a new candidate drug molecule for its anti-arthritic potential. Despite the positive results of studies published so far showing the potential for EP4 antagonism in treating RA, they have not mimicked the clinical situation where an EP4 antagonist would be used for therapeutic treatment as treatment has been initiated before bone damaging processes are occurring. Thus, we tested the anti-arthritic activity of ER-886046 in a mouse model of arthritis using both prophylactic and therapeutic dosing regimens and unlike earlier arthritis drug evaluation studies, we have not begun treatment until bone damage has reached measurable levels. We have used several clinically relevant imaging techniques and biomarkers to show that ER-886046 can reduce the progression of arthritis even when dosed therapeutically after bone damage has begun. Thus, this report demonstrates not only the therapeutic potential of a particular molecule, but shows how clinically meaningful markers and techniques can be used preclinically in combination to evaluate a candidate therapeutic’s potential.
Methods
Radioligand EP4 binding assay
The radioligand EP4 binding assay was performed using ChemiScreen recombinant human EP4 receptor membrane preparations from Millipore (Billerica, MA) or recombinant mouse EP4 receptor membrane preparations from GenScript (Piscataway, NJ) according to the manufacturer’s instructions. Briefly, membranes prepared from Chem-1 cells overexpressing human EP4 (Millipore) or U2OS cells overexpressing mouse EP4 (GenScript) were mixed with 1.8 nM [3H]-PGE2 and 5 μM unlabeled PGE2 in the presence or absence of various concentrations of ER-886046 in binding buffer (50 mM HEPES, pH 7.4, 5 mM MgCl2, 1 mM CaCl2, 0.2% BSA) in a nonbinding 96-well plate and incubated for 1 hr for human EP4 and 2 hr for mouse EP4 at room temperature. Prior to filtration, a GF/C 96-well filter plate was coated with 0.33% polyethyleneimine for 30 min, then washed with 50 mM HEPES, pH 7.4, 0.5% BSA. Binding reactions were transferred to the filter plate and washed 3 times with wash buffer. The plate was dried and radioactivity counted.
Cell-based EP4 antagonism assay
SE302 is a clone of the HEK293 cell line containing a cAMP response element (CRE) promoter that when activated drives secretion of placental-like alkaline phosphatase (PLAP). HEK293 cells express endogenous EP4 and show induction of PLAP in response to PGE2 and EP4 agonists, but not EP1, 2 or 3 agonists [5]. Cells were maintained in DMEM/F12 (50:50) (MediaTech, Manassas, VA) supplemented with 10% FBS (Tissue Culture Biologicals, Los Alamitos, CA) plus penicillin/streptomycin. When used for assays, cells were plated in a 96-well plate at 2x104 cells/100 μl/well in serum-free assay medium (DMEM/F12 supplemented with 0.1% BSA plus penicillin/streptomycin) and incubated for 4-6 hr. Cells were then stimulated with 3 ng/ml PGE2 in the presence or absence of various concentrations of test compound overnight and PLAP activity was measured by mixing 15 μl of culture supernatants with 75 μl of Lumi-phos (Lumigen, Inc., Southfield, MI) and 60 μl of assay buffer containing 8 mM MgSO4 in 0.1 mM carbonate-bicarbonate buffer pH 11 in a new 96-well black plate followed by incubation for 2 hr at room temperature. Luminescence was read with an Envision 2102 Multilabel reader.
CAIA model
All animal studies were performed with the approval of the the Eisai Institutional Animal Care and Use Committee in an AAALAC accredited animal facility. Mice were group-housed 5 mice per cage under controlled conditions with a constant temperature (21-23°C) and humidity (30-50%), a 10/14-hr light/dark cycle and ad libitum access to water and standard pelleted food. DBA/1 mice used for the prophylactic treatment experiments were males from Harlan and mice used in the therapeutic treatment studies were males from Jackson Labs. Mice were used at 10-11 weeks of age. The anti-collagen antibody cocktail was purchased from Chondrex Inc. (Redmond, WA) and used according to the manufacturer’s instructions. Briefly, mice were injected with the cocktail of anti-collagen antibodies i.v. Three days later the mice were injected with 50 μg of LPS (Sigma, St. Louis, MO) i.p. to trigger arthritis development. On day 10 the mice were given a second LPS injection to promote further arthritis development. Dosing was begun on day 4 for the prophylactic dosing regimen and on day 11 for the therapeutic dosing regimen after the mice had been imaged by SPECT on day 10 and stratified into groups based on their 99mTc-MDP uptake. For dosing, all compounds were formulated in 0.5% methyl cellulose and administered orally via gavage once per day for ER-886046 and prednisolone and twice per day for celecoxib. In the prophylactic regimen ER-886046 was dosed at 100 mg/kg and in the therapeutic regimen it was administered at 200 mg/kg while prednisolone was used at 1 mg/kg and celecoxib at 15 mg/kg. Mice were scored for clinical arthritis by 2 observers who were blinded to the group assignments. Each paw was scored on a scale of 0-4 based on signs of swelling and inflammation.
Luminol-based bioluminescence imaging
Mice were imaged for myeloperoxidase (MPO) activity in paws using a technique described previously [16]. Briefly, mice were injected with a luminol sodium salt solution (Sigma, St. Louis, MO) i.p. 200 mg/kg and then anesthetized using 1.5% isofluorane. Twelve minutes after the luminol injection mice were imaged for bioluminescence using the IVIS Spectrum imaging system (Caliper Lifesciences, Hopkington, MA). Bioluminescence in the hind paws was quantitated using the Living Image software program.
99mTc-MDP-SPECT imaging
A preliminary pilot study was used to generate time-activity curves showing the uptake kinetics of 99mTc-Medronate (MDP) in the CAIA mice. These data showed that MDP uptake reaches a steady-state by 50-60 minutes post-injection and remains constant between 1 and 3 hours post-injection. A multi-mouse bed (Minerve, France) was used to increase imaging throughput for this study by facilitating image-acquisition on 3 mice simultaneously. MDP was administered by i.v. injection via the lateral tail-vein. Mice were injected in groups of 3 with all 3 injections taking place within a span of 2-3 minutes. Subjects were awake during tracer administration and were returned to their cages after injection to serve an uptake period. Each subject received 1.6 mCi of 99mTc-Medronate in 200 μL. For each injection, the syringe containing 99mTc-MDP was assayed before and after injection using a CRC-25 dose calibrator (Capintec, Ramsey, NJ). The time of each assay was recorded along with the time of injection to allow accurate calculation of the injected dose. Anesthesia was induced using 3% isoflourane in oxygen (2 L/min) at 50 minutes-post injection. Anesthetized subjects were transferred to the animal bed on the NanoSPECT/CT® (Bioscan, Washington, DC) in the prone position. Anesthesia was maintained using 2% isofluorane for the duration of image acquisition.
A planar X-ray scan was used to set the scan range for CT and SPECT acquisitions. The scan range for image acquisition was set to include the hind paws and ankles only. CT and SPECT acquisition began 60 minutes post-tracer injection. The SPECT scan employed 24 projection angles, 200 second exposure time per projection, yielding a 20 minute scan time. SPECT data were reconstructed using HiSPECT software (Scivis Gmbh, Germany). A quantitative calibration was performed prior to the beginning of the study using a dedicated mouse-phantom filled with a known amount of 99mTc-Pertechnetate to facilitate absolute quantification of radioactivity measured in-vivo. The percent injected dose was calculated for each paw and used as the unit of measurement.
Histology and F4/80 immunohistochemistry
At the end of the study mice were euthanized and the hind paws fixed in formalin. Paws were decalcified and sections prepared and stained by H&E by HistoTox Labs (Boulder, CO). The sectioned paws were then evaluated by a trained veterinary pathologist. The scoring methodology used was as follows: the metatarsal-P1 joints on the hind paws were scored to generate a composite severity score: if 3 or more joints are affected the severity grade was multiplied by 2; if 2 or less joints were affected the severity grade was multiplied by 1. The severity grade score for each arthritis aspect was assessed using a standardized scoring system. Infiltration of F4/80 macrophages into paws was determined by immunohistochemistry (IHC) on the sectioned paws. Briefly, the sectioned slides were treated for heat induced epitope retrieval for 20 minutes at 95°C followed by blocking endogenous peroxidase activity for 10 min at room temperature. The primary antibody (anti-F4/80, AbD Serotec, Raleigh, NC) incubation was then performed followed by the secondary antibody incubation and chromagen application. A hematoxylin counterstain and coverslipping were then performed. The sections were scored by two observers blinded to the treatment groups on a scale of 0-4 for each paw based on the intensity and extensiveness of the staining.
Results
ER-886046 is a novel EP4 antagonist with drug-like properties
The molecule evaluated in this report is an advanced analog from a program that previously delivered other EP4 antagonist compounds [5]. The structure of the molecule (ER-886046) under investigation here is reported in Figure 1. The compound was found to inhibit the mouse and human EP4 receptors in binding assays with IC50 values of 371 nM and 29.3 nM respectively (Figure 1B) with a high degree of selectivity for EP4. Compound activity was also demonstrated in a cellular PGE2 induced cAMP reporter assay where it was found to have an IC50 of 13 nM against the human receptor (Figure 1C) in good agreement with the binding IC50. Results for pharmacokinetic studies performed in mice showed that ER-886046 has good pharmacokinetic properties including an oral bioavailability of greater than 31% and a half-life of more than 3 hr (data not shown). Thus, ER-886046 is a highly selective and potent EP4 antagonist with properties suitable for in vivo usage.
Figure 1.
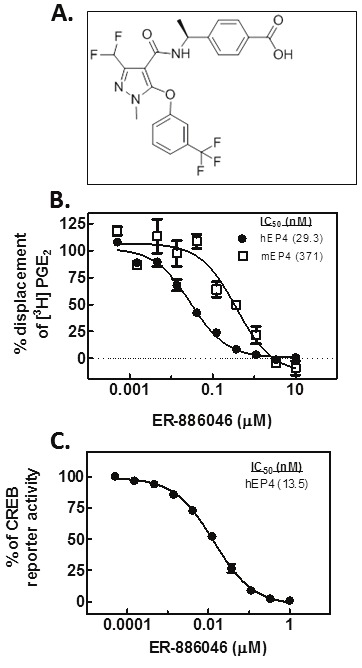
ER-886046. The chemical structure of ER-886046 is shown (A) along with the compound’s ability to block the binding of radiolabeled PGE2 to either mouse or human recombinant EP4 (B). The functional cellular activity of the compound was also evaluated and Figure 1C illustrates the ability of ER-886046 to block PGE2 activation of a reporter gene.
Evaluation of biomarkers in the CAIA model
We wished to test the ability of our compound to reduce disease in a mouse model of arthritis and chose the CAIA model for this purpose. An outline of the model’s procedures and timecourse for pathology is shown in Figure 2B. We used this model in combination with several imaging based technologies that are illustrated in Figure 2A. The timecourse for the biomarkers that could be tracked longitudinally were consistent with what pathology they were measuring. The increase in luminol bioluminescence peaked very early in the disease and progressively dissipated (Figure 3D). This likely reflects the rapid infiltration and subsequent disappearance of neutrophils. In contrast, MDP uptake was not seen until later in the disease (Figure 3E), as would be expected, as bone destruction does not begin until inflammation is well established. The clinical scoring timecourse (Figure 3A) is consistent with published reports and this readout is likely an indicator of inflammation and seems to be reflected in the luminol imaging. This is notable as clinical scoring is a typical readout for these drug evaluation experiments and might not be reflective of a blockade of bone damage which is clinically important. In general, there was good agreement in the results with the different bone damage biomarkers. MDP uptake, X-ray analysis, and histology all showed that bone damage occurs in this arthritis model and that compound treatment can block it. This seems to indicate that 99mTc-MDP SPECT may be a highly useful technique as it offers the advantage of being readily used longitudinally and is non-invasive. F4/80 IHC and histology analysis also were disease readouts that showed arthritis pathology and were responsive to ER-886046 treatment.
Figure 2.
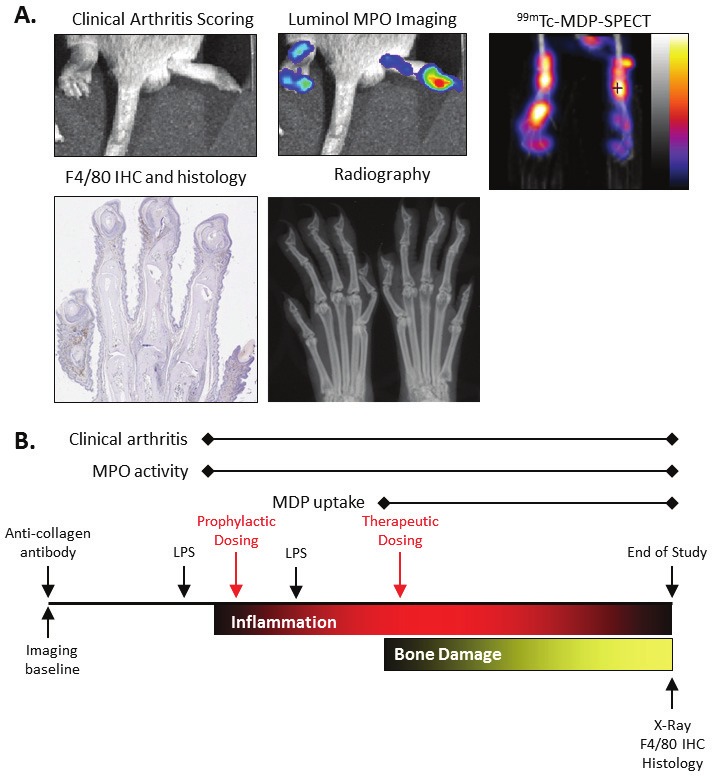
Example images of CAIA arthritis and experimental design. Various techniques were used to assess arthritis and associated damage in the CAIA model. Shown in panel A are pictures from arthritic mice illustrating the manifestation of the disease. Examples of images are shown for clinical arthritis scoring, luminol MPO bioluminescent imaging, 99mTc-MDP-SPECT imaging, F4/80 immunohistochemistry and histology, and X-ray analysis. In panel B the experimental design for the CAIA studies that were run are shown with the timing of the different procedures and biomarker measurements. Also shown is the relative timing for development of the inflammation and bone damage pathological aspects of arthritis in the CAIA model.
Figure 3.
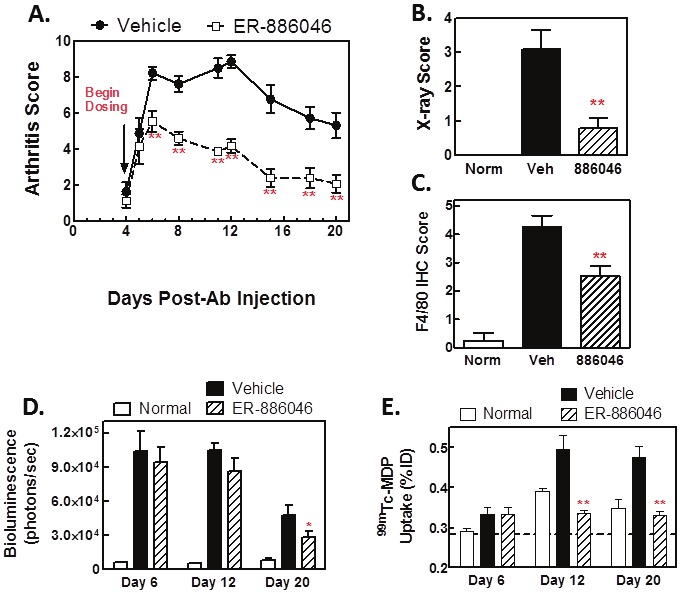
Prophylactic ER-886046 treatment decreases clinical arthritis and prevents bone damage. ER-886046 treatment was begun on day 4 at the onset of clinical arthritis and the effect on arthritic paws was followed over time by visual clinical scoring (A) and X-rays were collected on paws from mice at the termination of the study (B) and scored for bone destruction. After being X-rayed paws were fixed in formalin and then processed for IHC analysis for F4/80 (C) and the F4/80 staining was scored visually for intensity. In vivo imaging techniques were utilized at different timepoints during the study. Hind paws were imaged for bioluminescence using luminol for detection of MPO activity (D) and bone turnover in paws was detected using 99mTc-MDP-SPECT imaging (E) Shown are means ± SEM results from 2 experiments with 14 animals per group. *p<0.05 and **p<0.01 significantly different from vehicle treatment.
ER-886046 suppresses arthritis and bone damage
ER-886046 was first tested in the CAIA model with a prophylactic dosing regimen (Figure 3). For the purposes of this study we are defining prophylactic as before MDP-SPECT imaging detectable bone damage is occurring and therapeutic as treatment after an increase in MDP uptake has been detected. In these prophylactic studies compound treatment was begun on day 4, the day after arthritis development was initiated by the LPS injection.
When dosed prophylactically, the compound effectively prevented the development of clinical arthritis (Figure 3A). However, little effect was seen on MPO activity by luminol imaging in arthritic paws until late in disease (Figure 3D). Notably, ER-886046 very effectively blocked arthritic bone damage. It was found that ER-886046 blocked the increase in MDP uptake as measured by SPECT (Figure 3E) and also reduced the radiographically detectable bone damage (Figure 3B) and histological bone erosions (Table 1). The paw infiltration of inflammatory cells was found to be reduced by luminol imaging and this was confirmed by F4/80 IHC staining as ER-886046 significantly reduced F4/80 positive cells in paws at the end of the study (Figure 3B), indicating that macrophage infiltration was prevented. Histology analysis was also used for confirmation and (Table 1) revealed that ER-886046 could significantly reduce inflammation and cartilage destruction as well as bone damage. Overall, the biomarkers employed indicate that ER-886046 has significant anti-arthritic activity in mice when dosed prophylactically and is remarkably effective at preventing bone damage and other arthritis associated pathological processes.
Table 1.
Histology analysis of paws
| Therapeutic Treatment | Prophylactic Treatment | |||
|---|---|---|---|---|
| Histology scoringa | Veh | 886046 | Veh | 886046 |
| Inflammation | 2.3 ± 0.4 | 1.3 + 0.4 | 4.0 ± 0.6 | 2.3 ± 0.5 |
| Synovial Proliferation | 4.0 + 0.6 | 2.1 ± 0.5 | 4.3 ± 0.5 | 3.3 + 0.6 |
| Cartilage Damage | 2.5 ± 0.5 | 1.2 ± 0.5 | 4.2 ± 0.4 | 2.6 ± 0.6 |
| Bone Destruction | 2.5 ± 0.6 | 0.5 ± 0.3 | 4.8 ± 0.6 | 2.3 ± 0.5 |
Values are mean scores ± SEM.
Although a prophylactic (pre-bone damage) dosing regimen may be a reasonable evaluation of a compound’s anti-arthritic potential, we wished to test our compound in a therapeutic dosing regimen which intervenes late in the effector phase and is a more relevant evaluation of its clinical potential. We tested ER-886046 in a therapeutic regimen where arthritis was already established and bone damage was already underway (Figure 4) and used the RA treatment drugs celecoxib and prednisolone as comparators. In these studies dosing was begun on day 11 when clinical arthritis was nearly at its peak. Mice were imaged for 99mTc-MDP uptake by SPECT on day 10 to detect bone turnover and were stratified to treatment groups based on this measurement. Treatment was then initiated on day 11.
Figure 4.
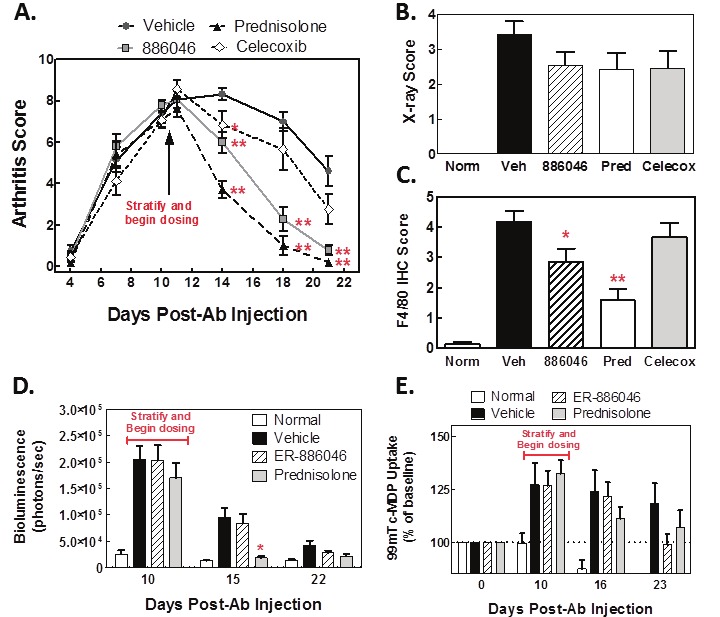
Therapeutic treatment with ER-886046 or standard of care drugs. Arthritis was induced in mice and allowed to develop to near its clinical peak as determined by visual clinical scoring (A). When SPECT detectable changes were noted (day 11), mice were stratified into groups and treatment with ER-886046, celecoxib or prednisolone was begun (A). X-rays were collected on paws from mice at the termination of the study (B) and the X-rays were scored for bone destruction. After being X-rayed paws were fixed in formalin and then processed for IHC analysis of F4/80 (C) and F4/80 staining was scored visually for intensity. Longitudinal in vivo imaging was performed throughout the study. Hind paws were imaged for bioluminescence using luminol for detection of MPO activity (D) and bone turnover in paws was detected using 99mTc-MDP-SPECT imaging (E). Shown are means ± SEM results from 2 experiments with 14 animals per group. *p<0.05 and **p<0.01 significantly different from vehicle treatment.
In the therapeutic dosing regimen both ER-886046 and prednisolone significantly reduced signs of clinical arthritis (Figure 4A) while celecoxib had a weaker effect. Likewise, prednisolone and ER-886046 effectively reduced F4/80+ macrophage infiltration (Figure 4C) while celecoxib was ineffective. This is likely to be a significant finding as the mouse F4/80 marker may be considered a surrogate for human CD68 macrophage staining and CD68 staining in synovial biopsies has been closely linked to treatment response in clinical trials [3]. However, all 3 drugs showed some activity in blocking radiographic bone damage (Figure 4B) (although without statistical significance). When we compared ER-886046 to prednisolone using the exploratory imaging techniques it was found that only prednisolone reduced MPO activity in paws as determined by luminol imaging (Figure 4D) (the limited throughput of the imaging techniques only allowed for comparison of two compounds). Both prednisolone and ER-886046 reduced (although without statistical significance) MDP uptake and the effect seemed to be more pronounced at the last timepoint, suggesting the compounds require time to be effective (Figure 4E). Histology analysis (Table 1) showed that there was a noteworthy reduction in bone destruction with ER-886046 and although ER-886046 also showed a trend toward reducing other histological readouts such as inflammation and cartilage damage, the effects on bone damage and synovial proliferation were most pronounced. These results show that ER-886046 performs well in a therapeutic setting and demonstrate the utility of employing a variety of biomarkers to effectively evaluate a candidate drug.
Discussion
Despite significant investments by the pharmaceutical industry in developing new RA therapeutics many patients are or become refractory to available treatments and this disease can still be considered an unmet medical need. We have aimed to address this need by development of an EP4 antagonist and present work herein characterizing a candidate drug. More broadly, we hoped to contribute to the arthritis drug development field by demonstrating the applicability of clinically relevant biomarkers to preclinical studies to hopefully make them more translatable.
One of the difficulties in using typical animal drug evaluation studies to predict efficacy in humans is that the clinical biomarkers and endpoints are lost in translation. In our studies we have employed several clinically used RA measurements to evaluate the activity of our candidate drug in the CAIA mouse model that is believed to replicate many aspects of the human effector phase of RA. This model generates disease via injection of an anti-collagen antibody cocktail followed by injections of LPS. The inflammatory response that follows antibody binding and LPS activation is innate immunity driven and best models the effector phase of RA [17] and likely involves processes involved in the pathogenesis of human RA such as antibody deposition and Fc receptor activation, recruitment of leukocytes to the joints and synovial proliferation, complement activation, angiogenesis, and production of a variety of inflammatory cytokines that combine to mediate joint damage [17].
In our studies a variety of readouts and biomarkers were used to monitor disease. Radiographic assessment of bone damage progression is currently the gold standard for clinically assessing RA treatment and we employed this endpoint measurement to show the effect of our compound on erosive bone damage. We also used 99mTc-MDP-SPECT imaging, which is clinically approved, to assess bone damage and evidence suggests that this technique is a sensitive tool that can be used for monitoring active arthritis in joints of RA patients [18]. By using this technique we were able to ascertain when erosive damage was beginning in our mice and could follow them longitudinally to determine the impact of drug treatment. This is a clinically relevant scenario and the use of 99mTc-MDP-SPECT imaging may be a more sensitive and responsive measurement of bone damage than X-ray, which takes a longer time for changes to manifest. In our studies ER-886046 demonstrated an ability to reduce bone damage. Given the clinical importance of reducing bone damage this is a significant finding for ER-886046. This suggests that EP4 antagonism may have important effects on bone damaging processes and further study on which cell types are affected by compound treatment in arthritic joints are warranted.
We have used other biomarkers and readouts to assess arthritis progression and to probe the mechanism of action of our compound. We not only used the classical visual scoring of clinical arthritis, but other newer techniques as well. We used luminol based bioluminescence imaging to detect MPO activity [16] and phagocyte infiltration of paws. Luminol has recently been shown to be a substrate for MPO that when metabolized leads to the production of light which can be measured in live animals using commercially available imaging systems [16]. We found that MPO activity is significantly elevated in arthritic paws early in the timecourse of CAIA and parallels the clinical signs of inflammation. However, treatment with ER-886046 gave a weak reduction in this readout. This is likely due to the fact that neutrophil infiltration is a very early event in the disease and compound treatment may require longer to affect this process. In contrast, prednisolone treatment showed a more robust ability to decrease MPO activity and this was also accompanied by a larger decrease in clinical scoring of paw inflammation. However, both compounds reduced the infiltration of F4/80 positive macrophages. This may be because the macrophages arrive later in the disease and treatment has already begun to reduce production of chemoattractants or because compound treatment is directly affecting macrophages. The reduction of these cells with compound treatment is an important observation as reduction in CD68 positive macrophages in RA joint biopsies has been associated with positive clinical responses in patients [3]. There may be effects on specific cell populations with ER-886046 treatment as there was a disconnect between MPO imaging, F4/80 IHC, and the histology inflammation scores. Furthermore, this suggests that neutrophils may not be required for bone destruction in this model.
The idea of treating RA by antagonism of EP4 is supported by a solid base of literature reports indicating that in preclinical models blocking EP4 activity can prevent arthritis development [5,11-15]. Cumulatively, the data here indicates that ER-886046 is therapeutically effective for reducing arthritis in the effector phase and suggests the mechanism of its activity. We compared our compound to the reference drugs prednisolone and celecoxib to try and validate the CAIA model’s ability to replicate the effects of the drugs in humans. Consistent with known clinical efficacy celecoxib appeared to have weaker effects than prednisolone as it did not effectively reduce arthritis scores or F4/80 macrophage infiltration. However, celecoxib did show a trend toward reducing radiographic bone damage, suggesting that possibly not all aspects of the human disease are translatable to the CAIA model. It is also important to note the significant reduction in efficacy when ER-886046 is dosed therapeutically instead of prophylactically. Even though there was a decrease in efficacy, it is important to evaluate candidate compounds by therapeutically dosing them as this is the more clinically relevant scenario. The anti-arthritic activity of ER-886046 shown here combined with its beneficial effects in multiple cell types relevant to RA pathogenesis suggests that it has the potential to be therapeutically beneficial.
The results presented here demonstrate how clinically relevant biomarkers not typically utilized in preclinical studies can be used to characterize an EP4 antagonist with drug-like properties that effectively blocks arthritis disease progression even when dosed therapeutically. We have employed clinically relevant methods and biomarkers to evaluate our compound and offer this as an example of a hopefully translatable program for assessing candidate therapeutics’ chances for successful RA treatment in humans. Although animal studies do not fully replicate human disease, we anticipate that employment of the biomarkers presented here will improve the predictive power of animal arthritis drug evaluation studies.
Acknowledgments
The authors would like to thank Sally Ishizaka for her help in preparing the manuscript and also Eva Skokanova, Wei Li, Kazuma Takase, Hiroe Hihara, Kazuyuki Fukushima, Toru Arai, Marc Lamphier, Dayong Qiu, Peter Bertinato, Christopher DesJardins, Edgar Schuck, Phil Saxton, Catherine Reardon, Lynn Chellarajan, Meng Ye, George Lai, Eric Williams, Nadia Farah, Robert Pelletier, and Donna Young for their contributions to experiments and discussion.
References
- 1.Combe B. Progression in early rheumatoid arthritis. Best Pract Res Clin Rheumatol. 2009;23:59–69. doi: 10.1016/j.berh.2008.11.006. [DOI] [PubMed] [Google Scholar]
- 2.Peterson JD, Labranche TP, Vasquez KO, Kossodo S, Melton M, Rader R, Listello JT, Abrams MA, Misko TP. Optical tomographic imaging discriminates between disease-modifying anti-rheumatic drug (DMARD) and non-DMARD efficacy in collagen antibody-induced arthritis. Arthritis Res Ther. 2010;12:R105. doi: 10.1186/ar3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haringman JJ, Gerlag DM, Zwinderman AH, Smeets TJ, Kraan MC, Baeten D, McInnes IB, Bresnihan B, Tak PP. Synovial tissue macrophages: a sensitive biomarker for response to treatment in patients with rheumatoid arthritis. Ann Rheum Dis. 2005;64:834–838. doi: 10.1136/ard.2004.029751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yao C, Sakata D, Esaki Y, Li Y, Matsuoka T, Kuroiwa K, Sugimoto Y, Narumiya S. Prostaglandin E2-EP4 signaling promotes immune inflammation through Th1 cell differentiation and Th17 cell expansion. Nat Med. 2009;15:633–640. doi: 10.1038/nm.1968. [DOI] [PubMed] [Google Scholar]
- 5.Chen Q, Muramoto K, Masaaki N, Ding Y, Yang H, Mackey M, Li W, Inoue Y, Ackermann K, Shirota H, Matsumoto I, Spyvee M, Schiller S, Sumida T, Gusovsky F, Lamphier M. A novel antagonist of the prostaglandin E(2) EP(4) receptor inhibits Th1 differentiation and Th17 expansion and is orally active in arthritis models. Br J Pharmacol. 2010;160:292–310. doi: 10.1111/j.1476-5381.2010.00647.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kabashima K, Sakata D, Nagamachi M, Miyachi Y, Inaba K, Narumiya S. Prostaglandin E2-EP4 signaling initiates skin immune responses by promoting migration and maturation of Langerhans cells. Nat Med. 2003;9:744–749. doi: 10.1038/nm872. [DOI] [PubMed] [Google Scholar]
- 7.Sakata D, Yao C, Narumiya S. Prostaglandin E2, an immunoactivator. J Pharmacol Sci. 2010;112:1–5. doi: 10.1254/jphs.09r03cp. [DOI] [PubMed] [Google Scholar]
- 8.De Jager PL, Jia X, Wang J, de Bakker PI, Ottoboni L, Aggarwal NT, Piccio L, Raychaudhuri S, Tran D, Aubin C, Briskin R, Romano S, Baranzini SE, McCauley JL, Pericak-Vance MA, Haines JL, Gibson RA, Naeglin Y, Uitdehaag B, Matthews PM, Kappos L, Polman C, McArdle WL, Strachan DP, Evans D, Cross AH, Daly MJ, Compston A, Sawcer SJ, Weiner HL, Hauser SL, Hafler DA, Oksenberg JR. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet. 2009;41:776–782. doi: 10.1038/ng.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Libioulle C, Louis E, Hansoul S, Sandor C, Farnir F, Franchimont D, Vermeire S, Dewit O, de Vos M, Dixon A, Demarche B, Gut I, Heath S, Foglio M, Liang L, Laukens D, Mni M, Zelenika D, Van Gossum A, Rutgeerts P, Belaiche J, Lathrop M, Georges M. Novel Crohn disease locus identified by genome-wide association maps to a gene desert on 5p13.1 and modulates expression of PTGER4. PLoS Genet. 2007;3:e58. doi: 10.1371/journal.pgen.0030058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Perdigones N, Martin E, Robledo G, Lamas JR, Taxonera C, Diaz-Rubio M, de la Concha EG, Lopez-Nevot MA, Garcia A, Gomez-Garcia M, Fernandez-Gutierrez B, Martin J, Urcelay E. Study of chromosomal region 5p13.1 in Crohn’s disease, ulcerative colitis, and rheumatoid arthritis. Hum Immunol. 2010;71:826–828. doi: 10.1016/j.humimm.2010.05.010. [DOI] [PubMed] [Google Scholar]
- 11.Honda T, Segi-Nishida E, Miyachi Y, Narumiya S. Prostacyclin-IP signaling and prostaglandin E2-EP2/EP4 signaling both mediate joint inflammation in mouse collagen-induced arthritis. J Exp Med. 2006;203:325–335. doi: 10.1084/jem.20051310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colucci J, Boyd M, Berthelette C, Chiasson JF, Wang Z, Ducharme Y, Friesen R, Wrona M, Levesque JF, Denis D, Mathieu MC, Stocco R, Therien AG, Clarke P, Rowland S, Xu D, Han Y. Discovery of 4-[1-[([1-[4-(trifluoromethyl)benzyl] -1H-indol-7-yl] carbonyl)amino] cyclopropyl] benzoic acid (MF-766), a highly potent and selective EP4 antagonist for treating inflammatory pain. Bioorg Med Chem Lett. 2010;20:3760–3763. doi: 10.1016/j.bmcl.2010.04.065. [DOI] [PubMed] [Google Scholar]
- 13.Okumura T, Murata Y, Taniguchi K, Murase A, Nii A. Effects of the selective EP4 antagonist, CJ-023,423 on chronic inflammation and bone destruction in rat adjuvant-induced arthritis. J Pharm Pharmacol. 2008;60:723–730. doi: 10.1211/jpp.60.6.0007. [DOI] [PubMed] [Google Scholar]
- 14.Clark P, Rowland SE, Denis D, Mathieu MC, Stocco R, Poirier H, Burch J, Han Y, Audoly L, Therien AG, Xu D. MF498 [N-{[4-(5,9-Diethoxy-6-oxo-6,8-dihydro-7H-pyrrolo[3,4-g] quinolin-7-yl)-3-m ethylbenzyl] sulfonyl}-2-(2-methoxyphenyl)acetamide] , a selective E prostanoid receptor 4 antagonist, relieves joint inflammation and pain in rodent models of rheumatoid and osteoarthritis. J Pharmacol Exp Ther. 2008;325:425–434. doi: 10.1124/jpet.107.134510. [DOI] [PubMed] [Google Scholar]
- 15.McCoy JM, Wicks JR, Audoly LP. The role of prostaglandin E2 receptors in the pathogenesis of rheumatoid arthritis. J Clin Invest. 2002;110:651–658. doi: 10.1172/JCI15528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gross S, Gammon ST, Moss BL, Rauch D, Harding J, Heinecke JW, Ratner L, Piwnica-Worms D. Bioluminescence imaging of myeloperoxidase activity in vivo. Nat Med. 2009;15:455–461. doi: 10.1038/nm.1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nandakumar KS, Holmdahl R. Antibody-induced arthritis: disease mechanisms and genes involved at the effector phase of arthritis. Arthritis Res Ther. 2006;8:223. doi: 10.1186/ar2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sahin M, Bernay I, Basoglu T, Canturk F. Comparison of Tc-99m MDP, Tc-99m HSA and Tc-99m HIG uptake in rheumatoid arthritis and its variants. Ann Nucl Med. 1999;13:389–395. doi: 10.1007/BF03164932. [DOI] [PubMed] [Google Scholar]