

Abstract
Insulin resistance is a major effect of burn injury and insulin receptor substrate–1 (IRS-1) plays an important role in signal transduction. Here, we explored the integrity of IRS-1 in muscle after burn injury. A murine model of severe burn injury was used to explore IRS-1 integrity/degradation in muscle and to map Ser/Thr phosphorylations which represent the trigger sites for degradation. The findings are: three C-terminal IRS-1 cleavage fragments were confirmed with tandem mass spectrometry; MWs 95, 44 and 42 kD. In sham burn animals the level of intact IRS-1 was 51.9 ng/g, whereas, total IRS-1 which includes degradation fragments and post-translationally modified protein was 196.7 ng/g. After burn, intact and total IRS-1 were reduced to 47.8 (92.1 % sham, p<0.05) and 86.9 ng/g (44.2 % sham, p<0.005). In contrast, ubiquitinated IRS-1 increased from 24.5 to 28.4 ng/g (15.9% increment, p< 0.05) in the burned mice. In cytosol, membrane and nuclear fractions, total IRS-1 was reduced by 89.8% (p<0.005), 25.8% (p<0.05) and 87.3% (p<0.005). To further evaluate the IRS-1 degradation pathway, SOCS-3 mRNA levels after burn injury were found to be increased by 35% (p<0.05), 110% (p<0.05) and 140% (p<0.005). However, phosphorylation of Ser473 and Thr308 of Akt1 were reduced to 26.2% (p<0.05) and 49.8% (p<0.005). We conclude: burn injury is associated with IRS-1 degradation via SOCS-3 and ubiquitin-mediated pathways and reduced subcellular levels of IRS-1, serve as molecular basis for burn injury induced alteration in insulin function.
Keywords: Insulin receptor substrate-1 degradation, tandem mass spectrometry, burn injury
Introduction
Insulin resistance and muscle wasting during the prolonged and intense inflammatory state following severe burn injury are associated with increased risks for infection, sepsis, and mortality. Clinical data indicate that a switch in resting metabolic rate occurs at burn injuries above 40% TBSA. Based on studies of 189 pediatric burn patients [1-4], it was demonstrated that the size of burn area determines the inflammatory and hypermetabolic response to injury; with the increase in serum glucose and insulin levels to 144 mg/dl and 32 ng/ml on day 8 after injury. Altered gene expression [5,6] and the release of various inflammatory cytokines [7,8] have significant impact on protein and substrate metabolism after burn injury [9-11]. Reduced insulin sensitivity and glucose intolerance, in spite of higher insulin levels, contribute to hyperglycemia, even in non-diabetic patients [12-18]. The signal for insulin effects on target cells starts with the binding of insulin to its receptor, leading to activation of insulin receptor tyrosine kinase, which phosphorylates insulin receptor substrate (IRS) proteins. Subsequently, these proteins function as docking platforms, for the two main signaling pathways, the phosphatidyl inositol 3-kinase (PI3k)-Akt/protein kinase (PKB) pathway and the Ras-mitogen-activated protein kinase (MAPK) pathway which are responsible for downstream regulation of insulin function [19]. Recent publications suggest that insulin resistance may be in part due to a phosphorylation-based negative-feedback in the two pathways: 1) Phosphorylated Ser/Thr residues of the IRS-1 PTB domain may simply uncouple IRS-1 from the insulin receptor b unit, terminating downstream signal transduction without changing IRS-1 protein integrity [20,21]. 2) Phosphorylated Ser/Thr sites may become proteolytic targets for CUL7 E3 ubiquitin ligase in a manner that depends on the mammalian target of rapamycin (mTOR) and p70 S6 kinase activities. Furthermore, Ser/Thr sites in the proximal C-terminal region of IRS-1 may provide multiple cleavage sites. Other studies have shown a role for SOCS-1 and SOCS-3 with the elongin BC ubiquitin ligase complex in IRS protein degradation [22,23]. Determination of the phosphorylation pattern of IRS-1 is essential for understanding the metabolic basis of many disease processes [24,25]. There are 50 to 70 potential phosphorylation sites in IRS-1, some of which up- and down-regulate insulin signaling [26-28]. To date, approximately 29 Ser/Thr phosphorylation sites in IRS1 have been reported from various in vivo and in vitro experiments [29-42]. Mapping all the phosphorylation sites of IRS-1 under different disease conditions remains a formidable if not impossible challenge. To the best of our knowledge, no systematic IRS-1 phosphorylation data associated with burn injury have been reported, neither the causative factors nor the cellular mechanisms for muscle wasting and insulin resistance during the severe inflammatory state induced by severe burn injury have been elucidated.
In this report, a murine model of burn injury was used for exploring IRS-1 protein integrity and the mechanism(s) for its degradation in skeletal muscle, which elucidate part of the mechanisms related to burn induced insulin resistance.
Materials and methods
Chemicals and assay kits
Methanol, acetonitrile (LC-MS Chromasolv, ACN), formic acid (FA), glacial acetic acid, LC-MS grade water, dithiotheretol (DTT) and [Glu1]-Fibrinopeptide B were purchased from Sigma Chemical Co. (St. Louis, MO, USA). SDS-PAGE ready gels (7.5%, #161-1100), Laemmli sample buffer (#161-0737) and Coomassie brilliant blue R250 (#161-0436) were obtained from BIO-RAD (Hercules, CA, USA). Trypsin digestion kits (#PP0100) were obtained from Sigma (St. Louis, MO, USA). IRS-1 (total) ELISA kit (KHO0511), Akt pThr308 ELISA kit (KHO0201) and pSer473 kit (KHO0111) were purchased from Invitrogen (Carlsbad, CA, USA). Anti-ubiquitin (rabbit polyclonal, Ab19247) and anti-C-terminal IRS-1 (rabbit polyclonal, Ab653) antibodies were purchased from Abcam (Cambridge, MA, USA). Anti-N-terminal IRS-1 (rabbit polyclonal, SC560) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). For phosphorylation tandem mass spectrometric analysis and cellular total IRS-1 measurements, cell lysis buffer (cat# 9803) was purchased from Cell Signaling (Beverly, MA, USA). Phenylmethanesulfonyl fluoride (PMSF, cat# P-7626) was purchased from Sigma. TRI reagent solution (cat# AM9738) was a product of Applied Biosystem (Framingham, MA, USA) for skeletal muscle RNA isolation. 1-bromo-3-chloropropane (cat# BP 151) was purchased from Molecular Research Center, Inc. (Cincinnati, OH, USA). Qproteome cell compartment kit used to obtain skeletal muscle subcellular fractions (Cat#: 37502, Qiagen) was obtained from Qiagen (Valencia, CA, USA). Protein G agarose (cat# 16-266) was obtained from Millipore (Billerica, MA, USA).
Mouse model of burn injury
The protocol for the studies was approved by the Massachusetts General Hospital and Shriners Hospital for Children Animal Care Committees. Our animal care facility is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care. Male CD-1 mice (Charles River Breeding Laboratories, Wilmington, MA, USA) weighing about 22-25 g were used. Sixteen animals were subjected to full thickness third degree burn injury under anesthesia (ketamine / xylazine). After clipping of the back hair, animals were placed in a template designed to expose 25% of their dorsum, and immersion of the exposed area in a water bath at 90°C for 9 second. After burn (n=15), the animals were immediately resuscitated with saline (2ml / mouse) by intraperitoneal injection. Buprenorphin (0.1 mg/kg, I.P.) was administered every 6-12 hours after injury. The sham group (n=8, matched for weight) was treated in the same manner as the burn group with the exception that they were exposed to warm water (36°C). All studies were performed with skeletal muscle excised from the animals on day 7 after either burn injury or sham burn treatment with the animals under deep anesthesia. The model is identical with the ones used previously in our laboratory which demonstrated insulin resistance in vivo [43].
Skeletal muscle preparation
Due to the limited sensitivity of sequence analysis using nanoLC-Q-TOF tandem mass spectrometry for very low abundant IRS-1 in muscle, whole body skeletal muscle remote from the burned area (~2.5-3.0 g/animal) was evaluated; fast and slow twitch muscles could not be studied independently. The muscle studied was a mixture of: trapezius, gluteus superficialis, rectus femorus, shoulder deltoid, serratus anterior, vastus medialis, semitendinosus, biceps femoris, adductor longus, gracilis and tricepts brachi, soleus and rectus abdominus. The muscle was harvested by careful dissection which excluded skin, bone and other non muscular tissues. Immediately after dissection, tissue samples were placed in liquid nitrogen.
Frozen tissue samples were immediately cut into pieces smaller than ~50 mg, and homogenized by 5 strokes over 30 seconds (full speed, model CTH-115, Cole Parmer). For tandem mass spectrometry, tissue samples were processed in freshly prepared cell signaling lysate buffer (5 ml per gram tissue) containing fresh PMSF (1mM) on ice. Homogenates were sonicated briefly in ice cold water, and centrifuged at 14,000 g for 10 minutes at 4°C. Supernatants were collected and stored at -80°C for further study.
Tissue supernatant for MS/MS analysis
The frozen supernatants were thawed and pretreated with protein G agarose (100 ml, 50% slurry) for 2 hrs at 4°C with end-over-end shaking. Protein G agarose beads bound with non-specific proteins were removed by centrifugation at 14,000 g for 30 seconds. Anti-IRS-1 antibody (Ab653, 2 ml) was added and the mixture was shaken at 4°C overnight. The immunocomplex was captured by adding 100 ml of washed protein G agarose beads at 4°C followed by shaking for 2 hrs. The beads were washed six times with ice-cold PBS and resuspended in 50 μl of 2X Laemmli sample buffer containing 5% (v/v) 2-mercaptoethanol followed by heating for 5 minutes at 95°C. The supernatants (30 μl) were applied to SDS-PAGE Ready gels (7.5%) and the gels were run at room temperature for 45 min. The gels were stained with Coomassie brilliant blue R-250 and the IRS-1 bands were excised as 1 X 1 mm pierces. In-gel trypsin digestion was performed according to the manufacture’s protocol (Sigma, #PP0100, 0.2 mg of trypsin in 70 ml of reaction buffer) at 37°C overnight. Formic acid (5 ml) was added to quench the digestion and stabilize the phosphorylation. Ten ml of each sample was injected via the full loop mode of the autoinjector into the mass spectrometer.
ELISA for determination of Intact, C-terminal and ubiquitinated IRS-1
Homogenate aliquots (100 μl) from muscle tissue harvested from 15 burned and 8 sham treated muscle tissues were diluted with Standard Diluent Buffer (400 μl) provided with the ELISA kits. IRS-1 standard calibration curves were prepared according to the manufacturer’s protocol. Diluted tissue lysates (100 μl) were pipetted into the 96-well plates. The ELISA plates were gently shaken to capture IRS-1 at 4°C overnight. Rabbit anti-ubiquitin antibody, rabbit anti-N-terminal IRS-1 antibody and rabbit anti-C-terminal IRS-1 antibody were diluted 500 fold using horseradish peroxidase (HR) diluents at room temperature. After six washings (0.4 ml Wash Buffer, gentle shaking for 30 seconds) at room temperature, rabbit anti-IRS-1 antibody provided with the IRS-1 ELISA kit together with the three detection antibodies (100 μl) were pipetted into each well. Detection was performed after gentle shaking with the detection antibodies at room temperature for 1 hour. After removal of excess detection antibodies, anti-rabbit IgG-HRP antibody was added and detected according to the manufacturer’s directions. The intact, C-terminal and ubiquitinated IRS-1 were identified separately with anti-N-terminal, anti C-terminal and anti-ubiquitin antibodies.
Quantification of IRS-1 C-terminal distribution in subcellular fractions
Mouse skeletal muscle tissue (20 mg) was homogenized with a TissueRuptor (Qiagen, Valencia, CA, USA) using a Qproteome cell compartment kit (Cat#: 37502, Qiagen) according to the manufacture’s protocol. IRS-1 C-terminal fragments in cytosolic, membrane, nuclear and cytoskeletal fractions were measured using an IRS-1 (total) ELISA kit. Aliquots (100 μl) of extracted subcellular fractions were directly used without acetone treatment.
Real-time RT-PCR analysis
Frozen tissue samples (~50 mg) were cut into small pieces in RNase-free tubes, and homogenized with 5 strokes (5 seconds each) with an Omni TH-tissue homogenizer in sterilized eppendorf tubes (2 ml) containing TRI reagent solution (cat# AM9738, 1 ml), Applied Biosystem, Framingham, MA, USA). RNA extraction was performed by the Ambion method. The average 260 nm/280 nm ratio was found to be 1.95, and RNA yield was ~1.2 μg/mg skeletal muscle. The quality of the isolated RNA was assessed with agarose gel electrophoresis. Samples were analyzed in triplicate using the comparative threshold cycle SYBR green method. GAPDH (F: 5’-AGG TCG GTG TGA ACG GAT TTG-3’, R: 5’-TGTAGACCATGTAGTTGAGGTCA-3’) was used as a mouse muscle house-keeping gene. PCR primers obtained from the MGH primer bank for mouse SOCS3 cDNA were used as F: 5’-CGA TGG CTT CTC AGA CGT G-3’, R: 5’-CAGCCCGCTTGTTGATGTTG-3’; F: 5’-ATG AAC GAC GTA GCC ATT GTG-3’, R: 5’-TTG TAG CCA ATA AAG GTG CCA T-3’: F: 5'-ATG GTC ACC CAC AGC AAG TTT-3’, R: 5'-TCC AGT AGA ATC CGC TCT CCT-3. Expression levels of target genes were corrected by normalization to the expression level of GAPDH, and relative expression levels were calculated: DCt=Ct(target gene)-Ct(GAPDH), DDCt=DCt(burn)- DCt(sham), targeted gene normalized to the endogenous reference is given by: 2-DDCt [44]. Using these primers, GAPDH mRNA levels were not significantly different between burned and sham treated mice 16.12 ±0.93 vs 15.62 ±2.98 (p=0.162, t-test).
Measurements of Akt1 pThr308 and pSer473
Muscle tissue lysates obtained with the Cell Signaling buffer as described above were further used for measurement of Akt pThr308 and pSer473 [18]. The phosphorylation levels were normalized to tissue weight. According to the manufacturer, one unit of phosphorylation standard is defined as the amount of Akt pThr308 or pSer473 derived from 500 pg or 100 pg of Akt protein, which is phosphorylated by MAPKAP 2 and PDK 1.
CapLC-MS/MS analysis of phosphorylated IRS-1 Se/Thr sites
The phosphorylated IRS-1 Ser/Thr sites were identified using a Waters CapLC-Q-TOFmicro system (Waters Corporation, Milford, MA, USA). An analytical column (75 mm I.D. X 150 mm, Vydac C18, 5 mm, 300 A, LC Packings, Dionex Company, San Francisco, CA) was used to connect the stream select module of the CapLC and the voltage supply adapter for ESI. After washing with mobile phase C (auxiliary pump, 0.1% formic acid in water/acetonitrile, 2% acetonitrile) for 2 minutes, the trapped peptides were back washed from the precolumn onto the analytical column using the 10-position stream switching valve. A linear gradient was used to elute the peptide mixture from mobile phase A (0.1% FA in water/ACN, 2% ACN) to mobile phase B (0.1% FA in ACN). The gradient was segmented as follow: isocratic elution with 2% solvent B for 3 min, 2-80 % solvent B from 3 to 45 min and 80 to 2% B from 45-50 min. The gradient flow rate was adjusted to ~160 nl/min. The electrospray voltage was set to ~ 3000 V to obtain an even ESI plume. Sample cone and extraction cone voltages were set at 45 and 3 V, respectively. The instrument was operated in positive ion mode with the electrospray source maintained at 90°C. The instrument was calibrated with synthetic human [Glu1]-Fibrinopeptide B (100 fmol/ml in acetonitrile/water = 10: 90, 0.1% formic acid, v/v) at an infusion rate of 1 ml/min in TOF MS/MS mode. The collision energy was set at 35 V. Instrument resolution for the [Glu1]-Fibrinopeptide B parent ion, m/z = 785.84, was found to be ~5000 FWHM. Neutral loss for p-Ser and p-Thr parent ion discovery was set at 97.977 ± 0.03 Da with CE 35 and 5 V, respectively. All data were acquired and processed using MassLynx 4.0 software.
Evaluation of phosphorylation sites
Sites of Ser/Thr phosphorylation were further verified by a three-step procedure: (1) Phosphorylated parent ion discoveries by centroid MS survey; the peptide mass tolerance was set at 0.2 Da for neutral loss; (2) Analysis of candidate parent ions with PepSeq of MassLynx V4.0 software to verify rat IRS-1 tryptic peptide sequences. Mercaptocysteine was searched as a fixed modification, whereas oxidation of methionine and phosphorylations were searched as variable modifications. (3) Confirmation of phosphorylated sites with the mass difference of 80.00 ± 0.15 Da (HPO4 moiety) between phosphorylated and non-phosphorylated y or b ions at S/N > 2.
Statistical analysis
The results were analyzed statistically by unpaired Student’s t- tests. All results were expressed as mean ± sem and differences with p-values of less than 0.05 were considered to be statistically significant.
Results
IRS-1 phosphorylations and integrity in muscle after thermal injury
The molecular weight pattern of IRS-1 in a representative 7.5% SDS-PAGE gel stained with Coomassie brilliant blue R250 is shown in Figure 1. Due to the similar molecular weights of rat and mouse IRS-1 and the availability at the time of our studies, we used rat IRS-1 as a standard. The position of intact recombinant rat IRS-1 (2 μg) with apparent MW 165 kDa on the gel is indicated in lane 1. No IRS-1 bands were visualized from muscle of sham treated animals (1 gram of muscle, lane 2) or muscle from burned animals (1 gram of muscle, lane 3). In contrast, Lane 4 shows several faint bands that were excised as indicated by arrows. Three C-terminal fragments with apparent SDS-PAGE at MWs of 95, 44 and 42 kDa were confirmed from the mouse skeletal muscle after burn injury as shown in Table 1.
Figure 1.
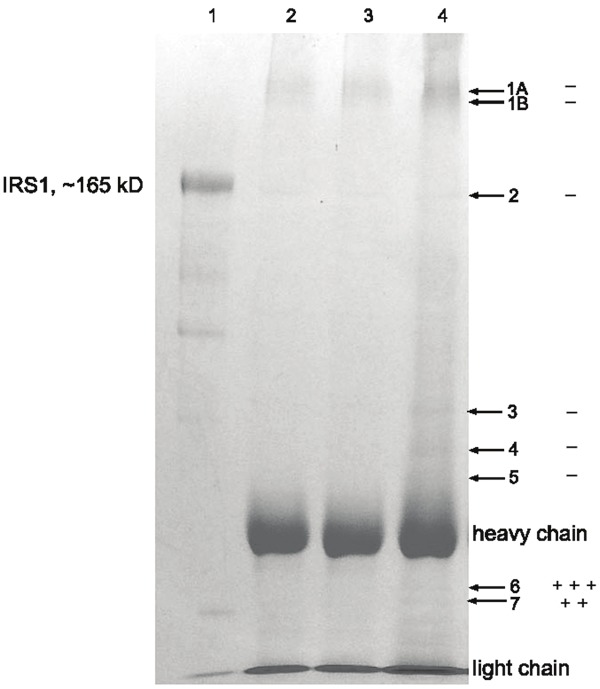
SDS-PAGE profile of IRS-1 degradation in skeletal muscle of burned mice. Antibody: rabbit polyclonal ab653 (immunogen: C-terminal 14 residues conjugated to KLH). Lane 1: recombinant rat IRS-1 standard (2 g), lane 2: IP from muscle lysates of sham treated mice (5 ml, 1 gram tissue), lane 3: IP from muscle lysates of burned mice (5 ml, 1 gram tissue), lane 4: IP from muscle lysates of burned mice (15 ml, 3 grams). Bands #6 and #7, located between the heavy (50 kDa) and light (25 kDa) chains of IgG, were verified by two IRS-1 C-terminal fragments with apparent SDS-PAGE MWs of 44 and 42 kDa, respectively. +: indicates one sequence match, -: indicates negative sequence match. Band #2 represents unreduced intact IgG (150 kDa). The apparent MW of recombinant rat IRS-1 on SDS-PAGE was found to be 165 kD (calc. MW 131 kD).
Table 1.
Tandem mass spectrometry (MS/MS) characterization of mouse skeletal muscle IRS-1 C-terminal fragments after burn injury
| Position | M+H+ | Sequence | Sequenced in SDS-PAGE bands |
|---|---|---|---|
| 1170-1180 | 1263.67 | SLNYIDLDLAK | 95 kD, 44 kD, 42 kD |
| 1156-1169 | 1363.61 | ESAPVCGAAGGLEK | 95 kD, 44 kD, 42 kD |
| 993-1010 | 1984.92 | QSYVDTSPVAPVSYADMR | 44 kD |
| 1135-1155 | 2242.11 | HSSASFNVWLRPGDLGGVSK | 95 kD |
In Figure 1, band 2 in lane 4 was not intact IRS-1 but rather unreduced IgG (150 kDa), which was further confirmed by MS/MS sequencing. Reduced IgG heavy chain (50 kDa) and light chain (25 kDa) chains provide MW markers to estimate the apparent MW of band 6 (three tryptic peptide matches) and band 7 (two tryptic peptide matches). MS/MS sequencing confirmed these bands as C-terminal fragments of mouse IRS-1. The apparent SDS-PAGE MWs of bands 6 and 7 were approximately 42 kDa and 44 kDa, respectively. The sizes of IRS-1 fragments were also estimated with SDS-PAGE and their sequences are listed in Table 1.
The MS/MS y ion profile of C-terminal tryptic peptide 993-QSYVDTSPVAPVSYADMR-1010 of mouse IRS-1 was identified in muscle from the burned mice in band 6 of Figure 1. Total of 9 y ions from y2 to y15 matched the peptide amino acid sequence as shown in Figure 2.
Figure 2.
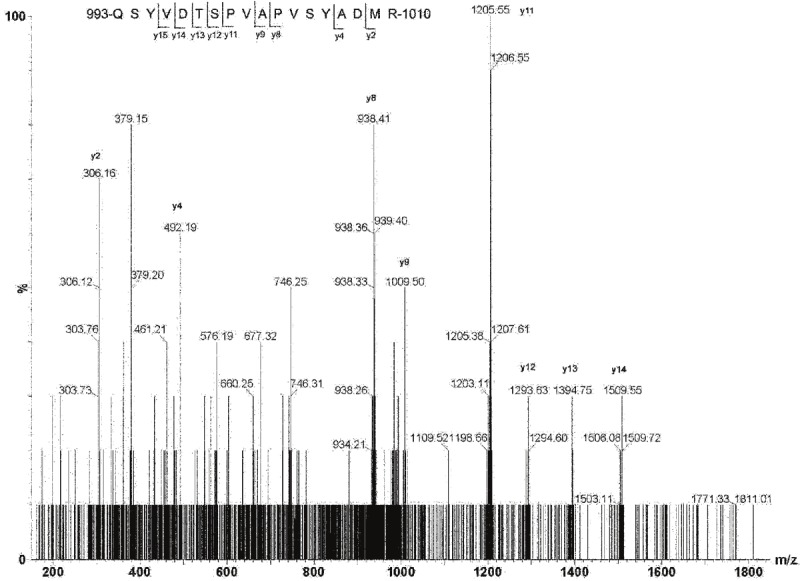
Q-TOF sequencing of mouse IRS-1 C-terminal peptide with YXXM motif. 993-QSYVDTSPVAPVSYADMR-1010 was identified in skeletal muscle from burned mice in band #6 in Figure 2. A total of 9 y ions from y2 to y15 matched the peptide amino acid sequence.
Subcellular distributions of IRS-1, C-terminal fragments and ubiquitinated IRS-1 after burn injury
Intact IRS-1 refers to the entire 1,233 amino acid sequence of the protein, whereas, total IRS-1 includes intact IRS-1 plus IRS-1 fragments which contain the c-terminal amino acid residues. These proteins were measured by ELISA antibodies which are specific to 14 C-terminal amino acids of IRS-1, N-terminal amino acid residues, and to all species of IRS-1 irrespective of post-translational modifications and integrity. The sub-cellular distribution of skeletal muscle IRS-1 after burn injury is illustrated in Figure 3. In sham burned animals, total cellular IRS-1 [intact + C-terminal fragments] in the membrane, cytosolic and nuclear compartments was 196.7 ng/g. The distribution pattern of IRS-1 demonstrated that cell membrane-bound IRS-1 (102.7ng/g) was almost equal to the sum of cytosolic (49.1 ng/g) and nuclear IRS-1 (44.9 ng/g). No IRS-1 was detected in the cytoskeletal compartment.
Figure 3.
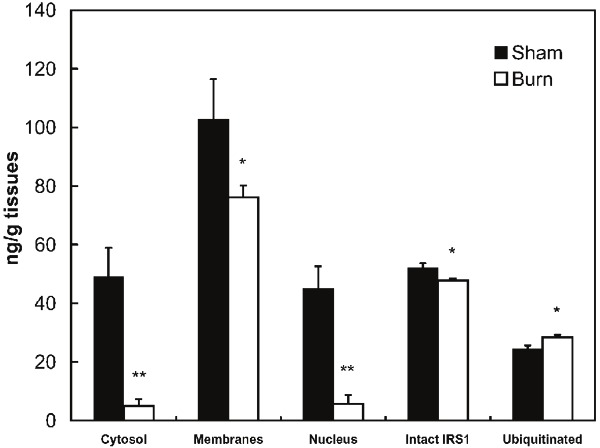
Quantitative (ELISA) measurement of IRS-1 integrity and sub-cellular distribution. IRS-1 was captured with anti-C-terminal mAb, and detected with anti-ubiquitin, anti-N-terminal, and anti-C-terminal rabbit polyclonal antibodies. For total intact and ubiquitinated IRS-1, skeletal muscle tissues were homogenized with Cell Signaling buffer and subcellular components were isolated according to the Qiagen protocol. Values represent mean ± sem for 15 burned and 8 sham treated mice. *: p<0.05, **: p<0.005.
Results with the anti-N-terminal antibody indicated that in sham burned animals about 13 % of total IRS-1 remained as intact protein, indicating that the majority of IRS-1 was present as C-terminal fragments. In contrast, after burn injury about 38% of total IRS-1 remained as intact protein. As also shown in Figure 3, burn injury altered levels of total IRS-1 and its subcellular distribution pattern. Total IRS-1 in the three sub-cellular compartments was reduced to 44.2% of sham burn, with a nearly 90% reduction of total IRS-1 in the sum of nuclear and cytosolic IRS-1 (p<0.01). The total intact cellular IRS-1 was decreased to 92.3% of sham burn levels (p<0.05). In contrast, there was a 15.9 % increase in ubiquitinated IRS-1, suggesting that ubiquitination may result in fragments that are not detectable with the C-terminal antibody that was used.
Messenger levels of IRS-1, SOCS-3 and involvement of ubiquitin-mediated cytokine-SOCS-3-Elongin-E3 Ligase in the IRS-1 degradation pathway
To determine whether the decrease in intact IRS-1 is occurring only at the protein level and to evaluate the two possible ubiquitin-mediated degradation pathways: PI3K/Akt1-Rheb-mTOR/S6K–CUL7 E3 ligase and cytokine-SOCS-3-Elongin-E3 ligase, quantitative real-time RT-PCR was performed to determine the levels of IRS-1, Akt1 and SOCS-3 mRNA; and the results are shown in Figure 4.
Figure 4.
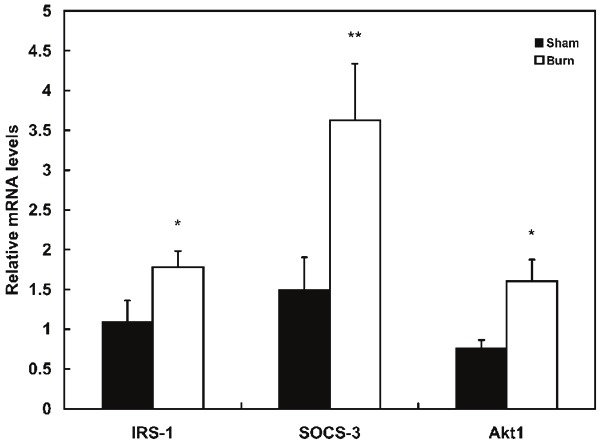
RT-PCR measurement of relative mRNA levels for IRS-1, SOCS-3 and Akt1. Frozen skeletal tissue samples (~50 mg) were homogenized (5 strokes of 5 sec. each with a Omni TH-tissue homogenizer) in TRI reagent solution. RNA extraction was performed according to standard procedures and RNA quality was assessed with agarose gel electrophoresis. Samples were analyzed in triplicate using the comparative threshold cycle SYBR green method. PCR primers obtained from MGH primer bank for mouse IRS1, Akt1 and SOCS3 cDNA were used as F: 5’-CGATGGCTTCTCAGACGTG-3’, R: 5’-CAGCCCGCTTGTTGATGTTG-3’; F: 5’-ATG AAC GAC GTA GCC ATT GTG-3’, R: 5’-TTG TAG CCA ATA AAG GTG CCA T-3’: F: 5’-ATGGTCACCCACAGCAAGTTT-3’, R: 5’-TCCAGTAGAATCCGCTCTCCT-3. GAPDH F: 5’-AGGTCGGTGTGAACGGATTTG-3’, R: 5’-TGTAGACCATGTAGTTGAGGTCA-3’ was used as a house-keeping gene for mouse muscle. Expression levels of the target genes were normalized to expression levels of GAPDH. Values represent mean ± sem for 15 burned and 8 sham treated mice. *: p<0.05, **: p<0.005.
These data demonstrate that in contrast to the decrease in cellular IRS-1, burn injury was associated a 35% increase (p<0.05) in expression of IRS-1 mRNA. This was accompanied by an approximately 2.4 fold increase in the expression of both SOCS-3 mRNA (p<0.005).
Figure 5 shows the ELISA results for measurements of the phosphorylation status of Akt1 residues Thr308 and Ser473; results are expressed as units per gram of tissue. The results clearly demonstrate that, in skeletal muscle of burned mice, phosphorylations of Akt1 at Thr308 and Ser473 are reduced to 50.2% by (p<0.005) and to 73.8 (p<0.05) respectively as compared to sham burned animals.
Figure 5.
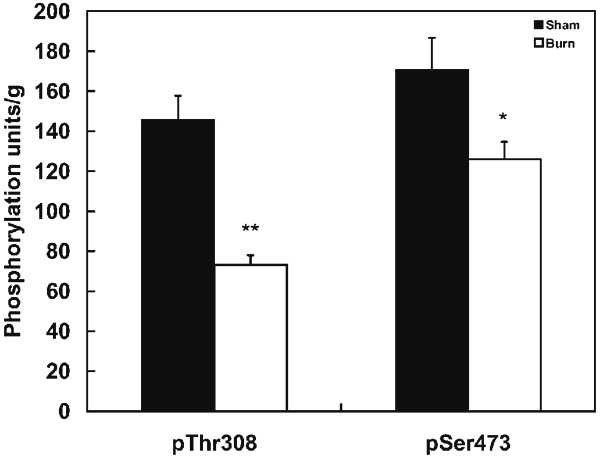
Impaired phosphorylation of Thr308 and Ser473 of Akt in skeletal muscle from burned mice. Skeletal muscle tissues from burned mice were homogenized with Cell Signaling buffer and phosphorylation levels of Thr308 and Ser473 in Akt1 were measured by ELISA. Calibration curves were prepared according to the manufacture’s protocol. The phosphorylation units obtained were normalized to tissue weight. Values represent mean ± sem for 15 burned and 8 sham treated mice. *: p<0.05, **: p<0.005
Discussion
More than 30 years has elapsed since the first report of burn induced insulin resistance by our laboratory [45,46], however the molecular mechanism(s) are still under investigation. The principal insulin receptor substrates, IRS1 in skeletal muscle and IRS2 in liver, play important roles in insulin signal transduction after binding to its receptors. The activated receptors promote phosphorylations on multiple tyrosine residues of IRS-1 and IRS-2. It subsequently promotes their binding to the Src homology 2 domains in various downstream signaling proteins, including the phosphatidylinositol 3kinase (PI 3kinase), Grb2, SHP2, and others. PI 3-kinase is activated through its association with IRS proteins and its phospholipid products promote the recruitment of serine kinases to the plasma membrane, where they are activated by the phosphorylation process. One of the membrane-associated kinases, Akt/PKB, phosphorylates multiple downstream effectors that promote diverse biological responses, including the regulation of gene expression, stimulation of glucose transport and the synthesis of proteins and glycogens, which affect cellular proliferation and survival. Furthermore, the mechanism(s) leading to muscle atrophy are also associated with two critical nodes in the insulin signaling pathways - at the initial docking protein, IRS-1, and the central signaling diversity kinase, Akt-1. Irreversible phosphorylations of these proteins are directly related to muscle catabolism. In severely burned patients, expression of ubiquitin specific proteases in skeletal muscle is increased by 2.5 fold, and in conditions of severe muscle wasting, the muscle specific ubiquitin-proteasome proteolytic activities (in particular MAFbx and MuRFI) were found to be increased 20-30 fold [47]. At the cellular level, it has been reported that insulin signaling was attenuated through two possible ubiquitin-mediated IRS-1 degradation pathways: PI3K/Akt1-Rheb-mTOR/S6K–CUL7 E3 ligase and cytokine-SOCS-3-Elongin-E3 ligase [48,49].
It has been reported that degradation of IRS-1 protein occurs through a mechanism involving insulin stimulated phosphorylaton at serine residues through the mTOR/S6K pathway [50]. The Ser/Thr phosphorylation of IRS-1 induces increased electrophoreretic mobility and subsequently triggers its degradation. This mobility shift is blocked by Wortmannin and rapamycin [51]. The proteasomal inhibitor epoxomicin and the lysosomal pathway inhibitor 3-methyladenine prevent IRS-1 degradation through mTOR/S6K 1 mediated IRS-1 serine phosphorylation and proteasomal degradation. [52] More specifically, IRS-1 has been identified as a proteolytic target of the degradation pathway of PI3K/Akt1-Rheb-mTOR/S6K–CUL7 E3 ligase [53]. The PI3K/Akt1-Rheb-mTOR/S6K–E3 ubiquitination pathway of IRS-1 degradation has been documented in obesity and NIDDM that are considered to be a chronic/low grade inflammatory states.
Our previous in vitro studies have demonstrated that IRS-1 C-terminal is cleaved at multiple sites after varying levels of insulin stress [54]. One of the major challenges is that intact IRS-1 is present at very low concentrations (~ 51.9 ng/gram tissues); even in tissues with the highest levels of IRS such as soleus muscle [55]. Therefore we used as much skeletal muscle as possible The present study demonstrated that burn injury was associated with a significant reduction of IRS-1 protein in major sub-cellular components of skeletal muscle, including the membrane, cytosolic and nuclear compartments. Using MS/MS we identified the phosphorylation sites which trigger the degradation of IRS-1 protein and the major breakdown fragments. IRS-1 plays important roles in cell transformation, cell differentiation and aging. IRS proteins can translocate to the cell nucleus, where they interact with nuclear and nuclear proteins. IRS-1 is preferentially nuclear in growing cells, such as cancer cells and it activates promoters of cell cycle progression genes. Losses of smaller cytosolic intact IRS-1 (7.7%) and larger nuclear intact IRS-1 (90%) on day 7 post burn suggests slowing of the cell cycle which could be associated with muscle wasting. These findings will help identify targets for developing strategies for the treatment of burn injury induced insulin resistance and muscle wasting.
The present study further revealed that the degradation of IRS-1 may be associated with increased expression levels of SOCS-3. Our study revealed the pathway for increased IRS-1 degradation after burn injury. There was an increase of ubiquitinated IRS-1 (Figure 3) associated with a 140% increment of SOCS-3 mRNA in burned as compared to sham treated animals. It has been reported that SOCS-3 protein and mRNA are significantly elevated within 4 hours after injury and persists for 8 days, while SOCS-1 and SOCS-2 levels remained unchanged or were undetectable [56]. It appears that a similar pattern of IRS-1 degradation occurs after severe burn injury via a high-grade inflammation induced, SOCS-3 mediated ubiquitin pathway.
As shown in Figure 4, burn injury was associated with a reduction in total intact IRS-1 and an increase in cytosolic ubiquitinated IRS-1. This was accompanied by an altered distribution of IRS-1 molecular fragments in the membrane-bound, cytosolic and nuclear compartments. The very low levels of IRS-1 in the cytosolic and nuclear compartments after burn suggest that membrane bound intact IRS-1 might be a secondary regulator after recruiting IRS-1 to the plasma membrane. Previous studies have documented the trafficking of IRS-1 from the cell membrane to the intracellular and nuclear compartments [57,58]. Nuclear localization of IRS-1 accounts for approximately 25% of total liver IRS-1 [59], and nuclear IRS-1 seems to be independent of Akt levels [60]. IRS-1 plays important roles in cell transformation, cell differentiation and aging. IRS proteins at the cell nucleus interacts with nuclear proteins. IRS-1 is preferentially nuclear in growing cells, such as cancer cells and activates promoters of cell cycle progression genes. Losses of smaller cytosolic intact IRS-1 (7.7%) and larger nuclear intact IRS-1 (90%) on day 7 post burn suggests slowing of the cell cycle which could be associated with muscle wasting [61]. The IRS-1 located in the cytosolic and nuclear compartments is most likely C-terminal fragments which could function as primary messengers to improve survival of the host and aid in wound healing after severe burn injury. Unfortunately, due to the small amount of skeletal muscle harvestable from mice, the low level of IRS-1 in muscle and the relatively limited sensitivity of the MS/MS sequencing technique we were not able to evaluate IRS-1 degradation independently in slow and fast twitch muscle fibers. This was an unavoidable limitation of the study.
It has also been reported that Akt1 activity is mainly determined by phosphorylation of C-terminal Ser473 (90%) which forms a salt bridge with N-terminal Lysine as well as phosphorylation of Thr308 (10%) in the kinase loop which forms a second salt bridge with Lys297. The present study demonstrated 26.2% and 49.8% reductions in phosphorylation at Ser473 and Thr308 of Akt-1 protein (Figure 5). Taking all these findings together, the present study suggests that burn induced IRS-1 degradation is, at least partially, mediated through cytokine-SOCS-3-Elongin-E3 ligase; since Akt1 kinase activity is significantly impaired after burn injury.
Conclusions
The present study confirmed a reduction of IRS-1 in muscle cells after burn injury, which is the consequence of increased degradation. We further defined the exact degradation fragments of IRS-1 and a high-grade inflammation induced, SOCS-3 mediated ubiquitin pathway which may be involved in the degradation process. The degradation of IRS-1 is also associated with impaired phosphorylation of Akt-1 in the down stream insulin signal transduction pathway. Both changes contribute to burn induced insulin resistance and muscle wasting. These findings at the molecular level will be extremely useful for identifying targets for developing drugs for treating insulin resistance and muscle wasting in severely burned patients.
Acknowledgments
This study was supported in part by US Public Health Grant # 3P50 GM21700-27A1 and a grant from Shriners Hospitals for Children Grant #8470. We would like to acknowledge Dr. Xinyuan Chen for acquisition and analysis of the RT-PCR data, Dr. Edward A. Carter and Ms. Florence Lin for their aid with the animal model and tissue processing and Ms. Florence Lin for additional assistance with the ELISA analysis.
Competing interest statement
The authors have nothing to disclose.
References
- 1.Jeschke MG, Mlcak RP, Finnerty CC, Norbury WB, Gauglitz GG, Kulp GA, Herndon DN. Burn size determines the inflammatory and hypermetabolic response. Crit Care. 2007;11:R90. doi: 10.1186/cc6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gauglitz GG, Halder S, Boehning DF, Kulp GA, Herndon DN, Barral JM, Jeschke MG. Post-burn hepatic insulin resistance is associated with endoplasmic reticulum (ER) stress. Shock. 2010;33:299–305. doi: 10.1097/SHK.0b013e3181b2f439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pereira C, Murphy K, Jeschke M, Herndon DN. Post burn muscle wasting and effects of treatments. Int J Biochem Cell Biol. 2005;37:1948–1961. doi: 10.1016/j.biocel.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 4.Evers LH, Bhavsar D, Mailander P. The biology of burn injury. Exp Dermatol. 2010;19:777–783. doi: 10.1111/j.1600-0625.2010.01105.x. [DOI] [PubMed] [Google Scholar]
- 5.Barrow RE, Dasu MR, Ferrando AA, Spies M, Thomas SJ, Perez-Polo JR, Herndon DN. Gene expression patterns in skeletal muscle of thermally injured children treated with oxandrolone. Ann Surg. 2003;237:422–428. doi: 10.1097/01.SLA.0000055276.10357.FB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dasu MR, Barrow RE, Herndon DN. Gene profiling in muscle of severely burned children: Age- and Sex-dependent changes. J Surgical Res. 2005;123:144–152. doi: 10.1016/j.jss.2004.07.248. [DOI] [PubMed] [Google Scholar]
- 7.Przkora R, Herndon DN, Finnerty CC, Jeschke MG. Insulin attenuates the cytokine response in a burn wound infection model. Shock. 2007;27:205–208. doi: 10.1097/01.shk.0000238069.84826.1b. [DOI] [PubMed] [Google Scholar]
- 8.Jeschke MG, Boehning DF, Finnerty CC, Herndon DN. Effect of insulin on the inflammatory and acute phase response after burn injury. Crit Care Med. 2007;35:S519–S523. doi: 10.1097/01.CCM.0000282027.10288.10. [DOI] [PubMed] [Google Scholar]
- 9.Pidcoke HF, Wade CE, Wolf SE. Insulin and burned patient. Crit Care Med. 2007;35:S524–S530. doi: 10.1097/01.CCM.0000278065.72486.31. [DOI] [PubMed] [Google Scholar]
- 10.Gauglitz GG, Herndon DN, Kulp GA, Meyer WJ 3rd, Jeschke MG. Abnormal insulin sensitivity persists up to three years in pediatric patient post-burn. J Clin Endocrinol Metab. 2009;94:1656–1664. doi: 10.1210/jc.2008-1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gauglitz GG, Herndon DN, Jeschke MG. Insulin resistance postburn: Underlying mechanisms and current therapeutic strategies. J Burn Care Res. 2008;2:683–694. doi: 10.1097/BCR.0b013e31818481ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khoury W, Klausner JM, Benabraham R, Szold O. Glucose control by insulin for critically ill surgical patients. J Trauma. 2004;57:1132–1138. doi: 10.1097/01.ta.0000141889.31903.9c. [DOI] [PubMed] [Google Scholar]
- 13.Carter EA, Burks D, Fischman AJ, White M, Tompkins RG. Insulin resistance in thermally-injured rats is associated with post-receptor alterations in skeletal muscle, liver and adipose tissue. Int J Mol Med. 2004;14:653–658. [PubMed] [Google Scholar]
- 14.Johan Groeneveld AB, Beishuizen A, Visser FC. Insulin: a wonder drug in the critically ill? Crit Care. 2002;6:102–105. doi: 10.1186/cc1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ikezu T, Okamato T, Yonezawa K, Tompkins RG, Martyn JA. Analysis of thermal injury-induced insulin resistance in rodents. J Biol Chem. 1997;272:25289–25295. doi: 10.1074/jbc.272.40.25289. [DOI] [PubMed] [Google Scholar]
- 16.White MF. Insulin signaling in health and disease. Science. 2003;302:1710–1711. doi: 10.1126/science.1092952. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Q, Carter EA, Ma BY, White M, Fischman AJ, Tompkins RG. Molecular mechanism(s) of burn-induced insulin resistance in murine skeletal muscle: role of IRS phosphorylation. Life Sci. 2005;77:3068–3077. doi: 10.1016/j.lfs.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 18.Sugita H, Kaneki M, Sugita M, Yasukawa T, Yasuhara S, Martyn JA. Burn injury impairs insulin-stimulated Akt/PKB activation in skeletal muscle. Am J Physiol Endocrinol Metab. 2005;288:585–591. doi: 10.1152/ajpendo.00321.2004. [DOI] [PubMed] [Google Scholar]
- 19.Taniguchi CM, Emanuelli B, Kahn C. Critical nodes in signaling pathways: insights into insulin action. Nat Rev Mol Cell Biol. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- 20.Gual P, Marchand-Brustel Y, Tanti JF. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie. 2005;87:99–109. doi: 10.1016/j.biochi.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 21.Zick Y. Insulin resistance: a phosphorylation-based uncoupling of insulin signaling. Trends Cell Biol. 2010;11:437–441. doi: 10.1016/s0962-8924(01)02129-8. [DOI] [PubMed] [Google Scholar]
- 22.Shi H, Tzameli I, Bjorbak C, Flier JS. Suppressor of cytokine signaling 3 is a physiological regulator of adipocytes insulin signaling. J Biol Chem. 2004;279:34733–34740. doi: 10.1074/jbc.M403886200. [DOI] [PubMed] [Google Scholar]
- 23.Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem. 2002;277:42394–42398. doi: 10.1074/jbc.C200444200. [DOI] [PubMed] [Google Scholar]
- 24.Dong X, Park S, Lin X, Copps K, Yi X, White MF. Irs1 and Irs1 signaling is essential for hepatic glucose homeostasis and systemic growth. J Clin Invest. 2006;116:101–114. doi: 10.1172/JCI25735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.White MF. IRS proteins and the common path to diabetes. Am J Physiol Endocrinol Metab. 2002;283:E413–E422. doi: 10.1152/ajpendo.00514.2001. [DOI] [PubMed] [Google Scholar]
- 26.Zick Y. Ser/Thr phosphorylation of IRS proteins: a molecular basis for insulin resistance. Sci Stke. 2005;268:pe4. doi: 10.1126/stke.2682005pe4. [DOI] [PubMed] [Google Scholar]
- 27.Danielsson A, Nystrom FH, Stralfors P. Phosphorylation of IRS1 at serine 307 and serine 312 in response to insulin in human adipocytes. Biochem Biophys Res Comm. 2006;342:1183–1187. doi: 10.1016/j.bbrc.2006.02.075. [DOI] [PubMed] [Google Scholar]
- 28.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384–387. doi: 10.1126/science.1104343. [DOI] [PubMed] [Google Scholar]
- 29.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 30.Yi Z, Luo M, Mandarino LJ, Reyna SM, Carroll CA, Weintraub ST. Quantification of phosphorylation of insulin receptor substrate-1 by HPLC-ESI-MS/MS. J Am Soc Mass Spectrom. 2006;17:562–567. doi: 10.1016/j.jasms.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 31.Hers I, Bell CJ, Poole AW, Jiang D, Denton RM, Schaefer E, Tavare JM. Reciprocal feedback regulation of insulin receptor and insulin receptor substrate tyrosine phosphorylation by phosphoinositide 3-kinase in primary adipocytes. Biochem J. 2002;368:875–884. doi: 10.1042/BJ20020903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amoui M, Craddock BP, Miller WT. Differential phosphorylation of IRS-1 by insulin and insulin-like growth factor 1 receptor in Chinese hamster ovary cells. J Endocrinol. 2001;171:153–162. doi: 10.1677/joe.0.1710153. [DOI] [PubMed] [Google Scholar]
- 33.Sun XJ, Crimmin DL, Myers MG Jr, Miralpeix M, White MF. Pleiotropic insulin signals are engaged by multisite phosphorylation of IRS-1. Mol Cell Biol. 1993;13:7418–7428. doi: 10.1128/mcb.13.12.7418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sykiotis GP, Papavassiliou AG. Serine phosphorylation of insulin receptor substrate-1: A novel target for the reversal of insulin resistance. Mol Endocrinol. 2001;15:1864–1869. doi: 10.1210/mend.15.11.0725. [DOI] [PubMed] [Google Scholar]
- 35.Sommerfeld MR, Metzger S, Stosik M, Tennagels N, Eckel J. In vitro phosphorylation of insulin receptor substrate 1 by protein kinase C-zeta: functional analysis and identification of novel phosphorylation sites. Biochemistry. 2004;43:5888–5901. doi: 10.1021/bi049640v. [DOI] [PubMed] [Google Scholar]
- 36.Liberman Z, Eldar-Finkelman H. Serine 332 phosphorylation of insulin receptor substrate-1 by glycogen synthase kinase-3 attenuates insulin signaling. J Biol Chem. 2005;280:4422–4428. doi: 10.1074/jbc.M410610200. [DOI] [PubMed] [Google Scholar]
- 37.De Fea K, Roth RA. Protein kinase C modulation of insulin receptor substrate-1 tyrosine phosphorylation requires serine 612. Biochemistry. 1997;36:12939–12947. doi: 10.1021/bi971157f. [DOI] [PubMed] [Google Scholar]
- 38.Li Y, Soos TJ, Li X, Wu J, DeGennaro M, Sun X, Littman DR, Birnbaum MJ, Polakiewicz RD. Protein kinase C z inhibits insulin signaling by phosphorylating IRS1 at serine 1101. J Biol Chem. 2004;279:45304–45307. doi: 10.1074/jbc.C400186200. [DOI] [PubMed] [Google Scholar]
- 39.Werner ED, Lee J, Hansen L, Yuan M, Shoelson SE. Insulin resistance due to phosphorylation of insulin receptor substrate-1 at serine 302. J Biol Chem. 2004;279:35298–35305. doi: 10.1074/jbc.M405203200. [DOI] [PubMed] [Google Scholar]
- 40.Le Marchand-Brustel Y, Gual P, Gremeaux T, Gonzalez T, Barres R, Tanti JF. Fatty acid-induced insulin resistance: role of insulin receptor substrate 1 serine phosphorylation in the retroregulation of insulin signaling. Biochem Soc Trans. 2003;31:1152–1156. doi: 10.1042/bst0311152. [DOI] [PubMed] [Google Scholar]
- 41.Sciocia M, Gumaa K, Kunjara S, Paine MA, Selvaggi LE, Rodeck CH, Rademacher TW. Insulin resistance in human preeclamptic placenta is mediated by serine phosphorylation of insulin receptor substrate-1 and -2. J Clin Endocrinol Metab. 2006;91:709–717. doi: 10.1210/jc.2005-1965. [DOI] [PubMed] [Google Scholar]
- 42.Gual P, Gremeaux T, Gonzalez T, Le Marchand-Brustel Y, Tanti JF. MAP kinase and mTOR mediate insulin-induced phosphorylation of insulin receptor substrate-1 on serine residues 307, 612 and 632. Diabetologia. 2003;46:1532–1543. doi: 10.1007/s00125-003-1223-4. [DOI] [PubMed] [Google Scholar]
- 43.Carter EA, Bonab AA, Goverman J, Paul K, Yerxa J, Tompkins RG, Fischman AJ. Evaluation of the antioxidant peptide SS31 for treatment of burn- induced insulin resistance. Int J Mol Med. 2011;28:589–94. doi: 10.3892/ijmm.2011.752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pfaffl MW. Quantification Strategies in Real-Time PCR. In: Bustin SA, editor. A-Z of Quantitative PCR. La Jolla, CA: International University Line; 2004. pp. 87–112. [Google Scholar]
- 45.Wolfe RR, Burke JF. Effect of burn trauma on glucose turnover, oxidation, and recycling in guinea pigs. Am J Physiol. 1977;233:E80–85. doi: 10.1152/ajpendo.1977.233.2.E80. [DOI] [PubMed] [Google Scholar]
- 46.Thomas R, Aikawa N, Burke JF. Insulin resistance in peripheral tissues after burn injury. Surgery. 1979;86:742–747. [PubMed] [Google Scholar]
- 47.Hasselgren PO. Ubiquitination, phosphorylation, and acetylation--triple threat in muscle wasting. J Cell Physiol. 2007;213:679–689. doi: 10.1002/jcp.21190. [DOI] [PubMed] [Google Scholar]
- 48.Tang X, Tang G, Ozcan S. Role of microRNAs in diabetes. Biochim Biophys Acts. 2008;1779:697–701. doi: 10.1016/j.bbagrm.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.La Rocca G, Shi B, Badin M, De Angelis T, Sepp-Lorenzino L, Baserga R. Growth inhition by microRNAs that target the insulin receptor substrate-1. Cell Cycle. 2009;8:2255–2259. doi: 10.4161/cc.8.14.9026. [DOI] [PubMed] [Google Scholar]
- 50.Haruta T, Uno T, Kawahara J, Takano A, Egawa K, Sharma PM, Olefsky JM, Kobayashi M. A rapamycin-sensitive pathway down-regulates insulin signaling via phosphorylation and proteasomal degradation of insulin receptor substrates-1. Mol Endocrinol. 2000;14:783–794. doi: 10.1210/mend.14.6.0446. [DOI] [PubMed] [Google Scholar]
- 51.Pederson TM, Kramer DL, Rondinone CM. Serine/Threonine phosphorylation of IRS-1 triggers its degradation: possible regulation by tyrosine phosphorylation. Diabetes. 2001;50:24–31. doi: 10.2337/diabetes.50.1.24. [DOI] [PubMed] [Google Scholar]
- 52.Mayer CM, Beisham DD. Central insulin signaling is attenuated by long-term insulin exposure via insulin receptor substrate-1 serine phosphorylation, proteasomal degradation, and lysosomal insulin receptor degradation. Endocrinology. 2010;151:75–84. doi: 10.1210/en.2009-0838. [DOI] [PubMed] [Google Scholar]
- 53.Xu X, Sarikas A, Dias-Santagata DC, Dolios G, Lafontant PJ, Tsai SC, Zhu W, Nakajima H, Nakajima HO, Field LJ, Wang R, Pan ZQ. The CUL7 E3 ubiquitin ligase targets insulin receptor substrate 1 for unbiquitin-dependent degradation. Mol Cell. 2008;30:403–414. doi: 10.1016/j.molcel.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu XM, Hamrahi VF, Tompkins RG, Fischman AJ. Effect of insulin levels on the phosphorylation of specific amino acid residues in IRS-1: implications for burn-induced insulin resistance. Int J Mol Med. 2009;4:531–538. doi: 10.3892/ijmm_00000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu C, Chen Y, Cline GW, Zhang D, Zong H, Wang Y, Bergeron R, Kim JK, Cushman SW, Cooney GJ, Atcheson B, White MF, Kraegen EW, Shulman GI. Mechanism by white fatty acids inhibits insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J Biol Chem. 2002;277:50230–50236. doi: 10.1074/jbc.M200958200. [DOI] [PubMed] [Google Scholar]
- 56.Ogle CK, Kong F, Guo X, Wells DA, Aosasa S, Noel G, Horseman N. The effect of burn injury on suppressions of cytokine signaling. Shock. 2000;14:392–398. [PubMed] [Google Scholar]
- 57.Baserga R. Insulin receptor substrate-1: A biomarker for cancer? Exp Cell Res. 2009;315:727–732. doi: 10.1016/j.yexcr.2008.09.017. [DOI] [PubMed] [Google Scholar]
- 58.Chen J, Wu A, Sun H, Drakas R, Garofalo C, Cascio S, Surmacz E, Baserga R. Functional significant of type 1 insulin-like growth factor-mediated nuclear translocation of the insulin receptor substrate-1 and b-catenin. J Biol Chem. 2005;280:29912–29920. doi: 10.1074/jbc.M504516200. [DOI] [PubMed] [Google Scholar]
- 59.Boyla JM, Gruppuso PA. Insulin receptor substrate-1 is present in hepatocyte nuclei from intact rats. Endocrinol. 2000;143:4178–4183. doi: 10.1210/en.2002-220321. [DOI] [PubMed] [Google Scholar]
- 60.Zhao Y, Biswas SK, McNulty PH, Kozak M, Jun JY, Segar L. PDGF-induced vascular smooth muscle cell proliferation is associated with dysregulation of insulin receptor substrates. Am J Physiol Cell Physiol. 2011;300:C1375–85. doi: 10.1152/ajpcell.00670.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Seol KC, Kim SJ. Nuclear matrix association of insulin receptor and IRS-1 by insulin in osteoblast-like UMR-106 cells. Biochem Biophy Res Commun. 2003;306:898–904. doi: 10.1016/s0006-291x(03)01046-5. [DOI] [PubMed] [Google Scholar]