

ABSTRACT
Coxiella burnetii is an intracellular pathogen that replicates within a lysosome-like vacuole. A Dot/Icm type IVB secretion system is used by C. burnetii to translocate effector proteins into the host cytosol that likely modulate host factor function. To identify host determinants required for C. burnetii intracellular growth, a genome-wide screen was performed using gene silencing by small interfering RNA (siRNA). Replication of C. burnetii was measured by immunofluorescence microscopy in siRNA-transfected HeLa cells. Newly identified host factors included components of the retromer complex, which mediates cargo cycling between the endocytic pathway and the Golgi apparatus. Reducing the levels of the retromer cargo-adapter VPS26-VPS29-VPS35 complex or retromer-associated sorting nexins abrogated C. burnetii replication. Several genes, when silenced, resulted in enlarged vacuoles or an increased number of vacuoles within C. burnetii-infected cells. Silencing of the STX17 gene encoding syntaxin-17 resulted in a striking defect in homotypic fusion of vacuoles containing C. burnetii, suggesting a role for syntaxin-17 in regulating this process. Lastly, silencing host genes needed for C. burnetii replication correlated with defects in the translocation of Dot/Icm effectors, whereas, silencing of genes that affected vacuole morphology, but did not impact replication, did not affect Dot/Icm translocation. These data demonstrate that C. burnetii vacuole maturation is important for creating a niche that permits Dot/Icm function. Thus, genome-wide screening has revealed host determinants involved in sequential events that occur during C. burnetii infection as defined by bacterial uptake, vacuole transport and acidification, activation of the Dot/Icm system, homotypic fusion of vacuoles, and intracellular replication.
IMPORTANCE
Q fever in humans is caused by the bacterium Coxiella burnetii. Infection with C. burnetii is marked by its unique ability to replicate within a large vacuolar compartment inside cells that resembles the harsh, acidic environment of a lysosome. Central to its pathogenesis is the delivery of bacterial effector proteins into the host cell cytosol by a Dot/Icm type IVB secretion system. These proteins can interact with and manipulate host factors, thereby leading to creation and maintenance of the vacuole that the bacteria grow within. Using high-throughput genome-wide screening in human cells, we identified host factors important for several facets of C. burnetii infection, including vacuole transport and membrane fusion events that promote vacuole expansion. In addition, we show that maturation of the C. burnetii vacuole is necessary for creating an environment permissive for the Dot/Icm delivery of bacterial effector proteins into the host cytosol.
Introduction
Coxiella burnetii is a Gram-negative facultative intracellular pathogen and the causative agent of the global zoonosis Q fever (1). Human Q fever is often discernible as an acute debilitating influenza-like illness. In rare cases, chronic disease can develop and lead to endocarditis in individuals with pre-existing valvular heart disease (2). In nature, C. burnetii infects vertebrates and arthropods. Typically, animals and humans are infected by inhalation of contaminated aerosols containing the bacteria that are shed into the environment by domestic ruminants as part of livestock operations (1). Due to its low infectious dose (fewer than 10 organisms), environmental stability, and the ease of aerosol dissemination, C. burnetii has been classified as a category B biological weapon agent by the Centers for Disease Control and Prevention (3).
Many intracellular bacteria reside in a vacuolar compartment that is modified by bacterial proteins to provide an environment suitable for replication (4, 5). This often involves specific strategies to evade fusion between the vacuole and lysosomes to prevent the acquisition of antibacterial proteins contained in this hydrolytic compartment. Coxiella burnetii is somewhat unique in that it must be delivered to an acidified lysosome-derived compartment in order to replicate. The organelle it creates is referred to as the C. burnetii-containing vacuole (CCV). The CCV expands considerably during the first 2 days of infection. Bacterial replication and expansion of the CCV continue over a period of several days, resulting in a mature compartment that often occupies the majority of the host cell cytoplasm (6). During infection, the CCV membrane acquires proteins of the endolysosomal system such as Rab7, LAMP-1 and LAMP-2, and the vacuolar type H+ ATPase. The CCV lumen has a pH of about 4.8, and this acidic environment is required to activate the metabolism of C. burnetii and initiate bacterial replication (7). Furthermore, the CCV stains positively for several proteins that are present on autophagosomes, consistent with the endocytic and autophagic pathways merging at a stage that occurs before creation of the mature vacuole containing C. burnetii (8–10).
Despite the clear overlap with the host endocytic pathway, several lines of evidence indicate that the CCV is distinct from a canonical phagolysosome and is indeed a unique bacterially derived compartment. The CCV is highly fusogenic and promotes unregulated homotypic fusion with endolysosomal vesicles (11, 12). At high multiplicities of infection, multiple CCVs present in the cell following entry fuse to form a single large and spacious vacuole, which is in contrast to what is observed with most other phagolysosomes (12). This process correlates with bacterial replication, and continued protein synthesis is essential for maintaining the fusogenic properties of the CCV (13, 14). Additionally, the CCV membrane is rich in cholesterol and inhibitors of cholesterol metabolism decrease C. burnetii replication (15, 16). In this regard, host processes that play a specific role in establishment of C. burnetii infection are of great interest; however, the molecular mechanisms governing biogenesis of the vacuole in which C. burnetii replicates have not been fully delineated. Processes that promote entry of the bacterium into the host cell, establishment and transport of the CCV, vacuole acidification, nutrient acquisition, vesicular fusion, and continued maintenance of the mature replicative compartment are all predicted to be important (17).
Coxiella burnetii encodes a Dot/Icm type IV secretion system analogous to the Legionella pneumophila Dot/Icm system (18, 19). Similar to L. pneumophila, a functional Dot/Icm system is required for establishment of a C. burnetii vacuole that supports replication (20, 21). Dot/Icm-translocated effector proteins of L. pneumophila have been characterized and act with or upon specific host cell molecules to modulate their function. Collectively, the effector repertoire exploits host membrane transport pathways to create a specialized vacuole that promotes L. pneumophila survival and replication (22, 23). Several studies have used L. pneumophila as a surrogate host to identify Dot/Icm-translocated effectors of C. burnetii (20, 24–28). Several of these effectors were further tested in C. burnetii and shown to be translocated by the C. burnetii Dot/Icm system (20, 21, 28) and have been found associated with host organelles and the CCV membrane (20, 24, 28). The Dot/Icm effector AnkG has been shown to block the pathogen-induced proapoptotic signaling pathway to prevent premature death of host cells during bacterial infection (26); however, the biochemical and biological function of most other C. burnetii effectors remains to be determined. Given the important role translocated effectors play in intracellular bacterial pathogenesis, it is expected that the effector repertoire of the C. burnetii Dot/Icm system targets additional host cell molecules involved in different stages of C. burnetii infection.
To gain a more complete understanding of host mechanisms required for C. burnetii infection, and to identify pathways that might be targeted by C. burnetii effectors, we conducted an unbiased genome-wide screen that used small interfering RNA (siRNA) technology to identify host determinants that are involved in the establishment of the CCV and replication of C. burnetii in mammalian host cells.
RESULTS
Evaluating host determinants required for C. burnetii infection using fluorescence microscopy.
Intracellular bacteria subvert host functions to create specialized vacuoles that support intracellular replication. We used a genome-wide RNA interference screen to identify host proteins required for infection of mammalian cells by C. burnetii. Because Rab7 function is important for C. burnetii replication in mammalian cells (9), the gene encoding Rab7 was silenced by siRNA in HeLa cells and fluorescence microscopy was used to measure the replication of C. burnetii after four days of infection to determine if a fluorescence-based screening approach would be sensitive enough to identify host factors important for C. burnetii replication. A significant decrease in the total number and size of C. burnetii fluorescent vacuoles was detected within a field of cells that had been treated with siRNA that targets the gene encoding Rab7A compared to mock transfection (Fig. 1A). Thus, we reasoned that a fluorescence-based siRNA screening approach could be used to identify additional host factors important for C. burnetii infection.
FIG 1 .

Genome-wide siRNA screen to identify host factors involved in C. burnetii infection. (A) Mock-transfected or Rab7A siRNA-treated HeLa cells were infected with C. burnetii and imaged at 4 days postinfection (p.i.). Shown is a 10× objective field of cells indicating immunolabeling of C. burnetii (green) and host nuclei with Hoechst (blue). The insets show closeups of a portion of the field. (B) Schematic representation of the screening procedure. HeLa cells were transfected with siRNA SMARTPools for 2 days and incubated with C. burnetii (Nine Mile strain phase II) for 4 days. Samples were fixed, immunostained, and imaged by automated microscopy as described here (see Materials and Methods). (C) Quantitative imaging of positive (mock infection)- and negative (mock infection plus chloramphenicol [chlor])-control phenotypes for C. burnetii replication. Samples were fixed and stained with Hoechst and a FITC-conjugated polyclonal antibody directed against C. burnetii. Segmented fluorescent images were analyzed using Metamorph software by quantifying the total numbers and sizes of the C. burnetii vacuoles in each image. (D) Pie chart based on GeneOntology (GO) index showing the biological process categories of host factors that were overrepresented within the population of siRNA screen hits that showed a defect in C. burnetii replication. Categories names are shown with the number of host factors identified within the category and the P value calculated using the binominal statistical test within the Panther classification system.
A high-throughput fluorescence-based genome-wide siRNA screen was carried out in C. burnetii-infected HeLa cells to identify specific host genes that are important for C. burnetii infection. A human whole-genome small interfering RNA (siRNA) SMARTPool library was used to silence individual host genes following transfection into individual wells of 384-well plates containing HeLa cells (Fig. 1B). Cells were infected with the C. burnetii Nine Mile phase II strain 2 days after gene silencing. The infected cells were incubated for 4 days and fluorescently stained using an antibody specific for C. burnetii. Automated microscopy was used to identify and enumerate C. burnetii vacuoles and to measure the area of the vacuoles in infected cells. Cells treated with the antibiotic chloramphenicol served as a negative control to determine the average size of vacuoles that were present in cells in the absence of bacterial replication, and untreated cells served as the positive control to determine the average size of vacuoles that contained replicating C. burnetii. In contrast to the untreated cells, large vacuoles containing replicating bacteria were not detected in the chloramphenicol-treated cells after the 4-day incubation period; however, small fluorescent objects of fewer than five pixels in size were detected and likely represent individual internalized bacteria (Fig. 1C). These control parameters were used to screen infected cells after gene silencing in the automated analysis and to validate phenotypes after primary screening.
Genome-wide siRNA screening identifies host genes involved in C. burnetii infection.
The genome-wide screen identified target genes in which silencing led to either an increase or decrease in the total number and/or size of C. burnetii vacuoles. Z values were calculated for each target to determine the number of standard deviations (SD) above or below the mean value obtained for the control wells (see Materials and Methods). Significant hits were filtered using a Z value that was higher than 2 or lower than −2 as the threshold. Wells having a low number of cells due to specific siRNA toxicity were excluded from further analysis (see Materials and Methods). For a complete list of the significant hits along with Gene Ontology (GO) index assignments, see Table S1 in the supplemental material. Pathway analyses of the genes identified in the screen were conducted using the Panther classification system (29), which revealed several statistically overrepresented categories of biological processes. Host targets that resulted in a decrease in C. burnetii vacuole numbers and/or size were identified in distinct categories that included endocytosis, intracellular protein transport, lipid transport, and lipid metabolism (Fig. 1D). Within this broad classification are host subcategories such as vesicle-mediated transport, lysosomal transport, cholesterol metabolism, apoptosis, cell cycle, cytoskeleton, and signal transduction (see Table S1 in the supplemental material). In addition, host factors predicted to influence entry of C. burnetii into nonphagocytic cells were identified within these categories and included regulators of cytoskeletal dynamics such as the Rho GTPases, members of the Arp2/3 complex, and the adaptor protein Crk (see Table S1 in the supplemental material) (30, 31).
Several candidate genes revealing a reduction in the average size of vacuoles containing C. burnetii were selected for further validation. Initial screening data suggested that silencing of the small GTPases Rab5A and Rab7A and of individual subunits of the vacuolar-type H+ ATPase (shown is subunit ATP6V1D) resulted in a phenotype characterized by a significant reduction in the size of vacuoles containing C. burnetii (Fig. 2A). The screen also implicated the retromer-associated vacuolar protein sorting (VPS) protein VPS35 and the class C VPS protein VPS16 as being important for replication of C. burnetii. These factors were validated through additional quantitative studies that demonstrated a considerable decrease in the total number of C. burnetii vacuoles detected per field, as normalized to the total number of nuclei (Fig. 2B).
FIG 2 .

Representative C. burnetii fluorescence phenotypes identified in the siRNA screen. (A to D) Representative fluorescent images (10×) of siRNA-treated cells showing (A) fewer and smaller or (C) more and/or larger C. burnetii vacuoles at 4 days p.i. compared to mock treatment and (B and D) quantification of the fluorescent image phenotypes. Shown are C. burnetii-stained vacuoles and Hoechst-stained cell nuclei. (A) Rab5A, Rab7A, VPS16, VPS35, and ATP6V1D SMARTPool-treated cells, and Z-values corresponding to a normalized vacuole count compared to mock treatment. chlor, chloramphenicol. (B) Quantification is represented as the normalized C. burnetii vacuole count (total vacuoles/total nuclei per image) graphed as a ratio of the siRNA-treated value (Treated) versus the average value from mock-transfected wells (Untreated). (C) RAB40C (larger vacuoles), USP10 (more and larger vacuoles), and STX17 (more vacuoles) SMARTPool-treated cells, and Z-values corresponding to normalized vacuole count/percentage of large C. burnetii vacuoles. (D) Quantification is represented as both the normalized C. burnetii vacuole count (light gray) and the percentage of large C. burnetii vacuoles (dark gray). Shown are representative graphs with results from a single experiment presented as means + standard deviations. (E) Inside-out staining of C. burnetii-infected VPS35, Rab7A, and STX17 siRNA knockdowns compared to mock-transfected cells. After siRNA transfection and incubation for 3 days, cells were infected at an approximate multiplicity of infection (MOI) of 100 or 1,000 with C. burnetii for 4 h and processed for inside-out staining as described here (see Materials and Methods). Fluorescent microscopy was used to determine the percentages of cells infected.
Another phenotypic category detected in the screen was an increase in the number and/or size of C. burnetii fluorescent vacuoles (Fig. 2C). Additional quantitation validated this phenotypic category. Depletion of the ubiquitin-specific peptidase USP10 resulted in an increase in the total number of C. burnetii vacuoles as well as an increase in the percentage of large-sized C. burnetii vacuoles (Fig. 2D). Depletion of the predicted GTPase Rab40C resulted in an increase in the overall area of C. burnetii vacuoles, although the total number of vacuoles did not change significantly. Finally, a subcategory of hits was discovered in which there was a significant increase in the total number of C. burnetii vacuoles within a field of cells that did not correlate with an appreciable change in vacuole size (Fig. 2D). One of the strongest hits in this subcategory was the gene encoding syntaxin-17 (STX17). Importantly, silencing of the genes encoding Rab7A, VPS35, and syntaxin-17 did not affect infection frequencies by C. burnetii, indicating that the phenotypes resulting from gene silencing are manifested at the intracellular stage of infection (Fig. 2E). Thus, this screen identified host genes important for C. burnetii replication in mammalian cells and host genes that may play a role in restricting vacuole dynamics to limit intracellular replication of C. burnetii.
VPS35 function is important for host cell infection by C. burnetii.
The host protein VPS35 was one of the newly identified host factors predicted to be required for C. burnetii replication. VPS35 functions in the retrograde transport of specific cargo proteins from endosomes to the trans-Golgi network (TGN) and has not been described previously in the development of the C. burnetii-containing vacuole (32). To control for off-target effects that can occur when using siRNA pools to silence host gene expression, we investigated the effects of the four different individual siRNA molecules contained in the pool used to silence VPS35 in the initial screen. These data revealed that gene silencing using all four single siRNA molecules gave a phenotype that was similar to the VPS35 pool phenotype and decreased the number of C. burnetii vacuoles that were visible in host cells after infection (Fig. 3A and B). Consistent with these data, at day 3 post-siRNA transfection, quantitative real-time PCR (qRT-PCR) showed at least a 10-fold reduction in VPS35 mRNA levels for three of the four individual siRNAs, which represent reductions that were similar to that obtained using the pooled siRNAs. In addition, a reduction in VPS35 protein levels was observed by immunoblot analysis (Fig. 3C). A reduction in the number of C. burnetii bacteria in the VPS35-silenced host cells was confirmed by the decrease in total fluorescence emission detected using live HeLa cells depleted for VPS35 that were infected with C. burnetii expressing mCherry over the course of 8 days of infection (21) (Fig. 3D). These data validate that VPS35 is required for successful C. burnetii infection and demonstrate that the reduction in C. burnetii vacuoles observed in the initial screen when VPS35 expression was silenced was not due to an off-target effect.
FIG 3 .

siRNA targeting host VPS35 decreases C. burnetii replication in HeLa cells. (A) Fluorescent C. burnetii-labeled (green) and Hoechst-labeled (blue) images for the 4 individual siRNA targeting VPS35 and the parent SMARTPool compared to mock-transfected cells, and corresponding Z-values for normalized vacuole count. (B) Quantification of the fluorescent image phenotypes for the VPS35 siRNA-treated cells (total vacuole count normalized to counts of nuclei). The data are graphed as a ratio of the siRNA-treated value (Treated) versus the average value from mock-transfected wells (Untreated). Shown is a representative graph with results from a single experiment presented as means + standard deviations. (C) qRT-PCR measuring VPS35 mRNA levels in HeLa cells 3 days posttransfection for VPS35 siRNA-treated cells. Data are normalized to GAPDH and shown as fold expression level compared to mock-transfected cells. Results of an immunoblot analysis of siRNA-transfected HeLa cell lysates 3 days posttransfection measuring VPS35 and calnexin total protein levels using specific antibodies are shown. VPS35 protein expression levels were quantified by optical densitometry from immunoblots and shown below the immunoblot as fold change compared to mock-transfected cells. (D) Total fluorescence measurement of live HeLa cells infected with C. burnetii expressing mCherry. Cells subjected to mock transfection or transfected with VPS35 siRNA were incubated for 3 days before infection. The same wells were measured for total fluorescence emission at 610 nm once a day for 8 days postinfection.
The retromer complex is involved in C. burnetii infection.
VPS35 interacts with VPS29 and VPS26, which comprise a core subcomplex of the eukaryotic retromer complex that functions in retrograde transport of protein cargo from endosomes to the TGN (32). VPS35 and VPS29 were identified in the initial screen as being host factors important for C. burnetii replication. The small-vacuole phenotype observed when the genes encoding VPS35 and VPS29 were silenced suggests that a functional retromer complex may be critical for successful infection of host cells by C. burnetii. Retromer-mediated recycling of transmembrane cargo is important for efficient maturation of endocytic compartments, which suggests that maturation of the CCV membrane also requires retromer-mediated transport events. Gene silencing and quantitative immunofluorescence microscopy were used to determine whether other components of the retromer complex were important for C. burnetii replication. Depletion of VPS29 or VPS35 results in the loss of core retromer functions (33). Treatment with individual siRNA directed toward VPS29 resulted in fewer detectable CCVs at day 4 postinfection (p.i.) and was comparable to the VPS35 knockdown result (Fig. 4A). In contrast, silencing of either VPS26A or VPS26B did not result in a detectable change in CCV numbers.
FIG 4 .
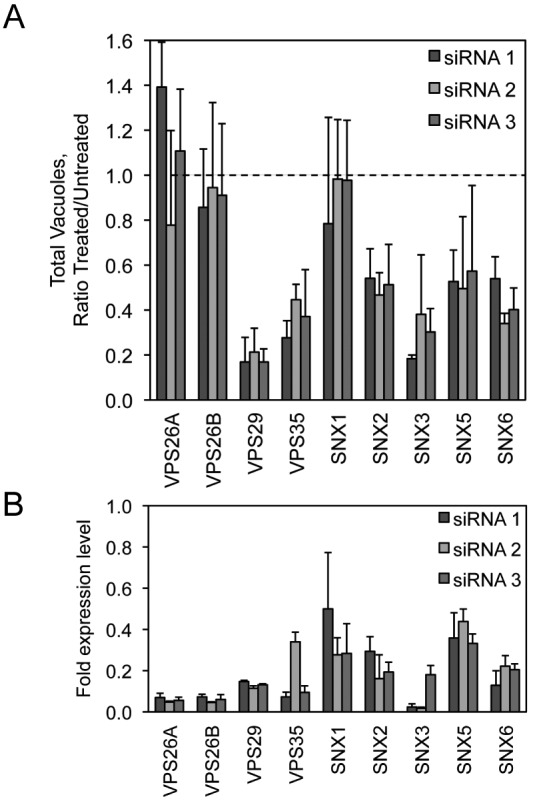
siRNA targeting the host retromer complex decreases C. burnetii replication in HeLa cells. (A) Quantification of C. burnetii::Tn mCherry fluorescent image phenotypes for cells treated with each of 3 different siRNA against individual members of the host retromer complex (VPS26A, VPS26B, VPS35, VPS29, SNX1, SNX2, SNX3, SNX5, and SNX6) compared to mock-transfected cells. The data are graphed as a ratio of the siRNA-treated value versus the average value from mock-transfected wells at 4 days p.i. for the total vacuole count normalized to counts of nuclei. (B) qRT-PCR measuring individual retromer component mRNA levels in HeLa cells 3 days posttransfection for each of 3 different siRNA targeting the corresponding retromer complex member. Data are normalized to GAPDH and shown as fold expression level compared to the mock-transfected cells.
The retromer complex is further composed of proteins that function as cargo adapters called sorting nexins, which include the proteins SNX1, SNX2, SNX3, SNX5, and SNX6. Depletion of sorting nexins interferes with the retrieval of specific cargo proteins from endosomal membranes by the retromer complex (33). Targeting of genes encoding the individual sorting nexins SNX2, SNX3, SNX5, and SNX6 using siRNA resulted in a phenotype in which the total number of C. burnetii vacuoles was significantly decreased at day 4 postinfection (Fig. 4A), comparable to the loss of VPS29 and VPS35. In contrast, depletion of the gene encoding SNX1 resulted in a phenotype in which the total number of C. burnetii vacuoles did not significantly change.
qRT-PCR using primer pairs specific to the individual retromer mRNAs showed that the mRNA levels were sufficiently depleted 3 days after siRNA transfection (Fig. 4B). Silencing of VPS26 or SNX1 did not yield significant differences in the number of the vacuoles containing C. burnetii, which could reflect that levels of gene silencing and/or protein depletion were not sufficient to reveal robust phenotypic differences in this assay. On the basis of these data, we conclude that VPS35 is required for infection of host cells by C. burnetii because a functional retromer complex is important for biogenesis of the vacuole in which this pathogen resides.
STX17 function is important for the homotypic fusion of C. burnetii vacuoles.
A phenotype revealed in the screen that was of special interest was the apparent increase in the number of vacuoles containing C. burnetii in cells where the gene encoding syntaxin-17 had been silenced (Fig. 2). The syntaxin-17 protein is a divergent member of the SNARE (Soluble NSF Attachment Protein Receptor) protein family (34). Although it is clear that SNAREs mediate membrane fusion events within the exocytic and endocytic pathways (35), whether syntaxin-17 is a bona fide SNARE has not been demonstrated and specific membrane transport processes requiring syntaxin-17 remain unknown. Thus, determining how syntaxin-17 affects biogenesis of vacuoles containing C. burnetii could provide novel insight into the function of this host factor.
A quantitative assay that measured the number of vacuoles present in cells infected with C. burnetii revealed that the majority of infected cells contained multiple vacuoles when STX17 was silenced using either an siRNA pool or the individual siRNA molecules from the pool (Fig. 5A). This was in contrast to mock-transfected cells where syntaxin-17 had not been silenced, where the majority of cells contained only a single CCV. Consistent with this finding, the individual siRNA molecules in the pool used to silence STX17 were effective at reducing STX17 mRNA levels and reducing syntaxin-17 protein levels (Fig. 5B). Direct examination of cells infected for 2 days with C. burnetii showed that silencing of STX17 resulted in multiple spacious vacuoles containing C. burnetii being maintained in a single cell. This was in contrast to control cells, where vacuoles established independently by C. burnetii that had been internalized by the same host cell ultimately coalesced into a single vacuole through homotypic fusion (Fig. 5C). Silencing of STX17 did not increase the rate of entry of C. burnetii into host cells as determined by measuring uptake through “inside-out” staining immediately after infection (Fig. 2E) and by measuring the percentage of infected cells as determined by the appearance of at least one spacious vacuole at day 2 postinfection (Fig. 5D). The multivacuole phenotype mediated by silencing of STX17 was validated using the single siRNA molecules specific for STX17. All four single siRNA molecules targeting STX17 resulted in a multivacuole phenotype following C. burnetii infection, whereas, single vacuoles were predominant in cells depleted for the related protein syntaxin-18 (STX18) (Fig. 5E) (36). Importantly, immunofluorescence microscopy confirmed that the vacuoles observed in cells depleted for STX17 and infected with C. burnetii were LAMP-1 positive, similar to the vacuoles in the mock-transfected cells (Fig. 6A). There was no evidence that the syntaxin-17 protein was directly recruited to vacuoles containing C. burnetii (Fig. 6B), suggesting that syntaxin-17 is indirectly involved in the homotypic fusion process. These data implicate syntaxin-17 as having an important role in the process that promotes homotypic fusion of vacuoles containing C. burnetii.
FIG 5 .

Depletion of syntaxin-17 increases the number of C. burnetii-containing vacuoles per cell. (A) Quantification of the fluorescent image phenotypes for the STX17 siRNA-treated cells. The data are graphed as a ratio of the siRNA-treated value versus the average value from mock-transfected wells (untreated). Shown are two phenotypic categories: total vacuole counts normalized to counts of nuclei (left) and the percentages of cells with large vacuoles (>50-pixel-sized objects) (right). Shown is a representative graph with results from a single experiment presented as means + standard deviations. (B) Left: results of an immunoblot analysis of siRNA-transfected HeLa cell lysates 3 days posttransfection measuring STX17 total protein levels using a STX17-specific antibody. The asterisk (*) represents a nonspecific band. STX17 protein expression levels were quantified by optical densitometry from immunoblots and shown below the immunoblot as fold change compared to mock-transfected cells. Right: qRT-PCR measuring STX17 mRNA levels in HeLa cells 6 days posttransfection for the 4 individual siRNA targeting STX17 and the parent SMARTPool. Data are normalized to GAPDH and shown as fold expression level compared to mock-transfected cells. (C) Fluorescent C. burnetii-stained images (10×) for STX17 SMARTPool-transfected HeLa cells compared to mock-transfected cells at 4 days p.i. (top images) and 40× phase-contrast images showing spacious C. burnetii vacuoles (arrowheads) for STX17 SMARTPool-transfected HeLa cells compared to mock-transfected cells at 2 days p.i. (bottom images). (D and E) Single-cell quantification of C. burnetii vacuole counts in STX17-transfected HeLa cells compared to STX18-transfected cells and mock-transfected cells at 2 days p.i. (D) The percentages of cells with one or more C. burnetii vacuoles were counted. (E) Subpopulations of infected cells from panel D that had two or more C. burnetii vacuoles per cell were counted.
FIG 6 .

Depletion of syntaxin-17 results in multiple C. burnetii vacuoles per cell. (A) Immunolabeling of STX17 SMARTPool-transfected HeLa cells compared to mock-transfected cells at 2 days p.i. with C. burnetii. Cells were immunostained for LAMP-1 (red) and C. burnetii (green). Cell nuclei and C. burnetii were Hoechst stained. (B) Ectopic expression of 3× Flag-STX17 in uninfected HeLa cells or HeLa cells persistently infected with C. burnetii. At 24 h posttransfection, cells were immunostained for calnexin (red) and 3× Flag (green). Cell nuclei and C. burnetii were Hoechst stained. Arrow = C. burnetii-containing vacuole.
The retromer pathway is important for delivery of C. burnetii effectors.
It remains unclear when and where the Dot/Icm system begins to translocate effectors during intracellular infection by C. burnetii. The prediction is that some of the host proteins important for C. burnetii infection confer activities needed for effector translocation by the Dot/Icm system. To determine whether host proteins identified in the siRNA screen are needed for delivery of effectors by the Dot/Icm system, we used a strain of C. burnetii producing a fusion protein called BlaM-77 that consisted of the effector protein CBU0077 having a β-lactamase enzyme fused to the amino-terminal region of the effector protein (20). The BlaM-77 fusion protein is delivered into host cells by the Dot/Icm system during infection by C. burnetii. Delivery is detected by the addition of a β-lactamase substrate, CCF4, carrying a fluorescence resonance energy transfer tag. Unprocessed CCF4 fluoresces at 535 nm; however, CCF4 that encounters cytoplasmic BlaM-77 is cleaved to a form that now fluoresces at 460 nm. The fluorescence ratio shift resulting from CCF4 cleavage is an indication of the amount of BlaM-77 protein translocated into the cytosol, which is measured using fluorescence microscopy or a spectrofluorimeter (20).
Because VPS35 and the retromer complex represent components in a pathway that has been newly identified as being important for C. burnetii infection, we set out to determine whether the retromer pathway is needed before or after the Dot/Icm system begins to deliver effector proteins. To this end, retromer components were silenced in HeLa cells before infection by C. burnetii producing BlaM-77. Coxiella burnetii producing a BlaM protein that lacks a Dot/Icm-dependent secretion signal necessary for translocation into the host cytosol was a negative control used to determine background fluorescence at 460 nm (Fig. 7A). Translocation of BlaM-77 in the silenced cells was assessed 24 h after infection. Translocation of BlaM-77 was abrogated in cells where VPS35 levels had been reduced by siRNA as determined by both fluorescence microscopy (Fig. 7A) and spectrofluorometry (Fig. 7B). Reducing VPS35 levels by gene silencing resulted in a defect in BlaM-77 translocation that was equivalent to the defect observed after reducing the levels of Rab7 by gene silencing. In contrast, silencing of STX17 had no effect on translocation of BlaM-77 (Fig. 7B). Although VPS16 was also found to be required for CCV formation (Fig. 2A; see also Table S1 in the supplemental material), we did not detect a translocation defect for BlaM-77 in cells where VPS16 had been silenced. Translocation of two other effectors was tested using the BlaM system, and these data also showed no defect in effector translocation and revealed that depletion of Rab7 in all cases reduced effector translocation, whereas depletion of VPS16 did not affect effector translocation (Fig. 3D). Thus, both VPS35 and Rab7 control important processes that precede Dot/Icm-dependent delivery of effector proteins into cells, whereas, syntaxin-17 and VPS16 are involved in host processes that occur after delivery of effectors by the Dot/Icm system.
FIG 7 .

The effect of siRNA depletion of host genes on translocation of a C. burnetii Dot/Icm effector protein. HeLa cells (Rab7A, VPS35, and STX17) treated with siRNA for 3 days were infected with C. burnetii expressing a plasmid carrying beta-lactamase (BlaM) alone or BlaM fused to the amino terminus of the C. burnetii Dot/Icm effector CBU0077 (20). At 24 h p.i., cells were incubated with the CCF4-AM substrate and assayed for translocation by (A) image analysis and (B) total fluorescence measurement. (A) The mean percentages of translocation-positive cells (lower right corner of images) were determined by visual observation of blue fluorescence emission at 460 nm indicative of CCF4-AM cleavage by translocated BlaM. (B) Translocation of BlaM and BlaM-CBU0077 was determined by measuring the change in the 460-nm/535-nm fluorescence emission ratio resulting from CCF4-AM cleavage. (C) BlaM-CBU0077 translocation from C. burnetii was measured in cells treated with each of 3 different siRNA targeting separate components of the retromer complex. (D) Left: translocation assay using 3 C. burnetii effectors (CBU0077, CBU1823, CBU1524) fused to BlaM. BlaM-effector translocation from C. burnetii was measured in cells treated with SMARTPool siRNA targeting Rab7A and VPS16. Right: qRT-PCR measuring Rab7A and VPS16 mRNA levels in HeLa cells 3 days posttransfection for SMARTPool siRNA targeting Rab7A and VPS16. Data are normalized to GAPDH and shown as fold expression levels compared to mock-transfected cells.
Because the BlaM-77 translocation assay appeared to be efficient at identifying genes important for maturation of the CCV to a compartment that supports effector translocation, this approach was used to further characterize the retromer components required for C. burnetii transport. A core retromer complex containing VPS35 and VPS29 was validated as being important for BlaM-77 translocation as indicated by defects resulting from nonoverlapping single siRNAs targeting the gene of interest (Fig. 7C). The acidotropic dye LysoTracker Red was used to test for vacuole acidification, and it was found that the number of CCVs that stained positive for LysoTracker Red in cells where VPS29 or VPS16 was silenced was similar to the number seen in control cells that were mock transfected (see Fig. S1 in the supplemental material). Thus, the defect observed in effector translocation in the VPS29-silenced cells does not correlate with a general defect in acidification of the CCV.
Because the retromer complex recruits specific sorting nexins to retrieve distinct cargo proteins from the endosomal pathway, the BlaM-77 translocation assay was used to determine if these sorting nexins were important for C. burnetii infection. Using single siRNA molecules to silence the genes encoding these proteins, SNX2 but not SNX1 was found to be important, as indicated by reduced BlaM-77 translocation levels (Fig. 7C). Additionally, silencing of the sorting nexins SNX5 and SNX6 resulted in reduced BlaM-77 translocation. A significant defect in BlaM-77 translocation was also observed when the gene encoding SNX3 was silenced. SNX3 protein is a unique retromer-associated sorting nexin that does not contain a BAR (Bin/Amphiphysin/Rvs) domain that senses membrane curvature. Thus, these data show that the retromer and most associated sorting nexins play important roles in stages of CCV maturation that are important for effector translocation (Fig. 7C).
DISCUSSION
In the present study, a human genome-wide siRNA screen was performed to uncover host proteins required for key events in C. burnetii infection. The siRNA screen validated the importance of several endocytic Rab proteins in the early stages of CCV biogenesis. Silencing of the genes encoding both Rab5 and Rab7 resulted in strong defects in C. burnetii replication as indicated by a reduction in the number and size of vacuoles after host cell infection. Our results are consistent with previous data showing that perturbing Rab7 function abrogates formation of a vacuole that supports C. burnetii replication (9). Similarly, silencing of components of the vacuolar ATPase required for acidification of endocytic compartments resulted in fewer and smaller CCVs, which is consistent with data showing that a mature acidified compartment is required for activation of C. burnetii metabolism and replication (7) and that preventing acidification of the CCV using bafilomycin A1 to inhibit the the V-ATPase function interferes with C. burnetii replication (37). In addition to components of the V-ATPase, it was found that silencing of the genes encoding the proteins CLN3 and CLCN5 interfered with CCV biogenesis, which is likely due to the role these host proteins have in regulating pH in endolysosomal compartments (38, 39). These data not only validate the importance of endocytic maturation in the biogenesis of the CCV but also provide confidence that nonbiased genome-wide siRNA screens will identify new host determinants that are involved in the CCV maturation process.
There is evidence that C. burnetii subverts the autophagy pathway during infection (9, 10). A large number of host genes are required for a functional autophagy system in mammalian cells, which means that there are a large number of targets that should have been identified in this siRNA screen if the autophagy pathway was critical for intracellular replication of C. burnetii. Thus, it was somewhat unexpected that the screen did not identify a significant number of genes encoding proteins essential for host autophagy if these host factors are indeed required for C. burnetii growth (see Table S1 in the supplemental material). This raises the possibility that host autophagy may not be essential for maturation of the CCV to a vacuole that supports replication and indicates that further research is needed to determine whether the association of autophagy proteins with the vacuole containing C. burnetii has functional significance.
The proteins VPS29 and VPS35 were identified as factors important for C. burnetii intracellular replication and were subsequently validated using individual siRNA molecules to reduce the levels of these core retromer components in host cells. The involvement of the retromer complex in C. burnetii replication has not been demonstrated previously, indicating that the genome-wide screen was successful at identifying a new host determinant important for C. burnetii infection. Because the silencing of genes encoding VPS29 and VPS35 did not affect the uptake of C. burnetii, the defect observed in replication indicates the CCVs were not progressing through the maturation pathway successfully. The retromer complex is involved primarily in selecting cargo proteins that are sorted by a retrograde pathway from endosomes to the Golgi apparatus. Host receptors sorted by the retromer include proteins such as the cation-independent mannose-6-phosphate receptor, which is involved in delivery of acid hydrolases to lysosomes (40). Thus, it is possible that perturbing this membrane transport pathway may interfere with the delivery of host factors into the lumen of the CCV that are needed for replication. Additionally, the retromer may be involved in removing proteins and membrane-associated determinants on the CCV that are detrimental to further maturation, which could delay progression of the CCV through the endocytic pathway.
Importantly, it was observed that disrupting VPS29 and VPS35 function interfered with the delivery of effector proteins into host cells by the C. burnetii Dot/Icm system. Effector protein delivery could be similarly blocked when vacuole biogenesis was perturbed by interfering with Rab7 function or by interfering with vacuole acidification. Given that the Dot/Icm system is essential for C. burnetii replication (20, 21), the defect in C. burnetii replication observed when retromer function is blocked may in large part be due to the vacuole not providing an environment that enables the Dot/Icm system to efficiently translocate effector proteins.
There is evidence that Rab7 is important for regulating the function of the retromer on endosomal membranes (41). Thus, Rab7 acquisition may represent a stage that precedes a retromer-dependent step in CCV maturation and suggests that one possible role for Rab7 function in CCV maturation is to promote retromer-mediated membrane-transport processes. Consistent with this hypothesis, the number of intracellular C. burnetii bacteria that were found in LysoTracker-positive vacuoles in the mock-transfected cells was similar to the number seen in cells where VPS16 or the VPS29 retromer had been silenced, which would be consistent with these factors acting downstream of Rab7 and suggest that the defect in replication observed in these siRNA-silenced cells is not the result of a gross defect in vacuole acidification. It remains possible that defects in retromer function or VPS16 activity delay the kinetics of CCV maturation or result in subtle differences in the pH of these vacuoles and that these effects may delay effector translocation or replication.
The VPS26-VPS29-VPS35 trimer associates with sorting nexin proteins to function in retrograde transport of vesicular-bound transmembrane cargo from endosomes to the TGN (42, 43). In mammalian cells, there are at least two different sorting nexin dimers associated with the VPS complex. Both the SNX1-SNX2 and SNX5-SNX6 dimers are composed of sorting nexins containing a BAR domain that senses membrane curvature, which leads to the assembly of a retromer complex that generates membrane tubules that retrieve cargo proteins from endocytic organelles. The SNX3 protein also associates with the VPS complex but does not have a BAR domain and is involved in retrieval of unique cargo proteins such as the Wnt receptor (44). Replication of C. burnetii was reduced when the gene encoding SNX2, but not SNX1, was silenced by siRNA, indicating that SNX2 is involved in the important role the retromer has in CCV biogenesis. Although depletion of SNX1 had no detectable effect on C. burnetii replication, this could reflect an inability to efficiently silence SNX1 or diminish SNX1 protein levels. Additionally, intracellular growth defects were detected after C. burnetii infection in cells where genes encoding other retromer-associated sorting nexins were silenced. Silencing of these genes resulted in more-robust defects in the translocation of BlaM-77 by C. burnetii during infection. Because the BlaM-77 translocation assay provides a quantitative measure for host processes that are important for early CCV maturation events, these data suggest that CCV biogenesis is retarded in the absence of SNX2, SNX3, SNX5, or SNX6. Thus, the strong intracellular growth defect observed in cells where the VPS complex is depleted likely results from interfering with multiple membrane transport pathways requiring different sorting nexins.
Lastly, VPS35 has also been shown to localize to mitochondrion-derived vesicles and thereby facilitate retromer-mediated vesicular transport of cargo between mitochondria and peroxisomes (45). Although the function of VPS35 in this capacity has not been fully characterized, it is plausible that the retromer participates in communication between mitochondria and peroxisomes that may result in regulation of peroxisome biogenesis or function. Several C. burnetii Dot/Icm effectors localize to mitochondria, including the CBU0077 effector protein (20). The role of mitochondria in C. burnetii pathogenesis is unknown; however, several molecules are shared within the mitochondria and endocytic networks (46), suggesting a similar overlap with C. burnetii transport. Thus, the retromer may function during C. burnetii infection both at the level of membrane sorting at the CCV and also through maintaining important membrane transport pathways at other locations that are indirectly important for C. burnetii intracellular replication.
In addition to identifying genes encoding host proteins important for early events in CCV maturation that enable intracellular replication, the genome-wide screen identified phenotypes that included larger or a greater number of vacuoles containing C. burnetii in the infected host cells. Silencing of the gene encoding syntaxin-17 resulted in one of the more interesting phenotypes in this category, which was defined as infected cells having a greater number of CCVs. It was determined that this phenotype was not due to a higher rate of infection, although even at a high multiplicity of infection, it is unusual to find infected cells that have multiple CCVs because of the high degree of homotypic fusion observed between CCVs in the same cell, which ultimately stimulates the coalescing of all CCVs to form one large vacuole. These data suggest that syntaxin-17 has a role in regulating the kinetics of homotypic fusion.
The syntaxin-17 protein is a divergent member of the SNARE family of proteins. SNARE proteins are involved in the fusion of intracellular vesicles. Identified in a yeast two-hybrid screen as a potential syntaxin-3 binding partner (34), the syntaxin-17 protein is found localized to the endoplasmic reticulum (ER) (47) and on vesicles that cycle between the ER and the ER-Golgi intermediate compartment (48). Importantly, this defect in homotypic fusion of CCVs was specific for syntaxin-17, as the phenotype was not observed in cells after silencing of the gene encoding syntaxin-18, which is a related ER-localized SNARE protein. Expression of syntaxin-17 in C. burnetii-infected HeLa cells reveals staining that overlaps with calnexin-positive structures indicative of ER; however, there is no detectable localization of syntaxin-17 to the CCV. This suggests that a syntaxin-17-dependent transport pathway or some other activity regulated by syntaxin-17 is needed for homotypic fusion of the CCV but that syntaxin-17 itself may not be directly involved in the homotypic fusion reaction. Because reducing syntaxin-17 levels in host cells did not affect either C. burnetii replication or translocation of BlaM-77, which is in contrast to what is observed when Rab7 or components of the retromer were reduced in cells, these data indicate that homotypic fusion occurs only after a mature vacuole is established that contains metabolically active C. burnetii capable of translocating effectors using the Dot/Icm system.
Finally, the genome-wide siRNA screen identified the class C vacuolar-protein-sorting protein VPS16 as being required for C. burnetii replication in HeLa cells, similar to the phenotypes seen for Rab7 and VPS35. However, unlike the Rab7 and retromer knockdowns, there was no corresponding defect observed in translocation of BlaM fusions to three different C. burnetii effectors. Based on homology to the yeast class C Vps protein, mammalian VPS16 is implicated in vesicle transport within the endolysosomal pathway and may mediate membrane docking and fusion events (49, 50). As with syntaxin-17, the data for VPS16 indicate that this protein functions only after a mature vacuole is established and therefore may have an important role in the C. burnetii vacuole biogenesis after effector translocation has occurred.
In conclusion, these data reveal that genome-wide siRNA screening can be used to identify host pathways important for distinct stages of C. burnetii intracellular infection. With the recent developments in genetic manipulation and axenic cultivation of C. burnetii (51), it should be possible to design new screening strategies that utilize C. burnetii producing fluorescent proteins or BlaM-effector fusions and to monitor infection over time after siRNA treatment to identify pathways important for infection, replication, effector translocation, and vacuole expansion.
MATERIALS AND METHODS
Reagents.
Chemicals were purchased from Sigma, unless otherwise noted. Restriction and molecular cloning enzymes were purchased from New England BioLabs.
Cell culture and RNA silencing.
HeLa 229 cells (ATCC) were maintained in DMEM (Dulbecco minimal Eagle’s medium) (Invitrogen) containing 5 to 10% fetal bovine serum (FBS) at 37°C in 5% CO2. Small interfering RNA (siRNA) duplexes were purchased from Dharmacon and diluted in 1× siRNA buffer (Thermo Scientific). siRNA transfections were carried out with Dharmafect-1 (Thermo Scientific) in serum-free DMEM according to the manufacturer’s protocol.
Bacterial strains.
Coxiella burnetii (Nine Mile strain in phase II) was propagated in HeLa cells in DMEM–5% FBS at 37°C in 5% CO2 or grown axenically in Acidified Citrate Cysteine Media (ACCM) at 37°C in 5% CO2 and 2.5% O2 as described previously (51). Axenically grown C. burnetii bacteria were quantified by qPCR using dotA-specific primers (6).
Genome-wide siRNA screen.
A small-interfering RNA (siRNA) SMARTPool library covering 21,121 genes of the human genome was purchased from Dharmacon and divided into aliquots in black clear-bottom 384-well plates (Corning) which were sealed and stored at −20°C until the time of transfection. Dharmafect-1 in serum-free DMEM was added to the siRNA in each well and allowed to form siRNA-lipid complexes. HeLa cells were then added to the wells at 2,000 cells per well. The final concentration of total siRNA per well was 50 nM. After 48 h at 37°C, the siRNA-transfected cells were infected with C. burnetii for 5 h, washed, and incubated for 4 days at 37°C, during which time the media were replaced on day 3 postinfection (p.i.). Wells were fixed with 4% paraformaldehyde (PFA) and immunostained with rabbit anti-C. burnetii polyclonal antibody (1:10,000 dilution), followed by secondary incubation with anti-rabbit Alexa Fluor 488-conjugated secondary antibody (Invitrogen) (1:2,000 dilution). DNA was stained with Hoechst 33342 (Invitrogen).
Data acquisition.
Automated microscopy was carried out using computer-assisted fluorescent imaging software (MetaMorph, v. 7.1; Molecular Devices Inc.). A Nikon TE 2000 computerized inverted fluorescence microscope fitted with an ORCA-ER digital CCD (charge-coupled device) (Hamamatsu) automatically captured focused fluorescent images of the infected cells from 4 different fields within each well across the entire 384-well plate. Images were captured in the DAPI (4',6-diamidino-2-phenylindole) channel to detect host cell nuclei and in the FITC (fluorescein isothiocyanate) channel to detect C. burnetii.
Data analysis.
The software calculates nuclear DNA/C. burnetii fluorescence ratios for each image. These were then compared to ratios from internal positive-control wells in each plate in which intracellular C. burnetii replication occurred in the absence of siRNA (mock) and from negative-control wells in which intracellular C. burnetii replication was prevented by the addition of chloramphenicol. Data from the individual wells were organized into phenotypic categories based on the relative C. burnetii fluorescence ratio compared to control wells. To assign statistical significance, Z values were calculated by subtracting the control (mock) population mean within a given 384-well plate from an individual (siRNA) raw score within the same plate and then dividing the difference by the control (mock) population standard deviation.
Several fluorescent images were further quantified for differences in vacuole size distributions using a custom MatLab (MathWorks, 2010a) algorithm. The original 8-bit images were converted to binary using Otsu’s thresholding method (52). Objects within the binary image, defined as having a Moore neighborhood value of 1, were split into large and small categories using a morphological opening algorithm (bwareaopen.m; Image processing Toolbox; MathWorks) with a constant threshold set by the user. The total numbers of large and small objects were acquired using a binary image processing algorithm (bwconncomp.m). To allow comparisons across images, both large and small objects were normalized by dividing by the total number of objects.
Validation.
For the validation experiments, 4 individual siRNAs specific to a single human mRNA target were used with the following GenBank accession numbers: VPS35 (NM_018206), VPS29 (NM_016226), VPS26A (NM_004896), VPS26B (NM_052875), SNX1 (NM_148955), SNX2 (NM_003100), SNX3 (NM_003795), SNX5 (NM_014426), SNX6 (NM_152233), STX17 (NM_017919), STX18 (NM_016930), RAB5A (NM_004162), RAB7A (NM_004637), ATP6V1D (NM_015994), and VPS16 (NM_022575).
For several validation experiments, C. burnetii expressing the mCherry marker from a transposon insertion was used to detect C. burnetii fluorescence in infected HeLa cells after fixation, as described above. In addition, live HeLa cells infected with C. burnetii Tn::mCherry in 96-well plates were measured for total fluorescence emission with a Tecan M1000 plate reader, using an excitation value of 587 nm and emission at 610 nm.
Real-time PCR.
HeLa cells were transfected with siRNA in 96-well plates and incubated for at least 3 days prior to cell lysis in the presence of DNase using a Cells-to-Ct kit (Ambion) per the manufacturer’s instructions. Quantitative real-time PCR was performed from cDNA generated with the kit using gene-specific primer pairs and iQ SYBR green Supermix using an iCycler iQ real-time PCR detection system (Bio-Rad). Gene expression levels were normalized to GAPDH (glyceraldehyde-3-phosphate dehydrogenase) expression using a GAPDH-specific primer pair.
Immunoblot analysis.
HeLa cells were transfected with siRNA in 24-well plates and incubated for at least 3 days prior to cell lysis in buffer containing 20 mM Tris (pH 7.4), 100 mM NaCl, 1 mM MgCl2, 1% Triton X-100, 1 mM PMSF (phenylmethylsulfonyl fluoride), 1 mM DTT (dithiothreitol), and protease inhibitor cocktail. Lysates were centrifuged and the supernatant separated by SDS-PAGE for immunoblot analysis using an anti-STX17 antibody (Sigma), anti-VPS35 antibody (Abcam), or anti-calnexin antibody (Enzo Life Sciences). Quantitation of immunoblots was obtained by optical densitometry using ImageJ software (http://imagej.nih.gov/ij/).
Cloning of STX17.
Human syntaxin-17 cDNA was purchased from Open Biosystems (Thermo Scientific), amplified by PCR, and introduced into the BamHI site of pcDNA 4/T0 carrying a 3× Flag marker under the control of the CMV (cytomegalovirus) promoter. The resulting plasmid was transfected using Effectene (Qiagen) into uninfected or persistently C. burnetii-infected HeLa cells. After 24 h incubation, the cells were processed for fluorescence microscopy.
Fluorescence microscopy.
Cells were seeded in 24-well plates with 12-mm-diameter coverslips, incubated for at least 24 h, and fixed with 4% PFA. Fixed cells were permeabilized with 0.05% saponin and processed for immunofluorescence microscopy. Samples were incubated with primary antibodies in 0.5% BSA (bovine serum albumin)-0.2% saponin at the concentrations indicated: anti-LAMP1 H4A3-C (Developmental Studies Hybridoma Bank) (1:300 dilution), anti-C. burnetii (1:10,000 dilution), anti-Flag M2 (Sigma) (1:1,000 dilution), and anti-calnexin (Enzo Life Sciences) (1:250 dilution). Samples were then incubated with secondary antibodies (Alexa Fluor 488 and 546; Invitrogen) (1:2,000 dilution). Bacterial and host cell DNA was labeled using Hoechst. Coverslips were mounted on slides using ProLong Gold (Invitrogen). Digital images were acquired with a Nikon Eclipse TE2000-S inverted fluorescence microscope using a 100×/1.4 numerical aperture objective lens and a Hamamatsu Photonics ORCA-ER camera controlled by IPLab imaging software.
Inside-out staining was used to differentiate extracellular from intracellular bacteria. First, extracellular bacteria were stained with rabbit anti-C. burnetii antibody in PBS (phosphate-buffered saline)-2% BSA followed by Alexa Fluor 594-conjugated anti-rabbit antibodies (Invitrogen). Cells were washed and permeabilized with saponin, and total bacteria were stained with mouse anti-C. burnetii followed by Alexa Fluor 488-conjugated anti-mouse antibodies. After washes in PBS, the coverslips were mounted onto glass slides using ProLong Gold. Bacterial and cell nuclei were stained with DAPI (Invitrogen).
For syntaxin-17 validation experiments, cells were transfected with siRNA and infected with C. burnetii as described above and were fixed at 48 h p.i. Cells were then coimmunostained using anti-LAMP-1 and anti-C. burnetii. Cells and CCVs were counted in 3 independent wells to determine the percentages of cells that were infected and the number of CCVs per cell.
For LysoTracker experiments, HeLa cells were transfected with siRNA, incubated for 3 days, and infected with C. burnetii. At 24 h p.i., cells were incubated in media containing 0.5 µM LysoTracker Red DND-99 (Molecular Probes) for 2 h at 37°C and were fixed. Cells were immunostained using antibodies specific for C. burnetii and tubulin to indicate cell morphology. As a control, growth media containing 0.1 µM bafilomycin A1 was added at 18 h p.i., 6 h prior to LysoTracker treatment.
BlaM translocation assay.
Construction of C. burnetii carrying the plasmid pJB-CAT-BlaM-effector (CBU0077, CBU1524, or CBU1823) and the resulting translocation have been described previously (20). siRNA transfection of HeLa cells was carried out prior to infection. siRNA was divided into aliquots in a black clear-bottomed 96-well dish (Corning), and transfections were carried out as described above using Dharmafect-1 transfection reagent and 1.2 × 104 HeLa cells per well. After 72 h incubation at 37°C, the siRNA-transfected cells were infected with C. burnetii phase II bacteria and incubated for 24 h at 37°C. Cells were loaded with the fluorescent substrate CCF4/AM, using a LiveBLAzer-FRET B/G loading kit (Invitrogen) with 15 mM probenecid, as described previously (20). After 2 h incubation at room temperature in the dark, the total fluorescence, using an excitation value of 415 nm and emission at 460 nm and 535 nm, was quantified using a Tecan M1000 plate reader. In addition, single cells were imaged by fluorescence microscopy. A total of 400 cells were counted in 3 independent wells to determine the percentages of cells that were BlaM positive.
SUPPLEMENTAL MATERIAL
Silencing of the retromer protein VPS29 does not alter trafficking of C. burnetii to a LysoTracker-positive vacuole. (A) Mock-transfected or siRNA-treated HeLa cells were infected with C. burnetii for 24 h and then incubated with media containing LysoTracker Red for 2 h before immunostaining with antibodies to C. burnetii (green) and tubulin (blue). Areas indicated with arrows are magnified in the boxes below the image. Bafilomycin-A1 treatment was used as a negative control for LysoTracker fluorescence. (B) Quantification of C. burnetii numbers per cells. (C) C. burnetii colocalization with LysoTracker Red. Data represent means ± SD; n = 30 infected cells per group. Download
Annotated gene list for significant siRNA screen hits. Human genes identified in the siRNA screen as having significantly different C. burnetii growth phenotypes after siRNA treatment compared to mock-treated cells are listed within two phenotypic categories: (a) total vacuole counts (normalized to total counts of nuclei) and (b) percent small vacuoles. Z values were assigned and represent a measure of the number of standard deviations an individual (siRNA-treated cells) phenotypic value is away from the mean of the mock-transfected controls. For either phenotypic category, a Z value higher than 2 indicates a significant enhancement over mock-transfected cells, while a Z value lower than −2 indicates a significant reduction. Hits are listed with National Center for Biotechnology Information (NCBI) reference sequence (RefSeq) accession numbers and were assigned GO indexes using the Panther classification system (http://www.pantherdb.org/).
Oligonucleotide sequences used in this study.
ACKNOWLEDGMENTS
We thank Isabelle Derré and members of the Roy laboratory for helpful advice. This research was supported by a Ruth L. Kirschstein National Research Service Award Individual Postdoctoral Fellowship (F32-AI0829272) to J.A.M. and by NIH grants (R01-AI064559 and Northeast Biodefense Center U54-AI057158-Lipkin) to C.R.R.
Footnotes
Citation McDonough JA, Newton HJ, Klum S, Swiss R, Agaisse H, Roy CR. 2013. Host pathways important for Coxiella burnetii infection revealed by genome-wide RNA interference screening. mBio 4(1):e00606-12. doi:10.1128/mBio.00606-12.
REFERENCES
- 1. Maurin M, Raoult D. 1999. Q fever. Clin. Microbiol. Rev. 12:518–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fenollar F, Fournier PE, Carrieri MP, Habib G, Messana T, Raoult D. 2001. Risks factors and prevention of Q fever endocarditis. Clin. Infect. Dis. 33:312–316 [DOI] [PubMed] [Google Scholar]
- 3. Madariaga MG, Rezai K, Trenholme GM, Weinstein RA. 2003. Q fever: a biological weapon in your backyard. Lancet Infect. Dis. 3:709–721 [DOI] [PubMed] [Google Scholar]
- 4. Alix E, Mukherjee S, Roy CR. 2011. Subversion of membrane transport pathways by vacuolar pathogens. J. Cell Biol. 195:943–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bhavsar AP, Guttman JA, Finlay BB. 2007. Manipulation of host-cell pathways by bacterial pathogens. Nature 449:827–834 [DOI] [PubMed] [Google Scholar]
- 6. Coleman SA, Fischer ER, Howe D, Mead DJ, Heinzen RA. 2004. Temporal analysis of Coxiella burnetii morphological differentiation. J. Bacteriol. 186:7344–7352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hackstadt T, Williams JC. 1981. Biochemical stratagem for obligate parasitism of eukaryotic cells by Coxiella burnetii. Proc. Natl. Acad. Sci. U. S. A. 78:3240–3244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Romano PS, Gutierrez MG, Berón W, Rabinovitch M, Colombo MI. 2007. The autophagic pathway is actively modulated by phase II Coxiella burnetii to efficiently replicate in the host cell. Cell. Microbiol. 9:891–909 [DOI] [PubMed] [Google Scholar]
- 9. Berón W, Gutierrez MG, Rabinovitch M, Colombo MI. 2002. Coxiella burnetii localizes in a Rab7-labeled compartment with autophagic characteristics. Infect. Immun. 70:5816–5821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gutierrez MG, Vázquez CL, Munafó DB, Zoppino FC, Berón W, Rabinovitch M, Colombo MI. 2005. Autophagy induction favours the generation and maturation of the Coxiella-replicative vacuoles. Cell. Microbiol. 7:981–993 [DOI] [PubMed] [Google Scholar]
- 11. Hechemy KE, McKee M, Marko M, Samsonoff WA, Roman M, Baca O. 1993. Three-dimensional reconstruction of Coxiella burnetii-infected L929 cells by high-voltage electron microscopy. Infect. Immun. 61:4485–4488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Howe D, Barrows LF, Lindstrom NM, Heinzen RA. 2002. Nitric oxide inhibits Coxiella burnetii replication and parasitophorous vacuole maturation. Infect. Immun. 70:5140–5147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Howe D, Melnicáková J, Barák I, Heinzen RA. 2003. Maturation of the Coxiella burnetii parasitophorous vacuole requires bacterial protein synthesis but not replication. Cell. Microbiol. 5:469–480 [DOI] [PubMed] [Google Scholar]
- 14. Howe D, Melnicákova J, Barák I, Heinzen RA. 2003. Fusogenicity of the Coxiella burnetii parasitophorous vacuole. Ann. N. Y. Acad. Sci. 990:556–562 [DOI] [PubMed] [Google Scholar]
- 15. Howe D, Heinzen RA. 2005. Replication of Coxiella burnetii is inhibited in CHO K-1 cells treated with inhibitors of cholesterol metabolism. Ann. N. Y. Acad. Sci. 1063:123–129 [DOI] [PubMed] [Google Scholar]
- 16. Howe D, Heinzen RA. 2006. Coxiella burnetii inhabits a cholesterol-rich vacuole and influences cellular cholesterol metabolism. Cell. Microbiol. 8:496–507 [DOI] [PubMed] [Google Scholar]
- 17. Voth DE, Heinzen RA. 2007. Lounging in a lysosome: the intracellular lifestyle of Coxiella burnetii. Cell. Microbiol. 9:829–840 [DOI] [PubMed] [Google Scholar]
- 18. Zamboni DS, McGrath S, Rabinovitch M, Roy CR. 2003. Coxiella burnetii express type IV secretion system proteins that function similarly to components of the Legionella pneumophila Dot/Icm system. Mol. Microbiol. 49:965–976 [DOI] [PubMed] [Google Scholar]
- 19. Zusman T, Yerushalmi G, Segal G. 2003. Functional similarities between the icm/dot pathogenesis systems of Coxiella burnetii and Legionella pneumophila. Infect. Immun. 71:3714–3723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Carey KL, Newton HJ, Lührmann A, Roy CR. 2011. The Coxiella burnetii Dot/Icm system delivers a unique repertoire of type IV effectors into host cells and is required for intracellular replication. PLoS Pathog. 7:e1002056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Beare PA, Gilk SD, Larson CL, Hill J, Stead CM, Omsland A, Cockrell DC, Howe D, Voth DE, Heinzen RA. 2011. Dot/Icm type IVB secretion system requirements for Coxiella burnetii growth in human macrophages. mBio 2:e00175-00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hubber A, Roy CR. 2010. Modulation of host cell function by Legionella pneumophila type IV effectors. Annu. Rev. Cell Dev. Biol. 26:261–283 [DOI] [PubMed] [Google Scholar]
- 23. Mukherjee S, Liu X, Arasaki K, McDonough J, Galán JE, Roy CR. 2011. Modulation of Rab GTPase function by a protein phosphocholine transferase. Nature 477:103–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pan X, Lührmann A, Satoh A, Laskowski-Arce MA, Roy CR. 2008. Ankyrin repeat proteins comprise a diverse family of bacterial type IV effectors. Science 320:1651–1654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Voth DE, Beare PA, Howe D, Sharma UM, Samoilis G, Cockrell DC, Omsland A, Heinzen RA. 2011. The Coxiella burnetii cryptic plasmid is enriched in genes encoding type IV secretion system substrates. J. Bacteriol. 193:1493–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lührmann A, Nogueira CV, Carey KL, Roy CR. 2010. Inhibition of pathogen-induced apoptosis by a Coxiella burnetii type IV effector protein. Proc. Natl. Acad. Sci. U. S. A. 107:18997–19001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Voth DE, Howe D, Beare PA, Vogel JP, Unsworth N, Samuel JE, Heinzen RA. 2009. The Coxiella burnetii ankyrin repeat domain-containing protein family is heterogeneous, with C-terminal truncations that influence Dot/Icm-mediated secretion. J. Bacteriol. 191:4232–4242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen C, Banga S, Mertens K, Weber MM, Gorbaslieva I, Tan Y, Luo ZQ, Samuel JE. 2010. Large-scale identification and translocation of type IV secretion substrates by Coxiella burnetii. Proc. Natl. Acad. Sci. U. S. A. 107:21755–21760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thomas PD, Kejariwal A, Campbell MJ, Mi H, Diemer K, Guo N, Ladunga I, Ulitsky-Lazareva B, Muruganujan A. 2003. PANTHER: a browsable database of gene products organized by biological function, using curated protein family and subfamily classification. Nucleic Acids Res. 31:334–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Misselwitz B, Dilling S, Vonaesch P, Sacher R, Snijder B, Schlumberger M, Rout S, Stark M, von Mering C, Pelkmans L, Hardt WD. 2011. RNAi screen of Salmonella invasion shows role of COPI in membrane targeting of cholesterol and Cdc42. Mol. Syst. Biol. 7:474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Pielage JF, Powell KR, Kalman D, Engel JN. 2008. RNAi screen reveals an Abl kinase-dependent host cell pathway involved in Pseudomonas aeruginosa internalization. PLoS Pathog. 4:e1000031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Seaman MN, McCaffery JM, Emr SD. 1998. A membrane coat complex essential for endosome-to-Golgi retrograde transport in yeast. J. Cell Biol. 142:665–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rojas R, Kametaka S, Haft CR, Bonifacino JS. 2007. Interchangeable but essential functions of SNX1 and SNX2 in the association of retromer with endosomes and the trafficking of mannose 6-phosphate receptors. Mol. Cell. Biol. 27:1112–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Steegmaier M, Yang B, Yoo JS, Huang B, Shen M, Yu S, Luo Y, Scheller RH. 1998. Three novel proteins of the syntaxin/SNAP-25 family. J. Biol. Chem. 273:34171–34179 [DOI] [PubMed] [Google Scholar]
- 35. Jahn R, Scheller RH. 2006. SNAREs—engines for membrane fusion. Nat. Rev. Mol. Cell Biol. 7:631–643 [DOI] [PubMed] [Google Scholar]
- 36. Hatsuzawa K, Hirose H, Tani K, Yamamoto A, Scheller RH, Tagaya M. 2000. Syntaxin 18, a SNAP receptor that functions in the endoplasmic reticulum, intermediate compartment, and cis-Golgi vesicle trafficking. J. Biol. Chem. 275:13713–13720 [DOI] [PubMed] [Google Scholar]
- 37. Heinzen RA, Scidmore MA, Rockey DD, Hackstadt T. 1996. Differential interaction with endocytic and exocytic pathways distinguish parasitophorous vacuoles of Coxiella burnetii and Chlamydia trachomatis. Infect. Immun. 64:796–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Novarino G, Weinert S, Rickheit G, Jentsch TJ. 2010. Endosomal chloride-proton exchange rather than chloride conductance is crucial for renal endocytosis. Science 328:1398–1401 [DOI] [PubMed] [Google Scholar]
- 39. Golabek AA, Kida E, Walus M, Kaczmarski W, Michalewski M, Wisniewski KE. 2000. CLN3 protein regulates lysosomal pH and alters intracellular processing of Alzheimer’s amyloid-beta protein precursor and cathepsin D in human cells. Mol. Genet. Metab. 70:203–213 [DOI] [PubMed] [Google Scholar]
- 40. Kornfeld S. 1992. Structure and function of the mannose 6-phosphate/insulinlike growth factor II receptors. Annu. Rev. Biochem. 61:307–330 [DOI] [PubMed] [Google Scholar]
- 41. Rojas R, van Vlijmen T, Mardones GA, Prabhu Y, Rojas AL, Mohammed S, Heck AJ, Raposo G, van der Sluijs P. 2008. Regulation of retromer recruitment to endosomes by sequential action of Rab5 and Rab7. J. Cell Biol. 183:513–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bonifacino JS, Hurley JH. 2008. Retromer. Curr. Opin. Cell Biol. 20:427–436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bonifacino JS, Rojas R. 2006. Retrograde transport from endosomes to the trans-Golgi network. Nat. Rev. Mol. Cell Biol. 7:568–579 [DOI] [PubMed] [Google Scholar]
- 44. Harterink M, Port F, Lorenowicz MJ, McGough IJ, Silhankova M, Betist MC, van Weering JR, van Heesbeen RG, Middelkoop TC, Basler K, Cullen PJ, Korswagen HC. 2011. A SNX3-dependent retromer pathway mediates retrograde transport of the Wnt sorting receptor Wntless and is required for Wnt secretion. Nat. Cell Biol. 13:914–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Braschi E, Goyon V, Zunino R, Mohanty A, Xu L, McBride HM. 2010. Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr. Biol. 20:1310–1315 [DOI] [PubMed] [Google Scholar]
- 46. McBride HM, Neuspiel M, Wasiak S. 2006. Mitochondria: more than just a powerhouse. Curr. Biol. 16:R551–R560 [DOI] [PubMed] [Google Scholar]
- 47. Steegmaier M, Oorschot V, Klumperman J, Scheller RH. 2000. Syntaxin 17 is abundant in steroidogenic cells and implicated in smooth endoplasmic reticulum membrane dynamics. Mol. Biol. Cell 11:2719–2731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Muppirala M, Gupta V, Swarup G. 2011. Syntaxin 17 cycles between the ER and ERGIC and is required to maintain the architecture of ERGIC and Golgi. Biol. Cell 103:333–350 [DOI] [PubMed] [Google Scholar]
- 49. Peterson MR, Emr SD. 2001. The class C Vps complex functions at multiple stages of the vacuolar transport pathway. Traffic 2:476–486 [DOI] [PubMed] [Google Scholar]
- 50. Huizing M, Didier A, Walenta J, Anikster Y, Gahl WA, Krämer H. 2001. Molecular cloning and characterization of human VPS18, VPS 11, VPS16, and VPS33. Gene 264:241–247 [DOI] [PubMed] [Google Scholar]
- 51. Omsland A, Cockrell DC, Howe D, Fischer ER, Virtaneva K, Sturdevant DE, Porcella SF, Heinzen RA. 2009. Host cell-free growth of the Q fever bacterium Coxiella burnetii. Proc. Natl. Acad. Sci. U. S. A. 106:4430–4434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Otsu N. 1979. A threshold selection method from gray-level histograms. IEEE Transact. Syst. Man Cybern. 9:62–66 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Silencing of the retromer protein VPS29 does not alter trafficking of C. burnetii to a LysoTracker-positive vacuole. (A) Mock-transfected or siRNA-treated HeLa cells were infected with C. burnetii for 24 h and then incubated with media containing LysoTracker Red for 2 h before immunostaining with antibodies to C. burnetii (green) and tubulin (blue). Areas indicated with arrows are magnified in the boxes below the image. Bafilomycin-A1 treatment was used as a negative control for LysoTracker fluorescence. (B) Quantification of C. burnetii numbers per cells. (C) C. burnetii colocalization with LysoTracker Red. Data represent means ± SD; n = 30 infected cells per group. Download
Annotated gene list for significant siRNA screen hits. Human genes identified in the siRNA screen as having significantly different C. burnetii growth phenotypes after siRNA treatment compared to mock-treated cells are listed within two phenotypic categories: (a) total vacuole counts (normalized to total counts of nuclei) and (b) percent small vacuoles. Z values were assigned and represent a measure of the number of standard deviations an individual (siRNA-treated cells) phenotypic value is away from the mean of the mock-transfected controls. For either phenotypic category, a Z value higher than 2 indicates a significant enhancement over mock-transfected cells, while a Z value lower than −2 indicates a significant reduction. Hits are listed with National Center for Biotechnology Information (NCBI) reference sequence (RefSeq) accession numbers and were assigned GO indexes using the Panther classification system (http://www.pantherdb.org/).
Oligonucleotide sequences used in this study.