

Abstract
The gonococcal transferrin receptor is composed of two distinct proteins, TbpA and TbpB. TbpA is a member of the TonB-dependent family of integral outer membrane transporters, while TbpB is lipid modified and thought to be peripherally surface exposed. We previously proposed a hypothetical topology model for gonococcal TbpA that was based upon computer predictions and similarity with other TonB-dependent transporters for which crystal structures have been determined. In the present study, the hemagglutinin epitope was inserted into TbpA to probe the surface topology of this protein and secondarily to test the functional impacts of site-specific mutagenesis. Twelve epitope insertion mutants were constructed, five of which allowed us to confirm the surface exposure of loops 2, 3, 5, 7, and 10. In contrast to the predictions set forth by the hypothetical model, insertion into the plug region resulted in an epitope that was surface accessible, while epitope insertions into two putative loops (9 and 11) were not surface accessible. Insertions into putative loop 3 and β strand 9 abolished transferrin binding and utilization, and the plug insertion mutant exhibited decreased transferrin-binding affinity concomitant with an inability to utilize it. Insertion into putative β strand 16 generated a mutant that was able to bind transferrin normally but that was unable to mediate utilization. Mutants with insertions into putative loops 2, 9, and 11 maintained wild-type binding affinity but could utilize only transferrin in the presence of TbpB. This is the first demonstration of the ability of TbpB to compensate for a mutation in TbpA.
Nearly all microorganisms require iron for growth and metabolism (29, 52). Human pathogens have the unique dilemma of proliferating in an extremely low-iron-content environment that is established and maintained by the human host. In order to combat these low iron concentrations (which may be as low as 10−18 M) (9), bacteria have developed a variety of iron acquisition mechanisms. One common way of acquiring iron is via the secretion of siderophores, which are high-affinity iron binding molecules that deliver the bound iron directly to the cell by means of a specific ferric-siderophore receptor. Two such receptors in Escherichia coli are FepA and FhuA, which transport the siderophores ferric enterobactin and ferrichrome, respectively. FepA and FhuA have been crystallized, and the three-dimensional structures were determined at high resolution (8, 22). Both of these proteins form pores in the outer membrane and are members of the TonB-dependent class of transporters (40, 49). The crystal structures revealed that both proteins are composed of two unique domains: the C-terminal β-barrel domain and the N-terminal globular plug domain. The β barrel of both proteins is composed of 22 transmembrane β strands. The N-terminal globular plug domain occludes the pore formed by the barrel and spans the outer membrane from the periplasm to the extracellular environment (for a review, see reference 21).
FepA and FhuA rely on TonB-derived energy to sequester iron from the environment. TonB, which interacts with ExbB and ExbD in the cytoplasmic membrane (1, 27), transduces energy from the proton motive force to TonB-dependent transporters located in the outer membrane (6, 48). These outer membrane receptors share several homologous domains, three of which are located within the globular plug domain, while the others are located within the β strands of the barrel (18, 30). The first domain, the TonB box, is located within the plug domain and has been shown by physical and genetic means to be important for interaction between TonB and the TonB-dependent receptors it services (11, 25, 32, 36).
There is no evidence that Neisseria gonorrhoeae produces any siderophores (53). Instead, this exclusive human pathogen can directly bind and utilize host iron binding proteins, such as transferrin, as the sole iron source (43). The gonococcal transferrin receptor is composed of two dissimilar proteins, transferrin-binding proteins A and B (TbpA and TbpB) (18). TbpA is a TonB-dependent receptor and putatively forms a pore in the outer membrane, like FepA and FhuA (18). TbpA is required for transferrin utilization and is responsible for removing the iron from transferrin and for transporting iron across the outer membrane in a TonB-dependent manner (16, 18). Unlike FepA and FhuA, TbpA has an accessory protein, TbpB, which is a lipidated, surface-exposed protein that binds transferrin independently of TbpA and which increases the efficiency of transferrin-iron uptake (2).
Although we previously proposed a hypothetical topology model of TbpA as a basis for guiding deletion mutagenesis studies (5), this model was completely computer generated and has never been tested experimentally. A limited deletion analysis was performed, the results of which implicated putative loops 4 (L4) and 5 (L5) as critical for both transferrin binding and utilization (5). However, this deletion study could not directly address whether these regions were surface exposed. Because TbpA is a candidate for incorporation into a vaccine to prevent neisserial infections (15, 20, 23, 38), elucidating surface-exposed and/or functional epitopes is of considerable importance. The epitope tagging approach has been instrumental in elucidating surface topology and structure-function relationships for several other outer membrane proteins, including LamB (12, 13, 37, 46), FepA (3), and FhuA (44, 45). In the present study, we inserted the hemagglutinin (HA) epitope (YPYDVPDYA) into TbpA as a surface probe and also as a means for the generation of site-directed mutants, with which the functional importance of these regions could be evaluated.
MATERIALS AND METHODS
Strains and media.
The gonococcal strains utilized in this study are described in Table 1. Gonococci were routinely propagated on GC medium base (Difco) with Kellogg's supplement 1 (31) and 12 μM Fe(NO3)3. Streptomycin was added to GC medium agar plates at a concentration of 100 μg/ml for selection of the streptomycin resistance phenotype. Gonococci were grown at 35°C in a 5% CO2 atmosphere. For growth under iron-stressed conditions, the gonococci were cultured from GC medium agar plates (without added iron) into liquid chelexed defined medium (CDM) (54) which was pretreated with Chelex-100 (Bio-Rad). CDM-agarose plates were supplemented with 30% iron-saturated human transferrin in order to assess each mutant's ability to utilize transferrin-bound iron (18). Mutant strains were grown on CDM-transferrin plates for 24 to 48 h. Plasmids (described in Table 1) were propagated in E. coli strain TOP10 (Table 1), which was routinely grown in Luria-Bertani medium with the addition of ampicillin (50 μg/ml) or kanamycin (50 μg/ml) (41).
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Phenotypea (genotype) or description | Source or reference |
|---|---|---|
| Strains | ||
| E. coli | ||
| TOP10 | F−mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 recA1 deoR araD139 Δ(ara-leu)7697 galU galK rpsL(Strr) endA1 nupG | Invitrogen |
| N. gonorrhoeae | ||
| FA19 | Wild type | 43 |
| FA6905 | TbpB− (ΔtbpB) | 19 |
| FA6815 | TbpA− TbpB− (tbpB::Ω) | 2 |
| FA6839 | FA19, Lbp− (lbpB::Ω) | 4 |
| MCV505 | L5HA(509) TbpA Lbp− (tbpA∇HA lbpB::Ω) | This study |
| MCV506 | L5HA(509) TbpA TbpB− Lbp− (tbpA∇HA lbpB::Ω ΔtbpB) | This study |
| MCV507 | PHA(110) TbpA Lbp− (tbpA∇HA lbpB::Ω) | This study |
| MCV508 | PHA(110) TbpA TbpB− Lbp− (tbpA∇HA lbpB::Ω ΔtbpB) | This study |
| MCV509 | L7HA(642) TbpA Lbp− (tbpA∇HA lbpB::Ω) | This study |
| MCV510 | L7HA(642) TbpA TbpB− Lbp− (tbpA∇HA lbpB::Ω ΔtbpB) | This study |
| MCV511 | L3HA(343) TbpA Lbp− (tbpA∇HA lbpB::Ω) | This study |
| MCV512 | L3HA(343) TbpA TbpB− Lbp− (tbpA∇HA lbpB::Ω ΔtbpB) | This study |
| MCV513 | T2HA(296) TbpA Lbp− (tbpA∇HA lbpB::Ω) | This study |
| MCV514 | T2HA(296) TbpA TbpB− Lbp− (tbpA∇HA lbpB::Ω ΔtbpB) | This study |
| MCV515 | L9HA(750) TbpA Lbp− (tbpA∇HA lbpB::Ω) | This study |
| MCV516 | L9HA(750) TbpA TbpB− Lbp− (tbpA∇HA lbpB::Ω ΔtbpB) | This study |
| MCV517 | L10HA(809) TbpA Lbp− (tbpA∇HA lbpB::Ω) | This study |
| MCV518 | L10HA(809) TbpA TbpB− Lbp− (tbpA∇HA lbpB::Ω ΔtbpB) | This study |
| MCV519 | L11HA(843) ΔAla847 TbpA Lbp− (tbpA∇HA lbpB::Ω) | This study |
| MCV520 | L11HA(843) ΔAla847 TbpA TbpB− Lbp− (tbpA∇HA lbpB::Ω ΔtbpB) | This study |
| MCV521 | β9HA(479) TbpA Lbp− (tbpA∇HA lbpB::Ω) | This study |
| MCV522 | β9HA(479) TbpA TbpB− Lbp− (tbpA∇HA lbpB::Ω ΔtbpB) | This study |
| MCV523 | β16HA(713) TbpA Lbp− (tbpA∇HA lbpB::Ω) | This study |
| MCV524 | β16HA(713) TbpA TbpB− Lbp− (tbpA∇HA lbpB::Ω ΔtbpB) | This study |
| MCV525 | T4HA(467) TbpA Lbp− (tbpA∇HA lbpB::Ω) | This study |
| MCV526 | T4HA(467) TbpA TbpB− Lbp− (tbpA∇HA lbpB::Ω ΔtbpB) | This study |
| MCV527 | L2HA(229) TbpA Lbp− (tbpA∇HA lbpB::Ω) | This study |
| MCV528 | L2HA(229) TbpA TbpB− Lbp− (tbpA∇HA lbpB::Ω ΔtbpB) | This study |
| Plasmids | ||
| pCR2.1-TOPO | (Kanr Ampr) | Invitrogen |
| pHSS6GCU | Vector containing gonococcal uptake sequence (Kanr) | 50 |
| pUNCH411 | pBS-SK(+) containing entire tbpA gene | 17 |
| pVCU509 | pCR2.1 containing tbpA gene amplified by PCR with primers oVCU100 and oVCU101; HA in L5 | This study |
| pVCU511 | pCR2.1 containing tbpA gene fragment amplified between primers Tfbp5 and Tfbp22; HA in P | This study |
| pVCU512 | pCR2.1 containing tbpA gene fragment amplified by PCR with primers Tfbp22 and Tfbp32; HA in L2 | This study |
| pVCU513 | pCR2.1 containing tbpA gene fragment amplified by PCR with primers Tfbp36 and Tfbp18; HA in L7 | This study |
| pVCU514 | pHSS6GCU containing EcoRI fragment from pVCU511 | This study |
| pVCU515 | pHSS6GCU containing EcoRI fragment from pVCU512 | This study |
| pVCU516 | pHSS6GCU containing EcoRI fragment from pVCU513 | This study |
| pVCU517 | pCR2.1 containing tbpA gene fragment amplified by PCR with primers Tfbp3 and Tfbp15; HA in L3 | This study |
| pVCU518 | pHSS6GCU containing EcoRI fragment from pVCU517 | This study |
| pVCU519 | pCR2.1 containing tbpA gene fragment amplified by PCR with primers Tfbp2 and Tfbp17; HA in T2 | This study |
| pVCU520 | pHSS6GCU containing EcoRI fragment from pVCU519 | This study |
| pVCU521 | pCR2.1 containing tbpA gene fragment amplified by PCR with primers Tfbp24 and oVCU7; HA in L9 | This study |
| pVCU522 | pCR2.1 containing tbpA gene fragment amplified by PCR with primers Tfbp24 and oVCU7; HA in L10 | This study |
| pVCU523 | pCR2.1 containing tbpA gene fragment amplified by PCR with primers Tfbp24 and oVCU7;HA in L11 | This study |
| pVCU524 | pCR2.1 containing tbpA gene fragment amplified by PCR with primers Tfbp13 and Tfbp28; HA in β9 | This study |
| pVCU525 | pCR2.1 containing tbpA gene fragment amplified by PCR with primers Tfbp24 and oVCU7; HA in β16 | This study |
| pVCU526 | pCR2.1 containing tbpA gene fragment amplified by PCR with primers Tfbp13 and Tfbp28; HA in T4 | This study |
| pVCU527 | pHSS6GCU containing EcoRI fragment from pVCU524 | This study |
| pVCU528 | pHSS6GCU containing EcoRI fragment from pVCU526 | This study |
Phenotype description includes the position of the last amino acid position prior to the HA insertion (in parentheses) after the loop number designation. ∇, insertion of HA. Strr, streptomycin resistant; Kanr, kanamycin resistant; Ampr, ampicillin resistant; Tf, transferrin.
Epitope insertion mutagenesis.
The sequence encoding the HA epitope (YPYDVPDYA) was inserted into approximately 1-kb segments of the tbpA gene essentially by the method of Horton et al. (28). Briefly, in the first part of this two-step PCR technique, upstream and downstream amplicons were amplified by using HA-encoding primers oVCU102 to oVCU125 and tbpA-specific primers (Table 2). The template for these reactions was a plasmid, pUNCH411, which contained the entire tbpA gene (Table 1). For the secondary PCR, the two amplicons from the primary PCR were mixed together along with the same tbpA-specific primers used in the primary PCR. The HA-encoding regions overlapped and primed one another in this second reaction. The final PCR product encoding the HA epitope was gel extracted (Qiagen) and cloned into pCR2.1-TOPO (Table 1). Plasmids pVCU509 to pVCU528 (Table 1) contained the epitope-tagged gene fragments and were propagated in E. coli strain TOP10 (Table 1). Mutated DNA fragments were sequenced to ensure preservation of the reading frame and the proper HA-encoding sequence. Every mutant constructed maintained the native flanking tbpA sequence, with the exception of the L11HA mutant, from which alanine 847 was deleted. No restorative mutagenesis was undertaken.
TABLE 2.
Oligonucleotides used in this study
| Primer name | Oligonucleotide sequencea | Target |
|---|---|---|
| HA-encoding primers | ||
| oVCU102 | GTC CGG GAC GTC GTA CGG GTA CGT CCT CGT CCC GCC | Plug |
| oVCU103 | CCG TAC GAC GTC CCG GAC TAC GCC GCG GGC AGC AGC GGC | Plug |
| oVCU112 | GTC CGG GAC GTC GTA CGG GTA GCT GCC GTC ATC AAC CGG | L2 |
| oVCU113 | CCG TAC GAC GTC CCG GAC TAC GCC AAG TAC GCC TAT TTC ATC GTT | L2 |
| oVCU114 | GTC CGG GAC GTC GTA CGG GTA GCC GTA TTT GTG GTT GCC | L3 |
| oVCU115 | CCG TAC GAC GTC CCG GAC TAC GCC GGA CTG TTT ACC AGC GGC | L3 |
| oVCU106 | GTC CGG GAC GTC GTA CGG GTA GTT GTT TTG AGG GGG CGT | L5 |
| oVCU107 | CCG TAC GAC GTC CCG GAC TAC GCC GGC AAA AAA ACC AGC CCC | L5 |
| oVCU116 | GTC CGG GAC GTC GTA CGG GTA TAT TTT ATC GCC CGA CCG C | L7 |
| oVCU117 | CCG TAC GAC GTC CCG GAC TAC GCC AAA GCC GTC AAA ATC GAT CC | L7 |
| oVCU128 | GTC CGG GAC GTC GTA CGG GTA GTC TGC GCG TTT TTT GAT GTC | L9 |
| oVCU129 | CCG TAC GAC GTC CCG GAC TAC GCC CGC ACC GAT ATT CAA TCA CAC | L9 |
| oVCU130 | GTC CGG GAC GTC GTA CGG GTA GCG GCT GTT GCC GTT GAG | L10 |
| oVCU131 | CCG TAC GAC GTC CCG GAC TAC GCC AAT ACA AAA GCC ACC GCG C | L10 |
| oVCU132 | GTC CGG GAC GTC GTA CGG GTA GTA AAC GCC GAC ATT TTT GTG | L11 |
| oVCU133 | CCG TAC GAC GTC CCG GAC TAC GCC AAC CGA TAT GCCb CCC GGC C | L11 |
| oVCU120 | GTC CGG GAC GTC GTA CGG GTA AAA ACG AAA ACC CGG GCG G | T2 |
| oVCU121 | CCG TAC GAC GTC CCG GAC TAC GCC GAA AAC AAA CGG CAC TAC ATC | T2 |
| oVCU122 | GTC CGG GAC GTC GTA CGG GTA GGC GGT ATC GAA GGA TTT TTT | T4 |
| oVCU123 | CCG TAC GAC GTC CCG GAC TAC GCC AAA ATC CGC CAC AAC CTG AG | T4 |
| oVCU134 | GTC CGG GAC GTC GTA CGG GTA GTA ACC GAG ATT CAC GCT CA | β9 |
| oVCU135 | CCG TAC GAC GTC CCG GAC TAC GCC GAC CGC TTC GGC TCT AAT C | β9 |
| oVCU124 | GTC CGG GAC GTC GTA CGG GTA ATT GAT GCC GGT AAT CCG C | β16 |
| oVCU125 | CCG TAC GAC GTC CCG GAC TAC GCC ATT TTG GGC AAA ATC GAT TGG | β16 |
| tbpA-specific primers | ||
| Tfbp2 | AGC GGC GCA TTG GCG GGC TC | |
| Tfbp3 | GGG GCG CAT CGG CGG TGC GG | |
| Tfbp5 | AAA ACA GTT GGA TAC CAT AC | |
| Tfbp13 | GGT CGT GGC TGT TCC GCC CG | |
| Tfbp15 | TAT AAG TAT TGT TGC CAA GA | |
| Tfbp17 | AAC CGA GAT TCA CGC TCA GG | |
| Tfbp18 | GCC ATA AGC TCT TGC AGG CG | |
| Tfbp22 | ATC GGC ATT GGT ATA GAC AT | |
| Tfbp24 | CCG CAC CCT GTC CTG GAA CG | |
| Tfbp28 | AAA TCC AGC CAG TCG GCA GG | |
| Tfbp32 | AAG CTC CGG CTA CTC GAT AC | |
| Tfbp36 | GCT TCC TCG CCG ATC CGC | |
| oVCU7 | CTC GAG GCT CTA GAA ACC CCA ACG CAG | |
| oVCU100 | ATG CAA CAG CAA CAT TTG TTC | |
| oVCU101 | GAA ACG GTG GGG ATT GTG |
Underlined sequence indicates the nucleotides that encode the HA epitope.
Mutagenic primer encoding alanine 847 deletion.
Gonococcal transformation.
Mutagenized tbpA gene fragments that did not include a naturally occurring gonococcal uptake sequence (10, 24) were subcloned into pHSS6GCU (50), which contains this sequence. Plasmids consisting of an epitope insertion in tbpA and a gonococcal uptake sequence were used to transform N. gonorrhoeae strains essentially by the method of Gunn and Stein (26), with the following modifications. Congression was used to provide a selectable marker for the transformation event. Chromosomal DNA from strain FA6839 (Table 1), which has an Ω cassette inserted into the lbpB gene (encoding lactoferrin-binding protein B) (4), was used in conjunction with the linearized plasmid DNA containing epitope-tagged tbpA. These donor DNAs were used simultaneously to transform piliated FA19 or FA6905 (ΔtbpB) (Table 1). Transformants were selected by streptomycin resistance (encoded by the Ω cassette), and a PCR screen was employed to identify the tbpA insertion mutants among the streptomycin-resistant transformants. In this way, we simultaneously generated HA insertion mutants that were incapable of expressing the Lbp system, with which TbpA-specific antibodies cross-react (18). This resulted in the creation of strains MCV505 to MCV528 (Table 1), all of which contained a tbpA gene mutagenized with the HA-encoding sequence. Segments of these chromosomal genes were PCR amplified and sequenced to ensure proper HA insertion and maintenance of the reading frame.
Protein detection by Western blotting.
Gonococcal strains were grown in liquid CDM to induce iron stress (54); after 4 h of growth, aliquots were removed and standardized to culture density. In order to detect the insertionally mutagenized TbpA protein, cells were pelleted and lysed with Laemmli solubilizing buffer (35). Five percent β-mercaptoethanol and 6 M urea were added, and then the samples were incubated at 4°C for at least 24 h. The urea-treated whole-cell lysates were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (35) without the inclusion of additional urea in the gel system. Proteins were transferred to polyvinylidene difluoride (Bio-Rad). Blots were blocked with Western blocking reagent (Roche Molecular Biosciences) and probed with an anti-HA-peroxidase high-affinity (3F10) monoclonal antibody (Roche Molecular Biosciences). The blots were developed with ECL Plus (Amersham Biosciences) and exposed to film. To detect TbpA, we treated the samples with 5% β-mercaptoethanol and boiled them for 2 min; no urea was added. TbpA was detected by Western blot analysis using a polyclonal antibody raised against full-length TbpA, as described previously (19). Alternatively, TbpA was detected with polyclonal antisera raised against defined regions of TbpA, corresponding to putative loops 4 and 5, as described previously (42).
Solid-phase anti-HA MAb binding assay.
Gonococcal strains were grown in liquid CDM for 4 h, standardized to culture density, and spotted onto nitrocellulose (Schleicher and Schuell). Blots were blocked with Western blocking reagent (Roche Molecular Biosciences) and probed with a monoclonal anti-HA monoclonal antibody (MAb) conjugated to peroxidase (Roche Molecular Biosciences) as described above. Blots were developed with Opti4CN (Bio-Rad).
Equilibrium-phase, transferrin-binding assay.
Equilibrium-phase, transferrin-binding assays were performed as described previously (19). Human transferrin (Calbiochem) was iodinated with 125I (Amersham) to a specific activity of 5 × 105 to 7 × 105 cpm/μg of Tf. Both iodinated transferrin and unlabeled competitor transferrin were quantitated using the bicinchoninic acid assay (Pierce). Capacity, Kd, and associated errors were calculated with Grafit software (Erithacus Software).
Protease accessibility assay.
Protease accessibility experiments were performed as described previously (19). Briefly, whole, iron-stressed gonococci were exposed to trypsin for 0, 10, 20, or 30 min and were then pelleted, lysed, and separated by SDS-PAGE. After proteins were transferred to nitrocellulose or polyvinylidene difluoride, the membrane was probed with polyclonal TbpA-specific antiserum, or alternatively with horseradish peroxidase-conjugated anti-HA MAb (Roche Molecular Biosciences) as described above.
RESULTS
Mutants express full-length, epitope-tagged TbpA protein.
In order to test the hypothetical TbpA topology model (5), we inserted the HA epitope into various regions of TbpA (Fig. 1). For the HA epitope mutagenesis, we chose as targets seven regions corresponding to putatively surface-exposed loops, two putative periplasmic turns, two predicted β strands, and the putative plug domain (Fig. 1). Using sequence data derived from comparisons between gonococcal TbpAs and the TbpAs of other human and animal pathogens (15), we targeted regions of some sequence diversity within this group of well-conserved proteins. We reasoned that this sequence variability or plasticity might allow the protein to accommodate the epitope insertion with relatively little disruption of structure. The HA mutations were generated by PCR, and the amplicons were cloned in E. coli. Subsequently, the wild-type gonococcal strain (FA19) and an isogenic tbpB mutant (FA6905) were transformed with these mutagenized tbpA gene fragments to facilitate analysis of the mutated alleles in single copy. For clarity, the gonococcal HA insertion mutants were named according to the putative topological feature into which the HA epitope was inserted. Insertions into the putative loops resulted in mutants prefixed with the letter L (L2HA, L3HA, L5HA, L7HA, L9HA, L10HA, and L11HA). Insertions into putative β strands resulted in mutants prefixed with the symbol β (β9HA and β16HA). Insertions into putative turns resulted in mutants prefixed with T (T2HA and T4HA). Finally, insertion into the putative plug region resulted in the mutant designated PHA (Fig. 1 and Table 1).
FIG. 1.

Hypothetical topological model of gonococcal TbpA. Schematic representation of the hypothetical two-dimensional model of gonococcal TbpA proposed previously (5). The horizontal dashed lines represent the boundaries of the outer membrane. The protein sequence is shown as the solid black line, with putative transmembrane β strands shown as vertical boxes. Putative extracellular loops are designated L1 through L11. The open circles represent six conserved cysteine residues. The locations of the HA epitopes inserted during the present study are indicated by dark gray boxes. The previously proposed trypsin cleavage sites are indicated with asterisks. The modified positions of accessible trypsin cleavage sites demonstrated by the present study are shown as black bars.
We determined that all mutants produced a full-length TbpA protein that was detectable with TbpA-specific polyclonal antiserum (Fig. 2A) (19). In addition, all of the mutagenized TbpA proteins were detectable with epitope-specific antisera (42) that recognized regions corresponding to putative loops 4 and 5 (Fig. 2B). When these polyclonal antibody reagents were used, there was no evidence of proteolysis or degradation products. Moreover, the mutant strains expressed similar amounts of the HA fusion proteins, as evaluated in the Western blot analysis. These results indicate that the mutagenized proteins are stably expressed by the gonococcus and that the insertion mutation did not result in dramatically altered expression characteristics.
FIG. 2.

Expression of epitope-tagged TbpA proteins. Iron-stressed gonococci were lysed, and whole-cell proteins were separated by SDS-PAGE. Following protein transfer to solid support, blots were probed with one of the following antibody preparations: polyclonal anti-TbpA serum (A), polyclonal serum specific for epitopes within putative loops 4 and 5 of TbpA (B), and high-affinity, anti-HA monoclonal antibody conjugated to peroxidase (C and D). Relative to the blot in panel C, the blot represented in panel D was prepared with twofold more protein antigen, as described in Materials and Methods. Each lane is labeled above according to the strain from which the whole-cell proteins were derived. Those lanes containing proteins isolated from the TbpB− derivatives are highlighted with the bracket. The approximate positions of molecular mass markers (MWM), in kilodaltons, are located to the left of each blot.
Each of the mutants also expressed the HA epitope, which was detectable with a monoclonal anti-HA antibody (Fig. 2C and D). As anticipated, the anti-HA MAb detected a TbpA derivative in each mutant strain; however, this antibody did not react uniformly with all the mutant TbpAs tested. Eight of the epitope insertion derivatives were readily detected with the anti-HA MAb (Fig. 2C); however, the other insertion derivatives (T2, T4, β9, and β16) required twofold more protein to be detectable by Western blot analysis (Fig. 2D). Despite these variations in MAb reactivity, all HA-tagged samples produced a full-length TbpA protein. This finding is in contrast to our previous observations with TbpA proteins that were tagged at various positions with the c-myc epitope. TbpA-c-myc fusions were either not detectable at all or were detectable only as proteolytic fragments by Western blot analysis (data not shown). These data indicate that the TbpA-HA insertions were expressed and stable in gonococci but that some aspects of primary or secondary structure might influence the level of epitope reactivity with the HA-specific MAb as detectable by Western blotting. We next sought to determine if the HA epitopes were accessible in native TbpA, as presented on the gonococcal outer membrane.
Determination of surface accessibility of TbpA-HA epitope fusions.
We analyzed the surface accessibility of the HA epitope insertions in TbpA by probing whole, iron-stressed gonococci, affixed to nitrocellulose, with the anti-HA MAb. We utilized this solid-phase antibody binding assay as a qualitative assessment of the ability of the anti-HA MAb to interact with the gonococcal cell surface. As shown in Fig. 3, the HA epitope was clearly accessible in mutants L2HA, L3HA, L5HA, L7HA, and L10HA, thus confirming the extracellular location of these areas of the protein, in support of the hypothetical topology model (Fig. 1). The HA tag in mutants L9HA, L11HA, T2HA, T4HA, β9HA, and β16HA were not accessible in this assay; therefore, the cellular location of these areas of TbpA cannot be confirmed. In contrast to the predicted reaction, the plug domain insertion was readily detected with the anti-HA MAb, suggesting that this region was at least partially surface exposed. Two of the epitope-tagged TbpA constructs (PHA and L3HA) expressed in a wild-type strain (TbpB+) demonstrated a decreased reaction with the anti-HA antibody compared to that of the isogenic tbpB mutant strain (Fig. 3). This observation is consistent with a close association between TbpA and TbpB on the bacterial surface and suggests that access to portions of TbpA can be occluded by TbpB expression.
FIG. 3.
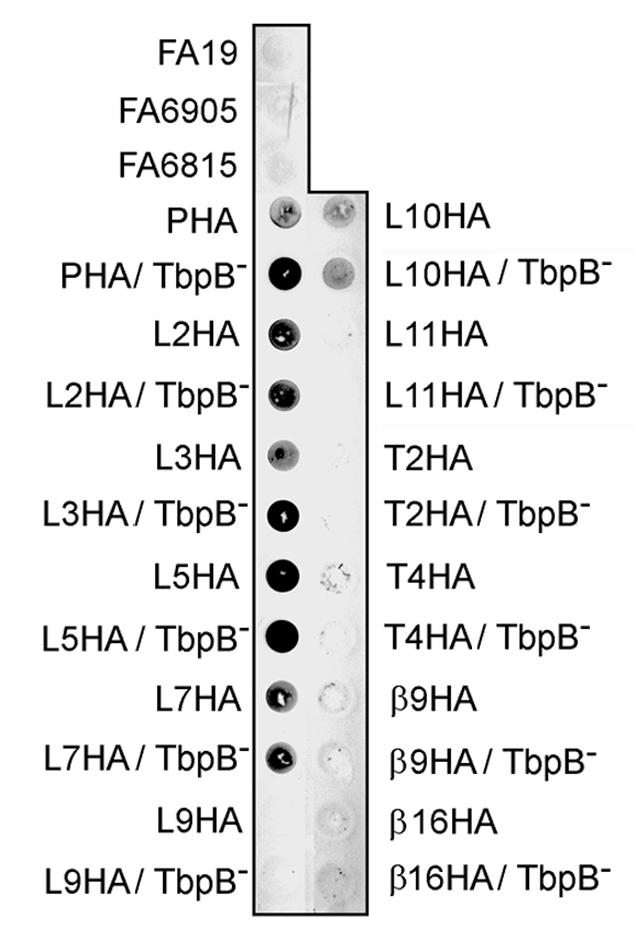
Solid-phase, anti-HA antibody binding. Whole, iron-stressed gonococcal cells were applied to a nitrocellulose membrane and probed with a high-affinity monoclonal anti-HA antibody. Dots are labeled to the left and right according to the strain that was applied to the membrane. The wild-type (FA19), TbpB− (FA6905), and TbpA− TbpB− (FA6815) strains served as negative controls since they do not contain the HA epitope.
Epitope insertions affect both binding and transferrin-iron utilization.
The first step in determining the functional impacts of these mutations was to test their transferrin-binding capability. We have previously suggested that solid-phase transferrin-binding assays are inadequate for quantitation of transferrin-binding parameters, as this assay underestimated the ligand binding attributable to gonococcal TbpB (19). In retrospect, it seems quite likely that the underestimation of TbpB-specific binding in the solid-phase assay was instead due to the poor saturation of the commercially available horseradish peroxidase-labeled transferrin used for this assay. In contrast, transferrin binding in the liquid-phase assays was evaluated with fully saturated transferrin used as the ligand, for which the TbpB protein has a marked preference (14, 19). This new interpretation notwithstanding, the solid-phase binding assay can be used to qualitatively evaluate interaction between whole cells and either transferrin (14, 19) or antibody (Fig. 3). However, only in equilibrium-phase binding assays can quantitative measures of affinity and receptor number (capacity) be discerned in the context of a whole, energized cell. Therefore, to evaluate the binding parameters of all of the HA insertion mutants, we performed equilibrium-phase transferrin-binding assays. Figure 4 shows representative examples of the isotherm plots generated from these assays, which are grouped according to the predicted location of the insertionally mutagenized region: plug domain (Fig. 4A), extracellular loops (Fig. 4B), and periplasmic turns and β strands (Fig. 4C). All of the isotherms were transformed into Scatchard plots by Grafit software, from which Kd and capacity values were calculated (Table 3). Many of the insertion mutations had no dramatic impact on the mutants' transferrin-binding abilities (Fig. 4 and Table 3). The PHA, L3HA, and β9HA mutants were exceptions. L3HA and β9HA insertion mutants demonstrated no detectable transferrin binding in this assay (Fig. 4B and C; Table 3). The plug insertion mutant demonstrated a decrease in affinity; the calculated Kd was approximately 10-fold higher than the wild-type value (Fig. 4A and Table 3). Quantitative Western blot analysis was used to evaluate the expression level of TbpA in the epitope insertion mutants. In the course of this analysis, the mutant strains, grown in triplicate on different days, were analyzed by Western blotting and compared against a standard curve of TbpA (data not shown). In this manner, the relative amount of TbpA expressed by each strain analyzed in the equilibrium-phase assays could be compared. Using this method, we determined that all strains evaluated in this assay expressed, on average, 70 to 150% of wild-type TbpA levels; thus, the changes in the apparent capacity were not a result of poor TbpA expression by the mutants. These results indicate that insertion into most putative loops did not dramatically alter ligand-binding functions; however, mutagenized regions in the L3HA, β9HA, and PHA mutants appear to play crucial roles in this binding process.
FIG. 4.

125I-transferrin binding isotherms. Whole, iron-stressed gonococci were mixed with various amounts of 125I-labeled human transferrin, and specific binding of the ligand was calculated by subtracting nonspecific binding (with excess competitor) from total binding. The amount of specific transferrin bound is reported on the y axis as nanograms of transferrin (Tf) bound per microgram of total cell protein (TCP). Isotherms are grouped for display according to the putative location of the epitope tag within TbpA: plug (A), extracellular loops (B), and periplasmic turns and β strands (C). Representative samples from each group are shown. The data shown represent the means and standard deviations generated from three independent binding experiments.
TABLE 3.
Affinity and capacity measurements generated from equilibrium-phase binding assays conducted with epitope insertion mutants
| Strain | Description of mutation | Kda (nM) | Capacitya (no. of receptors/ μg of TCPb) |
|---|---|---|---|
| FA6905 | Wild type | 10.4 ± 2.0 | 2.1 × 108 |
| MCV508 | PHA TbpA | 120.5 ± 74.8 | 3.3 × 108 |
| MCV528 | L2HA TbpA | 18.0 ± 5.3 | 1.8 × 108 |
| MCV512 | L3HA TbpA | ||
| MCV506 | L5HA TbpA | 14.8 ± 5.2 | 2.2 × 108 |
| MCV510 | L7HA TbpA | 8.7 ± 1.9 | 1.2 × 108 |
| MCV516 | L9HA TbpA | 20.6 ± 2.6 | 1.6 × 108 |
| MCV518 | L10HA TbpA | 5.8 ± 0.5 | 1.8 × 108 |
| MCV520 | L11HA TbpA | 28.6 ± 8.2 | 1.3 × 108 |
| MCV514 | T2HA TbpA | 6.6 ± 1.5 | 1.4 × 108 |
| MCV526 | T4HA TbpA | 21.3 ± 5.0 | 2.0 × 108 |
| MCV522 | β9HA TbpA | ||
| MCV524 | β16HA TbpA | 16.2 ± 6.6 | 7.8 × 107 |
Kd, capacity and error calculated with Grafit software.
TCP, total cellular protein determined by bicinchoninic acid assay.
To function in vivo, TbpA must mediate two separable phenomena: transferrin binding (assessed in the previous assay) and transferrin-iron utilization (evaluated in the experiments whose results are depicted in Fig. 5). On the basis of the results of binding and growth assays, the mutants can be separated into several distinct categories. The first category of mutants, including L5HA, L7HA, L10HA, T2HA, and T4HA, had no discernible phenotype, since there was no defect in terms of either binding or utilization of transferrin (Table 3 and Fig. 5). This result strongly reinforces the assertion that incorporation of small epitopes into TonB-dependent receptors is well tolerated and has little impact on the overall topology of the protein. In the second category of mutants, exemplified by L3HA and β9HA, no transferrin-binding capability was detected, and mutants in this category could not grow when they were provided with transferrin as the sole iron source. Insertion into the plug domain resulted in a mutant that had a 10-fold-higher Kd than that of the wild type and that could not grow with transferrin as the sole iron source (Table 3 and Fig. 5). This category of mutants was anticipated, since the inability to bind transferrin, or a severe decrease in binding affinity, would be expected to prevent initiation of the utilization process. The β16HA mutant was the lone representative of a third category; this mutant bound ligand at near-wild-type levels but could not grow with transferrin as the sole iron source (Table 3 and Fig. 5). The final category of mutants, represented by L2HA, L9HA, and L11HA, bound transferrin with wild-type affinity but could utilize transferrin only in the presence of TbpB (Fig. 5). All the other mutants maintained their growth phenotypes regardless of the presence of TbpB. Taken together, these results suggest that the transferrin-iron acquisition process can be genetically separated into several phases: initial interaction with ligand (defective in the L3HA mutant), internalization of iron (defective in the β16HA mutant), and cooperation between TbpA and TbpB to facilitate iron uptake (illustrated by the L9HA mutant).
FIG. 5.

Transferrin utilization growth assay. The ability of mutants to utilize transferrin as the sole iron source was evaluated by 24-h growth on CDM plates supplemented with 5 μM human transferrin (ca. 30% saturated with iron). The growth phenotype of each TbpA epitope insertion mutant was evaluated in a TbpB+ (shown as +) and in a TbpB− (shown as −) background. Each plate growth assay included a positive control strain expressing TbpA (TbpA+) and a negative control strain which did not express TbpA (TbpA−); neither control strain expressed TbpB.
TbpAs expressed by insertion mutants are surface exposed.
Several of the insertion mutants described in this study did not bind the anti-HA antibody in a dot blot assay—an outcome that may be the result of improper processing or surface presentation of mutagenized TbpAs. In order to evaluate whether the mutated proteins were appropriately surface exposed, we performed protease accessibility assays. By exposing whole, iron-stressed gonococci to low concentrations of trypsin for various lengths of time, we documented the surface presentation and overall topology of the mutagenized TbpAs. This assay has previously been used to examine surface exposure and conformation of the TbpA deletion mutants (5) and also to evaluate the conformations of TbpA and TbpB as a function of TonB-derived energy (16, 19). All of the TbpA proteins expressed by the insertion mutants generated in the present study were accessible to trypsin, as the full-length protein was proteolytically cleaved over the 30-min time course (Fig. 6 and data not shown). Despite the large number of predicted trypsin cleavage sites in TbpA, only two diagnostic cleavage fragments are detected in wild-type TbpA (5, 16, 19); the products generated by this cleavage have been designated T1 (95.2-kDa fragment) and T2 (54.9-kDa fragment). The results (Fig. 6 and data not shown) indicate that 4 of the 12 epitope insertions resulted in localized modifications in TbpA's surface accessibility. Exposure of L3HA and β9HA to trypsin resulted in the expected products, except that the L3 digestion products lacked T1 and the β9 products lacked T2. The L2 insertion mutant showed the same cleavage pattern as that seen in L3HA, and the β16 insertion mimicked the digestion pattern of the β9 insertion mutant (data not shown). Inaccessibility of some ordinarily accessible trypsin cleavage sites is consistent with localized impacts due to the insertion; however, digestion of the full-length protein in all of the mutants and the appearance of the other expected products suggests that these mutagenized proteins are exported properly and presented in a conformation that is at least similar to that of the wild-type protein.
FIG. 6.

Trypsin accessibility of epitope-tagged TbpA mutants. Iron-stressed gonococci were incubated with trypsin for 0, 10, 20, or 30 min before the protease was inactivated by the addition of aprotinin. The trypsinized cells were pelleted, lysed, and separated by SDS-PAGE. After transfer, the blots were probed with a polyclonal anti-TbpA serum. The duration of the trypsin incubation (in minutes) is shown above each lane; lanes labeled NT contain proteins that were not treated with trypsin before the addition of aprotinin. The approximate positions of molecular mass markers (MW), shown in kilodaltons, are located to the left of the blots. The previously characterized TbpA-derived trypsin digestion products, T1 (95.2 kDa) and T2 (54.9 kDa), are labeled on the right.
In the published model of TbpA, two protease accessibility sites were mapped according to the sizes of the fragments produced and the detection of these fragments with antibodies specific for peptides of TbpA (5, 16, 19). To continue our test of this model, we used the epitope tags to more closely map the trypsin cleavage sites. Detection of the HA epitope after trypsin exposure of the L5HA mutant revealed that the epitope was located on both the 95.2-kDa (T1) and the 54.9-kDa (T2) fragments (Fig. 7). This result indicates that the trypsin cleavage site in this mutant, resulting in the T2 fragment, was C terminal to the insertion point for the HA epitope (see Fig. 1), as opposed to N terminal to the HA insertion, as was previously suggested (5). If the previously identified trypsin cleavage site had resulted in the T2 fragment, T2 would not have been detectable with the anti-HA MAb in the course of this analysis. Detection of the epitope in the fragments produced by protease cleavage of the L10HA mutant revealed that the epitope was detectable only in full-length TbpA, not in T1 or T2 (Fig. 7). This result indicates that the cleavage site in this mutant, resulting in the T1 fragment, was N terminal to insertion in the L10HA mutant (Fig. 1), as opposed to C terminal, as was previously suggested (5). Both of these observations suggest that the actual trypsin cleavage sites are slightly different from those previously suggested, although it is also possible that insertion of the HA epitope altered the protease sensitivity of the mutated proteins, resulting in a modified digestion pattern.
FIG. 7.

Mapping of trypsin cleavage sites with the HA epitope tags. Iron-stressed gonococci were incubated with trypsin for 0, 10, 20, or 30 min before the protease was inactivated by the addition of aprotinin. The trypsinized cells were pelleted, lysed, and separated by SDS-PAGE. After transfer, the blots were probed with a monoclonal anti-HA antibody conjugated to peroxidase. The length of the trypsin incubation (in minutes) is shown above each lane; lanes labeled NT contain proteins that were not treated with trypsin before the addition of aprotinin. The approximate positions of molecular mass markers (MW), in kilodaltons, are located to the left of the blots. The previously characterized TbpA-derived trypsin digestion products, T1 (95.2 kDa) and T2 (54.9 kDa), are labeled on the right.
DISCUSSION
This study was designed to analyze the surface topology of gonococcal TbpA and resulted in the construction of 12 epitope insertion mutants, most of which accommodated the insertion with little detectable disruption of structure. Western blot analyses indicated that the insertionally mutagenized proteins were stably expressed and not subject to gross proteolytic breakdown. Furthermore, no growth anomalies were seen with any of the mutants except when provided with transferrin as the sole iron source. These results suggest a level of plasticity along the length of this well-conserved protein that allows TbpA to accommodate insertions with relatively little impact on structure or function. Results from a limited deletion analysis of TbpA (5) are in agreement with this assertion, and both studies support the theory that gonococcal TbpA, like other TonB-dependent receptors (3, 7, 33, 34, 39, 44, 47) is resilient to various types of mutations.
The hypothetical topology model of TbpA (Fig. 1) was originally based on computer predictions, diversity data, and a pairwise alignment with FepA (5, 15). Using an epitope tagging technique, we confirmed the surface exposure of loops 2, 3, 5, 7, and 10, and in so doing, we affirmed both the computer prediction methods upon which the model was based and several aspects of the model itself. Because of their confirmed surface exposure, these regions may be important for ligand interactions and furthermore might be reasonable targets for continued vaccine development efforts based on TbpA. In contrast, HA insertions into putative turns 2 and 4, putative β strands 9 and 16, and hypothetical loops 9 and 11 were all negative for surface exposure in this study. These negative results are difficult to interpret, as they could have been due to any of several different methodological or biological factors. One possibility is that an epitope is truly inaccessible by virtue of its periplasmic or transmembrane location. This was the prediction for the insertion mutants T2HA, T4HA, β9HA, and β16HA, and in fact, the accessibility assay yielded the anticipated negative reaction. The L9HA and L11HA mutants were also negative for surface exposure, which was contrary to the predictions from the hypothetical topology model. These results may reflect the fact that these regions of TbpA are actually membrane bound or periplasmically located. On the other hand, using the solid-phase accessibility assay, we could not differentiate between an epitope that is concealed below or within the membrane versus one that is simply unrecognizable due to primary or secondary structure constraints. Furthermore, the negative reactions could have arisen from steric hindrance of HA epitopes by other loops. For these reasons, all of the negative reactions in the surface accessibility assay should be interpreted cautiously.
Analysis of the PHA mutant suggests that the plug region of TbpA is surface exposed. Although it is in contrast to the hypothetical two-dimensional topology model (Fig. 1), this result might have been anticipated based upon the three-dimensional structure of the TonB-dependent transporters. The region into which the HA epitope in the PHA mutant was inserted is homologous to the NL2 region of the plug in FepA (8). The crystal structure indicates that this region of the plug in FepA extends outward into the extracellular milieu to a greater extent than does any other portion of the plug. Figure 8 compares the three-dimensional structure of the FepA plug to the predicted structure of the homologous region in TbpA. Due to the sequence conservation in this region, very similar secondary and tertiary structures are predicted. Moreover, based on these nearly identical structures, the position into which the HA epitope was inserted in the present study would be expected to be similarly located near the apex of the plug domain of gonococcal TbpA. This putative topology is consistent with the observation that the HA epitope fused after Ala110 of TbpA is surface exposed.
FIG. 8.

Comparison of the three-dimensional structure of the plug region of FepA with the predicted structure of the TbpA plug. The amino termini of the proteins are indicated by N. The carboxy termini are represented by C. The asterisk in the TbpA plug panel indicates the approximate position of Ala110 after the HA epitope was fused in the PHA mutant. β-Strand structure is indicated in yellow, α-helical structure is indicated in red, β-turn structure is indicated in blue, and unstructured sequence is shown in white. A sequence consisting of amino acids 1 to 162 of the processed form of gonococcal TbpA was aligned with the analogous sequence of FepA using the 3D-JIGSAW comparative modeling server.
The process of iron uptake from transferrin can be divided into four poorly defined steps: (i) transferrin binding, (ii) iron stripping, (iii) iron transport across the outer membrane, and (iv) apo-transferrin release. We evaluated the ability of the mutants we generated to bind transferrin as an indicator of the successful completion of step i. A mutant's ability to grow on transferrin as the sole iron source indicated completion of steps ii and iii. We discovered that most of the mutants (L5HA, L7HA, L10HA, T2HA, and T4HA) had no defect in the ability to either bind transferrin or utilize it as an iron source. Previous deletion data indicated that L5 contains residues essential for transferrin binding and utilization (5); however, critical residues within this loop were apparently not interrupted during the present insertion analysis, as the L5HA mutant bound and utilized transferrin in a wild-type manner. Masri and Cornelissen suggested that variable regions conceal functional domains located within L5 in order to protect the vulnerable functional residues from potentially detrimental antibodies (42). When designing the location of this epitope tag, we intentionally chose a variable region of L5. Our observation that the L5HA mutant was unaffected in ligand binding or transferrin-iron utilization is consistent with the hypothesis that variable, surface-exposed residues are not critical for the function of the receptor but may be important for immune evasion and camouflage of the underlying conserved binding epitopes. Furthermore, we observed that most insertions were readily accommodated by TbpA without drastic effects on binding or iron uptake. These well-tolerated insertions included some that were surface exposed (L5, L7, and L10) and others that were not detectable on the cell surface (T2 and T4). This finding validates the epitope insertion approach for mapping outer membrane topology and highlights the fact that these altered proteins must be properly expressed, exported, and situated in the membrane, despite the insertion.
Insertions into putative loops 2, 9, and 11 resulted in mutants that bound transferrin with wild-type affinity but could complete the iron acquisition process only in the presence of a functional TbpB protein. It was previously documented that TbpB can increase the efficiency of iron uptake from transferrin (2), but the mechanism by which it does so has not been addressed to date. This study gives the first indication that TbpB can compensate for a mutation in TbpA, resulting in a functional receptor. Because TbpB is believed to be peripherally exposed on the outer membrane, the compensatory function provided by this protein is likely mediated at the level of the cell surface, a location consistent with the hypothesis that the defect in these HA insertion mutants is exerted at the level of iron removal from transferrin. Furthermore, this compensatory behavior strongly supports the contention that TbpA and TbpB interact on the cell surface (16, 19) and could implicate putative loops 2, 9, and 11 as contributors to a TbpA-TbpB interaction.
Insertion at only two positions within TbpA resulted in abolition of transferrin-binding functions. Neither the L3HA nor the β9HA mutant was capable of transferrin binding, indicating that both mutants were blocked at the first phase of transferrin-iron utilization. These mutants were also unable to mediate the iron uptake process, which is not surprising since the initial step of the process could not be accomplished. Abolition of binding by insertion in L3 implicates this region as being important for transferrin binding. While deletion and expression analyses have previously identified putative loops 4 and 5 as critical binding domains, this is the first indication that epitopes in the region of the L3HA insertion are involved in transferrin binding. The binding defect seen in the β9 insertion mutant might be explained by defects in support that are normally imposed by the β strands. Mutations in amphipathic β strands, which interact to form the pore within outer membrane proteins, sometimes cause disruption of the protein's structure and function (46). Epitopes in the L3HA and β9HA mutants are in proximity to the previously identified L4 and L5 transferrin-binding domains, and the binding defects seen with these mutants may be attributable to an insertion-dependent disruption of structural support for or local topology of loops 4 and 5. It is also important that trypsin digestion of the L3 and β9 epitope insertions resulted in modified proteolytic patterns. While one digestion product was visualized in each case, the companion fragment, detected in the other mutants and in the wild-type strain, was missing. This result suggests localized disruptions in surface accessibility of trypsin cleavage sites and indicates that the negative binding and growth phenotypes should be interpreted cautiously. Taken together, the protease-accessible, binding-defective, and growth-incompetent phenotypes suggest either that L3 and β9 are directly involved in ligand binding or that insertion into these critical regions disrupts the local secondary structure in the binding domain, resulting in binding and subsequent uptake defects.
In contrast to the β9HA insertion mutant, the β16HA mutant retained ligand-binding functions. This mutant bound transferrin with wild-type affinity, albeit with a slightly lower capacity. This result suggests that the β16HA mutant may be defective either in stripping of iron from transferrin (step ii) or in transport of iron across the outer membrane (step iii). We were able to isolate revertants of β16HA after extended growth on plates containing transferrin as the sole iron source (data not shown). By PCR analysis, we confirmed that the HA insertion was retained in its original position in the revertant (data not shown). Isolation of this second-site revertant further supports the assertion that the original TbpA HA insertion mutants were not grossly altered in their structure.
The phenotype of the plug insertion mutant resembles those of the L3 and β9 insertions, except that ligand binding functions were diminished, not eliminated. This decreased binding affinity was correlated with an inability to grow on transferrin as the sole iron source, similar to results obtained when loop 8 was deleted (5). Since an insertion into the plug domain may cause defects resulting in an increase in flexibility of the rest of the protein, collateral impact on critical binding epitopes could be responsible for the decreased binding affinity seen with this mutant. On the other hand, Usher et al. found that the plug domain of FepA was able to bind its ligand, ferric enterobactin, without the β-barrel domain (51). If the analogous structure in TbpA also plays a direct role in ligand binding, insertion into the plug domain would be expected to negatively affect transferrin-binding affinity. Whether the impact of the plug insertion is direct or indirect, the result of this mutation was a nonfunctional transporter, in terms of iron acquisition from transferrin, implying that a high-affinity interaction between ligand and transporter is necessary for successful completion of phases ii (stripping) and/or iii (transport) of the uptake process.
In summary, we have created mutants that allowed us to identify surface-exposed domains of TbpA, thereby confirming in part the hypotheses set forth by our hypothetical two-dimensional topology model. Using these mutants, we implicated several regions of TbpA in the successful completion of the various stages of the transferrin-iron utilization process, including binding of transferrin and iron stripping and/or transport across the outer membrane. The observation that expression of TbpB can compensate for defined defects in TbpA, generating a functional receptor, further strengthens the argument that TbpA and TbpB operate together on the cell surface to mediate efficient iron acquisition from transferrin.
Acknowledgments
This work was supported by Public Health Service grant numbers AI39523 and AI47141 from the National Institute of Allergy and Infectious Diseases. M. K. Yost-Daljev was supported by the Training in Molecular Pathogenesis grant (number T32AI07617) from the National Institute of Allergy and Infectious Diseases.
We gratefully acknowledge Heather Masri, Greg Price, Amanda DeRocco, Tracey Hagen, and Heather Strange for critical reading of the manuscript.
Editor: J. T. Barbieri
REFERENCES
- 1.Ahmer, B. M., M. G. Thomas, R. A. Larsen, and K. Postle. 1995. Characterization of the exbBD operon of Escherichia coli and the role of ExbB and ExbD in TonB function and stability. J. Bacteriol. 177:4742-4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson, J. E., P. F. Sparling, and C. N. Cornelissen. 1994. Gonococcal transferrin-binding protein 2 facilitates but is not essential for transferrin utilization. J. Bacteriol. 176:3162-3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Armstrong, S. K., and M. A. McIntosh. 1995. Epitope insertions define functional and topological features of the Escherichia coli ferric enterobactin receptor. J. Biol. Chem. 270:2483-2488. [DOI] [PubMed] [Google Scholar]
- 4.Biswas, G. D., J. E. Anderson, C.-J. Chen, C. N. Cornelissen, and P. F. Sparling. 1999. Identification and functional characterization of the Neisseria gonorrhoeae lbpB gene product. Infect. Immun. 67:455-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boulton, I. C., M. K. Yost, J. E. Anderson, and C. N. Cornelissen. 2000. Identification of discrete domains within gonococcal transferrin-binding protein A that are necessary for ligand binding and iron uptake functions. Infect. Immun. 68:6988-6996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bradbeer, C. 1993. The proton motive force drives the outer membrane transport of cobalamin in Escherichia coli. J. Bacteriol. 175:3146-3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braun, V., H. Killmann, and R. Benz. 1994. Energy-coupled transport through the outer membrane of Escherichia coli: small deletions in the gating loop convert the FhuA transport protein into a diffusion channel. FEBS Lett. 346:59-64. [DOI] [PubMed] [Google Scholar]
- 8.Buchanan, S. K., B. S. Smith, L. Venkatramani, D. Xia, L. Esser, M. Palnitkar, R. Chakraborty, D. van der Helm, and J. Deisenhofer. 1999. Crystal structure of the outer membrane active transporter FepA from Escherichia coli. Nat. Struct. Biol. 6:56-63. [DOI] [PubMed] [Google Scholar]
- 9.Bullen, J. J., H. J. Rogers, and E. Griffiths. 1978. Role of iron in bacterial infection. Curr. Top. Microbiol. Immunol. 80:1-35. [DOI] [PubMed] [Google Scholar]
- 10.Burnstein, K. L., D. W. Dyer, and P. F. Sparling. 1988. Preferential uptake of restriction fragments from a gonococcal cryptic plasmid by competent Neisseria gonorrhoeae. J. Gen. Microbiol. 134:547-557. [DOI] [PubMed] [Google Scholar]
- 11.Cadieux, N., and R. J. Kadner. 1999. Site-directed disulfide bonding reveals an interaction site between energy-coupling protein TonB and BtuB, the outer membrane cobalamin transporter. Proc. Natl. Acad. Sci. USA 96:10673-10678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Charbit, A., J. C. Boulain, A. Ryter, and M. Hofnung. 1986. Probing the topology of a bacterial membrane protein by genetic insertion of a foreign epitope; expression at the cell surface. EMBO J. 5:3029-3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Charbit, A., J. Ronco, V. Michel, C. Werts, and M. Hofnung. 1991. Permissive sites and topology of an outer membrane protein with a reporter epitope. J. Bacteriol. 173:262-275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cornelissen, C. N. 2003. Transferrin-iron uptake by gram negative bacteria. Front. Biosci. 8:D836-D847. [DOI] [PubMed] [Google Scholar]
- 15.Cornelissen, C. N., J. E. Anderson, I. C. Boulton, and P. F. Sparling. 2000. Antigenic and sequence diversity in gonococcal transferrin-binding protein A. Infect. Immun. 68:4725-4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cornelissen, C. N., J. E. Anderson, and P. F. Sparling. 1997. Energy-dependent changes in the gonococcal transferrin receptor. Mol. Microbiol. 26:25-35. [DOI] [PubMed] [Google Scholar]
- 17.Cornelissen, C. N., G. D. Biswas, and P. F. Sparling. 1993. Expression of gonococcal transferrin-binding protein 1 causes Escherichia coli to bind human transferrin. J. Bacteriol. 175:2448-2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cornelissen, C. N., G. D. Biswas, J. Tsai, D. K. Paruchuri, S. A. Thompson, and P. F. Sparling. 1992. Gonococcal transferrin-binding protein 1 is required for transferrin utilization and is homologous to TonB-dependent outer membrane receptors. J. Bacteriol. 174:5788-5797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cornelissen, C. N., and P. F. Sparling. 1996. Binding and surface exposure characteristics of the gonococcal transferrin receptor are dependent on both transferrin-binding proteins. J. Bacteriol. 178:1437-1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Danve, B., L. Lissolo, M. Mignon, P. Dumas, S. Colombani, A. B. Schryvers, and M.-J. Quentin-Millet. 1993. Transferrin-binding proteins isolated from Neisseria meningitidis elicit protective and bactericidal antibodies in laboratory animals. Vaccine 11:1214-1220. [DOI] [PubMed] [Google Scholar]
- 21.Ferguson, A. D., and J. Deisenhofer. 2002. TonB-dependent receptors—structural perspectives. Biochim. Biophys. Acta 1565:318-332. [DOI] [PubMed] [Google Scholar]
- 22.Ferguson, A. D., E. Hofmann, J. W. Coulton, K. Diederichs, and W. Welte. 1998. Siderophore-mediated iron transport: crystal structure of FhuA with bound lipopolysaccharide. Science 282:2215-2220. [DOI] [PubMed] [Google Scholar]
- 23.Gorringe, A. R., R. Borrow, A. J. Fox, and A. Robinson. 1995. Human antibody response to meningococcal transferrin-binding proteins: evidence for vaccine potential. Vaccine 13:1207-1212. [DOI] [PubMed] [Google Scholar]
- 24.Graves, J. F., G. D. Biswas, and P. F. Sparling. 1982. Sequence-specific DNA uptake in transformation of Neisseria gonorrhoeae. J. Bacteriol. 152:1071-1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gudmundsdottir, A., P. E. Bell, M. D. Lundrigan, C. Bradbeer, and R. J. Kadner. 1989. Point mutations in a conserved region (TonB box) of Escherichia coli outer membrane protein BtuB affect vitamin B12 transport. J. Bacteriol. 171:6526-6533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gunn, J. S., and D. C. Stein. 1996. Use of a non-selective transformation technique to construct a multiply restriction/modification-deficient mutant of Neisseria gonorrhoeae. Mol. Gen. Genet. 251:509-517. [DOI] [PubMed] [Google Scholar]
- 27.Higgs, P. I., P. S. Myers, and K. Postle. 1998. Interactions in the TonB-dependent energy transduction complex: ExbB and ExbD form homomultimers. J. Bacteriol. 180:6031-6038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horton, R. M., Z. L. Cai, S. N. Ho, and L. R. Pease. 1990. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. BioTechniques 8:528-535. [PubMed] [Google Scholar]
- 29.Imbert, M., and R. Blondeau. 1998. On the iron requirement of lactobacilli grown in chemically defined medium. Curr. Microbiol. 37:64-66. [DOI] [PubMed] [Google Scholar]
- 30.Kadner, R. J. 1990. Vitamin B12 transport in Escherichia coli: energy coupling between membranes. Mol. Microbiol. 4:2027-2033. [DOI] [PubMed] [Google Scholar]
- 31.Kellogg, D. S., Jr., W. L. Peacock, Jr., W. E. Deacon, L. Brown, and C. I. Pirkle. 1963. Neisseria gonorrhoeae. I. Virulence genetically linked to clonal variation. J. Bacteriol. 85:1274-1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kenney, C. D., and C. N. Cornelissen. 2002. Demonstration and characterization of a specific interaction between gonococcal transferrin binding protein A and TonB. J. Bacteriol. 184:6138-6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Killmann, H., R. Benz, and V. Braun. 1996. Properties of the FhuA channel in the Escherichia coli outer membrane after deletion of FhuA portions within and outside the predicted gating loop. J. Bacteriol. 178:6913-6920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koebnik, R., and V. Braun. 1993. Insertion derivatives containing segments of up to 16 amino acids identify surface- and periplasm-exposed regions of the FhuA outer membrane receptor of Escherichia coli K-12. J. Bacteriol. 175:826-839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 36.Larsen, R. A., D. Foster-Hartnett, M. A. McIntosh, and K. Postle. 1997. Regions of Escherichia coli TonB and FepA proteins essential for in vivo physical interactions. J. Bacteriol. 179:3213-3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leclerc, C., A. Charbit, A. Molla, and M. Hofnung. 1989. Antibody response to a foreign epitope expressed at the surface of recombinant bacteria: importance of the route of immunization. Vaccine 7:242-248. [DOI] [PubMed] [Google Scholar]
- 38.Lissolo, L., G. Maitre-Wilmotte, P. Dumas, M. Mignon, D. Danve, and M.-J. Quentin-Millet. 1995. Evaluation of transferrin-binding protein 2 within the transferrin-binding complex as a potential antigen for future meningococcal vaccines. Infect. Immun. 63:884-890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu, J., J. M. Rutz, J. B. Feix, and P. E. Klebba. 1993. Permeability properties of a large gated channel within the ferric enterobactin receptor, FepA. Proc. Natl. Acad. Sci. USA 90:10653-10657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lundrigan, M. D., and R. J. Kadner. 1986. Nucleotide sequence of the gene for the ferrienterochelin receptor FepA in Escherichia coli. Homology among outer membrane receptors that interact with TonB. J. Biol. Chem. 261:10797-10801. [PubMed] [Google Scholar]
- 41.Maniatis, T., E. F. Fritsch, and J. Sambrook. 1982. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
- 42.Masri, H. P., and C. N. Cornelissen. 2002. Specific ligand binding attributable to individual epitopes of gonococcal transferrin binding protein A. Infect. Immun. 70:732-740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mickelsen, P. A., and P. F. Sparling. 1981. Ability of Neisseria gonorrhoeae, Neisseria meningitidis, and commensal Neisseria species to obtain iron from transferrin and iron compounds. Infect. Immun. 33:555-564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moeck, G. S., B. S. Bazzaz, M. F. Gras, T. S. Ravi, M. J. Ratcliffe, and J. W. Coulton. 1994. Genetic insertion and exposure of a reporter epitope in the ferrichrome-iron receptor of Escherichia coli K-12. J. Bacteriol. 176:4250-4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moeck, G. S., M. J. Ratcliffe, and J. W. Coulton. 1995. Topological analysis of the Escherichia coli ferrichrome-iron receptor by using monoclonal antibodies. J. Bacteriol. 177:6118-6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Newton, S. M., P. E. Klebba, V. Michel, M. Hofnung, and A. Charbit. 1996. Topology of the membrane protein LamB by epitope tagging and a comparison with the X-ray model. J. Bacteriol. 178:3447-3456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Newton, S. M. C., J. D. Igo, D. C. Scott, and P. E. Klebba. 1999. Effect of loop deletions on the binding and transport of ferric enterobactin by FepA. Mol. Microbiol. 32:1153-1165. [DOI] [PubMed] [Google Scholar]
- 48.Postle, K. 1993. TonB protein and energy transduction between membranes. J. Bioenerg. Biomembr. 25:591-601. [DOI] [PubMed] [Google Scholar]
- 49.Schoffler, H., and V. Braun. 1989. Transport across the outer membrane of Escherichia coli K12 via the FhuA receptor is regulated by the TonB protein of the cytoplasmic membrane. Mol. Gen. Genet. 217:378-383. [DOI] [PubMed] [Google Scholar]
- 50.Seifert, H. S., R. S. Ajioka, D. Paruchuri, F. Heffron, and M. So. 1990. Shuttle mutagenesis of Neisseria gonorrhoeae: pilin null mutations lower DNA transformation competence. J. Bacteriol. 172:40-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Usher, K. C., E. Ozkan, K. H. Gardner, and J. Deisenhofer. 2001. The plug domain of FepA, a TonB-dependent transport protein from Escherichia coli, binds its siderophore in the absence of the transmembrane barrel domain. Proc. Natl. Acad. Sci. USA 98:10676-10681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weinberg, E. D. 1997. The Lactobacillus anomaly: total iron abstinence. Perspect. Biol. Med. 40:578-583. [DOI] [PubMed] [Google Scholar]
- 53.West, S. E., and P. F. Sparling. 1985. Response of Neisseria gonorrhoeae to iron limitation: alterations in expression of membrane proteins without apparent siderophore production. Infect. Immun. 47:388-394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.West, S. E. H., and P. F. Sparling. 1987. Aerobactin utilization by Neisseria gonorrhoeae and cloning of a genomic DNA fragment that complements Escherichia coli fhuB mutations. J. Bacteriol. 169:3414-3421. [DOI] [PMC free article] [PubMed] [Google Scholar]