

Summary
Background
ZNF32 has been predicted to be a zinc finger protein and is involved in cell differentiation and tumor development, but its precise function is unknown. Specific monoclonal antibodies (mAbs) have been widely used in research and clinical diagnosis and treatments. Therefore, we established an anti-ZNF32 mAb to characterize this protein’s function.
Material/Methods
Peptide49–63, a specific small peptide of ZNF32, was chosen and the synthetic keyhole limpet hemocyanin (KLH)-peptide49–63 was used as an antigen to immunize mice. A mAb against peptide49–63 was generated by hybridoma technology, and hybridoma cells were screened by limiting dilution. The isoform of mAb-pZNF32-8D9 was identified by double agar diffusion. The sensitivity and specificity of the mAb and expressed levels of ZNF32 in various cells and tissues were identified by enzyme-linked immunosorbent assay (ELISA), immunocytochemistry, immunohistochemistry, and Western blotting.
Results
A stable anti-pZNF32-8D9 hybridoma secreting the anti-peptide49–63 mAb was established and the clone positive to the peptide49–63 in supernatant was 92% in ELISA. The mAb-pZNF32-8D9 is an immunoglobulin-1 that can be used for detecting the ZNF32 protein by immunocytochemistry, immunohistochemistry, and Western blotting and is highly sensitive and specific. We also found ZNF32 expressed at high levels in Jurkat and pulmonary squamous carcinoma cells, but it was not expressed in squamous epidermis cells.
Conclusions
mAb-pZNF32-8D9 can be used for the identification and expression of ZNF32. It might also provide a new tool for diagnostics or therapy for ZNF32-related diseases.
Keywords: peptide, ZNF32 protein, hybridomas, monoclonal antibody
Background
There have been many studies on zinc finger (ZNF) proteins. Most act as DNA-binding proteins [1] that play important roles in activities such as gene transcription [2], developmental control [3], and pathological processes such as cancer formation [4]. The gene encoding ZNF32 has been mapped to a human chromosome region on 10q23–24 [5]. Its full-length cDNA is 1111 bp, encoding 273 amino acids (GenBank GeneID: 7580). ZNF32 belongs to the Kruppel-like family, comprises 7 consecutive C2H2 ZNF motifs and is predicted to be a ZNF protein according to putative bioinformatics analysis. However, the function of ZNF32 is not known. To date there have been only 2 papers published on Zpf637 [6,7]. Based on the characterization of this gene and the possible nuclear location of its protein [7], ZNF32 might function as a transcription factor. We have demonstrated that Zfp637 promotes EMT6 cell proliferation in vitro[6] and inhibits C2C12 muscle differentiation [7]. This implies that Zfp637 expression is involved in cell differentiation and tumorigenesis.
Antibodies are very important tools for protein research. Polyclonal antibodies have been obtained from rabbits immunized with the synthetic keyhole limpet hemocyanin (KLH)-peptide49–63 fragment of ZNF32 [7]. However, the production of polyclonal antibodies needs to be done repeatedly, as the specificity and sensitivity of polyclonal antibodies from different batches are not stable and they have a limited production [8]. Therefore, we need to prepare monoclonal antibodies (mAbs). Mouse mAbs with high specificity have been used widely in basic research and clinical diagnostics and therapeutics. We needed to generate a mAb against ZNF32 to characterize its cellular localization, tissue distribution, and any protein-protein interactions. However, there are no commercially available antibodies for this protein.
Im the present study, we generated a mAb against peptide49–63 of ZNF32. We used KLH-peptide49–63 as an antigen to immunize mice, and then obtained a specific mAb using hybridoma technology. The mAb-pZNF32-8D9 from the hybridoma cells was highly specific and sensitive in detecting the peptide49–63 by enzyme-linked immunosorbent assay (ELISA) and could detect the native conformation of ZNF32 in human cells using immunocytochemistry. We also demonstrated expression profiling via immunohistochemistry, the binding ability of the mAb to both endogenous ZNF32 and to exogenous green fluorescent protein (GFP)-ZNF32 fusion protein in a denatured form by Western blotting. We also detected the expression of ZNF32 in human pulmonary squamous cell carcinoma and skin tissues with mAb-pZNF32-8D9.
Material and Methods
Reagents
The peptide sequence (49SFRREKLEQKSPESK63 (abbreviated as peptide49–63, GenBank GeneID: 7580) and KLH-peptide49–63 were synthesized by Abgent Biotechnology Co., Ltd. (Suzhou, P. R. China) with a purity of 90%. Polyethylene glycol (PEG, 1500) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Hypoxanthine-aminopterin-thymidine (HAT), hypoxanthine-thymidine (HT), fetal bovine serum (FBS) and newborn calf serum (NBCS) were purchased from Gibco (Invitrogen, Carlsbad, CA, USA). A goat anti-mouse immunoglobulin G (IgG)-horseradish peroxidase (HRP) conjugate was bought from Zhongshan (Beijing, P. R. China). Mouse IgG was bought from Dako Denmark A/S (Glostrup, Denmark). TurboFect™ in vitro transfection reagent was obtained from Fermentas.
Mice, cell lines, and tissues
BALB/c female mice at 6–8 weeks of age were bred and maintained under standard housing conditions in the animal facility of Sichuan University. All experiments in this study were performed in accordance with relevant national laws and animal welfare requirements. The mouse myeloma cell line SP2/0, human embryonic kidney cell line HEK293 and the human Jurkat T-cell leukemia cell line were obtained from the American Type Culture Collection (Manassas, VA, USA). Human lung squamous cell carcinoma cell line SK-MES-1 was purchased from DS Pharma Biomedical Co. Ltd. (Osaka, Japan) and maintained in RPMI-1640 medium (Gibco, Invitrogen) or DMEM medium (high glucose; Gibco) containing 10% FBS with 2 g/L sodium bicarbonate (NaHCO3), 10% NBCS, 100 units/mL penicillin, and 100 mg/mL streptomycin.
Five archived formalin-fixed, paraffin wax-embedded samples of pulmonary squamous cell carcinomas were obtained from the West China Hospital and 3 normal human preputial skin tissues were obtained from patients at the Pediatric Surgery Department of West China Hospital. This study and all procedures were approved by the Ethics Committee on Human Experimentation of the West China Hospital.
Immunization screening of sera from immunized mice
The antigen KLH-peptide49–63 (100 μg) and complete Freund’s adjuvant (0.5 mL) were injected into BALB/c mice at multiple subcutaneous sites for primary immunization. The subsequent injections were 50 μg of antigen in the same volume of incomplete Freund adjuvant at the 4th, 6th, and 8th weeks. One week after the third immunization, the mice were bled by cutting the tail tips and the serum was tested for its ability to bind to peptide49–63 by ELISA. Mice with the highest serum antibody titers to peptide49–63 were selected to be spleen donors for hybridoma production. A final injection of 50 μg antigen and 500 μL of incomplete Freund adjuvant was given to the donor mice. Three days later, the mice were euthanized and the spleens removed. Splenic cells were prepared for fusion and the sera of the donors were stored at −20°C for subsequent analysis.
ELISA
Microtiter plates were coated with 100 μL of peptide49–63 (500 ng/mL) in coating buffer (0.05 M Na2CO3−NaHCO3, pH 9.6) overnight at 4°C. Then plates were washed once with phosphate-buffered saline containing 0.05% Tween 20 (PBST) and blocked with 5% horse serum diluted with PBS for 30 min at 37°C to avoid nonspecific binding. The plates were washed once more and incubated with 100 μL/well of a diluted positive control (immunized mouse serum), hybridoma supernatants from each clone, a negative control (unimmunized mouse serum), or a blank control (RPMI-1640 with 10% NBCS) for 2 h at 37°C. After being washed 4 times, as before, plates were incubated with 100 μL/well goat anti-mouse IgG-HRP conjugate at a 1:8000 dilution for 1 h at 37°C. All plates were washed 4 times and then 100 μL/well TMB reaction solution (1 mg/100 μL in DMSO) was added in the dark. After incubation for 15 min at 37°C, the reaction was stopped with 50 μL/well 2 M H2SO4. The absorbance was measured at 450 nm on a Model 680 microplate reader (Bio-Rad Laboratories, K. K., Tokyo, Japan). ELISA values at least 5 times higher than the negative control were considered positive for identifying peptide49–63.
Cell fusion and selection
Spleen cells from the selected immunized mice were fused with SP2/0 cells at a 7:1 ratio in the presence of 50% (w/v) PEG [9]. Hybridomas (10 plates/fusion) were added dropwise to 96-well cell culture plates and cultured in selective medium (RPMI-1640 containing 20% FBS with 1×HAT) at 37°C in an atmosphere of 5% CO2 for 10–15 days and the selective medium was changed every 3 days. Supernatants were screened by ELISA and positive hybridomas were maintained in RPMI-1640 with 20% FBS with 1×HT for about 2 weeks, then culture medium was changed to RPMI-1640 with 10% NBCS. Hybridomas showing significant and specific response to peptide49–63 were subcloned several times by limiting dilution and ELISA. These clones were chosen for further study.
Hybridoma chromosomal analysis
Selected hybridomas were treated with RPMI-1640 plus 20% FBS containing Colcemid (Shanghai Haoran Biological Technology CO., Ltd, China, 0.2 μg/mL) for 2 h the day after incubation. The cells were collected and resuspended in 75 mmol/L KCl solution 37°C and held in a water bath for 30 min at 37°C. Carnoy’s fluid (methanol/acetic acid, 3/1, v/v) was added and the suspension was mixed and then stood for 30 min at room temperature. This protocol was repeated once more. Drops of suspension were pipetted onto ice-cold microscope slides, oven-dried, and then stained with 10% Giemsa for 20 min, washed with water, and allowed to dry in air. Complete chromosomes with clear contours distributed in the same horizontal plane with no overlapping were counted for cell line stability analysis.
Production and purification of the mAb
After 3 times limiting dilution, hybridoma 8D9-F4-E2-D6 cells were selected and named anti-pZNF32-8D9 cells. In our study, 2 methods were used to obtain the mAb. One was to obtain the mAb from the culture supernatant and the other was from mouse ascites fluid. In the first method, anti-pZNF32-8D9 cells were cultured in 175 cm2 culture flasks. Then the supernatants were collected until cell death, at about 7 days. MAb was purified from the culture supernatant using saturated ammonium sulfate (SAS) [10]. In the second method, anti-pZNF32-8D9 cells (2×106 cell/per mouse) were injected into the abdominal cavities of BALB/c mice pretreated with liquid paraffin oil. About 7 days later, ascites fluid was collected and clarified by centrifugation at 3000 g for 10 min and then initially purified using SAS. We further purified the mAb by affinity column coupling with the antigen peptide49–63, and it was named mAb-pZNF32-8D9.
Identification of MAb isotyping
MAb isotyping was performed by double agar diffusion testing as follows: A 1% agar sheet was prepared with 3 mm holes placed 3 mm apart as a plum-flower style; 10 μL of mAb-pZNF32-8D9 was placed into the middle hole and 10 μL PBS (control), 10 μL IgG, 10 μL IgG subtypes (IgG2a, IgG2b, IgG1, and IgG3) were added into the other holes arranged around the middle hole. The agar sheet was incubated at 37°C in a wet box overnight and was washed with 0.9% NaCl twice per day for 3 days. It was stained with Coomassie Brilliant Blue R-250 for 15 min and then decolorized with a bleaching solution. Pictures of the sheets were obtained using a Gel Doc EZ imager (Bio-Rad) and analyzed.
Preparation of cells over- or under-expressing ZNF32
HEK293 cells (1×106 per well) were plated in 6-well plates. For overexpression of ZNF32, 4 μg pEGFP-ZNF32 (we constructed the recombinant EGFP-ZNF32 fusion expression plasmid) was transfected into HEK293 cells by using TurboFect™ according to the manufacturer’s instructions (Fermentas). For knocking out the expression of ZNF32, 20 pmol of small interfering (si)RNA-ZNF32 was transfected into the cells using TurboFect™. The siRNA-ZNF32 was synthesized by GenePharma Co., Ltd (Shanghai, P. R. China) with the following sequences: sense, 5′-GGG CCA AAG GCA AUC UUG UdTdT-3′; anti-sense, 5′-ACA AGA UUG CCU UUG GCC CdTdT-3′. After 48 h, the total RNA of the cells was extracted using the TAKARA kit (Takara, Dalian, China) for ZNF32 mRNA measurements. Proteins from the cells were extracted for Western blot analysis.
The expression levels of ZNF32 mRNA in HEK293 cells, HEK293-ZNF32 cells (transfected with pEGFP-ZNF32), HEK293-pEGFP cells (transfected with pEGFP-N1), HEK293-siZNF32 cells (transfected with siRNA-ZNF32), and HEK293-siNC cells (transfected with siRNA-negative control) were analyzed using real-time polymerase chain reaction (PCR). Briefly, cDNAs were synthesized using the Prime Script™ RT Reagent Kit (Takara). Real-time PCR was performed and was analyzed to measure mRNA expression using an IQ5 Multi-color Real-time PCR Detection System (Bio-Rad, Hercules, CA, USA). The gene-specific primer sequences used were as follows: ZNF32 forward: 5′-AGA ATG TAG CGT TCT TCA ATG TC-3′; reverse, 5′-CCT GTA GTG TCT TCG AAT CTG G-3′. GAPDH forward: 5′-ACC ACA GTC CAT GCC ATC AC; reverse: 5′-TCC ACC ACC CTG TTG CTG TA-3′. The relative mRNA level of each sample for each gene was normalized to the mRNA level of GAPDH, a housekeeping gene. The target product was detected using the SYBR® Premix Ex Taq II kit (Takara) according to the manufacturer’s instructions. The relative expression of ZNF32 was analyzed using the Gene Expression Macro software (version 1.1; Bio-Rad).
Western blotting
Jurkat, SK-MES-1, HEK293, HEK293-ZNF32, and HEK293-siZNF32 cells were selected for Western blot analysis. The cells were collected and lysed with general lysis buffer (BioTeke Corporation, Beijing, P. R. China) for 20 min on ice. Aliquots of 20 μg of cell lysate proteins were loaded onto 12.6% polyacrylamide gels, separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). The membrane was blocked with 10% skim milk in PBST for 1 h at 37°C. It was then washed with PBST and incubated with our mAb (dilution: 1:1000) overnight at 37°C, and then washed in TBST. The membrane was then incubated with HRP-conjugated goat anti-mouse IgG (Sigma-Aldrich) at room temperature for 1 h. Finally, after 3 washes, the films were developed using Immobilon™ Western Chemiluminescent HRP Substrate (Millipore). Protein levels were analyzed by scanning blots on a Gel Doc EZ imager (Bio-Rad).
Immunocytochemistry and immunohistochemistry
Jurkat and SK-MES-1 cell lines were chosen for immunocytochemistry. Cells were prepared on slides and washed with PBS, fixed in cold acetone for 5 min, and rinsed thoroughly. For immunohistochemistry, archived tissue samples were fixed in 10% neutral buffered formalin embedded in paraffin wax; 5 μm sections were prepared and pretreated with 1.7% hydrogen peroxide in ethanol for 30 min [11]. After being blocked with 5% horse serum in PBS at room temperature for 1 h, the cells or tissue slides were incubated with mAb-pZNF32-8D9 (1:100) overnight at 4°C. Mouse IgG was used as a negative control. Then, the slides were washed with PBS and incubated with a diluted (1:1000) HRP-conjugated goat anti-mouse IgG for 1 h at 37°C. Diaminobenzidine was used as a substrate chromogen and slides were counterstained with hematoxylin.
Results
Immunization of mice and screening of hybridomas
Because production of whole proteins is complex and the cost of whole protein synthesis as immunizing antigen is too high, we chose a small peptide49–63 from ZNF32 as an antigen. Peptide49–63 was predicted as a surface epitope and a specific region of the ZNF32 protein by Abgent Biotechnology Co., Ltd. Because the peptide alone injected into mice could not generate antibodies because of its low immunogenicity, KLH as a carrier was conjugated to peptide49–63 to immunize 3 BALB/c mice [12]. The ELISA result of antisera titers among the mice immunized with KLH-peptide49–63 showed that the sensitivity from mouse #2 (the highest dilution of anti-peptide49–63 positive serum: 1:256 000) was better than the others, thus we chose the spleen from this mouse for fusion.
At 10–20 days after cell fusion, different cell supernatants from hybridoma clones were screened using ELISA. There were 62 positive hybridoma clones from 372 screened. We chose 6 clone cells for the first time-limiting dilution and then, from the first time-limiting dilution, we chose 4 clones with high values for the second time-limiting dilution. After 3-fold dilution, the hybridoma clone 8D9-F4-E2-D6 was proved to be the clone positive for the peptide49–63 with a purity of 92% in ELISA. We chose these cells for antibody production and named them anti-pZNF32-8D9 cells. During these processes, all clone cells showing high ELISA values were frozen as soon as possible to avoid the loss of chromosomes.
Hybridoma chromosomal analysis
Chromosomes are easily lost during hybridoma culture, especially in the early phase. To analyze the stability and source of anti-pZNF32-8D9 hybridoma cells, we checked the chromosome numbers. Cells were treated with Colcemid to select those in metaphase. We chose 100 chromosome preparations from the anti-pZNF32-8D9 cells (Figure 1); the chromosome numbers in single cells ranged from 92 to 98. As there are 40 chromosomes in the mouse and 62–68 in SP2/0 cells, these chromosome numbers in hybridoma cells are normal. The chromosome numbers among the 100 cells are relatively concentrated and chromosomes in each single cell are also more concentrated.
Figure 1.
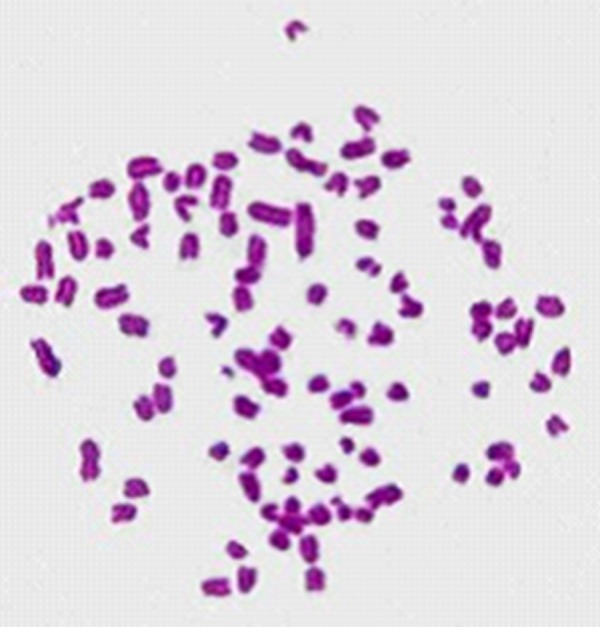
Chromosomes of a single anti-pZNF32-8D9 hybridoma cell. This is representative of 100 hybridoma chromosome sets counted (40×). Chromosomal morphology was clear and the chromosomes in single cells were relatively well concentrated.
Immunoglobulin subclasses of mAb-pZNF32-8D9
The IgG subclasses of the mAbs were determined using a double agar diffusion assay. Antibodies from different sources showed different results: the antibody isotype was only IgG1 from the culture supernatant of anti-pZNF32-8D9 cells (Figure 2A); the antibody components from the ascites of anti-pZNF32-8D9 cells were IgG1, IgG2a, and IgG2b; and the serum from immunized mice contained all IgG types (Figure 2A). Figure 2A also shows that the concentrations of mAbs from blood and ascites fluids were much higher than from culture supernatants, but the purity was lower. Therefore, to increase the recovery of mAbs, the supernatants should be concentrated and the mAb should be purified from the ascites. Here the mAb was obtained from ascites and then purified by using beads conjugated with ZNF32. The mAb-pZNF32-8D9 was purely IgG1 after purification (Figure 2B).
Figure 2.
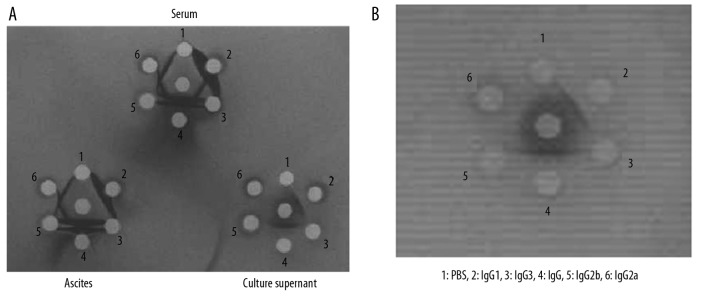
The isotype analysis of mAb-pZNF32-8D9. The immunoglobulin subclasses of the mAb-pZNF32-8D9 were determined using a double agar diffusion assay. (A) mAbs from three different sources showed different results. mAb isotypes from mouse serum contained all IgG subtypes. mAb isotypes from ascites fluids included all except the IgG3 isotype. From the culture supernatant the isotype was only IgG1. (B) The isotype analysis of the ascites fluid mAbs affinity-purified in columns using ZNF32 peptide49–63.
The valency of mAb-pZNF32-8D9 in ELISA
To identify the valency of mAb-pZNF32-8D9 in ELISA, we measured the effects on peptide49–63 using different concentrations of this mAb. As shown in Figure 3A, the responses ranged from 20 to 160 ng/ml; the lowest concentration of mAb-pZNF32-8D9 for a positive response was 10 ng/ml. We recommend 40 ng/ml as a working concentration of mAb-pZNF32-8D9. The minimum concentration of peptide49–63 detected was 0.1 ng/ml when 4 ng/ml mAb-pZNF32-8D9 was used (Figure 3B). We recommend 1 ng/mL of peptide49–63 as a suitable concentration for the antigen-positive control in ELISA. Thus, mAb-pZNF32-8D9 was sensitive for detecting peptide49–63 in this ELISA assay.
Figure 3.

The valency of mAb-pZNF32-8D9 shown by ELISA. (A) The effects of different concentrations of mAb-pZNF32-8D9 when 1 ng/mL peptide49–63 was coated. (B) The concentration of peptide49–63 detected with 40 ng/mL mAb-pZNF32-8D9.
Identification of proteins labeled with mAb-pZNF32-8D9 by Western blotting
ZNF32 is an endogenous protein and Jurkat and SK-MES-1 cells showed positive expression of this protein using real-time PCR (Figure 4A), so it was used in this study as a sample for endogenous protein. As shown in Figure 4B, the endogenous binding of mAb-pZNF32-8D9 in Jurkat and SK-MES-1 cells was at about 31 kDa. This is correct, as ZNF32 has a molecular weight (MW) of 31 kDa according to its protein sequence predicted by ExPASy (http://expasy.org/). To further identify the specificity of mAb-pZNF32-8D9, we prepared HEK293-ZNF32 and HEK293-siZNF32 cells. Figure 4a shows that ZNF32 mRNA was overexpressed in HEK293-ZNF32 and the expression was knocked down in HEK293-siZNF32 cells. The mAb-pZNF32-8D9 could bind both to endogenous ZNF32 and to the exogenous GFP-ZNF32 fusion protein (MW 56 kDa) in HEK293-ZNF32 cells. Moreover, there was decreased expression of ZNF32 in HEK293-siZNF32 cells displayed by Western blot analysis with mAb-pZNF32-8D9 (Figure 4B).
Figure 4.
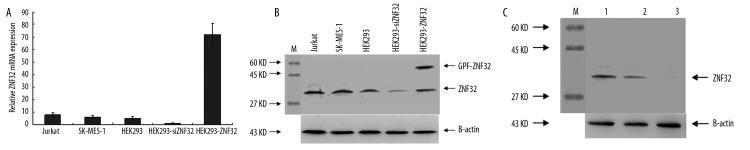
The binding ability of mAb-pZNF32-8D9 to ZNF32. (A) The expression of ZNF32 mRNA in HEK293 cells, HEK293-ZNF32 cells (transfected with pEGFP-ZNF32), HEK293-pEGFP cells (transfected with pEGFP-N1), HEK293-siZNF32 cells (transfected with siRNA-ZNF32), and HEK293-siNC cells (transfected with siRNA-negative control) were analyzed using real-time PCR. (B) The ZNF32 expression levels in Jurkat, SK-MES-1, HEK293, HEK293-ZNF32, and HEK293-siZNF32 cells were detected with mAb-pZNF32-8D9 using western blotting. (C) The ZNF32 expression of HEK293 was detected by mAb-pZNF32-8D9 (1) or mAb-pZNF32-8D9 pre-incubated with peptide49–63 at dilutions of 1:50 (2) or 1:500 (3) in western blot analysis. M means the marker scaned from PVDF membrane.
To further confirm the binding specificity of mAb-pZNF32-8D9, we preincubated mAb-pZNF32-8D9 with peptide49–63 at dilutions of 1:50 or 1:500 and then used them as primary antibodies to detect ZNF32 in HEK293 cells by Western blot. Figure 4C shows that the bands were weakened gradually by preincubating mAb-pZNF32-8D9 with peptide49–63. This suggests that the binding activity of mAb-pZNF32-8D9 was blocked by peptide49–63.
The expression of ZNF32 in cells and tissues detected by immunocytochemistry and immunohistochemistry
To analyze the location of ZNF32 in cells, we detected its expression in Jurkat and SK-MES-1 cells by immunocytochemistry. As shown in Figure 5, brown ZNF32 immunostaining was located in the cytoplasm and nucleoli, whereas the cell membranes were unstained. Further, we observed the expression of ZNF32 in pulmonary squamous cell carcinoma tissues and skin tissues by immunohistochemistry using mAb-pZNF32-8D9. As shown in Figure 6, the squamous carcinoma cells were positive, but only a few basal cells were positive in preputial skin tissues.
Figure 5.
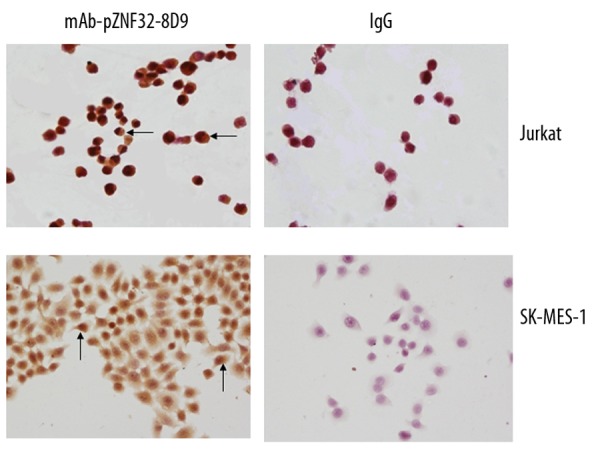
Immunostaining for ZNF32 using mAb-pZNF32-8D9 or mouse IgG in Jurkat and SK-MES-1 cells. The cytoplasm and nucleoli were positive (40×). Arrowheads indicate positive cells.
Figure 6.
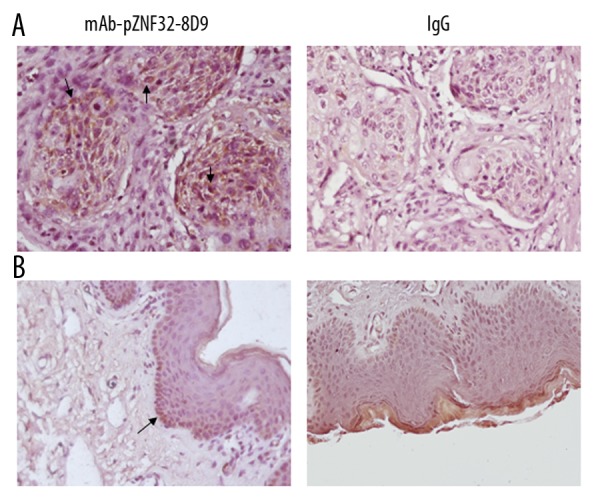
Immunostaining for ZNF32 using mAb-pZNF32-8D9 or mouse IgG in human tissues. (A) lung squamous carcinoma cells were positive. (B) preputial skin squamous epithelium cells were negative. Arrowheads indicate positive cells.
Discussion
Antibody is a critical tool for research on function and distribution of proteins. ZNF32 is predicted to be a zinc finger protein and may be involved in cell differentiation and tumorigenesis, but the function of ZNF32 remains to be determined. However, there are no commercially available antibodies for ZNF32. In the present paper we describe the generation, isolation and characterization of the monoclonal antibody mAb-pZNF32-8D9 specific for ZNF32, which is produced by hybridoma anti-pZNF32-8D9 cells.
By several limiting dilutions, a hybridoma clone (anti-pZNF32-8D9 cells) was obtained and proved to be the clone positive for the peptide49–63, with a purity of 92% on ELISA. The chromosome numbers for single cells ranged from 92 to 98 in the 100 cells counted. It suggests that the antibody-secreting hybridoma cells were formed from fused spleen and SP2/0 cells in a stable manner. The double agar diffusion assay showed that the mAb isotypes varied between sources. Antibodies from the cells’ supernatant were relatively pure and could be obtained easily. We generate these mAbs from cells via small-scale cell culture, but we could increase the recovery by large-scale culture. At present, we have obtained purified mAbs from ascites fluids by using peptide49–63 affinity chromatography.
ELISA measurement revealed that mAb-pZNF32-8D9 was sensitive for detecting peptide49–63, as the minimum concentration of peptide49–63 detected was 0.1 ng/ml. The endogenous binding of mAb-pZNF32-8D9 of Jurkat and SK-MES-1 cells in Western blotting was at about 31 kDa, as predicted. In addition, the decrease expression of ZNF32 in HEK293-siZNF32 cells, which were knocked down with siZNF32, can be detected out by Western blotting, as the bind of ZNF32 was weaken. The both expression of endogenous ZNF32 and exogenous GFP-ZNF32 fusion protein in HEK293-ZNF32 cells transfected with GFP-ZNF32 fusion protein plasmid could be detected by mAb-pZNF32-8D9. Moreover, the binding activity of mAb-pZNF32-8D9 was blocked by peptide49–63; in other words, mAb-pZNF32-8D9 combines specifically with this peptide sequence. The detection of ZNF32 from cells by Western blotting confirmed that mAb-pZNF32-8D9 specifically recognizes this protein. Thus, Western blotting results obtained in the present study suggest that mAb-pZNF32-8D9 is potentially useful for identifying both endogenous and exogenous ZNF32.
The distribution of a protein usually relate to its function. Immunocytochemistry or immunohistochemistry can reflect the location of protein. mAb-pZNF32-8D9 was successfully used to identify the expression of ZNF32 in cells and tissues. We detected expression ZNF32 in Jurkat and SK-MES-1 cells by immunocytochemistry. The results revealed ZNF32 immunostaining was located in the cytoplasm and nucleoli, coinciding with the predicted function of ZNF32 as a transcription factor. Interestingly, we observed the positive expression of ZNF32 in the squamous carcinoma cells of pulmonary squamous cell carcinoma tissues and a few basal cells of skin tissues by immunohistochemistry using mAb-pZNF32-8D9. In our previous research we found that ZNF32 promotes EMT6 cell proliferation in vitro[6]. Therefore, these results further demonstrate that ZNF32 is associated with tumorigenesis.
Conclusions
The mAb-pZNF32-8D9 prepared in the present study demonstrates that mAb-pZNF32-8D9 can be used in laboratory studies of ZNF32. The chromosomal assays of the anti-pZNF32-8D9 cells showed that they are stable. Double agar diffusion assays confirmed that the isotype of the mAb from this cell line was IgG1. We can obtain high purity mAbs from both supernatant and ascites fluids, but we recommend the latter source because it is easy to obtain high amounts. ELISA results demonstrated that mAb-pZNF32-8D9 is sensitive at binding to the ZNF32 peptide and Western blotting revealed a high specificity. ZNF32 was demonstrated to be present in the cytoplasm and nucleoli by immunocytochemistry and ZNF32 appears to be associated with cell proliferation. Thus, the mAb-pZNF32-8D9 anti-peptide49–63 can be used in research on the cell functions of ZNF32. As the pulmonary squamous cell carcinoma tissues were immunopositive, but preputial skin tissue was negative when using this mAb, we expect that this mAb may be used in clinical fields such as carrier detection, diagnosis, or therapy for cancer.
Acknowledgments
We thank Wei Dapeng (Department of Immune, Basic and Forensic Medicine, Sichuan University) for assistance in some techniques.
Footnotes
Source of support: This study was supported by grant 31070675 from the National Natural Science Foundation of China and by Sichuan Province Foundation (2010JY0055) from the Science and Technology Department of Sichuan Province
References
- 1.Bray P, Lichter P, Thiesen HJ, et al. Characterization and mapping of human genes encoding zinc finger proteins. Proc Natl Acad Sci USA. 1991;88(21):9563–67. doi: 10.1073/pnas.88.21.9563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Del Rio M, Molina1 F, Bascoul-Mollevi C, et al. Gene expression signature in advanced colorectal cancer patients select drugs and response for the use of leucovorin, fluorouracil, and irinotecan. J Clin Oncol. 2007;25(7):773–80. doi: 10.1200/JCO.2006.07.4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Middelbos IS, Vester BM, Karr-Lilienthal LK, et al. Age and Diet Affect Gene Expression Profile in Canine Skeletal Muscle. PLoS One. 2009;4(2):e4481. doi: 10.1371/journal.pone.0004481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu Q, Sung AH, Chen Z, et al. Feature Selection and Classification of MAQC-II Breast Cancer and Multiple Myeloma Microarray Gene Expression Data. PLoS One. 2009;4(12):e8250. doi: 10.1371/journal.pone.0008250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cannizzarro LA, Aronnson MM, Thiesen HJ. Human zinc finger gene ZNF23 (KOX16) Maps to a zinc finger gene cluster on chromosome 16q22 and ZNF32 (KOX30) to chromosome 10q23-24. Hum Genet. 1993;91(4):383–85. doi: 10.1007/BF00217362. [DOI] [PubMed] [Google Scholar]
- 6.Li K, Ren JJ, Zhang J, et al. Construction of eukaryotic expression plasmid for a novel zinc finger protein gene Zfp637 and its effect on breast carcinoma cell EMT6. Sichuan Da Xue Xue Bao Yi Xue Ban. 2009;40(4):575–603. [PubMed] [Google Scholar]
- 7.Li K, Zhang J, Ren JJ, et al. A novel zinc finger protein Zfp637 behaves as a repressive regulator in myogenic cellular differentiation. J Cell Biochem. 2010;110(2):352–62. doi: 10.1002/jcb.22546. [DOI] [PubMed] [Google Scholar]
- 8.Crosnier C, Staudt N, Wright GJ. A rapid and scalable method for selecting recombinant mouse monoclonal antibodies. BMC Biol. 2010;8:76. doi: 10.1186/1741-7007-8-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256(5517):495–97. doi: 10.1038/256495a0. [DOI] [PubMed] [Google Scholar]
- 10.Becker C, Hoschützky H, Rist W, et al. Generation of monoclonal antibodies against human regulatory T cells. J Immunol Methods. 2010;353(1–2):62–70. doi: 10.1016/j.jim.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 11.Lee SH, Lillehoj HS, Park MS, et al. Development and characterization of mouse monoclonal antibodies reactive with chicken CD80. Comp Immunol Microbiol Infect Dis. 2011;34(3):273–79. doi: 10.1016/j.cimid.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 12.Kim M, Yun CH, Park SK, et al. Production of polyclonal antibody against peptide antigens using polystyrene bead as a carrier. Biotechnol Lett. 2007;29(11):1735–40. doi: 10.1007/s10529-007-9463-x. [DOI] [PubMed] [Google Scholar]