

Summary
Background
Detection and quantification of adenoviruses (AdVs) causing life-threatening complications are important abilities in recognition of infection and management of immunocompromised patients. Due to the rapid increase in the number of known AdV types, most commercial tests for detection and identification of AdVs are outdated.
Material/Methods
We designed an improved, easier and faster real-time quantitative polymerase chain reaction (RQ-PCR) method for detection and quantification of 54 types of human AdVs. A wide validation effort was undertaken to ensure confidence in highly sensitive and specific detection of AdVs in compromised patients. The validation process included evaluation of the method’s suitability and reliability for use in routine diagnostics.
Results
Due to high sensitivity (9.2×102 copies/ml) and broad dynamic range (7 log) we are able to detect specific viral DNA in large amounts of cell-free body fluids. The new assay is characterized by high precision and low variation within and between individual virus tests (CV=0.036%, CV=1.29%), low bias error (4%) and no cross-reactivity with other pathogens.
Conclusions
The implementation of this new assay in clinical and laboratory practice provides a rapid, reliable and less laborious method for detection and monitoring of AdV replication in immunocompromised patients. Moreover, it offers the ability to distinguish between active and latent infection and assess treatment efficiency.
Keywords: adenoviruses, infection, molecular diagnostics, RQ-PCR
Background
Increasing use of RQ-PCR in clinical diagnostics in recent years has made this technique an essential tool in laboratory detection of many infections [1,2]. RQ-PCR outrivals classic PCR due to many advantages, including improved speed, sensitivity, lower intra- and inter-assay variability, and increased specificity. The use of probes allows for precise measurement and monitoring of viral DNA replication in clinical samples [3]. Due to rapid expansion of knowledge about new AdV types (type 54 characterized in 2009), most commercial tests for detection of AdV infections are outdated. Previously used tests could detect and/or quantify only several types of AdVs considered as the most common or dangerous pathogens. Nevertheless, AdV infections in patients with an impaired immune system may be life-threatening, affecting many organs and entire systems [4,5]. Because the availability of virus detection tests is crucial for clinical management of infections in recipients of progenitor cells, many clinical laboratories elaborate their own assays. In recent years, innovative molecular techniques for viral DNA detection have also been implemented. In 2010, Fujimoto et al. described the new Hyper-PCR method that allows for rapid amplification within 17 min [6]. The Luminex xTAG respiratory viral panel FAST assay, presented by Takao et al. (2011), is based on suspension microarray technology that enables the detection of a large number of targets in a single reaction [7]. Although these methods have great sensitivity and specificity, they still detect only several of the 54 known AdV types. Moreover, application of these technologies demands specific instrumentation not available in most routine laboratories. Hence, we report a specific and rapid RQ-PCR method for detection of 54 types of AdVs and precise quantification of viral load in clinical samples. Previously, we had used the method characterized elsewhere [8] but it was too costly and labor intensive to permit its wide application in the clinical setting. Therefore we attempted to elaborate a new assay for virus detection and monitoring of infection progress.
Material and Methods
Control virus strains
Five control virus prototype strains of different types (18, 21, 15, 4 and 41) representing groups A-F (except C) were obtained from the American Type Culture Collection (ATCC) (Manassas, USA), and AdV type 5 (C) was kindly provided by the Institute of Immunology and Experimental Therapy (Wrocław, Poland). These strains were inoculated into A549 and Graham 293 cells and cultured. DNA extracted from 200 μl of viral lysates using QIAamp Blood Mini Kit (Qiagen GmbH, Hilden, Germany) served as a positive control and as a template for specificity evaluation (according to Clinical and Laboratory Standards Institute recommendations MM6-A) [9]. The concentration of viral DNA was determined by means of NanoPhotometer (Implen, Munich, Germany). DNA of viruses other than AdVs (cytomegalovirus [CMV], Epstein-Barr virus [EBV], BK virus [BKV], hepatitis B virus [HBV]) extracted from positive clinical samples of known concentration was used for non-specificity testing.
Primers and probes
The aim of this study was to design oligonucleotides binding all known types of human AdVs. The heterogeneity of this family made the primer design complicated. By means of the ClustalW2 tool (www.ebi.ac.uk/Tools/clustalw2/index.html) we compared the hexon gene sequences of all human AdVs according to sequences available in NCBI to establish the most conservative regions. Specific oligonucleotides were selected with Primer Express Software v. 2.0 (Applied Biosystems, Foster City, USA), according to their recommendations (Table 1). We applied RQ-PCR using 1 multiplex reaction consisting of 4 primers and 1 probe (5-oligo method) (Table 2) (patent pending, no. P.392974). Due to slight hexon gene sequence variations between AdVs there was a need to introduce 2 degenerate primers (ADVF and ADVRII) and 1 degenerate nucleotide in the probe sequence. These degenerations were situated in a central part of the sequences, not disturbing specific hybridization to the target DNA. We used FAM-NFQ-MGB TaqMan probes to increase the Tm and facilitate the specificity of binding between the probe and AdV DNA. All nucleotides were synthesized and purified by Applied Biosystems. Before experimental testing, MFEprimer and BLAST alignment software were used for verification of oligonucleotides’ complementarity to DNA sequences of AdVs. Moreover, the homology between oligonucleotides and DNA of relevant organisms (human, viruses, bacteria, fungi, protozoa) was screened. None of the primer and probe combinations presented significant homology to DNA other than AdV.
Table 1.
Nucleotide sequences of the primers and probe and their complementary for hexon gene regions in several AdV serotypes: 31 (A), 11 (B), 5 (C), 10 (D), 4 (E), 41 (F), 52 (G), 54 (non-classified)).
| Group | AdV type | ADVF | Probe | ADVRI | ADVRII | ADVRIII |
|---|---|---|---|---|---|---|
| (Consensus sequence) | (Consensus sequence) | (Consensus sequence) | ||||
| A | caggacgcy*tcggagtacctga | tggtgcagtty*gcccg | acccacgatgtgaccaccg | gccaccgav**acgtacttcagc | gccacggacacctacttcacc | |
| AdV12 | -----t---------------- | ---------------- | --------------------- | |||
| AdV18 | -----t--------a------- | ---------------- | --------------------- | |||
| AdV31 | ---------------------- | ---------------- | --------------------- | |||
| B | AdV3, AdV7, AdV11, AdV14, AdV16, AdV21, AdV34, AdV35, | -----t---------------- | ---------------- | ------------------- | ||
| AdV50 | -----t---------------- | ---------------- | --------------------- | |||
| C | AdV1, AdV2, AdV5, AdV6 | ---------------------- | ---------------- | --------------------- | ||
| D | AdV8-AdV10, AdV13, AdV15, AdV17, AdV19, AdV20, AdV22-AdV30, AdV32, AdV33, AdV36-AdV38, AdV42-AdV49, AdV51, AdV53 | ---------------------- | ---------------- | --------------------- | ||
| AdV39 | ---------------------- | ---------------- | ---------t----------- | |||
| E | AdV4 | ---------------------- | ---------------- | ------------------- | ||
| F | AdV40 | ---------------------- | ---------------- | -----------c--------- | ||
| AdV41 | ---------------------- | -------a-------- | --------------------- | |||
| G | AdV52 | ---------------------- | -c-------------- | --------------------- | ||
| Non-cl*** | AdV54 | ---------------------- | ---------------- | --------------------- |
Y = T/C;
V = G/C/A;
Non-cl, non-classified; NCBI GI numbers of AdVs: AdV1|33330439; AdV2|9626158; AdV3|197944726; AdV4|51527264; AdV5|58177684; AdV6|74146110; AdV7|56160876; AdV8|202957860; AdV9|190340974; AdV10|74146114; AdV11|197944766; AdV12|9626621; AdV13|74146116; AdV14|74146108; AdV15|74146118; AdV16|57116031; AdV17|190356556; AdV18|74146104; AdV19|74146120; AdV20|74146122; AdV21|190356564; AdV22|74146124; AdV23|74146126; AdV24|74146128; AdV25|74146130; AdV26|134141818; AdV27|74146134; AdV28|74146136; AdV29|74146138; AdV30|74146140; AdV31|74146106; AdV32|74146142; AdV33|74146144; AdV34|190356590; AdV35|56160914; AdV36|74146146; AdV37|74146148; AdV38|74146150; AdV39|74146152; AdV40|9626553; AdV41|199589312; AdV42|74146154; AdV43|74146156; AdV44|74146158; AdV45|74146160; AdV46|62177315; AdV47|74146164; AdV48|134141802; AdV49|88810171; AdV50|74146170; AdV51|74146168; AdV52|124375632; AdV53|205363480; AdV54|255958147.
Table 2.
Specification and concentrations of primers and probe used in novel real-time PCR assay.
| Primers/Probe | Sequence (5′-3′) | AdV types | Product size (bp) | GenBank No | Nucleotide position 5′-3′ | Final conc. (nM) |
|---|---|---|---|---|---|---|
| ADVF Probe ADVRI |
caggacgcy*tcggagtacctga tggtgcagtty*gcccg cggtggtcacatcgtgggt |
3 | 124 | AY599836 | 18466–18589 | |
| 4 | 124 | AY458656 | 18310–18433 | |||
| 7 | 124 | GQ478341 | 18470–18593 | |||
| 11 | 124 | AF532578 | 18306–18429 | 300 | ||
| 14 | 124 | AY803294 | 18303–18426 | 250 | ||
| 16 | 124 | AY601636 | 18501–18624 | 900 | ||
| 21 | 124 | AB330102 | 52–175 | |||
| 34 | 124 | AY737797 | 18295–18418 | |||
| 35 | 124 | AY271307 | 18308–18431 | |||
| ADVF Probe ADVRII |
caggatgcy*tcggagtacctga tggtgcagtty*gcccg gctgaagtacgtv**tcggtggc |
1 | 69 | AF534906 | 18912–18980 | |
| 2 | 69 | J01917 | 18889–18957 | |||
| 5 | 69 | AY339865 | 18893–18961 | |||
| 6 | 69 | HQ413315 | 18890–18958 | |||
| 8 | 69 | AB448769 | 17827–17895 | |||
| 9 | 69 | AJ854486 | 17841–17909 | |||
| 10 | 69 | DQ149615 | 52–120 | |||
| 13 | 69 | DQ149616 | 52–120 | |||
| 15 | 69 | AB562586 | 17861–17929 | |||
| 17 | 69 | AF108105 | 17835–17903 | |||
| 19 | 69 | AB448774 | 17842–17910 | |||
| 20 | 69 | DQ149619 | 52–120 | |||
| 22 | 69 | FJ619037 | 17843–17911 | |||
| 23 | 69 | DQ149621 | 52–120 | |||
| 24 | 69 | DQ149622 | 52–120 | |||
| 25 | 69 | DQ149623 | 52–120 | |||
| 26 | 69 | EF153474 | 17836–17904 | |||
| 27 | 69 | DQ149625 | 52–120 | |||
| 28 | 69 | FJ824826 | 17828–17896 | 300 | ||
| 29 | 69 | AB562587 | 17847–17915 | 250 | ||
| 30 | 69 | DQ149628 | 52–120 | 900 | ||
| 32 | 69 | DQ149629 | 52–120 | |||
| 33 | 69 | DQ149630 | 52–120 | |||
| 36 | 69 | GQ384080 | 17827–17895 | |||
| 37 | 69 | DQ900900 | 17856–17924 | |||
| 38 | 69 | DQ149633 | 52–120 | |||
| 39 | 69 | DQ149634 | 52–120 | |||
| 40 | 69 | L19443 | 17694–17762 | |||
| 41 | 69 | DQ315364 | 17649–17717 | |||
| 42 | 69 | DQ149635 | 52–120 | |||
| 43 | 69 | DQ149636 | 52–120 | |||
| 44 | 69 | DQ149637 | 52–120 | |||
| 45 | 69 | DQ149638 | 52–120 | |||
| 46 | 69 | AY875648 | 17838–17906 | |||
| 47 | 69 | DQ149640 | 52–120 | |||
| 48 | 69 | EF153473 | 17820–17888 | |||
| 49 | 69 | DQ393829 | 17842–17910 | |||
| 51 | 69 | DQ149642 | 52–120 | |||
| 52 | 69 | DQ923122 | 17395–17463 | |||
| 53 | 69 | FJ169625 | 17611–17679 | |||
| 54 | 69 | AB448770 | 17759–17827 | |||
| ADVF Probe ADVRIII |
caggatgcy*tcggagtacctga tggtgcagtty*gcccg ggtgaagtaggtgtccgtggc |
12 | 67 | AC000005 | 17791–17857 | 300 |
| 18 | 67 | GU191019 | 17817–17883 | 250 | ||
| 31 | 67 | AM749299 | 17626–17692 | 300 | ||
| 50 | 67 | AY37798 | 18511–18577 |
The multiplex real-time PCR reaction was composed with 4 primers (one forward: ADVF and three reverse primers: ADVRI, ADVRII, ADVRIII) and one probe creating three sets of oligonucleotides for detection of all known types of human adenoviruses.
Y = T/C;
V = G/C/A.
Clinical samples
DNA from AdV-positive samples of whole blood (EDTA), plasma, urine and stool (n=203) obtained from 57 consecutive patients undergoing hematopoietic stem cell transplantation (HSCT) at the local Pediatric Bone Marrow Transplantation Unit between 2007 and 2009 was used in comparison of 2 testing methods in the clinical trial. Patients consisted of 36 male and 21 female recipients aged from 1 to 44 years (median, 9 years). The patients were tested for AdV infection on a regular basis after transplantation; therefore clinical samples were collected in different periods after transplantation. DNA was extracted from clinical samples using spin columns of the QIAamp Blood Mini Kit, QIAamp Viral RNA Mini Kit and the QIAamp DNA Stool Mini Kit from Qiagen, according to the manufacturer’s instructions. This study was approved by the Bioethics Committee of Medical University in Wroclaw, Wroclaw, Poland.
Screening PCR
Viral load in clinical samples was assessed by means of sensitive screening PCR (data not shown). Briefly, PCR was performed in a total volume of 20 μl, containing HotStarTaq Master Mix Kit (Qiagen), 25 nM forward and reverse primers [10], 2 μl of distilled water and 6 μl of target DNA. The amplification was performed according to the following protocol: 95°C for 15 min, 34 cycles: 91°C for 1min 30 sec, 58°C for 1min 30 sec and 72°C for 2 min 30 sec followed by 72°C for 10 min. PCR products were visualized in 3% agarose gel. Reference strains of adenoviruses obtained from ATCC were used as positive controls and distilled water served as a negative control. Subsequently, all positive samples were evaluated with 2 compared quantitative methods.
RQ-PCR
RQ-PCR was performed using the 7500 Real-Time PCR System (Applied Biosystems). The reaction mixture consisted of 1 × TaqMan Universal Master Mix (Applied Biosystems), 2 primers in final concentration of 0.3 μM, 2 primers of 0.9 μM and 1 fluorescent probe (0.25 μM concentration of FAM probe), 1.68 μl of 10 × Exo IPC Mix (exogenous internal positive control) (Applied Biosystems), 5 μl of target DNA and H2ODEPC to a final volume of 24 μl. The amplification protocol was as follows: 90°C for 2 min, 95°C for 10 min, 40 cycles: 95°C for 15 s and 60°C for 1 min. Five μl of 50 × Exo IPC DNA (Applied Biosystems) was added into a lysis buffer prior to extraction of DNA from the clinical sample and was co-amplified in the multiplex reaction as IPC in order to monitor the integrity of the DNA and the presence of PCR inhibitors. To reduce the possibility of false-positive results, AmpErase UNG and dUTP (Applied Biosystems) were used. Each run included at least 2 no template controls (NTC), no amplification controls (NAC) and 3 different positive controls (PC, viral DNA). Data were collected and analyzed using Sequence Detection System software v. 1.4 from Applied Biosystems.
Standard curve
Routine diagnostics of adenoviral infections in patients after HSCT requires the determination of viral proliferation in a patient. Preparation of exogenous standard curves for quantification of viral load in clinical samples was based on 10-fold serial dilutions of spectrophotometrically quantified standards (amplicons were cloned and amplified in a pGEM-T Easy Plasmid Vector System [Promega, Madison, USA]). Viral DNA was derived from reference strains as representatives of species A-F and as targets for PCR in a multiplex reaction. Plasmid DNA was purified with QIAGEN Plasmid Midi Kit and checked by restriction enzyme cutting. Subsequently, the plasmid concentration and purity were determined using a NanoPhotometer (Implen GmbH). The viral copy number was calculated based on the known molecular weight of the plasmids and amplicons. Ten-fold serial dilutions were amplified in triplicate for construction of standard curves. The standard curves were generated by plotting threshold cycle (Ct) against the log of standard concentration using Sequence Detection System v.1.4. Due to high similarity of standard curves allowing reliable quantification of viral load throughout the tested range, we selected 1 standard curve (based on AdV5, ranging from 3.7×100 to 3.7×107 copies/μl; y=−3.52(x) + 37.59) as a representative for multiplex PCR. Ten-fold serial dilutions of AdV5 standard also served as a target for sensitivity, linearity and dynamic range evaluation. The standard curve also provided information on the amplification efficiency.
Reference RQ-PCR assay
The results of the 5-oligo method were compared with the outcomes of the previously used reference method (13-oligo method) developed in our laboratory but based on specific primers and probes described elsewhere [8]. Briefly, amplification was performed on a 7500 Real-Time PCR System (Applied Biosystems) by using TaqMan-based chemistry. Fifty-one types of AdVs were detected in 2 multiplex reactions, ACF and BDE (for appropriate AdV subgroups), based on the standard protocol. TaqMan Exo IPC (VIC probe) from Applied Biosystems was added during the DNA extraction step (5 μl/200 μl of lysate) and was co-amplified in the multiplex reaction. To reduce the risk of sample contamination, the AmpErase system was used. Each run included at least 2 NTCs and 3 different PCs.
Analytical verification
Before a new quantitative molecular test is implemented in clinical practice, its performance characteristics under laboratory conditions must be assessed. As part of analytical verification, laboratories must determine the assay’s analytical sensitivity, specificity, accuracy, precision, and measurement range of the overall process to determine when changes in the quantity of the analyte are due to inherent test error before clinical relevance is considered. Therefore, to ensure the reliability of the method, a wide verification effort was undertaken in our study, according to Clinical and Laboratory Standards Institute and PN-EN ISO/IEC 17025: 2001 recommendations [9,11]. The validation procedures and statistical methods were also taken from references 5, 8, and 12. For accuracy assessment, the Jouden’s method (modified) was used [13].
Results
Characterization of the assay
The heterogeneity of adenoviruses necessitated the use of a multiplex assay containing more than 1 set of primers and probes. We applied 1 forward and 3 reverse primers formulating 3 primer pairs and 1 fluorescent probe covering all types of human AdVs. Evaluation of oligonucleotide complementarity by means of MFEprimer software revealed only 1 mismatch in 1 primer sequence in 12 types and 2 mismatches in 1 primer in 1 type, but in a part of the sequence not decreasing the specificity and sensitivity of virus detection. We also revealed 1mismatch in the probe sequence in AdV41 and AdV52.
Sensitivity and specificity
Serially diluted plasmid (3.7×107–3.7×10−1 copies/μl) was repeatedly (5 times) amplified by RQ-PCR based on the above protocol. The Ct values were plotted against serial dilutions, generating the standard curve. The sensitivity of the assay was determined by the lowest standard dilution repeatedly detected in the reaction. The lower detection limit allowing for reliable detection was as low as 9.2×102 copies/ml or per gram of clinical sample (SD, 0.38; CV, 0.011). Occasionally, it was possible to detect 9.2×101 copies/ml. We also experimentally determined the oligonucleotides binding in the presence of AdV target DNA of types representing 6 subgroups of human AdVs by using the same primers and probes in PCR (Figure 1) and RQ-PCR. The new set of oligonucleotides specifically binds all AdVs of examined species. The potential cross-reactivity with other viruses was evaluated by using EBV-, CMV-, BKV- and HBV-positive samples of known concentration (EBV, range 3.1×101–1.5×105; CMV, range 2.1×102–7.0×103; BKV, range 8.6×101–7.9×107; HBV, range 8.5×101–6.5×107 copies/ml). No cross-reactivity was noted with these viruses, considered as among the most frequent viral infections after HSCT. Inhibition was excluded by co-amplification of IPC.
Figure 1.
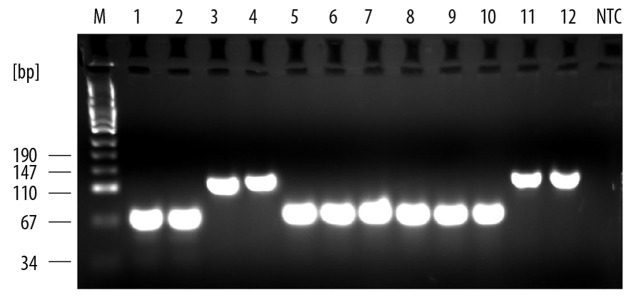
Specificity of primers for AdV DNA. Specificity was checked by means of PCR reactions in the presence of AdV DNA of types 18, 21, 5, 15, 41, 4 representing six subgroups of human adenoviruses- lines 1,2; 3,4; 5,6; 7,8; 9,10; 11,12 respectively. M – molecular mass marker. NTC – no template control.
Dynamic range and linearity
Serially diluted standard plasmid of AdV5 was used to estimate the dynamic range of the 5-oligo RQ-PCR assay. Regression analysis showed a high correlation coefficient (R2=0.998) for the range 9.2×102–9.2×109 copies/ml. It confirms a strong correlation between Ct values and copy number in clinical samples. Our method also offers quite a wide dynamic range that covers 7 log of adenoviral nucleic acid. In reference to the theoretical maximum, the new assay also offers quite high amplification efficiency − 1.92 (96%).
Intra- and inter-assay variability
The validation process also included determination of intra- and inter-assay variability of the 5-oligo method. Three control samples of known concentrations covering the expected range of viral concentrations in clinical samples (1.6×109, 1.6×106 and 1.6×103 copies/ml) were tested 11 times to evaluate variation of the test (data not shown). The coefficient of variation (CV) was very low for intra-assay variability − 0.023%, 0.043% and 0.044%, respectively. The inter-assay variability was assessed by examining 11 DNA aliquots in independent assays and it was as follows: 0.56%, 0.64% and 1.036%. Interestingly, in comparison to the 13-oligo method, we achieved much lower total intra- and inter-assay variability (0.036% vs. 0.67%; 0.74% vs. 1.29%, respectively).
The next aim of the experimental part of the research was to estimate the accuracy of the assay. We used 3 control samples − 2 of unknown concentrations of viral DNA and 1 sample created by mixing both previous control samples in a 1:1 ratio. All controls were amplified repeatedly in the same run. The mean copy number in the third control sample was compared to the calculated result (mean of 2 previous control samples) to estimate bias (4.2%). This experiment revealed that the 5-oligo method is accurate, allowing for monitoring of viral replication with bias as low as 4%. The summary of the real-time PCR validation results is given in Table 3.
Table 3.
Summary of the real-time PCR validation results.
| Assay characteristics | Validation results |
|---|---|
| Limit of quantification (LOQ) | 9.2×102 copies/ml or gram (SD, 0.38; CV, 0.011) |
| Primers and probe complementarity to the AdV DNA | Pass (to all AdV types) |
| Specificity | No cross-reactivity to DNA other than AdV |
| Linearyty | 9.2×102–9.2×109copies/ml or g, R2=0.998 |
| Dynamic range | At least 7 log |
| Accuracy | 96% (% recovery) |
| Total intra-assay variability | CV (%)=0.036 |
| Totao inter-assay variability | CV (%)=0.075 |
Comparison of methods
Viral loads in clinical samples were estimated using sensitive screening PCR and the 2 compared (5-oligo and 13-oligo) methods, according to conditions outlined in the Materials and Methods section. We analyzed only AdV-positive samples (n=203) assessed by means of screening PCR. We were able to detect AdV DNA with both quantitative methods in only 25% of positive samples (Table 4). Both methods revealed poor total sensitivity in comparison to the qualitative test. The quantitative results of the 5-oligo and 13-oligo methods overlapped by 13%. We observed that 58% of results obtained by means of the 5-oligo method were higher by about 1.9 log than the results of the 13-oligo method (data not shown). The 5-oligo method allowed for detection of 48% of positive samples and about 23% of samples that were not revealed by the 13-oligo method. As a result of this examination, we were able to detect more positive samples (more than 8%) of low copy number (<103 copies/ml) in comparison to the 13-oligo assay due to higher sensitivity of the 5-oligo assay (9.2×102vs. 1.0×103 copies/ml).
Table 4.
Comparison of two molecular methods in clinical samples evaluation (n=203).
| 13-oligo method | 5-oligo method | Total | |
|---|---|---|---|
| Positive | Negative | ||
| Positive | 24.6% (n=50) | 23.1% (n=47) | 47.7% (n=97) |
| Negative | 15.7% (n=32) | 36.4% (n=74) | 52.1% (n=106) |
| Total | 40.3% (n=82) | 59.5% (n=121) | |
Discussion
We report a multiplexed, real-time PCR assay capable of detecting and quantifying the entire range of human AdVs. The novel assay consists of 5 oligonucleotides, including only 1 probe. Primer and probe design allowing for amplification of the conserved region of the AdV genome was crucial for reliable and sensitive detection of all types of human AdVs. The low degree of sequence homology between AdVs belonging to the different subgroups [8,14] has been the main cause of difficulty in designing oligonucleotides. This heterogeneity of adenoviruses necessitated the use of a multiplex assay containing more than 1 set of primers and probes.
Implementation of the new 5-oligo method in clinical practice would provide many benefits. It offers reduction of costs and higher throughput of laboratory diagnostics. In comparison to the 13-oligo method, which uses 8 primers and 5 probes, the 5-oligo assay is less expensive by over 60%. Besides, using 1 multiplex reaction is less laborious and time-consuming, allowing the number of tests to be increased. Other diagnostic tests such as cell culture and serologic tests are unsuitable as they are require much time and labor due to deficiency of the immune system in patients after HSCT and a high seroprevalence in children [5]. Moreover, recombination between co-infecting types cannot be detected with these methods. RQ-PCR also allows the monitoring of viral load in clinical samples as a marker of disease progression. Some authors have described a correlation between number of viral copies and clinical presentation of infection [15–17]; hence use of a quantitative assay offers the possibility for distinction between active and latent infection and evaluation of treatment efficiency.
In comparison to the 13-oligo method, we achieved quite similar sensitivity to ACF reaction (9.0×102 copies/ml) and higher sensitivity in comparison to BDE reaction (1.0×103 copies/ml). Sensitive detection of AdVs in immunocompromised patients is crucial for detection of infection at the early stage. Prompt detection and early initiation of appropriate anti-viral therapy in high-risk patients is crucial for successful outcome. Moreover, based on results of previous studies, monitoring of peripheral blood samples potentially containing a small number of viral copies a few days before the active infection may have predictive value in surveillance of infection development [18]. Taking into account that adenoviruses belonging to B, D, E subgroups are common causes of infectious complications like acute respiratory diseases, keratoconjunctivitis, hemorrhagic cystitis, encephalitis, meningitis in these patients, sensitive detection of these types of AdVs by means of our method, at the early stage of infection, is of great importance for the success of anti-viral therapy. The potential cross-reactivity with viruses considered as among the most frequent viral infections after HSCT was evaluated by use of positive clinical samples. No cross-reactivity was observed, guaranteeing specific detection of adenovirus infections in the patients. Regression analysis showed a high correlation coefficient of the 5-oligo method, slightly better in comparison to the 13-oligo method (0.998 vs. 0.993); it confirms a strong correlation between Ct values and copy number in clinical samples. Our method offers a wide dynamic range that covers a broad range of adenoviral nucleic acid concentrations and did not differ greatly from the 13-oligo method (6 and 8 log). The quite high amplification efficiency (96%) was slightly lower than efficiency of the 13-oligo method (99%). Interestingly, in comparison to the 13-oligo method, we achieved much lower total intra- and inter-assay variability, probably due to well-optimized and better reaction conditions. The accuracy of the new method was lower than the 13-oligo-ACF reaction (4% vs. 0.02%, respectively) and similar to the BDG reaction (4% vs. 3.4%, respectively). In most molecular techniques involved in virus replication monitoring, the bias of this size does not change the result of the total copy number in the clinical material by more than 1 log and therefore has only a slight influence on treatment strategies referring to viral load in the clinical sample.
Evaluation of viral load in clinical samples positive for adenovirus with the 2 compared methods revealed poor total sensitivity of both methods in comparison to the qualitative test. This might be a result of greater detectability (as low as 90 copies/ml) of classic PCR used for classification of clinical samples as positive or negative in screening examinations. We also observed that more than half of results obtained by means of the 5-oligo method were higher by about 1.9 log than the results of the 13-oligo method. This might be due to higher sensitivity of the 5-oligo method, mainly for AdVs from B, D, E and G groups, which might be common causes of infection in the study group. We were able to detect more positive samples (more than 8%) of low copy number (<103 copies/ml) in comparison to the 13-oligo assay due to higher sensitivity of the new assay. Because quantification of viral load in clinical samples by means of the 13-olio assay was carried out after screening testing, but before evaluation with the 5-oligo method, in our opinion the lower detectability of the 13-oligo assay was not associated with possible degradation of DNA due to subsequent freezing and thawing of the sample. Poor total sensitivity of the 5-oligo method in comparison to the screening test is not a major problem because we were able to exclude cases of latency not requiring the administration of anti-viral drugs. Moreover, there is substantial evidence that adenoviral infections with a low number of viral particles are not life-threatening and may be defeated by a patient restoring the immune system without harmful therapy [19,20].
Due to the rapid increase in the number of new types of AdV, most commercial tests for detection of AdVs are outdated. In our research, we designed primers and probe complementary to the highly conserved region of the virus genome; hence, we were able to detect 54 types of human AdVs. It is also very possible that these oligonucleotides will allow for detection of new types, thus far not described. In our laboratory, the results were available very quickly, with positive results ready, on average, 3 hours after testing. This quite short time of analysis is important for early diagnosis and monitoring of AdV infection in immunocompromised patients, who often die due to AdV complications.
Conclusions
The development of an assay for detection and quantification of the AdVs with a high degree of genetic heterogeneity presented a great diagnostic challenge. We have reported a powerful new RQ-PCR method for sensitive and reliable detection and quantification of 54 types of human AdVs. This homogeneous technique guarantees high throughput and reduces need for hand labor during laboratory diagnostics. The test is inexpensive and reproducible. An extensive validation effort was undertaken to assess the method’s suitability and reliability for use in routine diagnostics. The development of this molecular assay holds promise for use as a routine and rapid screening test for the presence of AdV infection in other groups of immunocompromised patients. Moreover, implementation of the multiplex reaction and the new oligo set involving only 1 fluorescent probe provides a great opportunity to apply this improved test in automated platforms for routine diagnostics in the future.
Footnotes
Source of support: This research was Sponsored by the Polish Ministry of Science and Higher Education (University of Medicine in Wroclaw grant no. ST-701)
References
- 1.Zawilinska B, Kopec J, Szostek S, et al. Lymphotropic herpesvirus DNA detection in patients with active CMV infection – a possible role in the course of CMV infection after hematopoietic stem cell transplantation. Med Sci Monit. 2011;17(8):CR432–41. doi: 10.12659/MSM.881904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tomaszewska A, Nasilowska-Adamska B, Dzieciatkowski T, Marianska B. Simultaneous human herpesvirus 6-associated encephalitis and Guillain-Barré syndrome in a patient after matched unrelated donor haematopoietic stem cell transplantation. Arch Med Sci. 2010;6:288–90. doi: 10.5114/aoms.2010.13912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mackay IM, Arden KE, Nitsche A. Real-time PCR in virology. Nucleic Acids Res. 2002;30:1292–305. doi: 10.1093/nar/30.6.1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bil-Lula I, Ussowicz M, Rybka B, et al. Adenovirus in urine sample – high predictive value in development of haemorrhagic cystitis. Bone Marrow Transplant. 2010;45(Suppl):S218. [Google Scholar]
- 5.Watzinger F, Suda M, Preuner S, et al. Real-time quantitative PCR assays for detection and monitoring of pathogenic human viruses in immunosuppressed pediatric patients. J Clin Microbiol. 2004;42:5189–98. doi: 10.1128/JCM.42.11.5189-5198.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fujimoto T, Konagaya M, Enomoto M. Novel high-speed real-time PCR method (Hyper-PCR): results from its application to adenovirus diagnosis. Jpn J Infect Dis. 2010;63:31–35. [PubMed] [Google Scholar]
- 7.Takao S, Hara M, Okazaki T, Suzuki K. Simultaneous multiple assay (Luminex xTAG respiratory viral panel FAST assay) efficacy in human respiratory virus detection. Kansenshogaku Zasshi. 2011;85:31–36. doi: 10.11150/kansenshogakuzasshi.85.31. [DOI] [PubMed] [Google Scholar]
- 8.Ebner K, Suda M, Watzinger F, Lion T. Molecular detection and quantitative analysis of the entire spectrum of human adenoviruses by two-reaction real-time PCR assay. J Clin Microbiol. 2005;43:3049–53. doi: 10.1128/JCM.43.7.3049-3053.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clinical and Laboratory Standards Institute. MM6-A. 28 Vol. 23. Pennsylvania, USA: Quantitative Molecular Methods for Infectious Diseases; Approved Guideline. [Google Scholar]
- 10.McDonough M, Kew O, Hierholzer JC. PCR detection of human adenoviruses. In: Persing DH, Smith TF, Tenover FC, White TJ, editors. Diagnostic Molecular Microbiology: Principles and Applications. Washington, D.C: American Society for Microbiology; 1993. pp. 389–93. [Google Scholar]
- 11.Wendt U, Rogulski J. Validation of testing methods as an element of quality asssurance system in medical laboratories. Diagn Lab. 2003;39:73–81. [in Polish] [Google Scholar]
- 12.Konieczka P, Namieśnik J. Validation of an analytical procedures. In: Konieczka P, Namieśnik J, editors. Evaluation and quality control of analytical measurements. Warszawa: Wydawnictwa Naukowo-Techniczne; 2007. pp. 225–300. [in Polish] [Google Scholar]
- 13.Westgard JO. Basic method validation. 3rd edition. Madison: Westgard QC, Inc; 2008. pp. 123–35. [Google Scholar]
- 14.Gu Z, Belzer SW, Gibson CS, et al. Multiplexed, real-time PCR for quantitative detection of human adenovirus. J Clin Microbiol. 2003;41:4636–41. doi: 10.1128/JCM.41.10.4636-4641.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Claas EC, Schilham MW, de Brouwer CS, et al. Internally controlled real-time PCR monitoring of adenovirus DNA load in serum or plasma of transplant recipients. J Clin Microbiol. 2005;43:1738–44. doi: 10.1128/JCM.43.4.1738-1744.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schilham MW, Claas EC, van Zaane W, et al. High levels of adenovirus DNA in serum correlate with fatal outcome of adenovirus infection in children after allogeneic stem-cell transplantation. Clin Infect Dis. 2002;35:526–32. doi: 10.1086/341770. [DOI] [PubMed] [Google Scholar]
- 17.Teramura T, Naya M, Yoshihara T, et al. Adenoviral infection in hematopoietic stem cell transplantation: early diagnosis with quantitative detection of the viral genome in serum and urine. Bone Marrow Transplant. 2004;33:87–92. doi: 10.1038/sj.bmt.1704320. [DOI] [PubMed] [Google Scholar]
- 18.Bil-Lula I, Ussowicz M, Rybka B, et al. PCR diagnostics and monitoring of adenoviral infections in hematopoietic stem cell transplantation recipients. Arch Virol. 2010;155:2007–15. doi: 10.1007/s00705-010-0802-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hale GA, Rochester RJ, Heslop HE, et al. Hemorrhagic cystitis after allogeneic bone marrow transplantation in children: clinical characteristics and outcome. Biol Blood Marrow Transplant. 2003;9:698–705. doi: 10.1016/s1083-8791(03)00269-6. [DOI] [PubMed] [Google Scholar]
- 20.Walls T, Hawrami K, Ushiro-Lumb I, et al. Adenovirus infection after pediatric bone marrow transplantation: is treatment always necessary? Clin Infect Dis. 2005;40:1244–49. doi: 10.1086/429235. [DOI] [PubMed] [Google Scholar]