

Summary
Background
Ventricular septal defect (VSD) is the most prevalent type of congenital heart disease and is a major cause of substantial morbidity and mortality in infants. Accumulating evidence implicates genetic defects, especially in cardiac transcription factors, in the pathogenesis of VSD. However, VSD is genetically heterogeneous and the genetic determinants for VSD in most patients remain to be identified.
Material/Methods
A cohort of 230 unrelated patients with congenital VSD was included in the investigation. A total of 200 unrelated ethnically matched healthy individuals were recruited as controls. The entire coding region of GATA4, a gene encoding a zinc-finger transcription factor essential for normal cardiac morphogenesis, was sequenced initially in 230 unrelated VSD patients. The available relatives of the mutation carriers and 200 control subjects were subsequently genotyped for the presence of identified mutations.
Results
Four heterozygous missense GATA4 mutations of p.Q55R, p.G96R, p.N197S, and p.K404R were identified in 4 unrelated patients with VSD. These mutations were not detected in 200 control individuals nor described in the human SNP database. Genetic analysis of the relatives of the mutation carriers showed that in each family the mutation co-segregated with VSD.
Conclusions
These findings expand the mutation spectrum of GATA4 linked to VSD and provide new insight into the molecular etiology responsible for VSD, suggesting potential implications for the genetic diagnosis and gene-specific therapy for VSD.
Keywords: ventricular septal defect, transcription factor, genetics
Background
Congenital heart disease is the most prevalent form of developmental abnormality in newborns, with an estimated prevalence of 1%, and is the leading non-infectious cause of infant mortality, with more than 29% of neonates who die of a birth defect having a cardiovascular deformity [1]. Congenital cardiovascular anomaly is clinically classified into at least 18 different types, with many additional anatomic variations, of which ventricular septal defect (VSD) is the most common type. VSD occurs in 30–60% of all children, with various kinds of congenital cardiovascular deformations, and accounts for 14–16% of birth defects that require an invasive procedure within the first year of life [1–3]. Congenital VSD can occur by itself or in combination with other cardiac malformations such as atrial septal defect, tetralogy of Fallot, and patent ductus arteriosis [1,2,4,5]. Regardless of other potential conditions that accompany VSD, isolated moderate-to-large VSD with persistent left-to-right shunting of blood may give rise to cardiac enlargement, ventricular dysfunction, pulmonary hypertension, Eisenmenger’s syndrome, delayed fetal brain development, arrhythmias, and even sudden cardiac death in the absence of surgical or catheter-based repair [6–13]. Despite the high incidence and the significant association of VSD with substantial morbidity and mortality in human, the etiology of VSD remains largely unclear [14,15].
Abnormally developed interventricular septum is implicated in a heterogeneous, complex pathogenic process, for which both environmental risk factors and genetic defects may be responsible [2,14–16]. Recently, growing evidence points to a crucial role of the zinc finger transcription factor GATA4 in the cardiogenesis [17,18]. The human GATA4 gene maps to chromosome 8p23.1-p22 and comprises 7 exons encoding a protein of 442 amino acids [19]. It is expressed throughout embryonic development and also in the adult heart [17,18]. Therefore, GATA4 has been one of prime candidate genes in identifying the molecular components underlying structural congenital heart defects. Presently, over 22 germline mutations in the coding exons of the GATA4 gene have been identified in patients with a wide variety of congenital heart anomalies, including VSD, atrial septal defect, atrioventricular septal defect, tetralogy of Fallot, and endocardial cushion defect [20–33]. Nevertheless, VSD is genetically heterogeneous and the heritable determinants leading to VSD in the majority of cases are still to be identified.
Material and Methods
Study participants
A cohort of 230 unrelated patients with VSD, including 205 cases with apparently sporadic VSD and 25 index patients with familial VSD, was identified among a Chinese Han population. Subjects were evaluated by individual and familial history, review of the medical records, complete physical examination, 12-lead electrocardiogram (ECG) and two-dimensional transthoracic echocardiography with color flow Doppler. All patients had a classic form of VSD, with a defect diameter of >3 mm and nearly all patients underwent cardiac catheterization and, if required, cardiac surgery. The available family members of the probands harboring identified GATA4 mutations were enrolled and evaluated by medical history, medical records, physical examination, ECG and echocardiography with color flow Doppler. A total of 200 ethnically matched unrelated healthy individuals, recruited from the general population, were used as controls to screen for likely mutations in GATA4. Peripheral venous blood specimens from VSD patients, available relatives of the mutation carriers, and control individuals were prepared. The study protocol was reviewed and approved by the local institutional ethics committee and written informed consent was obtained from all participants or their guardians prior to investigation.
Genetic studies
Genomic DNA from all participants was extracted from blood lymphocytes with Wizard Genomic DNA Purification Kit (Promega). The candidate gene GATA4 was screened initially in 230 unrelated patients with VSD. Genotyping GATA4 in the available relatives of an index patient carrying an identified mutation and the 200 ethnically matched unrelated healthy control individuals was conducted subsequently. The referential genomic DNA sequence of GATA4 was derived from GenBank (accession No. NC_000008). By the aid of on-line Primer 3 software (http://frodo.wi.mit.edu), the primer pairs used to amplify the coding exons and exon/intron boundaries of GATA4 by polymerase chain reaction (PCR) were designed, as shown in Table 1. The PCR was carried out using HotStar Taq DNA Polymerase (Qiagen) on a PE 9700 Thermal Cycler (Applied Biosystems), with standard conditions and concentrations of reagents. Amplified products were analyzed on 1% agarose gels stained with ethidium bromide and purified with QIAquick Gel Extraction Kit (Qiagen). Both strands of each PCR product were sequenced with a BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) under an ABI PRISM 3130 XL DNA Analyzer (Applied Biosystems). The sequencing primers were the same as previously designed for amplification of specific regions. The DNA sequences were viewed and analyzed with the DNA Sequencing Analysis Software v5.1 (Applied Biosystems). The variant was validated by re-sequencing an independent PCR-generated amplicon from the subject and met our quality control thresholds with a call rate >99%.
Table 1.
The intronic primers to amplify the coding exons and exon-intron boundaries of GATA4.
| Exon | Forward primer (5′ to 3′) | Reverse primer (5′ to 3′) | Amplicon (bp) |
|---|---|---|---|
| 2-a | GAT, CTT, CGC, GAC, AGT, TCC, TC | GTC, CCC, GGG, AAG, GAG, AAG | 458 |
| 2-b | GCT, GGG, CCT, GTC, CTA, CCT | AAA, AAC, AAG, AGG, CCC, TCG, AC | 554 |
| 3 | GGG, CTG, AAG, TCA, GAG, TGA, GG | GAT, GCA, CAC, CCT, CAA, GTT, CC | 437 |
| 4 | GAG, ATC, TCA, TGC, AGG, GTC, GT | GCC, CCT, TCC, AAA, TCT, AAG, TC | 390 |
| 5 | TCT, TTC, TCG, CTG, AGT, TCC, AG | GGG, ATG, TCC, GAT, GCT, GTC | 379 |
| 6 | GCC, ATC, CCT, GTG, AGA, ACT, GT | GAG, GGT, AGC, TCA, CTG, CTT, GC | 444 |
| 7 | AAG, TGC, TCC, TTG, GTC, CCT, TC | TTC, CCC, TAA, CCA, GAT, TGT, CG | 479 |
Multiple sequence alignments
The multiple GATA4 protein sequences across species were aligned using the online program of CLUSTALW (http://www.bioinformatics.nl/tools/clustalw.html/).
Results
Characteristics of the study subjects
A cohort of 230 unrelated patients with VSD was enrolled and clinically evaluated in contrast to a cohort of 200 ethnically matched unrelated healthy individuals as controls. None of them had apparent traditional risk factors for VSD. The baseline clinical characteristics of the 230 unrelated patients with VSD are summarized in Table 2.
Table 2.
Clinical characteristics of the 230 unrelated patients with ventricular septal defects.
| Number or mean value | Percentage or range | |
|---|---|---|
| Male: female | 122: 108 | 53: 47 |
| Age at the diagnosis of VSD (year) | 3.4 | 1–16 |
| Age at the present study (year) | 5.2 | 1–20 |
| Positive family history | 25 | 11 |
| Distribution of different types of VSDs | ||
| Subarterial | 9 | 4 |
| Perimembranous | 183 | 80 |
| Atrioventricular canal | 10 | 4 |
| Muscular | 28 | 12 |
| Prevalence of VSDs with other defects | ||
| Isolated VSD | 215 | 93 |
| VSD and ASD | 9 | 4 |
| VSD and ASD and PDA | 2 | 1 |
| VSD and ASD and DORV | 2 | 1 |
| VSD and PDA | 4 | 2 |
| VSD and PS | 2 | 1 |
| Incidence of arrhythmias | ||
| Atrioventricular block | 3 | 1 |
| Atrial fibrillation | 2 | 1 |
| Treatment | ||
| Surgical repair | 141 | 61 |
| Transcatheter closure | 83 | 36 |
| Follow-up | 6 | 3 |
VSD – ventricular septal defect; ASD – atrial septal defect; PDA – patent ductus arteriosus; DORV – double outlet right ventricle; PS – pulmonary stenosis.
GATA4 mutations
Direct sequencing of the coding exons of the GATA4 gene was conducted after PCR amplification of genomic DNA from the 230 unrelated VSD patients. Four heterozygous missense mutations in GATA4 were identified in 4 out of 230 patients. The total population prevalence of GATA4 mutations based on the cohort patients was approximately 1.74%. Specifically, displacement of adenine by guanine in the second nucleotide of codon 55 of the GATA4 gene (c.164A>G), equivalent to the replacement of glutamine by arginine at amino acid 55 (p.Q55R), was identified in the proband from family 1. Substitution of adenine for guanine in the first nucleotide of codon 96 of the GATA4 gene (c.286G>A), predicting the transition of glycine to arginine at amino acid position 96 (p.G96R), was identified in the index patient from family 2. A change of adenine into guanine in the second nucleotide of codon 197 of the GATA4 gene (c.590A>G), corresponding to the transversion of asparagine to serine at amino acid residue 190 (p.N197S), was identified in the proband from family 3. A GATA4 sequence variation of c.1211A>G, resulting in the conversion of lysine into arginine at amino acid 404 (p.K404R), was identified in the proband from family 4. The sequence chromatograms showing the detected heterozygous GATA4 mutations in comparison with control sequences are shown in Figure 1. The variants were not detected in 200 control individuals and they are not described in the human SNP database (http://www.ncbi.nlm.nih.gov/SNP/). Genetic scan of the available family members of the mutation carriers demonstrated that in each family the variant was present in all affected family members, but absent in unaffected family members who were tested. Analysis of the pedigrees showed that each mutation co-segregated with VSD in the family with complete penetrance. The pedigree structures of the families are illustrated in Figure 2. The phenotypic characteristics and results of genetic screening of the affected pedigree members are listed in Table 3.
Figure 1.
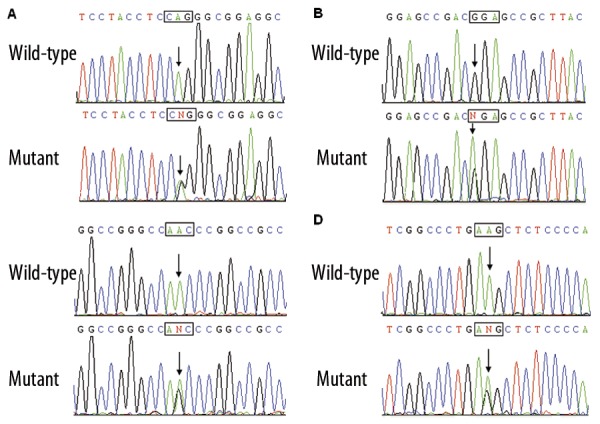
Sequence chromatograms of GATA4 in index patients and controls. The arrow indicates the heterozygous nucleotides of A/G (A), G/A (B), A/G (C), and A/G (D), in the probands from families 1, 2 3, and 4, respectively (mutant) or the homozygous nucleotides of A/A (A), G/G (B), A/A (C), and A/A (D), in the corresponding control individuals (wild-type). The square denotes the nucleotides comprising a codon of GATA4.
Figure 2.

Pedigree structures of the families with ventricular septal defect. Families are designated as family 1, family 2, family 3, and family 4. Family members are identified by generations and numbers. Squares indicate male family members; circles, female members; closed symbols, affected members; open symbols, unaffected members; arrow, proband; “+”, carriers of the heterozygous mutations; and “−”, non-carriers.
Table 3.
Phenotypic characteristics and status of the GATA4 mutations in the affected pedigree members.
| Subject Information | Phenotypes | Genotypes | |||||
|---|---|---|---|---|---|---|---|
| Identity | Gender | Age at time of study (years) | Age at diagnosis of VSD (years) | VSD (mm) | Other structural defects | AVB | Mutations |
| Family 1 | Q55R | ||||||
| I-2 | F | 30 | 12 | 5 | − | +/− | |
| II-1 | F | 5 | 3 | 13 | − | +/− | |
| Family 2 | G96R | ||||||
| I-1 | M | 36 | 18 | 6 | ASD | + | +/− |
| II-1 | M | 7 | 7 | 3 | − | +/− | |
| II-3 | F | 1 | 1 | 7 | PDA | − | +/− |
| Family 3 | N197S | ||||||
| I-1 | M | 32 | 7 | 11 | PS | − | +/− |
| II-1 | F | 2 | 2 | 15 | − | +/− | |
| Family 4 | K404R | ||||||
| II-1 | M | 2 | 2 | 8 | − | +/− | |
M – male; F – female; VSD – ventricular septal defect; ASD – atrial septal defect; PDA – patent ductus arteriosus; PS – pulmonary stenosis; AVB – atrioventricular block. + indicates present and – denotes absent.
Multiple alignments of the GATA4 protein sequences across species
A cross-species alignment of multiple GATA4 protein sequences showed that the affected amino acids of Q55, G96, N197, and K404 were completely conserved among mammals, as shown in Figure 3, suggesting that these amino acids are functionally important.
Figure 3.

Alignment of multiple GATA4 protein sequences across species. The altered amino acids of Q55, G96, N197, and K404 are completely conserved among mammals.
Discussion
In the present study, 4 novel heterozygous missense mutations of GATA4 were identified in 4 families with congenital VSD. In each family, the mutation was present in all the affected family members alive but absent in unaffected relatives tested and 400 normal chromosomes from a matched control population. A cross-species alignment of GATA4 protein sequences demonstrates that the altered amino acids were highly conserved evolutionarily. Therefore, it is very likely that mutated GATA4 is involved in the pathogenesis of VSD in these families.
GATA transcription factors are a family of transcription factors characterized by the ability to bind to the consensus DNA sequence ‘GATA’. To date, 6 members of the GATA family have been identified in vertebrates, of which GATA4, GATA5 and GATA6 are expressed mainly in the developing heart and in several endodermal lineages [34]. Structurally, GATA4 comprises 2 transcriptional activation domains (TAD1, amino acids 1–74; TAD2, amino acids 130–177), 2 zinc finger domains (ZF1, amino acids 215–240; ZF2, amino acids 270–294), 1 nuclear localization signal (NLS, amino acids 295–324), and 1 C-terminus (C-ter, amino acids 325–442) [35]. The 2 TADs are essential for the transcriptional activity of GATA4. The C-terminal ZF1 is required for DNA sequence recognition and binding to the consensus motif, while the N-terminal ZF2 is responsible for sequence specificity and stability of protein-DNA binding. The NLS sequence is associated with the subcellular trafficking and distribution of GATA4. The C-terminus is a regulator of the transcriptional activity of GATA4 [35]. The GATA4 mutation p.Q55R identified in this study is located in TAD1, and thus may be expected to exert direct influence on the transactivating activity of GATA4. The other 3 GATA4 mutations, including p.G96R and p.N197S located in the neighbor of TADs and p.K404R located in the C-terminus, may indirectly influence the transcriptional activity of GATA4 by altering the molecular conformation.
Our results are supported by the reports of other GATA4 mutations associated with congenital cardiac septal defects. Presently, at least 13 out of 22 germline mutations identified in the coding region of the GATA4 gene (p.H28Y, p.A66T, p.P163S, p.E216D, p.G296S, p.G296R, p.Q316E, p.S377G, p.V380M, p.P407Q, p.A411V, p.D425N, and p.H436Y) have been associated with isolated or syndromic VSD, showing that although GATA4 mutations underlie a long list of cardiac developmental aberrations, one of the most common phenotypes ascribed to mutations of the GATA4 gene is VSD [20–33]. However, the prevalence of GATA4 mutations varies significantly in different cohorts of individuals with congenital heart diseases. According to the 12 reports on the prevalence of GATA4 mutations in different cohorts of patients with VSD, the detection frequencies of GATA4 mutations are 12.50% (2/16) [23], 10.00% (5/50) [29], 6.90% (2/29) [22], 3.74% (4/107) [26], 3.23% (1/31) [30], 2.47% (12/486) [28], 1.67% (2/120) [24], 1.48% (2/135) [31], 0.80% (5/628) [25], 0.49% (1/205) [27], 0.48% (1/210) [33], and 0% (0/99) [36], and the prevalence in the compound population is 1.75% (37/2116). Similar to these findings, the mutational prevalence in our VSD cohort was 1.74% (4/230), suggesting that GATA4 mutations are an uncommon cause of congenital cardiovascular defects. Additionally, based on known mutation sites, no mutation hot spots exist within the GATA4 gene and the penetrance of different GATA4 mutations differs considerably, which may be ascribed to genetic backgrounds, environmental modifiers, epigenetic regulations, or even phenotypic ascertainment bias. Furthermore, the remarkable genetic heterogeneity of VSD was supported by a failure to detect mutations in nearly 98% of our cohort patients. Hence, the contribution of genes other than GATA4 to VSD pathogenesis appears likely.
Mutations in other transcription factors associated with cardiogenesis, such as NKX2-5 [37,38], TBX5 [39,40], TBX20 [41,42], and GATA6 [43,44], have also been identified in patients with VSD. In addition, mutations in cardiac structural proteins as troponin I type 3 (TNNI3) and alpha myosin heavy chain (MYH6) were identified in VSD patients [45,46]. Therefore, genetic analysis of these candidate genes in our cohort patients with VSD is warranted. However, these VSD-associated genes have also been reported to result in other cardiac or even extracardiac defects that underlie the clinical heterogeneity and the suggestive roles of the established genotype-phenotype relations of these genes. Interestingly, NKX2-5 mutations were mainly reported to cause atrial septal defect and atrioventricular block, implying the essential role of NKX2-5 in the morphogenesis of the heart and in the construction of the cardiac conduction system [47–49]. Moreover, TBX5 mutations were predominantly found to cause Holt-Oram Syndrome, which is clinically characterized by upper limb and heart defects. Despite the variable clinical manifestations, upper limb abnormalities were always present, highlighting the pivotal role of TBX5 in the development of both heart and upper limbs [50]. In the present study, no patients had extracardiac defects. A compound phenotype of VSD and atrioventricular block was observed in 3 out of 230 patients. Genetic screening of NKX2-5 in the 3 patients was performed, but no non-synonymous variants were found, largely excluding the possibility of NKX2-5 as a cause for VSD combined with atrioventricular block in these patients.
Association of compromised GATA4 with increased susceptibility to VSD has been demonstrated in animal experiments. In the embryonic hearts of knock-down chicks generated by using small interfering RNAs targeted to GATA4, the bilateral myocardial rudiments failed to travel to the midline, resulting in the formation of 2 separate hearts in lateral positions, an anomaly of cardia bifida [51]. In mice, GATA4 is one of the earliest transcription factors expressed in developing cardiac cells and continues to be expressed abundantly in cardiomyocytes throughout the life of mice [52]. Homozygous GATA4-deficient mice died between day 7.0 to 9.5 and analysis of the GATA4-null embryo substantiated the lethal failure to form a linear heart tube [53,54]. Transgenic mice expressing GATA4 mutants demonstrated a wide variety of cardiac malformations, including septal defects, right ventricular hypoplasia, endocardial cushion defect, tetralogy of Fallot, double outlets of the right ventricle, and cardiomyopathy, similar to the anomalies seen in humans [53,54]. Taken together, these experimental results from animals suggest that GATA4 mutations underlie a wide variety of congenital cardiac abnormalities, including VSD in humans.
Conclusions
The findings link novel GATA4 mutations to VSD and provide additional insight into the molecular etiology associated with VSD, suggesting potential implications for the prophylaxis and therapy of VSD.
Acknowledgements
We are indebted to the participants for their dedication to the study.
Footnotes
Source of support: The National Natural Science Fund of China (81070153, 30570768, and 30700776), the Natural Science Fund of Shanghai, China (10ZR1433100, 10ZR1428000), the Key Program of Shanghai Basic Research (10JC1414000, 10JC1414001, and 10JC1414002), and the National Basic Research Program of China (2010CB912604)
References
- 1.Lloyd-Jones D, Adams R, Carnethon M, et al. American Heart Association Statistics Committee and Stroke Statistics Subcommittee: Heart disease and stroke statistics--2009 update: a report from the American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Circulation. 2009;119:e21–181. doi: 10.1161/CIRCULATIONAHA.108.191261. [DOI] [PubMed] [Google Scholar]
- 2.Minette MS, Sahn DJ. Ventricular septal defects. Circulation. 2006;114:2190–97. doi: 10.1161/CIRCULATIONAHA.106.618124. [DOI] [PubMed] [Google Scholar]
- 3.Al-Jarallah AS. Down’s syndrome and the pattern of congenital heart disease in a community with high parental consanguinity. Med Sci Monit. 2009;15(8):CR409–12. [PubMed] [Google Scholar]
- 4.Orun UA, Bilici M, Yilmaz O, et al. Aortic coarctation with Down syndrome. Med Sci Monit. 2011;17(1):LE1. doi: 10.12659/msm.881316. [DOI] [PubMed] [Google Scholar]
- 5.Benatar A, Decraene T, Feenstra A. Ruptured sinus of Valsalva aneurysm in a child with Down syndrome: a rare cardiac anomaly. Med Sci Monit. 2010;16(11):CS135–37. [PubMed] [Google Scholar]
- 6.Sommer RJ, Hijazi ZM, Rhodes JF., Jr Pathophysiology of congenital heart disease in the adult: part I: Shunt lesions. Circulation. 2008;117:1090–99. doi: 10.1161/CIRCULATIONAHA.107.714402. [DOI] [PubMed] [Google Scholar]
- 7.McQuillen PS, Miller SP. Congenital heart disease and brain development. Ann N Y Acad Sci. 2010;1184:68–86. doi: 10.1111/j.1749-6632.2009.05116.x. [DOI] [PubMed] [Google Scholar]
- 8.Walsh EP. Interventional electrophysiology in patients with congenital heart disease. Circulation. 2007;115:3224–34. doi: 10.1161/CIRCULATIONAHA.106.655753. [DOI] [PubMed] [Google Scholar]
- 9.Walsh EP, Cecchin F. Arrhythmias in adult patients with congenital heart disease. Circulation. 2007;115:534–45. doi: 10.1161/CIRCULATIONAHA.105.592410. [DOI] [PubMed] [Google Scholar]
- 10.Jaworski R, Irga N, Haponiuk I, et al. Candidemia in children after complex congenital heart defects surgery treated with caspofungin – our own experience and a review of literature. Med Sci Monit. 2011;17(5):PH35–39. doi: 10.12659/MSM.881751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yap SC, Harris L. Sudden cardiac death in adults with congenital heart disease. Expert Rev Cardiovasc Ther. 2009;7:1605–20. doi: 10.1586/erc.09.153. [DOI] [PubMed] [Google Scholar]
- 12.Trojnarska O, Gwizdała A, Katarzyński S, et al. Evaluation of exercise capacity with cardiopulmonary exercise testing and BNP levels in adult patients with single or systemic right ventricles. Arch Med Sci. 2010;6:192–97. doi: 10.5114/aoms.2010.13893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baghdady Y, Hussein Y, Shehata M. Vascular endothelial growth factor in children with cyanotic and acyanotic and congenital heart disease. Arch Med Sci. 2010;6:221–25. doi: 10.5114/aoms.2010.13899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jenkins KJ, Correa A, Feinstein JA, et al. American Heart Association Council on Cardiovascular Disease in the Young. Noninherited risk factors and congenital cardiovascular defects: current knowledge: a scientific statement from the American Heart Association Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115:2995–3014. doi: 10.1161/CIRCULATIONAHA.106.183216. [DOI] [PubMed] [Google Scholar]
- 15.Pierpont ME, Basson CT, Benson DW, Jr, et al. American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young. Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. 2007;115:3015–38. doi: 10.1161/CIRCULATIONAHA.106.183056. [DOI] [PubMed] [Google Scholar]
- 16.Pemberton VL, McCrindle BW, Barkin S, et al. Report of the National Heart, Lung, and Blood Institute’s Working Group on obesity and other cardiovascular risk factors in congenital heart disease. Circulation. 2010;121:1153–59. doi: 10.1161/CIRCULATIONAHA.109.921544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pikkarainen S, Tokola H, Kerkelä R, Ruskoaho H. GATA transcription factors in the developing and adult heart. Cardiovasc Res. 2004;63:196–207. doi: 10.1016/j.cardiores.2004.03.025. [DOI] [PubMed] [Google Scholar]
- 18.Peterkin T, Gibson A, Loose M, Patient R. The roles of GATA-4, -5 and -6 in vertebrate heart development. Semin Cell Dev Biol. 2005;16:83–94. doi: 10.1016/j.semcdb.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 19.White RA, Dowler LL, Pasztor LM, et al. Assignment of the transcription factor GATA4 gene to human chromosome 8 and mouse chromosome 14: Gata4 is a candidate gene for Ds (disorganization) Genomics. 1995;27:20–26. doi: 10.1006/geno.1995.1003. [DOI] [PubMed] [Google Scholar]
- 20.Garg V, Kathiriya IS, Barnes R, et al. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424:443–47. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- 21.Okubo A, Miyoshi O, Baba K, et al. A novel GATA4 mutation completely segregated with atrial septal defect in a large Japanese family. J Med Genet. 2004;41:e97. doi: 10.1136/jmg.2004.018895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sarkozy A, Conti E, Neri C, et al. Spectrum of atrial septal defects associated with mutations of NKX2.5 and GATA4 transcription factors. J Med Genet. 2005;42:e16. doi: 10.1136/jmg.2004.026740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hirayama-Yamada K, Kamisago M, Akimoto K, et al. Phenotypes with GATA4 or NKX2.5 mutations in familial atrial septal defect. Am J Med Genet A. 2005;135:47–52. doi: 10.1002/ajmg.a.30684. [DOI] [PubMed] [Google Scholar]
- 24.Nemer G, Fadlalah F, Usta J, et al. A novel mutation in the GATA4 gene in patients with Tetralogy of Fallot. Hum Mutat. 2006;27:293–94. doi: 10.1002/humu.9410. [DOI] [PubMed] [Google Scholar]
- 25.Tomita-Mitchell A, Maslen CL, Morris CD, et al. GATA4 sequence variants in patients with congenital heart disease. J Med Genet. 2007;44:779–83. doi: 10.1136/jmg.2007.052183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rajagopal SK, Ma Q, Obler D, et al. Spectrum of heart disease associated with murine and human GATA4 mutation. J Mol Cell Cardiol. 2007;43:677–85. doi: 10.1016/j.yjmcc.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Posch MG, Perrot A, Schmitt K, et al. Mutations in GATA4, NKX2.5, CRELD1, and BMP4 are infrequently found in patients with congenital cardiac septal defects. Am J Med Genet A. 2008;146A:251–53. doi: 10.1002/ajmg.a.32042. [DOI] [PubMed] [Google Scholar]
- 28.Zhang W, Li X, Shen A, et al. GATA4 mutations in 486 Chinese patients with congenital heart disease. Eur J Med Genet. 2008;51:527–35. doi: 10.1016/j.ejmg.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 29.Chen MW, Pang YS, Guo Y, et al. GATA4 mutations in Chinese patients with congenital cardiac septal defects. Pediatr Cardiol. 2010;31:85–89. doi: 10.1007/s00246-009-9576-1. [DOI] [PubMed] [Google Scholar]
- 30.Chen Y, Mao J, Sun Y, et al. A novel mutation of GATA4 in a familial atrial septal defect. Clin Chim Acta. 2010;411:1741–45. doi: 10.1016/j.cca.2010.07.021. [DOI] [PubMed] [Google Scholar]
- 31.Peng T, Wang L, Zhou SF, Li XT. Mutations of the GATA4 and NKX2.5 genes in Chinese pediatric patients with non-familial congenital heart disease. Genetica. 2010;138:1231–40. doi: 10.1007/s10709-010-9522-4. [DOI] [PubMed] [Google Scholar]
- 32.Liu XY, Wang J, Zheng JH, et al. Involvement of a novel GATA4 mutation in atrial septal defects. Int J Mol Med. 2011;28:17–23. doi: 10.3892/ijmm.2011.638. [DOI] [PubMed] [Google Scholar]
- 33.Wang J, Fang M, Liu XY, et al. A novel GATA4 mutation responsible for congenital ventricular septal defects. Int J Mol Med. 2011;28:557–64. doi: 10.3892/ijmm.2011.715. [DOI] [PubMed] [Google Scholar]
- 34.Brewer A, Pizzey J. GATA factors in vertebrate heart development and disease. Expert Rev Mol Med. 2006;8:1–20. doi: 10.1017/S1462399406000093. [DOI] [PubMed] [Google Scholar]
- 35.Posch MG, Perrot A, Berger F, Ozcelik C. Molecular genetics of congenital atrial septal defects. Clin Res Cardiol. 2010;99:137–47. doi: 10.1007/s00392-009-0095-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang L, Tumer Z, Jacobsen JR, et al. Screening of 99 Danish patients with congenital heart disease for GATA4 mutations. Genet Test. 2006;10:277–80. doi: 10.1089/gte.2006.10.277. [DOI] [PubMed] [Google Scholar]
- 37.Schott JJ, Benson DW, Basson CT, et al. Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science. 1998;281:108–11. doi: 10.1126/science.281.5373.108. [DOI] [PubMed] [Google Scholar]
- 38.Benson DW, Silberbach GM, Kavanaugh-McHugh A, et al. Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J Clin Invest. 1999;104:1567–73. doi: 10.1172/JCI8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Basson CT, Bachinsky DR, Lin RC, et al. Mutations in human TBX5 cause limb and cardiac malformation in Holt-Oram syndrome. Nat Genet. 1997;15:30–35. doi: 10.1038/ng0197-30. [DOI] [PubMed] [Google Scholar]
- 40.Li QY, Newbury-Ecob RA, Terrett JA, et al. Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat Genet. 1997;15:21–29. doi: 10.1038/ng0197-21. [DOI] [PubMed] [Google Scholar]
- 41.Kirk EP, Sunde M, Costa MW, et al. Mutations in cardiac T-box factor gene TBX20 are associated with diverse cardiac pathologies, including defects of septation and valvulogenesis and cardiomyopathy. Am J Hum Genet. 2007;81:280–91. doi: 10.1086/519530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu C, Shen A, Li X, Jiao W, Zhang X, Li Z. T-box transcription factor TBX20 mutations in Chinese patients with congenital heart disease. Eur J Med Genet. 2008;51:580–87. doi: 10.1016/j.ejmg.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 43.Maitra M, Koenig SN, Srivastava D, Garg V. Identification of GATA6 sequence variants in patients with congenital heart defects. Pediatr Res. 2010;68:281–85. doi: 10.1203/PDR.0b013e3181ed17e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin X, Huo Z, Liu X, et al. A novel GATA6 mutation in patients with tetralogy of Fallot or atrial septal defect. J Hum Genet. 2010;55:662–67. doi: 10.1038/jhg.2010.84. [DOI] [PubMed] [Google Scholar]
- 45.Yang SW, Hitz MP, Andelfinger G. Ventricular septal defect and restrictive cardiomyopathy in a paediatric TNNI3 mutation carrier. Cardiol Young. 2010;20:574–76. doi: 10.1017/S1047951110000715. [DOI] [PubMed] [Google Scholar]
- 46.Granados-Riveron JT, Ghosh TK, Pope M, et al. Alpha-Cardiac myosin heavy chain (MYH6) mutations affecting myofibril formation are associated with congenital heart defects. Hum Mol Genet. 2010;19:4007–16. doi: 10.1093/hmg/ddq315. [DOI] [PubMed] [Google Scholar]
- 47.Pashmforoush M, Lu JT, Chen H, et al. Nkx2-5 pathways and congenital heart disease; loss of ventricular myocyte lineage specification leads to progressive cardiomyopathy and complete heart block. Cell. 2004;117:373–86. doi: 10.1016/s0092-8674(04)00405-2. [DOI] [PubMed] [Google Scholar]
- 48.Prall OW, Menon MK, Solloway MJ, et al. An Nkx2-5/Bmp2/Smad1 negative feedback loop controls heart progenitor specification and proliferation. Cell. 2007;128:947–59. doi: 10.1016/j.cell.2007.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moskowitz IP, Kim JB, Moore ML, et al. A molecular pathway including Id2, Tbx5, and Nkx2-5 required for cardiac conduction system development. Cell. 2007;129:1365–76. doi: 10.1016/j.cell.2007.04.036. [DOI] [PubMed] [Google Scholar]
- 50.Cerbai E, Sartiani L. Holt-oram syndrome and atrial fibrillation: opening the (T)-box. Circ Res. 2008;102:1304–6. doi: 10.1161/CIRCRESAHA.108.178079. [DOI] [PubMed] [Google Scholar]
- 51.Zhang H, Toyofuku T, Kamei J, Hori M. GATA-4 regulates cardiac morphogenesis through transactivation of the N-cadherin gene. Biochem Biophys Res Commun. 2003;312:1033–38. doi: 10.1016/j.bbrc.2003.11.019. [DOI] [PubMed] [Google Scholar]
- 52.Heikinheimo M, Scandrett JM, Wilson DB. Localization of transcription factor GATA-4 to regions of the mouse embryo involved in cardiac development. Dev Biol. 1994;164:361–73. doi: 10.1006/dbio.1994.1206. [DOI] [PubMed] [Google Scholar]
- 53.Kuo CT, Morrisey EE, Anandappa R, et al. GATA4 transcription factor is required for ventral morphogenesis and heart tube formation. Genes Dev. 1997;11:1048–60. doi: 10.1101/gad.11.8.1048. [DOI] [PubMed] [Google Scholar]
- 54.Molkentin JD, Lin Q, Duncan SA, Olson EN. Requirement of the transcription factor GATA4 for heart tube formation and ventral morphogenesis. Genes Dev. 1997;11:1061–72. doi: 10.1101/gad.11.8.1061. [DOI] [PubMed] [Google Scholar]