

Summary
This paper presents a new, recently formulated theory, which concerns the etiopathological process of autoimmune diseases. This theory takes into account the existence in the human genome, since approximately 40 million years, of so-called human endogenous retroviruses (HERVs), which are transmitted to descendants “vertically” by the germ cells. It was recently established that these generally silent sequences perform some physiological roles, but occasionally become active and influence the development of some chronic diseases like diabetes, some neoplasms, chronic diseases of the nervous system (eg, sclerosis multiplex), schizophrenia and autoimmune diseases. We present a short synopsis of immunological processes involved in the pathogenesis of autoimmune diseases, such as molecular mimicry, epitope spreading and activation of the superantigen. We then focus on experimental findings related to systemic lupus erythematosus, rheumatoid arthritis, Sjögren’s syndrome and some diseases of hepar and otorhinal tissues. We conclude the outline of this new model of the development of chronic diseases and indicate the conclusions important for the teaching of the basis of pathology.
Keywords: autoimmune diseases, systemic lupus erythematosus, human endogenous retroviruses, rheumatoid arthritis
Background
In recent years evidence has accumulated that supports a significant relationship between the so-called Human Endogenous Retroviruses (HERV) and the development of different autoimmune diseases [1–4], which suggests we need to change our understanding of the pathogenesis of these chronic diseases, including diabetes, some neoplasms and chronic diseases of the nervous system like sclerosis multiplex and even schizophrenia [5–10].
In this article we present a new, unusual model of the development of chronic diseases, especially the most common autoimmunological diseases.
Until recently, a simple, teaching model of pathogenesis recognized diseases caused by infectious agents, by chemical, physical and mechanical effects of the external environment, and pathogenesis involving genetic factors.
Some diseases are determined genetically by changes at the level of chromosomes (eg, Down syndrome), or by exactly known “point” changes in the structure of specific genes, such as in the case of hemophilia. Many diseases are undoubtedly genetically determined, as evidenced by epidemiological studies carried out on homozygous twins; however, the pathogenetic mechanisms are complex and, until now, unclear.
The statistical comparisons concerning the incidence of some diseases, made on groups of homozygotic twins, show that schizophrenia, manic-depressive illness and multiple sclerosis are genetically caused diseases. If one of the so-called ‘monozygotic twins’ has schizophrenia or manic-depressive illness, there is a probability of about 50% that the second twin will also have the same disease. The analogous figure for multiple sclerosis is 30% and for type 1 diabetes approximately 40%. In discussing the pathogenesis of these diseases, it is usually understood that the onset of these diseases is affected by changes in many genes, although it was not possible to demonstrate precisely the regions of the genome containing the errors influencing these diseases. People studying genetics as a basic field important for the understanding of pathogenic mechanisms have long been acquainted with facts related to some types of repeated nucleotide sequences, present in the human genome, which are not genes [11] (Figure 1).
Figure 1.
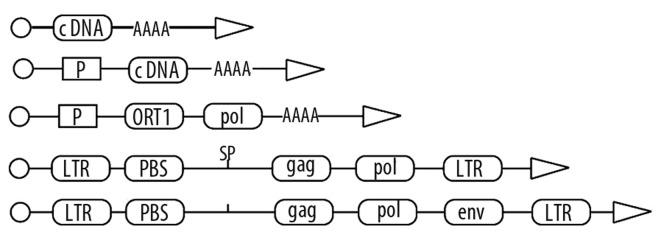
Types of retro elements: pseudogene, retrogen, retropozon, retrotranspozon, retrovirus. The meaning of symbols is as follows.: LTR-long terminal repeats, P-promoter, AAAA-adenine polynucleotide chain, ORF1-Open Reading Frame, PBS-tRNA primer binding region (primer binding site), gag-the gene encoding the structural proteins, pol-gene encoding reverse transcriptase, env-the gene encoding the envelope proteins.
The human genome includes about 3 billion base pairs, organized into 23 pairs of chromosomes. Only about 5% of the DNA polynucleotides consist of structural genes, coding for the synthesis of certain proteins. According to the authors of the “Human Genome Project” and Craig Venter, who also sequenced the whole human genome, there are only about 30 000 to 50 000 structural genes.
Non-coding polynucleotides constitute approximately 80% of the total genome. One group of these is called the pseudogenes, which are damaged, non-acting versions of known genes. Textbooks of genetics also distinguish between so-called “moderately and frequently repeated sequences” and “sequences unique or in small number of copies” [11]. Among the repeated sequences, distinction is made between “short interspersed nuclear elements” (SINE) and the “long interspersed nuclear elements” (LINE).
The most familiar SINE element is the “Alu sequence”, which is a “family” of polynucleotides (sequences quite similar), with an average length of 250 bp; approximately 700 000 copies are present in the genome. LINE elements are longer. For example, the well-known L1 LINE element has a length of 6500 bp and 60 000 copies are present in the genome. Classical genetics textbooks emphasize that LINE elements usually have the capacity of transposition, meaning that these elements have the ability to move and change their location in the genome by using reverse transcriptase [11]. Relatively recently, still other remarkable repeated sequences were discovered in the human genome.
Human Endogenous Retroviruses
Human Endogenous Retrovirus-HERVs are polynucleotide sequences representing the complete structure of the virus [12,13]. Their structure consists of 2 long-terminal repeats (LTR). Between them there are the gene encoding the structural proteins (gag), the genes encoding reverse transcriptase, protease, ribonuclease, and integrase (pol), the gene encoding the envelope proteins (env) and also the starter tRNA binding region (the primer binding site-PBS) and so-called packaging signal (Ψ) – which are important for the function of the virus.
Human endogenous retroviruses (HERVs) were discovered in the 1980. They represent about 8% of the human genome [12,13] and are present in about 450 000 copies. They can be classified into 200 different groups and subgroups. HERVs were integrated to germ-line cells of human ancestors. It is estimated that such insertions have occurred 30 million years ago [12,13]. The presence of HERVs in the human genome forms the basis for an argument for an alternative theory of the evolution [14,15].
A specific type of HERVs can occur in the genome in a single copy (eg, HRES-1), or can be repeated up to 1000 times (eg, HERVs-H). The greatest concentration of HERVs sequences is found in chromosomes Y, X, 4 and 20.
Human endogenous viruses are one of the so-called retro-elements, which are nucleotide sequences that can move within the genome through a mechanism of “rewriting” their composition to RNA sequence and incorporation of its “DNA equivalent” in another location of the genome by action of reverse transcriptase. Retrogenes, retroposons and retrotransposons are 3 types of retro-elements found in the human genome [11,12].
Recently, the exogenous factors affecting the expression of HERVs have been determined, their interactions with exogenous retroviruses were examined, and the impact of HERVs on the organization, functioning and evolution of the human genome has been considered [12–14].
Data on the Activity of Human Endogenous Retroviruses
The majority of HERVs is not expressed, due to the numerous inactivating mutations in the areas of reading frames. These viruses are also frequently silenced by epigenetic mechanisms (DNA methylation). Some of the HERVs are active, however, and their expression is regulated by many different factors. Some types of viruses HERVs are activated by X-rays and ultraviolet light. This occurs in patients with psoriasis. Pro-inflammatory cytokines, including IFN-alpha, TNF-alpha and IL-1a and IL-1b, and glucocorticoids can act as factors inducing activity of HERVs. Their expression is highest in the placenta tissue and endocrine glands (hypothalamus, male testes) [16–19].
Other factors influencing the activity of HERVs are products of exogenous viruses. For example, Epstein-Barr virus enhances the transcription of the Env gene HERVs-K18 located in the intron of the gene CD48 [7]. In vitro studies showed that HSV-1 induces expression of HERVs-W in human neuroblastoma cells. This virus also causes expression of HERVs-K. Cytomegalovirus (CMV) is activated by HERVs-K. The influenza virus causes increased levels of Env protein HERVs in neuroepithelial cells [12].
Some of the HERVs are present in genes evolving rapidly with a high incidence of mutations, as occurs in genes involved in immune response processes.
Surprisingly, the products of the expression of HERVs also have effects on physiological functioning and development of tissues. Fang Li et al. wrote as follows: “A prominent exception, however, is the ERVWE1 locus on chromosome 7q. This locus harbors a member of the HERV-W family with an open reading frame in the env gene that encodes a protein denoted syncytin. This protein is highly expressed in the syncytiotrophoblast layer of the human placenta and appears to have been functionally adopted by the human host for fusion of trophoblast cells and thus contributing to the formation of the syncytiotrophoblast layer” [20–22]. Thus, HERVs are helpful for the proper formation of the placenta and are involved in the suppression of rejection of fetal tissues. HERVs also interfere with exogenous viruses through interference with their receptors or the formation of antisense mRNA and are also related to the development of certain types of cancer [6,7,18].
Short Synopsis of the Structure and Function of the Human Immune System
The human body is constantly influenced by factors which could harm it. These include toxins, bacteria, fungi, parasites and viruses. The purpose of the immune system is to defend against the constant stream of possible infections and toxins.
There are 2 branches of the immune system, called the acquired immune system and the innate immune system.
The innate immune system protects the body without special preparation. It consists of physical barriers formed by skin, mucous membranes, saliva, flushing action of urine and tears, and stomach acid. Another part of the innate immune system is some elements present in the blood – phagocytes like neutrophils, macrophages, natural killer cells and complement cascade.
The acquired immune system needs to be primed before it can work and it is only effective after it has seen a possible infective agent.
The acquired immune system is composed of 2 subsystems: T-cell immunity (cell-mediated immunity) and B-cell immunity (humoral immunity).
Cell-mediated immunity is realized by 3 kinds of lymphocytes: helper T-cells, cytotoxic T-cells, and regulatory T-cells (T reg) (formerly termed suppressor T-cells).
B-cell immunity acts after exposure to the antigen and is realized by “tuned” lymphoblasts, which transform themselves into plasma cells or clonal B-cells. Plasma cells produce antibodies. Clonal B-cells constitute immune memory. Antibodies can activate complement cascade.
T-cells work together with B lymphocytes. The function of the helper T-cell is essential in the transition from detecting an invader to launching a defense against it.
Also important is the cooperation of macrophages, cells which metaphorically “eat” invaders.
Antibodies are molecules secreted by the B-cells and plasma cells. They are immunoglobulins, which latch onto invaders and neutralize them in various ways and also trigger the production of cytokines. Cytokines are important molecules, components of the immune system. They are molecular signals emitted by lymphocytes and other cells. Cells of the immune systems interact by means of cytokines or interleukins (Il).
T lymphocytes have receptors embedded in their membranes (TCR). These receptors are composed of 2 proteins connected together. They are similar to immunoglobulins, but are smaller. It is important to realize that T lymphocytes bind not just an antigen, but a complex of an antigen associated with a HLA protein. HLA proteins are molecules present in most cells of the body, which allows to take a bits of the antigen and hold it outside the cell so that it can checked by T-cells. The contacts between T-cells and other cells are usually performed in cooperation with some specific surface proteins (Figure 2).
Figure 2.
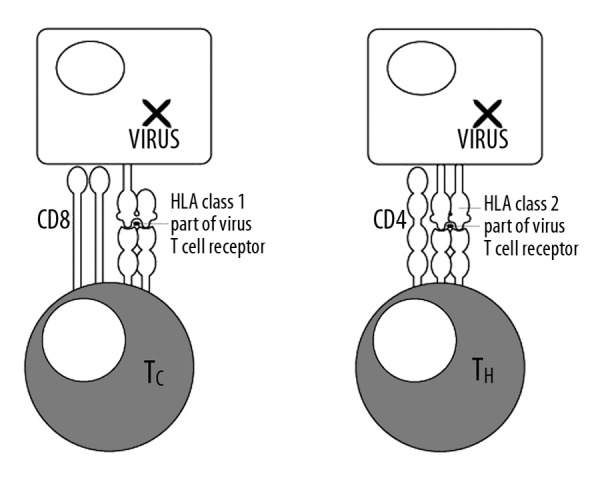
Cooperation of cytotoxic and helper T lymphocytes with HLA proteins of class I and class II of antigen presenting cells (APC).
Human Leukocyte Antigen
The human leukocyte antigen (HLA) system is the major histocompatibility complex (MHC) in humans. The group of genes from the locus on chromosome 6 contains a large number of genes related to immune system function in humans. This group of genes encodes cell-surface antigen-presenting proteins.
The HLA genes are the human versions of the MHC genes that are found in most vertebrates. HLA class I antigens present peptides from inside the cell, including viral peptides. Foreign antigens attract killer T-cells, also called CD8-positive or cytotoxic T-cells, which destroy cells. HLA class II antigens present antigens from outside of the cell to T-lymphocytes. These particular antigens stimulate T-helper cells to multiply, and these T-helper cells then stimulate antibody-producing B-cells to produce antibodies to that specific antigen (Figure 2).
So HLA (Human Leukocyte Antigen) is a class of proteins which is found on the surface membrane of cells, and which are “presenting” possible antigen to T- and B-cells. They are also present inside a cell, and have a groove in which they can attach little bits of protein. These bits of protein will have come from outside the cell or could be products of the cell’s own genes. Once a bit of protein or other possible antigen is attached, the HLA moves out and protrudes from the outside of the cell, so that a T-cell can come along and check the molecule that is being presented.
The T-cells, in turn, check whether the molecule is self or foreign, and will pass over it or react to its presence, either destroying the cell, or raising the alarm about the foreign substance by emitting cytokines.
T-cells
T-cells can be distinguished from other types of lymphocyte by the presence of a special receptor on their cell surface, called T-cell receptors (TCR). The shape and function of the T-cell receptor is variable. There are millions of different types of T-cell receptors in the body, and each of them is specialized to recognize different “foreign” matter. There are also receptors that would attack the body’s own proteins and cells but these are checked and destroyed in the thymus, where the T-cells mature, before the T cells are let loose in the body.
Helper T-cells
The helper T-lymphocytes are crucial to the function of the immune system. They assist other white blood cells in maturation of B-cells into plasma cells and B memory cells, and activation of cytotoxic T-cells and macrophages. Helper T-cells circulate throughout the body and check for antigens that might be signs of invasion by viruses or bacteria. They interface with HLA class II protein on other cells (antigen presenting cells-APCs), checking what kind of substance HLA class II is presenting. If the helper T-cell is stimulated by contact with antigen, it responds by cell division, as well as producing lymphokines and chemokines. Lymphokines and chemokines are chemical messages that transfer signals to other cells. Cell division results in more and more activated helper T-cells.
The helper T-cells are the main cells that carry the CD4 surface protein. It is the main type of cell targeted by HIV. The virus can destroy the majority of CD4+ cells in the human immune system and the immune system is then paralyzed.
Cytotoxic T-cells
The cytotoxic T-cells are known also as killer T-cells or CD8+ T-cells, as they express the CD8 glycoprotein at their surface. They destroy virally infected cells and tumor cells, and are also implicated in transplant rejection. Viral and other foreign peptides are processed by the MHC class I, which is present on the surface of nearly every cell of the body. A piece of foreign antigen is protruded to the outside of the cell by HLA protein. A cytotoxic T-cell then binds to such a cell by an interaction between CD8, TCR (T-cell receptor) and HLA. If the peptide presented to the cytotoxic T-cell by HLA fits the cleft in the TCR, then a chemical signal is triggered within the T cell, which causes it to attack and destroy the infected cell. Through IL-10, adenosine and other molecules secreted by regulatory T-cells, the CD8+ cells can be inactivated to an anergic state, which prevent autoimmune diseases.
Memory T-cells
Memory T-cells are a subset of antigen-specific T-cells that persist long-term after an infection has resolved. They quickly expand to a large numbers of T-cells upon re-exposure to the antigen. They constitute the immune system’s “memory” of past infections.
Regulatory T-cells (T reg Cells)
T reg cells formerly known as suppressor T-cells, are important for the maintenance of immunological tolerance. Their major role is to block T-cell-mediated immunity toward their own cells. Two major classes of CD4+ regulatory T cells have been described – the naturally occurring T reg cells and the adaptive T reg cells. Naturally occurring T reg cells can be distinguished from other T-cells by the presence of an intracellular molecule called FoxP3.
CD4 Cell surface protein
CD4 is a glycoprotein found mainly on surface of the mature helper T cells, macrophages and on some cytotoxic T cells. About 65% of T-cells in the blood are CD4+ with CD4 protein protruding from their membrane. Mature T-cells have CD4 or CD8, but not both. CD4 is a member of the immunoglobulin superfamily. The external part of the protein has 4 “domains” and these are the essential part of the protein composed from 433 amino acids.
CD4 cooperates with HLA protein of different cells to allow helper T-cells to check for ‘suspicious’ proteins of these cells. Normal CD4+ counts are between 500 and 1600, but a person with HIV/AIDS commonly has a CD4+ count of less than 200.
CD8 cell surface protein
CD8 is also a cell surface protein found predominantly on T-cells. CD8 is ordinarily found on the surface of killer T-cells, as opposed to CD4, which is ordinarily found on the surface of helper T-cells. CD8 cooperates with HLA class I, which allows the T-cell receptor protein to dock to HLA-I and check the antigen that HLA is presenting. If the shape of the T-cell receptor matches the antigen that is being presented, then a sequence of signals is triggered inside the T-cell which causes it to kill the cell to which it is attached.
T-cell receptor
The T-cell receptor exists as a complex of several proteins. TCR is composed of 2 separate peptide chains, which are produced from the independent T-cell receptor alpha and beta (TCRα and TCRβ) genes. TCR interfaces with major histocompatibility complex protein (HLA) to “feel” the shape of a protein presented by MHC.
Immunological Phenomena and Concepts Related to Pathogenetic Mechanisms of Autoimmune Diseases
Immunological tolerance
Tolerance is the ability of the normal immune system to recognize and respond to foreign antigens, but not to self antigens. Autoimmunity is evoked when this tolerance to self antigen is broken. The tolerance of a person is formed already in the fetus. During this period of life, maternal-fetal tolerance exists.
The pre-T-cells leave the bone marrow, where they were synthesized. They move to the thymus, where the maturation of T-cells occurs and the first step of T-cell tolerance is realized. Within the thymus, pre-T-cells encounter various self and foreign antigens and pre-T-cells undergo a double selection process. They must be positively selected and should avoid negative selection. A T-cell is positively selected when it binds with low affinity to self-MHC receptors. Other T-cells are eliminated by apoptosis. Cells that survive positive selection, but bind strongly to self-antigens, are negatively selected and are also eliminated by induction of apoptosis. This negative selection is known as clonal deletion.
T-cell tolerance must be developed further in the periphery, because there is only a limited repertoire of antigen that T-cells can encounter within the thymus. This same positive and negative selection mechanism is applied in peripheral tissues. This is known as clonal anergy. Another mechanism of T-cell tolerance is known as active suppression.
Autoimmunity
Autoimmune diseases are characterized by long, asymptomatic prodromal periods. The initiating events causing the loss of self tolerance occur long before the disease clinically manifests; therefore, it is difficult to recognize the initiating factors, which remain largely unknown.
Autoimmunity results from of a loss of immunological tolerance, which is the ability for an individual to discriminate between self and non-self. In another words, it is the failure to recognize self antigens as “self”.
Foreign agents are pathogens like bacteria and viruses that can induce autoimmunity by polyclonal activation of B- or T-cells or increased expression of the class I or II molecules of the major histocompatibility complex (MHC). There are some different ways in which a pathogen can cause an autoimmune response. The pathogens may contain a protein that acts as a mitogen to encourage cell division, thus causing intensified production of B- or T-cell clones. A pathogenic protein may act as a superantigen (see below), which can induce rapid polyclonal activation of B- or T-cells. Pathogens can also cause the release of cytokines, resulting in the activation of B- or T-cells, or they can alter macrophage function. Pathogens may also expose B- or T-cells to cryptic determinants that induce the molecular mimicry mechanism.
Superantigens (SAgs)
These are a class of antigens that cause non-specific activation of T-cells. This activation induces polyclonal T-cell activation and massive cytokine release. SAgs can be produced by viruses, mycoplasma, and bacteria. Compared to a normal antigen-induced T-cell response, where about 0.001% of the body’s T-cells are activated, the SAgs are capable of activating up to 20% of the body’s T-cells.
The large number of activated T-cells secretes large amounts of cytokines, especially TNF-alpha. When TNF-alpha is released in the blood in high doses it can cause severe and life-threatening symptoms, including shock and multiple organ failure.
Typical antigens are captured by the antigen presenting cell (APC), which processes them and places them within the histocompatibility complex (MHC) class II on their surface. In this form the antigen is presented to T lymphocytes. In the case of spatial matching of antigen, MHC II, and T-cell receptor (TCR), the Tc lymphocyte is activated (Figure 1). This means that in case of detecting the presence of a foreign protein, the activation occurs and a precise, specific T-cell clone multiplies. In the case of the presence of a superantigen, unlike the typical situation described above, SAg activates T lymphocytes in a nonspecific manner, creating a kind of “short circuit” between the MHC II and β-chain of T-cell receptors (Figures 3 and 4), a non-specific activation and division of numerous clones of T-cells, although the place of presentation of antigen on MHC II remains empty. Normally, 5–10% of all cells have receptors containing the β chain, but in the presence of SAgs this percentage grows rapidly due to the expansion of stimulated cells. Stimulated cells produce cytokines, especially interleukin-4 and interleukin-13. Staphylococcal enterotoxins also stimulate release of histamine and leukotrienes, which are mediators of allergic inflammation.
Figure 3.
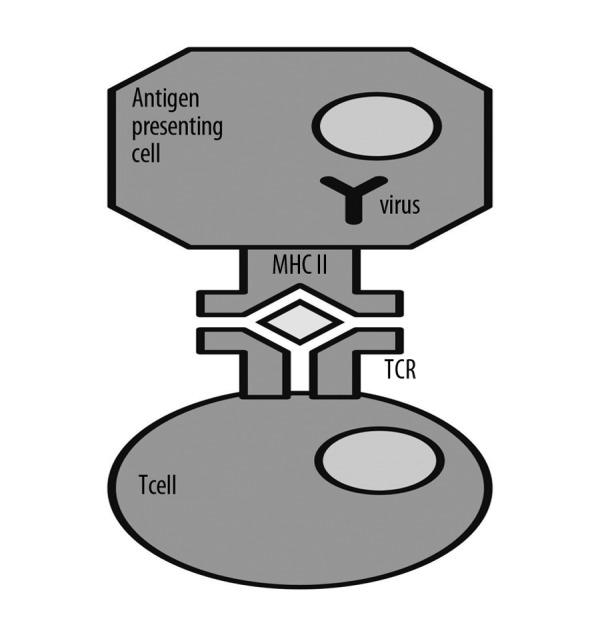
Classical immune process of presenting a foreign antigen by a Antigen Presenting Cell (APC) by means of Major Histocompatibility Complex of class II (MHC II) to a T cell, which has T Cell Receptor (TCR).
Figure 4.
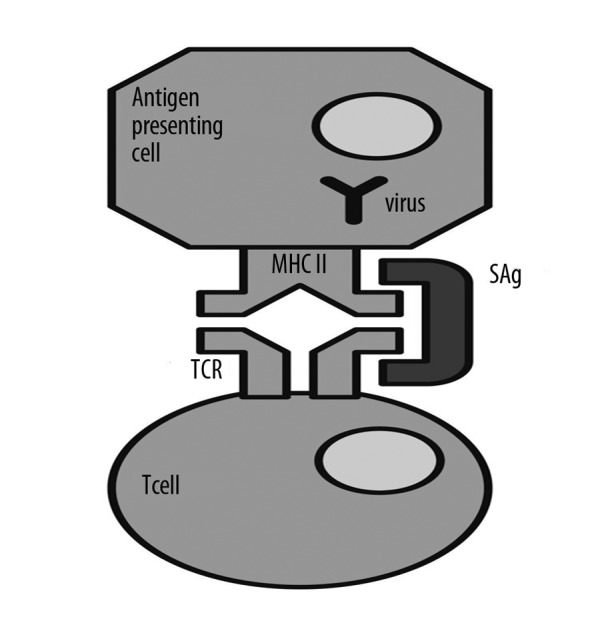
Comparison of the classical response to the antigens with the action of the superantygens. APC-antigen presenting cell, MHC II-major histocompatibility complex class II, SAg-superantigen, α, β-chains- fragments of T-cell receptor (TCR).
Molecular mimicry
There are several different ways in which autoimmunity can occur – one of which is molecular mimicry. Molecular mimicry happens when similar sequences of polynucleotides exist in different genes, or similar sequences of amino acids are found in the protein products of these genes.
Either the linear amino acid sequence or the conformational, structural fit of the immunodominant epitope may be shared between the pathogen and host. This causes “cross-reactivity” between self antigens of the host and immunodominant epitopes of the pathogen. An autoimmune response is then generated against the epitope.
Appropriate calculations show that the probability of very similar amino acid sequence of proteins, both within the pathogen and host tissues, is very small. But there is a possibility that the immune cells treat some similar sequences as identical on the basis on a very similar spatial structure of these proteins (Figure 5). The phenomenon of molecular mimicry is illustrated in Figure 6.
Figure 5.
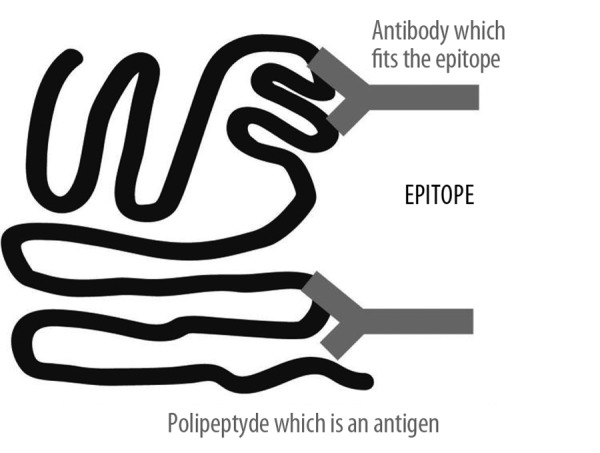
The are two kinds of epitope. The crucial region of the protein can be characterized by a similar amino acid sequence or the epitope can be formed on the basis of a similar spatial structure of the protein, which is an antigen.
Figure 6.
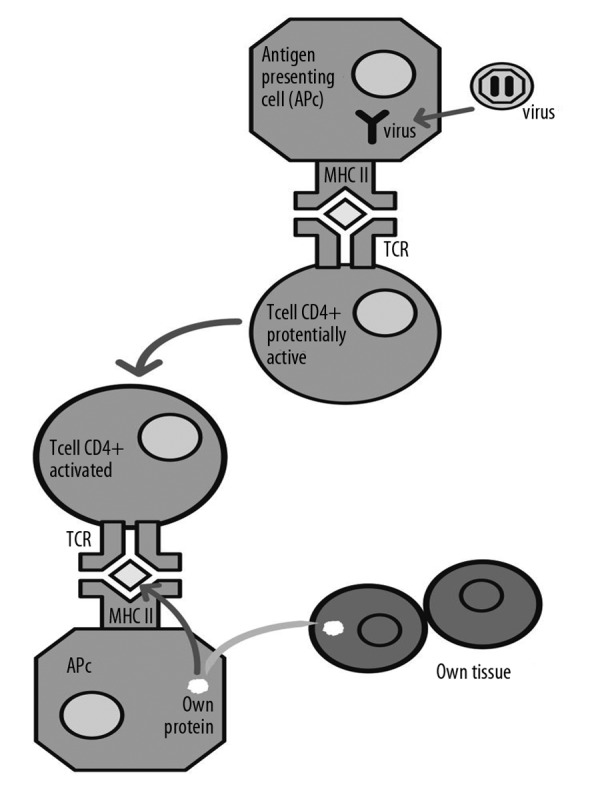
Intuitive illustration of the phenomena of molecular immune mimicry.
Epitope spreading or epitope drift
This phenomenon occurs when the immune reaction is induced not only by the primary the primary epitope but also by other epitopes. In contrast to molecular mimicry, the other epitopes need not be structurally similar to the primary one. Epitope spreading allows recognition of multiple antigenic targets. It involves non-cross-reactive epitopes, distinct from the inducing epitope, becoming major targets of an ongoing immune response. Epitope spreading involves an increased presentation of cryptic epitopes or cryptic antigens with action of MHC II molecules or cross-priming onto MHC I molecules. Subsequently, reactive T-cell and antibody repertoires diversify, resulting in an enhanced overall immune response. Epitope spreading is important pathogenetic mechanism involved in the development of autoimmune diseases.
Epitope modification or cryptic epitope exposure
This pathogenetic mechanism of the autoimmune diseases does not result from a defect in the hematopoietic system. The disease results from the exposure of cryptic N-glycan (polysaccharide) linkages common to lower eukaryotes and prokaryotes on the glycoproteins of mammalian non-hematopoietic cells and organs. This exposure of phylogenetically primitive glycans activates some mammalian innate immune cell receptors and induces a chronic sterile inflammatory state. In the presence of the chronic, inflammatory cell damage, the self-tolerance is lost and increased autoantibody production occurs. This route to autoimmune diseases may underlie various degenerative disease states.
Experimental data related to involvement of HERVs in the pathogenesis of autoimmune chronic diseases
Data indicating the involvement of HERV’s in etiopathogenesis were found in relation to some cancers, insulin-dependent diabetes mellitus, nervous system diseases (especially multiple sclerosis), schizophrenia and autoimmune rheumatic and connective tissue diseases. Some mechanisms linked to HERVs could give rise to autoimmune diseases, such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA) and Sjögren’s syndrome.
Krzysztalowska-Wawrzyniak et al. investigated the prevalence of HERV-K113 in patients with SLE, RA and in healthy subjects in the Polish population. Their data revealed statistically significant differences in the insertion frequencies of HERV-K113 between the groups of SLE and RA patients vs. healthy controls [23].
Freimanis et al. tried to clarify the role of HERVs in development of rheumatoid arthritis [3]. Initial stages of their investigation demonstrated the potential of Gag, Pol and Env proteins of HERVs to generate B-cell epitopes, based upon sequence analysis. A Gag1 sequence (KPR) was identified as an elicitor of anti-HERV antibodies, which also showed potential for cross-reactivity with host proteins through molecular mimicry. This epitope was thus selected for further investigation. The investigators scanned potential autoantigens implicated in autoimmunity for sequence identity with retroviral epitopes. The peptides modeling shared retroviral epitopes were synthesized. They developed a novel real-time polymerase chain reaction (PCR) assay to accurately quantify levels of HERV-K (HML-2) gag expression. They demonstrated that both serological and molecular assays showed significant increases in HERV-K (HML-2) gag activity in RA patients compared to the control group. The PCR assay identified significant up-regulation in HERV-K mRNA levels in RA patients compared to the inflammatory group and the healthy control group. They also proved that the exogenous viral protein expression and proinflammatory cytokines exert modulatory effects over HERV-K (HML-2) transcription. They concluded that RA patients exhibited significantly elevated levels of HERV-K (HML-2) gag activity compared to the control group.
The autoimmune diseases are often accompanied by different forms of cancer. Increased occurrences of malignancies in patients with established RA have been published [24]. Recent investigations have shown anti-HERV antibodies in patients with breast cancer, melanoma, germ cell tumors and lymphomas [6]. Whether such malignancies predispose these patients to develop autoimmunity is an intriguing question., HERVs probably play a role in the disease pathology of these patients.
The sequence of polynucleotides forming HERV-K exhibits polymorphism. It is possible that this polymorphism is responsible for the occurrence of different forms of RA [25]. HERV-K has been reported to exhibit increased expression of superantigens in the juvenile form of RA [26].
Baudino et al. examined murine systemic lupus erythematosus (SLE) as a model of the pathogenesis of the human equivalent of this autoimmune disease [27,28]. Murine SLE is an autoimmune disorder characterized by B-cell hyperactivity leading to the production of various autoantibodies and subsequent development of glomerulonephritis. Among the principal targets of autoantibodies produced in murine SLE are nucleic acid-protein complexes (eg, chromatin) and the envelope glycoprotein gp70 of endogenous retroviruses. Recent studies revealed that the receptors TLR7 and TLR9 are involved in the activation of autoreactive B-cells [28]. The regulation of autoimmune responses against endogenous retroviral gp70 by TLR7 suggests the involvement of endogenous retroviruses in this autoimmune response.
Perl and Fernandez found that the HRES-1 is a kind of HERV that encodes a 28-kD nuclear autoantigen and a 24-kD small GTP-ase, termed HRES-1/Rab4. HRES-1/p28 is a target of cross-reactive antiviral antibodies, whereas HRES-1/Rab4 regulates the surface expression of CD4 [29]. They concluded that HERVs can be considered as the molecular link between the human genome and environmental factors influencing the pathogenesis of SLE. HERV proteins may trigger lupus through structural and functional molecular mimicry, whereas the accumulation of HERV-derived polynucleotides stimulates interferon and anti-DNA antibody production.
Moyes et al. examined a group of 96 patients with Sjogrens’ syndrome. They found the HERV-K113 polynucleotide sequence in 15% of these patients. The prevalence of HERV-K113 determined by them in patients from the United Kingdom was 54.1% [30].
Prokop and Jagodzinski, employing the dot blot hybridization technique, observed in the group (n=79) of patients with systemic sclerosis (SSc) a conserved retroviral pol sequence in 34.6% of them and the antinuclear antibodies (ANA) positive in 40.7% of this group. They suggest that the presence of retroviral sequence in serum of SSc patients may correlate with development of autoimmune response directed against U1-70 kDa polypeptide antigen [31].
Discussion of possible molecular mechanisms related to HERVs, intermediating in the pathogenesis of autoimmune diseases
It is said that some diseases are determined genetically. There are diseases whose pathogenesis is caused by changes on the level of chromosomes or by exactly known “point” changes in the structure of specific genes, such as in the case of hemophilia. The frequent explanation of pathogenesis of many diseases, undoubtedly conditioned genetically, is that the disease is caused by many changes in different loci.
A very general question arises, however, when we consider the diseases conditioned genetically, in the context of the existence of HERVs. We should take into account the fact that HERVs were incorporated into human genome 40 million years ago. So in fact, we could say that it is not relevant whether an adverse change is located within the sequence representing one of the HERVs or rather it is within the structural part of the genome. But it seems that that is relevant, because HERVs’ sequences have the property of transposition. Displacement of such retroelement can cause pathogenic changes in very sensitive fragments of the genome. HERVs cause the occurrence of some diseases to be “random”. HERVs sequences are also often characterized by unfavorable polymorphism. Is of great importance that HERVs sequences are located in the same loci, which are located the genes responsible for the formation of MHC.
Renaudineau studied the role of inappropriate epigenetic changes in the development of autoimmune diseases [32]. Epigenetics signifies stable and heritable changes in gene expression without changes in the genetic code. There are some arguments that this kind of process can promote autoimmunity. The first clue is that inhibition of DNA methyl transferases (DNMTs) induces systemic lupus erythematosus (SLE) in animals. Moreover, defects in DNA methylation characterize lymphocytes from SLE, synoviocytes from rheumatoid arthritis, and neural cells from multiple sclerosis patients. It has also been shown that DNA hypomethylation of T- and B-cells correlates with reduced DNMT efficacy and histone acetylation in SLE. Once a gene promoter has been demethylated, the gene recovers its capacity to be transcribed (eg, genes for cytokines, activation receptors on cells, and endogenous retroviruses). Autoimmunity is believed to develop when genetically predisposed individuals encounter epigenetic modifications in response to environmental factors.
Obermayer-Straub and Manns emphasized that chronic infections with hepatitis C virus (HCV) are associated with various autoimmune manifestations, including mixed cryoglobulinemia, membranoproliferative glomerulonephritis, autoimmune thyroid diseases, sporadic porphyria, cutanea tarda and B-cell lymphoma. According to these authors, hepatitis C is also suspected to be associated with autoimmune hepatitis. They remark that anti-HIAP autoantibodies (HIAP-Human A type retroviral particles) are seen in primary biliary cirrhosis [33].
Some researchers consider the role of superantigen in the development of chronic diseases. Schubert proposed the outline of these investigations as early as 2001 [34]. He expressed the assumptions that very similar aspects of molecular and cellular pathology concern some chronic diseases treated by otolaryngologists and pulmonologists, including sinusitis, rhinosinusitis, allergic fungal sinusitis, allergic bronchopulmonary aspergillosis, nasal polyp, and chronic eosinophilic-lymphocytic respiratory mucosal inflammatory disorders like hypertrophic sinus disease and chronic severe asthma [30]. Schubert formulated the hypothesis that microbial T-cell superantigen production, persistence, and host-responsiveness are the fundamental components that unify the pathogenesis of all these diseases. Superantigen amplification of immune response is the proposed mechanism for disease induction and maintenance.
Wang et al. in 2008 reported that Staphylococcal exotoxins, acting as superantigens, activate the beta chains of T-cell receptors (TCR-V-beta), with subsequent massive proliferation and corresponding excursion of gene spectra, which contribute to the etiology of chronic rhinosinusitis with nasal polyps [35].
Punkosdy et al. emphasized that regulatory T-cells (T reg) play critical roles in the modulation of immune responses to infectious agents [36]. He demonstrated that chronic, but not acute, infection of mice with lymphocytic choriomeningitis virus results in a marked expansion of Foxp3(+) T reg, which is dependent on retroviral superantigen (sag) genes encoded in the mouse genome. They found that Sag-dependent T reg expansion was MHC class II-dependent, CD4-independent, and required dendritic cells; thus they postulated the link between certain infectious agents that cause host immune responses and endogenous, retroviral Sag-dependent activation and expansion of T reg [36].
Sawcer et al. conducted a genome-wide association study (GWAS), considering the pathogenesis of multiple sclerosis. They demonstrated that immunologically relevant genes are significantly overrepresented and are close to the identified loci, which particularly implicates T-helper-cell differentiation [37].
Balada et al. emphasized that HERVs could cause disease by reason of their retroelements capacity. They can move about in the genome and can be inserted next to certain genes, whose expression would be consequentially altered [1,2]. Their genetic sequences may be totally or partially integrated into genes that regulate the immune response [1,2].
According to Balada, disease could also be triggered when HERV-encoded proteins are expressed. These proteins could be considered as “foreign” and could trigger B-cells to produce antibodies against them, which, in turn, might cross-react with other proteins of our bodies [1]. This mechanism is called “molecular mimicry” [1,38,39]. Some researchers point out that HERV-proteins may act as superantigens [35,36].
Some environmental agents can induce the expression of HERVs, which can be evoked by ultraviolet light and several chemical agents. Such activation is mediated by altering the structure without modifying their nucleotide composition, which is possible when the methylation pattern is changed [40]. Balada maintains that the epigenetic changes observed in the course of systemic lupus erythematosus (SLE) or cancer could be the result of the activation of some of the retroelements present in our genome, which could have a direct or indirect role on the initiation and clinical evolution of certain chronic diseases [1,2].
Conclusions
It is still unclear whether HERVs are the triggering agent or are simply up-regulated as a consequence of inflammation and immune responses occurring in the course of different autoimmune diseases. This may be clarified when researchers consider the timing of HERV increases in relation to disease development. It should be also considered whether particular HERV polymorphisms play an important role in specific subsets of predisposed patients.
Recent discoveries of the role of HERVs enable a better understanding of the pathogenetic mechanisms of chronic diseases.
Knowledge of the existence and activity of HERVs aids understanding of the functioning of the human genome and contributes to theory of the mechanisms of evolution. This is so important that it should be incorporated into teaching programs of clinical disciplines.
Footnotes
Source of support: Self financing
References
- 1.Balada E, Ordi-Ros J, Vilardell-Tarrés M. Molecular mechanisms mediated by human endogenous retroviruses (HERVs) in autoimmunity. Rev Med Virol. 2009;19:273–86. doi: 10.1002/rmv.622. [DOI] [PubMed] [Google Scholar]
- 2.Balada E, Vilardell-Tarrés M, Ordi-Ros J. Implication of human endogenous retroviruses in the development of autoimmune diseases. Int Rev Immunol. 2010;29:351–70. doi: 10.3109/08830185.2010.485333. [DOI] [PubMed] [Google Scholar]
- 3.Freimanis G, Hooley P, Ejtehadi HD, et al. A role for human endogenous retrovirus-K (HML-2) in rheumatoid arthritis: investigating mechanisms of pathogenesis. Clin Exp Immunol. 2010;160:340–47. doi: 10.1111/j.1365-2249.2010.04110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Perl A, Fernandez D, Telarico T, Phillips PE. Endogenous retroviral pathogenesis in lupus. Curr Opin Rheumatol. 2010;22:483–92. doi: 10.1097/BOR.0b013e32833c6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marguerat S, Wang WY, Todd JA, Conrad B, et al. Association of human endogenous retrovirus K-18 polymorphisms with type 1 diabetes. Diabetes. 2004;53:852–54. doi: 10.2337/diabetes.53.3.852. [DOI] [PubMed] [Google Scholar]
- 6.Contreras-Galindo R, Kaplan MH, Leissner P, et al. Human endogenous retrovirus K (HML-2) elements in the plasma of people with lymphoma and breast cancer. J Virol. 2008;82:9329–36. doi: 10.1128/JVI.00646-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Romanish MT, Cohen CJ, Mager DL. Potential mechanisms of endogenous retroviral-mediated genomic instability in human cancer. Semin Cancer Biol. 2010;20:246–53. doi: 10.1016/j.semcancer.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 8.Christensen T. HERVs in neuropathogenesis. J Neuroimmune Pharmacol. 2010;5:326–35. doi: 10.1007/s11481-010-9214-y. [DOI] [PubMed] [Google Scholar]
- 9.Perron H, Lang A. The human endogenous retrovirus link between genes and environment in multiple sclerosis and in multifactorial diseases associating neuroinflammation. Clin Rev Allergy Immunol. 2010;39:51–61. doi: 10.1007/s12016-009-8170-x. [DOI] [PubMed] [Google Scholar]
- 10.Lewis DA. Retroviruses and the pathogenesis of schizophrenia. Proc Natl Acad Sci. 2001;98:4293–94. doi: 10.1073/pnas.081075898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Winter PC, et al. Genetics. Viva Books Prt. Ltd; UK: 2003. [Google Scholar]
- 12.Zwolińska K. Retroviruses-derived sequences in the human genome. Human endogenous retroviruses (HERVs) Postępy Hig Med Dośw. 2006;60:637–52. [in Polish] [PubMed] [Google Scholar]
- 13.Löwer R, Löwer J, Kurth R. The viruses in all of us: characteristics and biological significance of human endogenous retrovirus sequences. Proc Natl Acad Sci USA. 1996;93:5177–84. doi: 10.1073/pnas.93.11.5177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Martínez Barrio A, Ekerljung M, Jern P, et al. The first sequenced carnivore genome shows complex host-endogenous retrovirus relationships. PLoS One. 2011;6(5):e19832. doi: 10.1371/journal.pone.0019832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Purdom G. Human Endogenous Retroviruses (HERVs) – Evolutionary “Junk” or God’s Tools? Available from URL: http://www.answersingenesis.org/articles/2006/12/19/human-endogenous-retroviruses.
- 16.Kjellman C, Sjogren HO, Salford LG, Widegren B. HERV-F (XA34) is a full-length human endogenous retrovirus expressed in placental and fetal tissues. Gene. 1999;239:99–107. doi: 10.1016/s0378-1119(99)00372-8. [DOI] [PubMed] [Google Scholar]
- 17.Mi S, Lee X, Li X, et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature. 2000;403:785–89. doi: 10.1038/35001608. [DOI] [PubMed] [Google Scholar]
- 18.Yi JM, Kim HM, Kim HS. Expression of the human endogenous retrowirus HERV-W family in various human tissues and cancer cells. J Gen Virol. 2004;85:1203–10. doi: 10.1099/vir.0.79791-0. [DOI] [PubMed] [Google Scholar]
- 19.Yi JM, Kim HS. Molecular Phylogenetic Analysis of the Human Endogenous Retrovirus E (HERV-E) Family in Human Tissues and Human Cancers. Genes Genet Syst. 2007;82:89–98. doi: 10.1266/ggs.82.89. [DOI] [PubMed] [Google Scholar]
- 20.Li F, Nellåker C, Yolken RH, Karlsson H. A systematic evaluation of expression of HERV-W elements; influence of genomic context, viral structure and orientation. BMC Genomics. 2011;12:22. doi: 10.1186/1471-2164-12-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Black SG, Arnaud F, Palmarini M, Spencer TE. Endogenous retroviruses in trophoblast differentiation and placental development. Am J Reprod Immunol. 2010;64:255–64. doi: 10.1111/j.1600-0897.2010.00860.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dupressoir A, Heidmann T. Endogenous retroviruses in trophoblast differentiation and placental development. Med Sci (Paris) 2011;27:163–69. doi: 10.1051/medsci/2011272163. [DOI] [PubMed] [Google Scholar]
- 23.Krzysztalowska-Wawrzyniak M, Ostanek M, Clark J, et al. The distribution of human endogenous retrovirus K-113 in health and autoimmune diseases in Poland. Rheumatology (Oxford) 2011;50:1310–14. doi: 10.1093/rheumatology/ker022. [DOI] [PubMed] [Google Scholar]
- 24.Bernatsky S, Ramsey-Goldman R, Clarke A. Malignancy and autoimmunity. Curr Opin Rheumatol. 2006;18:129–34. doi: 10.1097/01.bor.0000209423.39033.94. [DOI] [PubMed] [Google Scholar]
- 25.Moyes D, Griffiths DJ, Venables PJ. Insertional polymorphisms: a new lease of life for endogenous retroviruses in human disease. Trends Genet. 2007;23:326–33. doi: 10.1016/j.tig.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 26.Sicat J, Sutkowski N, Huber BT. Expression of human endogenous retrovirus HERV- K18 superantigen is elevated in juvenile rheumatoid arthritis. J Rheumatol. 2005;32:1821–31. [PubMed] [Google Scholar]
- 27.Baudino L, Yoshinobu K, Morito N, et al. Role of endogenous retroviruses in murine SLE. Autoimmun Rev. 2010;10:27–34. doi: 10.1016/j.autrev.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 28.Santiago-Raber ML, Baudino L, Izui S. Emerging roles of TLR7 and TLR9 in murine SLE. J Autoimmun. 2009;33:231–38. doi: 10.1016/j.jaut.2009.10.001. [DOI] [PubMed] [Google Scholar]
- 29.Perl A, Fernandez D, Telarico T, Phillips PE. Endogenous retroviral pathogenesis in lupus. Curr Opin Rheumatol. 2010;22(5):483–92. doi: 10.1097/BOR.0b013e32833c6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moyes DL, Martin A, Sawcer S, et al. The distribution of the endogenous retroviruses HERV-K113 and HERV-K115 in health and disease. Genomics. 2005;86:337–41. doi: 10.1016/j.ygeno.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 31.Prokop J, Jagodzinski PP. Identification of retroviral conserved pol sequences in serum of mixed connective tissue disease and systemic sclerosis patients. Biomed Pharmacother. 2004;58:61–64. doi: 10.1016/j.biopha.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 32.Renaudineau Y, Youinou P. Epigenetics and autoimmunity, with special emphasis on methylation. Keio J Med. 2011;60:10–16. doi: 10.2302/kjm.60.10. [DOI] [PubMed] [Google Scholar]
- 33.Obermayer-Straub P, Manns MP. Hepatitis C and D, retroviruses and autoimmune manifestations. J Autoimmun. 2001;16:275–85. doi: 10.1006/jaut.2000.0488. [DOI] [PubMed] [Google Scholar]
- 34.Schubert MS. A superantigen hypothesis for the pathogenesis of chronic hypertrophic rhinosinusitis, allergic fungal sinusitis, and related disorders. Ann Allergy Asthma Immunol. 2001;87:181–88. doi: 10.1016/S1081-1206(10)62222-3. [DOI] [PubMed] [Google Scholar]
- 35.Wang M, Shi P, Yue Z, et al. Superantigens and the expression of T-cell receptor repertoire in chronic rhinosinusitis with nasal polyps. Acta Otolaryngol. 2008;128(8):901–8. doi: 10.1080/00016480701760122. [DOI] [PubMed] [Google Scholar]
- 36.Punkosdy GA, Blain M, Glass DD, et al. Regulatory T-cell expansion during chronic viral infection is dependent on endogenous retroviral superantigens. Proc Natl Acad Sci USA. 2011;108(9):3677–82. doi: 10.1073/pnas.1100213108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sawcer S, Hellenthal G, Pirinen M, et al. Genetic risk and a primary role for cell- mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–19. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ogasawara H, Kageyama M, Yamaji K, Takasaki Y. The possibility that autoimmune disease can be induced by a molecular mimicry mechanism between autoantigen and human endogenous retrovirus. Lupus. 2010;19:111–13. doi: 10.1177/0961203309106767. [DOI] [PubMed] [Google Scholar]
- 39.Sasaki N, Ogawa Y, Iinuma C, et al. Human endogenous retrovirus-R Env glycoprotein as possible autoantigen in autoimmune disease. AIDS Res Hum Retroviruses. 2009;25:889–96. doi: 10.1089/aid.2009.0048. [DOI] [PubMed] [Google Scholar]
- 40.Blank M, Shoenfeld Y, Perl A. Cross-talk of the environment with the host genome and the immune system through endogenous retroviruses in systemic lupus erythematosus. Lupus. 2009;18:1136–43. doi: 10.1177/0961203309345728. [DOI] [PubMed] [Google Scholar]