

Summary
Background
Enteric glia cells (EGCs) are essential for the integrity of the bowel. A loss of EGCs leads to a severe inflammation of the intestines. As a diminished EGC network is postulated in Crohn’s disease (CD), we aimed to investigate if EGCs could be a target of apoptosis during inflammation in CD, which can be influenced by Brain derived neurotrophic factor (BDNF).
Material/Methods
GFAP, BDNF and cCaspase-3 were detected in the gut of patients with CD. Primary EGC cultures were established and cultivated. Tyrosine receptor kinase (TrkB) receptors on these cells were investigated by western blot and immunofluorescence. Rate of apoptosis was induced by tumor necrosis factor (TNF-α) and interferon (IFN-γ). Apoptosis was determined by a fluorometric caspase 3/7 activation assay after preincubation of these cells with BDNF or neutralizing anti-BDNF antibodies.
Results
Mucosal GFAP-positive EGCs undergo apoptosis revealed by cCaspase-3 in the gut of patients with CD expressing BDNF highly. The combination of TNF-α and IFN-γ was able to induce apoptosis in primary EGCs, whereas these factors alone did not. Brain derived neurotrophic factor (BDNF) attenuate glia cell apoptosis to a small extent, but neutralizing antibodies against BDNF dramatically increased apoptosis.
Conclusions
Mucosal EGC apoptosis is an important finding in the gut of patients with CD. Proinflammatory cytokines, which are highly increased in CD, induce EGC apoptosis, whereas the neurotrophin BDNF might be protective for EGC. Since EGCs are implicated in the maintenance of the enteric mucosal integrity, EGC apoptosis may contribute to the pathophysiological changes in CD.
Keywords: enteric glia, BDNF, Crohn’s disease
Background
Crohn’s disease (CD) is characterized by a high increase of proinflammatory cytokines, e.g. tumor necrosis factor α (TNF-α) and Interferon γ (IFN-γ) [1]. Besides genetic and immune aspects there are some new insights in the regulation of the epithelial barrier during inflammation in CD [2].
It has been shown that enteric glia cells (EGCs) play an important role in the maintenance of the mucosal integrity in the context of CD [2–5]. Autoimmune targeting or an induced knock-out of the EGCs in mice leads to a fulminant hemorrhagic jejunoileitis, suggesting a protective role of EGCs in the gut [6–10]. A diminished EGC network has been shown in patients with CD [8,9], and has been postulated in other reports [7,10].
Own observations indicate that primary EGC are able to produce high amounts of neurotrophic factors and neurotrophins in response to inflammatory stimuli [10]. Moreover, we demonstrated that glial-derived neurotrophic factor (GDNF) is able to protect colonic epithelial cells from receptor-mediated apoptosis, that GDNF is highly upregulated in the gut mucosa during CD [10,11] and that EGCs are source for several factors like endothelins [12]. EGCs secrete transforming growth factor β (TGF-β) [13] that promotes intestinal barrier function [14] and release nitric oxide metabolite S-nitrosogluthatione (GSNO), a novel potent inducer of intestinal barrier function in human colon [15].
The EGC population, which is responsible for the mucosal upregulation of GDNF, TGF-β, GSNO and neurotrophins, are in close proximity to the enteric epithelial cells and belonged to the mucosal plexus [15]. Therefore, we hypothesized that the mucosal glial plexus plays an important role in the maintenance of the mucosal integrity by protecting the gut epithelial lining from apoptosis. Alterations in this putative mucosal protecting system would lead to a more severe course of CD. One possibility for an alteration of EGC function could be due to EGC apoptosis in the context of CD, but the knowledge about the susceptibility of EGC to apoptosis or antiapoptotic mechanisms in this cell population is rare.
Cellular markers suggested that the EGCs share some properties with astroglial cells in the central nervous system, but on the other hand some features of these cells showed similarities to microglia [4,5]. Therefore, we believe that EGCs represent a cell population with functions and properties that may be distinct from their central nervous system relations. Therefore, the characterisation of mucosal EGC is an important goal for the understanding of their role in the pathophysiology of CD.
In this paper, we aimed to investigate EGC apoptosis as a pathophysiological feature in CD and look for the mechanisms that may lead to EGC apoptosis in the context of gut inflammation, and the possible role of brain-derived neurotrophic factor (BDNF) in the protection of these cells from apoptosis under this inflammatory setting.
Material and Methods
Human tissue
The patients enrolled in the study gave their informed consent and the study was approved by the local ethics committee. The diagnosis of CD was established by common criteria (16). Biopsies from inflamed colon were taken from 20 patients with CD (10 female/10 male; mean age 34 years; range 29 to 41 years). Biopsies were taken during colonoscopy. The mean duration of CD at the time of biopsy was 4.5 years. No patients received biologics. 6 CD patients were treated with azathioprine, 3 patients with 6-Mercaptopurin one patient with MTX and 4 patients with budesonide. The other patients were not treated at the time of study and had only less clinical signs of activity.
Colon biopsies from healthy controls were taken from 26 patients, which underwent a routine screening colonoscopy (12 female/14 male; mean age 56 years; range 51 to 60 years). Tissue glial fibrillary acidic protein (GFAP), BDNF and c-Caspase-3 levels were measured by immunofluorescence.
Indirect immunofluorescence
Tissue biopsies were deparaffinized and permeabilized with phosphate buffered saline (PBS)/0.1% Triton X100. Antigen retrieval was performed by boiling the slides in 0.01 M trisodium citrate buffer, pH 6, for 10 min. Sections were then preincubated with 10% normal goat serum containing 0.2% Triton X-100 overnight at 4°C to block nonspecific binding. Slides were then incubated over night at 4°C with Antibodies against GFAP (Pharmingen, San Diego, USA, mouse, 1:100), BDNF (Abcam, USA, rabbit, 1:100), TrkB (Abcam, USA, rabbit, 1:200) and rabbit anticleaved caspase-3 (1:200, Cell Signaling Technology).
After washing in PBS/0,1% Tween 20, the slides were incubated with the appropriate secondary antibodies: Cy3 coupled goat anti-mouse IgG (DPC Biermann, Bad Nauheim, Germany) and Alexa 488 coupled goat anti-rabbit IgG (Sigma). Dilutions of the secondary antibodies were 1:800. After washing in PBS, slides were embedded in glycerol gelatine. Control labelling was performed with omission of the first antibodies to ensure that there was no unspecific labelling of cells.
Tissue-biopsies were analyzed using a Leica confocal laser-scanning microscope. Single optical sections were recorded under conditions, which exclude cross-activation of the individual signal channels.
Histological analysis
Three Immunolabelled biopsies of each of the 20 CD and 26 control persons were used for evaluating the expression of GFAP, BDNF, TrkB and cCaspase-3 in EGCs. Five immunolabeled sections of each colon of patients with CD or controls were used for evaluating the density of GFAP+, BDNF+, TrkB+ and cCaspase-3+ cells in colon sections. The mean ±SEM of mucosal GFAP+ EGCs was calculated semiquantitatively by numbers in 6 segments of each section from 5 patients with CD and 5 controls. The evaluation was performed in ablinded fashion by 2 raters. Interrater reliability ranged from 0.91 to 0.97 (P<0.0001).
Dissociated EGC cultures
Newborn rats (Wistar strain) were decapitated. The intestines were removed and the myenteric plexus was isolated as previously described [10]. In brief, small intestines were rinsed in sterile minimal essential medium (MEM) with 25 mM HEPES buffer (Gibco Life Technologies, California USA). The muscle layer containing the myenteric plexus was stripped from the mucosa. The tissue was incubated in a collagenase solution (CL type II, Gibco, 1 mg/ml) in Hanks balanced salt solution (Gibco) for 1.5 h at 37°C. The disintegrated tissue was vortexed and the isolated parts of the myenteric plexus were stored in MEM on ice. The collected pieces were incubated in trypsin (0.1 mg/ml, Gibco) for 15min at 37°C and then centrifuged. Trypsinisation was stopped by addition of fetal calf serum (Gibco). Then cells were plated on polyornithine coated (0.5 mg/ml, Sigma, Schnelldorf, Germany) coverslips and were topped with 450 μl DMEM-F-12 (Gibco). The cultures were kept in a humidified atmosphere of 95% air/5%CO2 at 37°C. At day 3, the culture consisted of approximately 98% of EGCs.
Indirect immunofluorescence
The EGCs of the myenteric plexus of each animal were pooled and cultured on 20 different coverslips. Cell cultures were fixed and permeabilized with 90% methanol, 10% acetic acid cooled down to −40°C. After washing in PBS cells were blocked with 1% bovine serum (Sigma) for 40 min, anti-Trk B (Pharmingen, mouse) antibodies were incubated for 1 h at room temperature. Immunofluorescence staining was done by secondary antibodies: goat anti-mouse IgG (cy3, Sigma). Control experiments were performed under omission of the first antibody in order to exclude unspecific staining of the primary antibody. After immunostaining, coverslips were mounted cell side down on microscope slides using moviol. Cell cultures were analyzed using a Leica confocal laser-scanning microscope.
Apoptosis in EGC cultures induced by tumor necrosis factor-α and interferon-γ
Rat primary EGC of the myenteric plexus was isolated as described above. For induction of apoptosis, the glia was seeded in 48 well plates. After reaching confluence, the cells were washed twice with PBS, and thereafter 180 μl Dulbecco’s minimal essential medium (DMEM) was added, containing 100 ng/ml interferon-γ (IFN-γ), 100 ng/ml TNF-α, both or vehicle. Then, the cultures were incubated with or without BDNF 100 ng/ml, or vehicle over a period of 40 hours. In case of the addition of neutralizing antibody against BDNF, the cells were preincubated for 1 hour with anti-BDNF 2 μg/ml. For detection of caspase 3/7 activity in the EGC cultures, the caspase substrate Rhodamin 110 was added in the relation 1:1 to the wells according to the instructions of the manufacturer of the assay (ApoOne Homogenous caspase 3/7 Assay, Promega, Germany), and the wells were vortexed at room temperature over 2 minutes at 400 rpm. After additional 2 hours at room temperature, the wells were vortexed again, followed by flurometric measurement of the caspase 3/7 activity according to the manufacturers recommendations. In brief, the stimulation wavelength was 485/20 nm, the emission wavelength was 528/20 nm. Every condition was repeated 6 times, the results are indicated as mean values±SD.
Western blot analysis of Trk B in primary EGC cultures of the myenteric plexus
Western blot analysis was performed according to standard protocols as described elsewhere [10]. In brief, primary enteric rat glia was seeded in 10 cm diameter culture dishes, and after reaching confluence rinsed with PBS three times. Thereafter, the cells were lysed and prepared as described before [10]. The antibody for the detection of TrkB was purchased from R&D Systems, Germany. For western blotting, it was used at a concentration of 1:25.
Data analysis
All data given in the text and figures are expressed as mean values ±SD. The data were analyzed using non-parametric two-tailed Mann-Whitney U test with p<0.05 considered as an indicator of significance.
Results
CD exhibit EGC apoptosis associated with capsase-3 activation and BDNF secretion
Considering the role of EGC for the integrity of the gut, we investigated whether apoptosis of EGC is increased in the gut of CD patients. Cleaved caspase-3 immunoreactivity was not detected in controls (Figure 1C,D), but its immunoreactivity was detected in the mucosal biopsies of patients with CD, principally in GFAP-immunopositive cells (Figure 1A–C). These findings indicated that in vivo chronic inflammation of the gut leads to EGC injury/apoptosis, likely mediated by cleavage of the effector caspases-3. Through their involvement in neurotransmitter uptake/release and production of neurotrophic factors [9], EGCs are crucial for maintaining a metabolically homeostatic environment for the mucosa environment. Hence, we investigated the effect of EGC injury on the mucosal neurochemical microenvironment by analyzing the BDNF secretion in the mucosa of CD patients and controls. BDNF could not be detected in controls (Figure 1J–L) but was highly expressed in mucosal EGCs of patients with CD (Figure 1G–I). This finding underlined the effect of chronic gut inflammation EGC mucosal microenvironment. Furthermore in the inflamed intestines of patients with CD TrkB receptors could be detected in GFAP-positive EGCs (Figure 1M–O).
Figure 1.
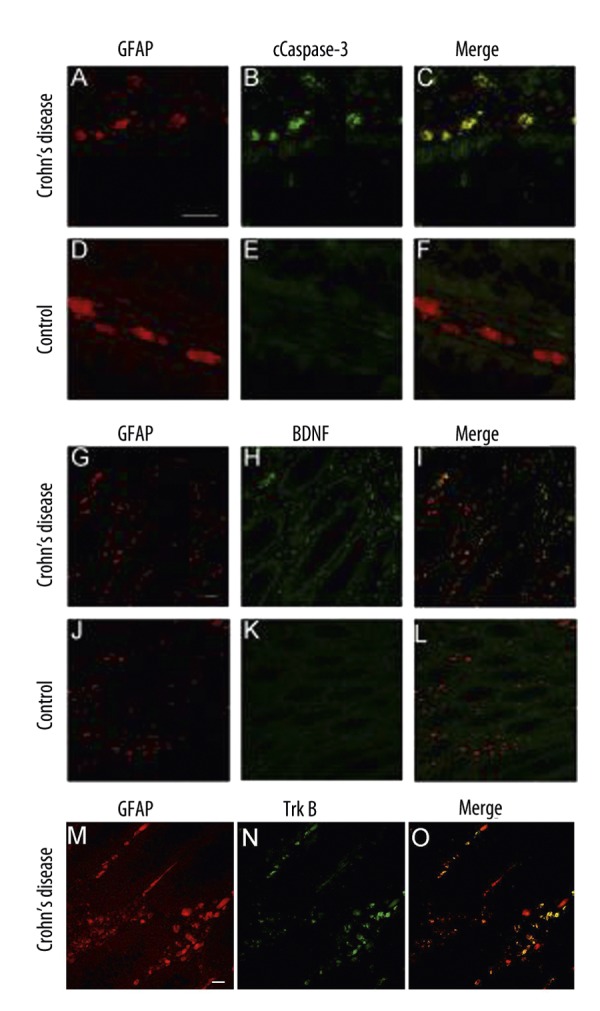
Biopsies of the inflamed colon of patients suffering from CD and controls were double immunolabelled with anti GFAP (A,D,G,J red) and cCaspase-3 (B,E green) or anti BDNF (H,K green) antibodies and were analyzed by optical sectioning using a confocal microscope. Both antigens, GFAP (A) and c-Caspase-3 (B) can be detected at high levels in the intestinal wall of the inflamed colon of CD (A,B). The merged images (C) reveal an almost complete overlap of both immunoreactivities (yellow). Only few GFAP-positive cells (D) display no cCaspase-3 immunoreactivity (E) in the control section (F). Although a high immunoreactivity of BDNF (H) in GDNF-positive cells (G,I) in the biopsies of patients with CD can be detected, in control biopsies GFAP-positive EGCs (J), which are positioned in the mucosal plexus in close vicinity to the epithelium of the colon, show no BDNF secretion (K,L). GFAP-positive EGCs (M) express TrKB receptors (N) in the inflamed colon of patients with CD (M–O). (Scale bars, 50 μm).
Semiquantitative Analysis of GFAP+, BDNF+, TrkB+ and cCaspase-3+ cells in the Mucosal Plexus
The numbers of GFAP+, BDNF+, TrkB+ and cCaspase-3+ cells cells per square millimeter in colon sections of patients with CD and control sections were counted. In cross-sections of the colon of patients with CD we found 34±6 GFAP+ cells/mm2, >20 BDNF+ cells/mm2, >20 cCaspase-3+ cells/mm2, whereas only about one third GFAP+ cells, BDNF+ cells and nearly no cCaspase-3+ cells were counted in control sections (9±5/mm2; 5±3/mm2, 2±2/mm2, respectively; Figure 2A–C).
Figure 2.

Semiquantification of GFAP-positive (A), BDNF-positive (B) and cCaspase-3-positive (C) cells in cross-sections of patients with CD and controls. Whereas >30 GFAP-positive cells/mm2, >20 BDNF-positive cells/mm2, >20 cCaspase-3-positive cells/mm2 are found in CD colon walls, only one third or less of this number is observed under control conditions. Significantly different from control (P<0.05).
Neurotrophin receptor expression on EGCs
Next we investigated if EGCs might also express BDNF receptors establishing an autocrine loop. Indirect immunofluorescence demonstrated expression of the BDNF specific neurotrophin receptor TrkB (Figure 3A). Western blot analysis corroborated these findings (Figure 3B).
Figure 3.
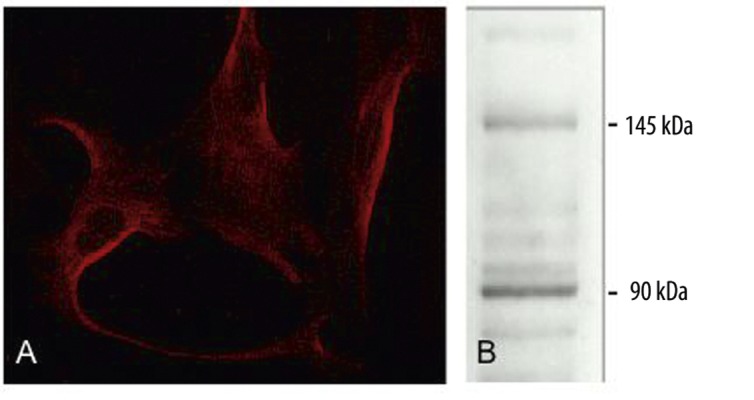
Detection of TrkB receptor in primary EGC culture. Indirect immunofluorescence (A) and Western blot (B) of the TrkB receptor. The molecular weight (kDa) of the full length protein, as well as the truncated form is shown on the right of the western blot figures.
Apoptosis in EGC and the influence of BDNF
We then tested in cultured EGCs, if the addition of BDNF could attenuate apoptosis induced by cytokines. Incubation of cultured primary EGCs with TNF-α or IFN-γ alone did not led to significantly increased apoptosis in these cells. However, the combination of both factors increased the activation of caspase 3/7 by two fold. Addition of BDNF slightly attenuated the apoptosis in EGCs, however this difference was not statistically significant (Figure 4). If the cells were preincubated with a neutralizing antibody against BDNF, the enteric glia was highly more susceptible to apoptosis, as the caspase 3/7 activity further increased to more than 3.6 fold over controls, and about 80% above the apoptosis rate induced in these cells by the combination of TNF-α and IFN-γ (Figure 5). Since EGCs secrete high amounts of BDNF under inflammatory conditions in CD, these data suggest an autocrine loop of BDNF mediated apoptosis attenuation in EGCs, which may disrupted or altered in CD.
Figure 4.

Activation of caspase 3/7 in primary EGC cultures. Fluorometric detection of caspase 3/7 activity after incubation of the EGC cultures with TNF-α (100 ng/ml), IFN-γ (100 ng/ml) and BDNF (100 ng/ml), alone or in combination as described above. The bars indicate mean values (6 fold detection) ±SD, normalized to controls. * Significantly different from control (P<0.05).
Figure 5.

Activation of caspase 3/7 activity in primary EGC cultures. Fluorometric detection of caspase 3/7 activity after incubation of the EGC cultures with TNF-α (100 ng/ml), IFN-γ (100 ng/ml), neutralizing anti-BDNF-antibody (2 μg/ml), alone or in combination as described above. The bars indicate the mean values (6 fold detection) ±SD, normalized to controls. * Significantly different from control (P<0.05).
Discussion
In this paper, we could show that apoptosis of mucosal EGCs are part of the pathophysiology in CD and that primary EGCs are susceptible to undergo apoptosis under inflammatory conditions. The combination of TNF-α and IFN-γ was able to induce apoptosis in cultured EGCs. However, as we have recently shown, these factors are also able to induce the secretion of high amounts of neurotrophic factors and neurotrophins.
Mucosal integrity might be important for remission in CD [2,5,17]. Several data implicate an essential role of mucosal EGC for an intact epithelial barrier [2,5,7,18,19]. In animal models a reduction of the EGC network led to a collapse of the epithelial lining causing a severe inflammation of the gut [9]. In CD a diminished EGC network is postulated and an inflammation within the nerval plexus with damage of the glial structures is associated with early clinical recurrence of CD and could be regarded in studies searching for new treatment strategies in the immediate postoperative period [18]. Our data shed light on possible molecular mechanisms underlying these clinical observations.
Because high levels of proinflammatory cytokines are present in the intestinal wall during acute flares in CD [1] we investigated, whether the cytokines TNF-α and IFN-γ could induce apoptosis in EGCs. Interestingly, the cytokines TNF-α and IFN-γ alone showed no relevant increase of EGC apoptosis, but the combination of these cytokines like in the intestines of CD patients led to a dramatic increase of apoptosis in these cells.
Neurotrophic factors and neurotrophins play a protective role for glial structures in the CNS. Also in the gut GDNF acts anti-apoptotic on intestinal epithelial cells [10,20].
Therefore, we looked for the role of neurotrophins in regulating EGC apoptosis. First we showed that EGC express the receptor TrkB, which could implicate a functional role of BDNF in these cells. As EGCs are one of the main sources of neurotrophic factors and neurotrophins in the gut [10], autocrine effects of these cells are imaginable. EGC cultures were incubated with BDNF to protect the cells for apoptosis. Nevertheless BDNF showed no relevant change in EGC apoptosis. Inflammatory conditions in the intestines of patients with CD lead to a dramatically increase of BDNF expression in EGCs, which is demonstrated in this study. Therefore, our cultured EGCs may be saturated with neurotrophins and neurotrophic factors and additional doses of BDNF might have no effect on induction of apoptosis.
However, after incubation of EGC cultures with neutralizing anti-BDNF antibodies the apoptosis rates of EGCs were dramatically increased. This implicates an autocrine protective pathway of BDNF for EGC during gut inflammation. A reduction of the secretion of BDNF increases the rates of EGC apoptosis in the inflamed gut. As a diminished EGC network is postulated for CD [8,9] it could be speculated that the mucosa of these patients is characterized by reduced amounts of BDNF. The loss of the protective agent BDNF during acute inflammatory flares might lead to a high increase of EGC apoptosis, which is triggered by the characteristic proinflammatory cytokines TNF α and IFN g [1]. Thus the disruption of the EGC network reduces the factors for regulating the epithelial barrier function, e.g. GDNF, GSNO, TGF-β, highly. The result is a leakage of the epithelial lining, which allows the translocation of luminal antigens (e.g., components of bacteria) into the mucosa, where inflammatory processes and release of proinflammatory cytokines are initiated, again. This vicious cycle might be part of the puzzle in the pathophysiology of CD.
This notion is supported by the observation that acute flares in CD may be triggered by impairment of the epithelial lining [5,16,21]. And the growth factors, like epidermal growth factor (EGF) were used as an effective treatment for inflammatory bowel disease [22] and promote intestinal mucosal healing [23]. Besides GDNF, an anti-apoptotic agent for epithelial cells, BDNF might be an important neurotrophin to protect for EGC apoptosis.
Conclusions
Therefore, it could be speculated that, besides EGF [23], the neurotrophic factor GDNF and the neurotrophin BDNF could represent an interesting cocktail as new therapeutic approach in inflammatory bowel disease.
Abbreviations
- BDNF
brain derived neurotrophic factor
- CD
Crohn’s disease
- DMEM
Dulbecco’s minimal essential medium
- EGC
enteric glia cell
- GDNF
glial-derived neurotrophic factor
- GFAP
glial fibrillary acidic protein
- GSNO
S-nitrosogluthatione
- IFN-γ
Interferon γ
- PBS
phosphate buffered saline
- MEM
minimal essential medium
- TGF-β
transforming growth factor β
- TNF-α
tumor necrosis factor α
Footnotes
Source of support: Departmental sources
References
- 1.Schreiber S, Nikolaus S, Hampe J, et al. Tumour necrosis factor alpha and interleukin 1beta in relapse of Crohn’s disease. Lancet. 1999;353:459–61. doi: 10.1016/S0140-6736(98)03339-X. [DOI] [PubMed] [Google Scholar]
- 2.Neunlist M, Van Landeghem L, Bourreille A, et al. Neuro-glial crosstalk in inflammatory bowel disease. J Intern Med. 2008;263:577–83. doi: 10.1111/j.1365-2796.2008.01963.x. [DOI] [PubMed] [Google Scholar]
- 3.Von Boyen GB, Reinshagen M, Steinkamp M, et al. Gut inflammation modulated by the enteric nervous system and neurotrophic factors. Scand J Gastroenterol. 2002;37:621–25. doi: 10.1080/00365520212498. [DOI] [PubMed] [Google Scholar]
- 4.Von Boyen GB, Reinshagen M, Steinkamp M, et al. Enteric nervous plasticity and development: dependence on neurotrophic factors. J Gastroenterol. 2002;37:583–88. doi: 10.1007/s005350200093. [DOI] [PubMed] [Google Scholar]
- 5.Cabarrocas J, Savidge T, Liblau R. Role of enteric glial cells in inflammatory bowel disease. Glia. 2003;41:81–93. doi: 10.1002/glia.10169. [DOI] [PubMed] [Google Scholar]
- 6.Bush TG, Savidge TC, Freeman TC, et al. Fulminant jejuno-ileitis following ablation of enteric glia in adult transgenic mice. Cell. 1998;93:189–201. doi: 10.1016/s0092-8674(00)81571-8. [DOI] [PubMed] [Google Scholar]
- 7.Bush TG. Enteric glial cells. An upstream target for induction of necrotizing enterocolitis and Crohn’s disease? Bioessays. 2002;24:130–40. doi: 10.1002/bies.10039. [DOI] [PubMed] [Google Scholar]
- 8.Cornet A, Savidge TC, Cabarrocas J, et al. Enterocolitis induced by autoimmune targeting of enteric glial cells: a possible mechanism in Crohn’s disease? Proc Natl Acad Sci USA. 2001;98:13306–11. doi: 10.1073/pnas.231474098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.von Boyen GB, Schulte N, Pflüger C, et al. Distribution of enteric glia and GDNF during gut inflammation. BMC Gastroenterol. 2011;11:3. doi: 10.1186/1471-230X-11-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Von Boyen GB, Steinkamp M, Geerling I, et al. Proinflammatory cytokines induce neurotrophic factor expression in enteric glia: a key to the regulation of epithelial apoptosis in Crohn’s disease. Inflamm Bowel Dis. 2006;12:346–54. doi: 10.1097/01.MIB.0000219350.72483.44. [DOI] [PubMed] [Google Scholar]
- 11.Steinkamp M, Geerling I, Seufferlein T, et al. Glial-derived neurotrophic factor regulates apoptosis in colonic epithelial cells. Gastroenterology. 2003;124:1748–57. doi: 10.1016/s0016-5085(03)00404-9. [DOI] [PubMed] [Google Scholar]
- 12.von Boyen GB, Degenkolb N, Hartmann C, et al. The endothelin axis influences enteric glia cell functions. Med Sci Monit. 2010;16(6):BR161–67. [PubMed] [Google Scholar]
- 13.Neunlist M, Aubert P, Bonnaud S, et al. Enteric glia inhibit intestinal epithelial cell proliferation partly through a TGF-beta1-dependent pathway. Am J Physiol Gastrointest Liver Physiol. 2007;292:G231–41. doi: 10.1152/ajpgi.00276.2005. [DOI] [PubMed] [Google Scholar]
- 14.Savidge TC, Newman P, Pothoulakis C, et al. Enteric glia regulate intestinal barrier function and inflammation via release of S-nitrosoglutathione. Gastroenterology. 2007;132:1344–58. doi: 10.1053/j.gastro.2007.01.051. [DOI] [PubMed] [Google Scholar]
- 15.Von Boyen GB, Steinkamp M, Reinshagen M, et al. Proinflammatory cytokines increase glial fibrillary acidic protein expression in enteric glia. Gut. 2004;53:222–28. doi: 10.1136/gut.2003.012625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nikolaus S, Schreiber S. Diagnostics of inflammatory bowel disease. Gastroenterology. 2007;133:1670–89. doi: 10.1053/j.gastro.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 17.Shanahan F. Inflammatory bowel disease: immunodiagnostics, immunotherapeutics, and ecotherapeutics. Gastroenterology. 2001;120:622–35. doi: 10.1053/gast.2001.22122. [DOI] [PubMed] [Google Scholar]
- 18.Sokol H, Polin V, Lavergne-Slove A, et al. Plexitis as a predictive factor of early postoperative clinical recurrence in Crohn’s disease. Gut. 2009;58:1218–25. doi: 10.1136/gut.2009.177782. [DOI] [PubMed] [Google Scholar]
- 19.Van Landeghem L, Mahé MM, Teusan R, et al. Regulation of intestinal epithelial cells transcriptome by enteric glial cells: impact on intestinal epithelial barrier functions. BMC Genomics. 2009;10:507. doi: 10.1186/1471-2164-10-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Savidge TC, Sofroniew MV, Neunlist M. Starring roles for astroglia in barrier pathologies of gut and brain. Lab Inves. 2007;87:731–36. doi: 10.1038/labinvest.3700600. [DOI] [PubMed] [Google Scholar]
- 21.Irvine EJ, Marshall JK. Increased intestinal permeability precedes the onset of Crohn’s disease in a subject with familial risk. Gastroenterology. 2000;119:1740–44. doi: 10.1053/gast.2000.20231. [DOI] [PubMed] [Google Scholar]
- 22.Sinha A, Nightingale J, West KP, et al. Epidermal growth factor enemas with oral mesalamine for mild-to-moderate left-sided ulcerative colitis or proctitis. N Engl J Med. 2003;349:350–57. doi: 10.1056/NEJMoa013136. [DOI] [PubMed] [Google Scholar]
- 23.Van Landeghem L, Chevalier J, Mahé MM, et al. Enteric glia promote intestinal mucosal healing via activation of focal adhesion kinase and release of proEGF. Am J Physiol Gastrointest Liver Physiol. 2011;300:G976–87. doi: 10.1152/ajpgi.00427.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]