

Abstract
Objectives
Improvements in survival for children with sickle cell disease (SCD) during the last 30 years have been well established. Whether similar improvements for adults with the disease have occurred is unknown. We investigated mortality rates for children and adults with SCD.
Methods
We used the National Center for Health Statistics multiple-cause-of-death files to examine age at death and calculate mortality rates from 1979 to 2005. We examined trends in mortality rates using negative binomial regression, and we examined age at death using t-tests and linear regression.
Results
We identified 16,654 sickle cell-related deaths. Mean age at death was significantly different for males (33.4 years, 95% confidence interval [CI] 33.0, 33.7) than for females (36.9 years, 95% CI 36.5, 37.4). In a regression model controlling for gender, the mean age at death increased by 0.36 years for each year of the study. The median age at death in 2005 was 42 years for females and 38 years for males. The overall mortality rate increased 0.7% (p<0.001) each year during the time period studied. The adult (>19 years of age) mortality rate increased by 1% (p<0.001) each year during the time period studied. The pediatric mortality rate decreased by 3% (p<0.001) each year during the time period studied.
Conclusions
These data confirm prior findings of a significant decrease in mortality for children with SCD. The mortality rate for adults appears to have increased during the same time period. It seems unlikely that this increase is due merely to an influx of younger patients surviving to adulthood and may reflect a lack of access to high-quality care for adults with SCD.
Sickle cell disease (SCD) is a common genetic disorder with potentially devastating consequences for those afflicted. It affects approximately 90,000–100,000 Americans, the majority of whom are African American.1,2 In the last 50 years, survival has improved dramatically for people with SCD in the United States. Their average life expectancy in the 1970s was <20 years of age. By the early 1990s, the Cooperative Study of Sickle Cell Disease estimated a median life expectancy of those with sickle cell anemia, the most severe form of the disease, of 42 years of age for males and 48 years of age for females.3 This dramatic improvement has been attributed to several interventions in early childhood, including widespread newborn screening programs, the use of penicillin prophylaxis, and the use of pneumococcal vaccination.4 Several studies have demonstrated decreasing mortality rates over time for children with SCD.4,5 Mortality rates in adults have not been reported recently.
In the last 15 years, hydroxyurea has become available for the treatment of SCD. This medication has demonstrated efficacy to reduce morbidity and mortality in SCD;6,7 therefore, there is an expectation that a reduction in mortality rates should occur with its widespread use. However, recent studies suggest that hydroxyurea is being underused8 and that the expected benefits of this therapy are not being realized at the population level.
The purpose of this study was to use a large population-based dataset to describe age at death and possible contributors to death in individuals with SCD. In addition, we investigated mortality rates for adults with SCD during the last 26 years, and determined whether mortality rates for this population have changed in the era of hydroxyurea use.
METHODS
We extracted age at death, underlying cause of death (COD), and contributing CODs for people in the U.S. with SCD from 1979 through 2005 from the National Center for Health Statistics multiple-cause-of-death (MCOD) databases.9 The MCOD files contain data from all death certificates filed in the U.S. Each certificate in the dataset includes the age at death, underlying COD, and a list of up to 20 conditions thought to contribute to the underlying COD. We identified deaths in people with SCD during the time period under study using International Classification of Diseases Ninth Revision (ICD-9)10 codes (282.41, 282.42, 282.60–64, 282.68, and 282.69) and ICD 10th Revision (ICD-10)11 codes (D570–D578) found in either the underlying COD or anywhere within the list of up to 20 diagnoses that contributed to the COD. We excluded records with the codes for sickle cell trait (282.5, D573) from analyses.
Analyses were not stratified by sickle genotype due to concerns regarding the accuracy of the coding. We restricted our analysis of the potential contributors to the underlying COD to illnesses known to be associated with mortality in SCD. These conditions included acute chest syndrome, renal disease, pulmonary hypertension, stroke, and infection.3,12 In addition, we investigated whether overdose of prescribed narcotics was also a significant contributor to COD.13 When evaluating the COD, the analysis reported in this study was restricted to those conditions identified in the MCOD files as the underlying COD. We conducted sensitivity analyses that suggested that the relative prevalence we observed for the different underlying CODs would not change had we expanded our scope to include the contributory CODs as well as the underlying COD. Therefore, we restricted our analyses to the underlying COD for the sake of simplicity and ease of interpretation.
We used logistic regression analyses to assess potential changes in the likelihood of the different specific conditions under study appearing as the underlying COD over time. To assess changes in mean age at death over time, we used multiple linear regression, t-tests, Mann Whitney, and F tests to determine the bivariate and multivariate relationships between age at death and gender, year of death, and underlying COD. We considered p<0.05 values to be significant. We examined the distribution of age at death, as well as the distribution of the residuals from regression analyses of age at death, using the Shapiro-Wilk test, which demonstrated significant non-normality. Robust and resistant regression methods were used as needed to account for slight departures from the assumptions of linear regression analysis and the presence of outliers. As these analyses led to the same conclusions as the ordinary linear regression, the results of the ordinary linear regressions are reported for ease of interpretation.
We examined trends in SCD mortality rates over time (1979–2005) using negative binomial regression. Mortality rates were calculated overall and stratified by pediatric/adult status (age ≤19 years = pediatric, age >19 years = adult). The African American population (overall and by pediatric/adult status) served as our denominator, and all rates are presented as deaths per 100,000 African Americans. We obtained our denominators for each year from the intercensal population estimates made available through the U.S. Census.14,15
Following the overall analysis, and to assess potential nonlinear trends in the mortality rates over time, we conducted analyses using linear spline terms. Visual inspection of graphical analyses suggested a nonlinear change in the trend in mortality rates during the early 1990s. To examine this trend, we used negative binomial regression models placing linear spline terms at different periods throughout the early 1990s. The best fitting model was selected using Akaike's Information Criterion.15
Finally, we conducted a subanalysis examining trends in mortality rates for the period 1992–2005 to assess potential changes in the SCD mortality rate restricted to the era of hydroxyurea use. We placed a linear spline term at the year 1998 to compare trends in mortality rates for the seven years before/up to Food and Drug Administration (FDA) approval of hydroxyurea (1992–1998) with trends in mortality rates for the seven years after FDA approval of hydroxyurea (1999–2005). We used Stata® version 10.016 to perform all statistical analyses.
RESULTS
We identified 16,654 sickle cell-related deaths during the period of study, 1979–2005. Table 1 shows the demographic characteristics by the most prevalent underlying CODs. The age range was 0–107 years. The female who was reported as being 107 had ICD-9 codes for SCD (unspecified) and coronary artery disease. The median age at death during the entire time period was significantly lower for males (33 years, interquartile range [IQR] = 20) than for females (37 years, IQR=23) (p<0.001 for both). The median age at death in 2005 (our most recent year of study) was 42 years (IQR=23) for females and 38 years (IQR=18) for males (data not shown).
Table 1.
Demographic characteristics of people with sickle cell disease who died from the top 14 underlying causes of death: U.S., 1979–2005a
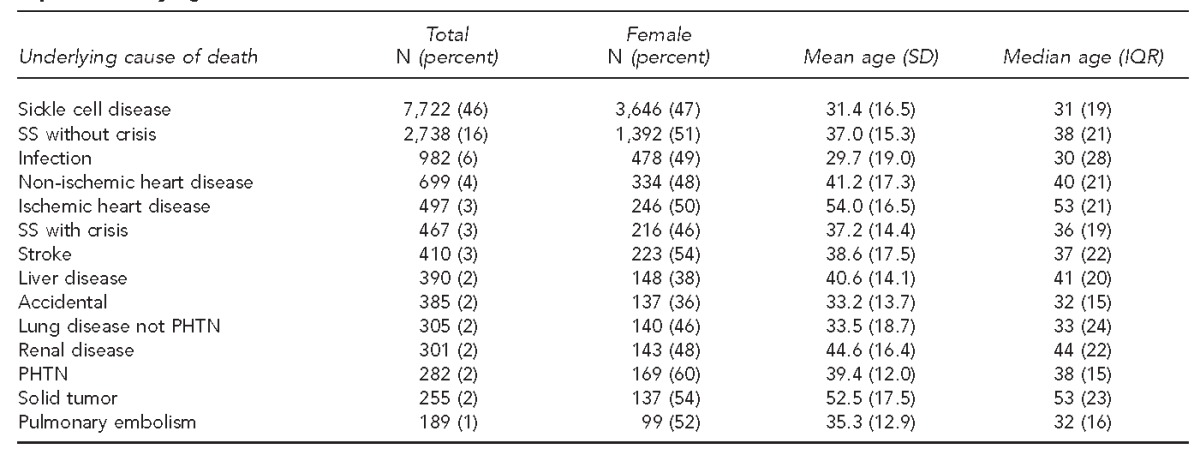
aThe table represents 93.8% of all deaths during this time period. All other diagnoses were present in <1% of deaths.
SD = standard deviation
IQR = interquartile range
SS = homozygous S
PHTN = pulmonary hypertension
Sixty-five percent of the death certificates listed SCD as the underlying COD. The top five most common diagnoses (not including SCD) that were listed as the underlying COD were infectious disease (6%), non-ischemic heart disease (4%), ischemic heart disease (3%), stroke (3%), and liver disease (2%). The odds of the COD being infection or stroke decreased by 2% and 1%, respectively, each year during the time period studied. In a regression model of age at death over time and controlling for gender, we found that the mean age at death increased by 0.36 years for each year of study. Table 2 shows how the known sickle cell comorbidities were associated with age at death. People with infection listed as the underlying COD were 5.17 years younger, on average, than people for whom infection was not listed as the underlying COD. Conversely, people with an underlying COD of stroke, pulmonary hypertension, or renal disease were older, on average, than those without those conditions listed as the underlying COD.
Table 2.
Linear regression model for correlates of age at death for people with sickle cell disease: U.S., 1979–2005

ap<0.001
bp<0.05
Based on U.S. Census data estimates, the African American population grew in size from 26,072,963 to 39,067,463 during the time period studied. The overall sickle cell mortality rate calculated from this Census data increased by 0.7% (p<0.001) each year during the time period studied. This rate represents an 18.2% increase in mortality rate during the 26 years studied. A graph of the smoothed mortality rates for adults and children over time are shown in the Figure. The adult (>19 years of age) mortality rate increased by 1% (p<0.001) each year during the time period studied. The pediatric mortality rate decreased by 3% (p<0.001) each year during the time period studied. In examining the graph of mortality rate, there appears to be a change in the adult mortality rate sometime in the early 1990s. This apparent nonlinearity was assessed using a spline term at 1994. We found that prior to 1994, the mortality rate increased by 2.6% (p<0.001) each year; however, from 1994 on, the mortality rate decreased by 0.9% each year (p=0.007). The adult mortality rate in 2005—2.5 per 100,000 African American population—never returned to the baseline value seen in 1979—2.2 per 100,000 African American population. The apparent decreasing trend does seem sensitive to the presence of the year 2005 data point, however, as removal of this data point from the analysis negates the statistical significance seen in this trend.
Figure.
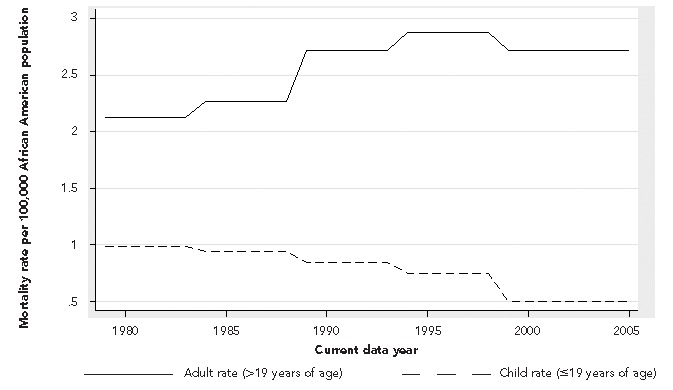
Mortality rates for adults and children with sickle cell disease: U.S., 1979–2005
In examining the potential impact of the use of hydroxyurea on mortality rates, we compared rates using spline terms for the seven years before and up to the FDA approval of hydroxyurea (1992–1998) with the seven years of data we had after the approval of hydroxyurea (1999–2005). We found that there was no significant change in mortality rates between these two time periods (incidence rate ratio [IRR] = 1.00, 95% CI 0.99, 1.02 for 1992–1998 vs. IRR=0.99, 95% CI 0.96, 1.01 for 1999–2005).
DISCUSSION
Our data suggest that, unlike in children, the mortality rate for adults with SCD in the U.S. increased during the time period studied, with little impact at the population level seen from the introduction of hydroxyurea. Several prior publications have demonstrated an improved survival rate for children with SCD. Quinn et al.5 examined deaths in the Dallas Newborn Cohort and demonstrated that the overall incidence rate for deaths per 100 patients decreased from 0.67 in children from the 1983–1990 era to 0.15 in the 2000–2007 era. Yanni et al.4 compared mortality rates for children using the MCOD files, as we did in this study. They compared mortality rates for children during the period 1983–1986 with rates during the period 1999–2002. They demonstrated that the relative rate of mortality dropped 68% for children 0–3 years of age, 39% for children 4–9 years of age, and 24% for children 10–14 years of age between the two time periods. Our data are consistent with those findings and demonstrate a decrease in mortality rate for children from the late 1970s through 2005. Along with the finding of a decrease in childhood mortality, we found a decrease over time in the odds of infection being listed as the underlying COD. This finding is consistent with, and would seem to support, the widely held view that interventions such as penicillin prophylaxis and vaccination provided to people with SCD in early childhood have played an important role in decreasing childhood mortality and preventing life-threatening infections.
While comprehensive care for children with SCD has been linked to improved survival, the lack of comprehensive care for adults with SCD17 may explain the overall increased mortality rate for this population seen during the entire study period. During this same time period, the age-adjusted death rates for the entire African American population steadily decreased from 1,314.8 per 100,000 population in 1980 to 1,016.5 per 100,000 population in 2005.18 While we found an overall increased mortality rate for adults with SCD comparing the end of our period of study with the beginning, it is interesting to note that the trend in adult SCD mortality rate did appear to start decreasing in 1994. It is difficult to identify any intervention in the early 1990s for adults that might easily explain this trend. In addition, the mortality rate for adults with SCD never returned to the baseline value seen in 1979. And if we take out the final year of data, the significance of the downward trend in adult mortality is no longer seen. It is likely too early to tell whether the apparent decrease seen is real or an artifact of an outlying late data point. Unlike the interventions for children, only one intervention has been expected to influence outcomes for adults with SCD and that is the use of hydroxyurea. Although there have not been any randomized studies showing the effect of hydroxyurea on survival, there are observational studies suggesting that the use of hydroxyurea may decrease mortality.7,19 Nevertheless, we found no change in mortality rates during the period before and after hydroxyurea was approved by the FDA. This finding may reflect poor use of this drug by this population or it may be that it is too early to see the effect using death certificate data.
A number of factors might be contributing to the overall increase in mortality rates for adults with SCD that we observed in this study. One hypothesis is that the increase in adult mortality rate is due in part to an increase in the number of children with SCD who are living to adulthood. While this is certainly a distinct possibility, it must be noted that the interventions that are thought to have improved childhood mortality, such as newborn screening and penicillin prophylaxis, all occur early in life. Given this fact, one would predict that interventions implemented in the early 1980s reducing pediatric mortality would result in a significant increase in the adult sickle cell population 20 years later. Yet, the trend seen in the Figure demonstrates an increase in mortality rate starting in the mid-1980s.
Another hypothesis is that changes in our overall approach to inpatient SCD care may have had an unintended detrimental effect on SCD mortality rates. The average length of hospital stays for SCD has been decreasing over time while the rate of hospital admissions has been increasing.20 Shorter hospital stays, however, may be associated with increased hospital visits for SCD, especially increased hospital readmissions. It is clear that hospital readmissions represent a significant number of the total hospital admissions for this patient population.21 Ballas and Lusardi22 have shown that patients with SCD who were readmitted within one week of discharge had a significantly higher mortality rate than other patients admitted with SCD. Houston-Yu et al.23 have shown that individuals with SCD who have the highest rates of hospitalization also have higher mortality rates. Increasing hospitalizations and readmissions, therefore, may explain some of the increase in mortality rates for adults that we have observed in this study.
The fact that the median age at death for people with SCD appears to be unchanged from that reported by Platt et al. in the early 1990s is of additional concern. Especially troubling is the fact that the median age at death reported in this study is for people with all genotypes of SCD, as we did not analyze the death certificates by separate genotypes. In their 1994 publication of the Cooperative Study of Sickle Cell Disease cohort, Platt and colleagues showed that for individuals with hemoglobin SC genotype, the median age at death was 60 years of age for males and 68 years of age for females, and it was those with the most severe form of SCD, homozygous SS genotype, that had a median age at death in their fifth decade of life.3 It may be that greater survival with SCD may introduce an inherent bias in these data. That is, if the sickle cell population lives longer, it may become less likely for a death certificate to include SCD as a COD. Were this to happen relatively frequently, our analyses would miss that percentage of patients who had SCD but were thought to die from causes unrelated to this underlying disease. This consequence may be especially true for individuals with the less severe forms of SCD, such as SC or S-Beta thalassemia. However, if greater survival with SCD truly introduced the biases we hypothesize in this study, the implication for our findings is that we would underestimate the overall mortality rate for people with SCD.
Limitations
This study was subject to several limitations. One limitation was that death certificates lack details and in 11% of cases, the only diagnosis on the certificate was SCD, thereby limiting the interpretation of the true immediate COD. In addition, the validity of our administrative mortality data depends upon the accuracy of the physician diagnosis and the use of appropriate coding of CODs. In calculating mortality rates, we used denominators based on the estimated number of all African Americans and not specific numbers of individuals with SCD, as these numbers were not available. There were also limitations in the intercensal population estimates provided by the U.S. Census that we used to estimate the African American population. Although errors in these estimates are less likely to occur when looking at data for larger populations (state or county level), more discrepancies have been noted when the data are stratified by race.24 Finally, as these data only contained age at death, we were unable to comment on life expectancy. We could only make comments on changes in the ages at death for those who died. As we did not have information about people who were alive during the time period under study, we could not comment on changes in life expectancy for the sickle cell population as a whole over time.
CONCLUSIONS
As has been described in a number of prior studies, we demonstrated a decrease in mortality rate for children with SCD during a 26-year period. What has not been shown previously, however, is that mortality rates for adults with SCD increased overall during the same time period. Although the mortality rate for adults appears to be decreasing in the tail end of the time period under study, the overall mortality rates for adults remain above the baseline rates of 1979. While it is impossible to know why there has not been a clear decrease in mortality rates for adults with SCD, we do know that there are interventions, including the provision of comprehensive care,25,26 which are not universally available for adults and may decrease hospitalization rates, thereby impacting mortality rates. Additional longitudinal studies of patients are required to have better estimates of mortality rates and life expectancy for this population.
Footnotes
Sophie Lanzkron's work was supported by grant #K23HL083089 from the National Heart, Lung, and Blood Institute (NHLBI). Carlton Haywood's work was supported by grant #K01HL108832-01 from the NHLBI. The findings and conclusions in this article are those of the authors and do not necessarily represent the views of the NHLBI.
As the authors used a publicly available deidentified database, they were not required to obtain Institutional Review Board approval.
REFERENCES
- 1.Hassell KL. Population estimates of sickle cell disease in the U.S. Am J Prev Med. 2010;38(4 Suppl):S512–21. doi: 10.1016/j.amepre.2009.12.022. [DOI] [PubMed] [Google Scholar]
- 2.Brousseau DC, Panepinto JA, Nimmer M, Hoffmann RG. The number of people with sickle-cell disease in the United States: national and state estimates. Am J Hematol. 2010;85:77–8. doi: 10.1002/ajh.21570. [DOI] [PubMed] [Google Scholar]
- 3.Platt OS, Brambilla DJ, Rosse WF, Milner PF, Castro O, Steinberg MH, et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death. N Engl J Med. 1994;330:1639–44. doi: 10.1056/NEJM199406093302303. [DOI] [PubMed] [Google Scholar]
- 4.Yanni E, Grosse SD, Yang Q, Olney RS. Trends in pediatric sickle cell disease-related mortality in the United States, 1983-2002. J Pediatr. 2009;154:541–5. doi: 10.1016/j.jpeds.2008.09.052. [DOI] [PubMed] [Google Scholar]
- 5.Quinn CT, Rogers ZR, McCavit TL, Buchanan GR. Improved survival of children and adolescents with sickle cell disease. Blood. 2010;115:3447–52. doi: 10.1182/blood-2009-07-233700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steinberg MH, McCarthy WF, Castro O, Ballas SK, Armstrong FD, Smith W, et al. The risks and benefits of long-term use of hydroxyurea in sickle cell anemia: a 17.5 year follow-up. Am J Hematol. 2010;85:403–8. doi: 10.1002/ajh.21699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Voskaridou E, Christoulas D, Bilalis A, Plata E, Varvagiannis K, Stamatopoulos G, et al. The effect of prolonged administration of hydroxyurea on morbidity and mortality in adult patients with sickle cell syndromes: results of a 17-year, single-center trial (LaSHS) Blood. 2010;115:2354–63. doi: 10.1182/blood-2009-05-221333. [DOI] [PubMed] [Google Scholar]
- 8.Brawley OW, Cornelius LJ, Edwards LR, Gamble VN, Green BL, Inturrisi CE, et al. NIH consensus development statement on hydroxyurea treatment for sickle cell disease. NIH Consens State Sci Statements. 2008;25:1–30. [PubMed] [Google Scholar]
- 9.Centers for Disease Control and Prevention (US). Multiple cause of death data, 1979-2005. [cited 2012 Oct 16]. Available from: URL: http://wonder.cdc.gov/mcd.html.
- 10.Centers for Disease Control and Prevention (US). International classification of diseases, ninth revision. [cited 2012 Oct 15]. Available from: URL: http://www.cdc.gov/nchs/icd/icd9.htm.
- 11.Centers for Disease Control and Prevention (US). International classification of diseases, tenth revision. [cited 2012 Oct 15]. Available from: URL: http://www.cdc.gov/nchs/icd/icd10.htm.
- 12.Powars DR, Chan LS, Hiti A, Ramicone E, Johnson C. Outcome of sickle cell anemia: a 4-decade observational study of 1056 patients. Medicine (Baltimore) 2005;84:363–76. doi: 10.1097/01.md.0000189089.45003.52. [DOI] [PubMed] [Google Scholar]
- 13.Webster LR, Cochella S, Dasgupta N, Fakata KL, Fine PG, Fishman SM, et al. An analysis of the root causes for opioid-related overdose deaths in the United States. Pain Med. 2011;12(Suppl 2):S26–35. doi: 10.1111/j.1526-4637.2011.01134.x. [DOI] [PubMed] [Google Scholar]
- 14.Centers for Disease Control and Prevention (US). Bridged-race population estimates (vintage 2006) [cited 2012 Oct 16]. Available from: URL: http://wonder.cdc.gov/bridged-race-v2006.html.
- 15.Long JS. Regression models for categorical dependent variables using Stata. 2nd ed. College Station (TX): Stata Press; 2006. [Google Scholar]
- 16.StataCorp. Stata®: Release 10.0. College Station (TX): StataCorp; 2007. [Google Scholar]
- 17.Grosse SD, Schechter MS, Kulkarni R, Lloyd-Puryear MA, Strickland B, Trevathan E. Models of comprehensive multidisciplinary care for individuals in the United States with genetic disorders. Pediatrics. 2009;123:407–12. doi: 10.1542/peds.2007-2875. [DOI] [PubMed] [Google Scholar]
- 18.National Center for Health Statistics (US). Hyattsville (MD): NCHS; 2011. [cited 2012 Oct 16]. Health, United States, 2010: with special feature on death and dying. Also available from: URL: http://www.cdc.gov/nchs/data/hus/hus10.pdf. [PubMed] [Google Scholar]
- 19.Steinberg MH, Barton F, Castro O, Pegelow CH, Ballas SK, Kutlar A, et al. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA. 2003;289:1645–51. doi: 10.1001/jama.289.13.1645. [DOI] [PubMed] [Google Scholar]
- 20.Lanzkron S, Haywood C, Jr, Segal JB, Dover GJ. Hospitalization rates and costs of care of patients with sickle-cell anemia in the state of Maryland in the era of hydroxyurea. Am J Hematol. 2006;81:927–32. doi: 10.1002/ajh.20703. [DOI] [PubMed] [Google Scholar]
- 21.Brousseau DC, Owens PL, Mosso AL, Panepinto JA, Steiner CA. Acute care utilization and rehospitalizations for sickle cell disease. JAMA. 2010;303:1288–94. doi: 10.1001/jama.2010.378. [DOI] [PubMed] [Google Scholar]
- 22.Ballas SK, Lusardi M. Hospital readmission for adult acute sickle cell painful episodes: frequency, etiology, and prognostic significance. Am J Hematol. 2005;79:17–25. doi: 10.1002/ajh.20336. [DOI] [PubMed] [Google Scholar]
- 23.Houston-Yu P, Rana SR, Beyer B, Castro O. Frequent and prolonged hospitalizations: a risk factor for early mortality in sickle cell disease patients. Am J Hematol. 2003;72:201–3. doi: 10.1002/ajh.10305. [DOI] [PubMed] [Google Scholar]
- 24.Phipps AI, Clarke CA, Ereman RR. Impact of intercensal population projections and error of closure on breast cancer surveillance: examples from 10 California counties. Breast Cancer Res. 2005;7:R655–60. doi: 10.1186/bcr1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Frempong T, Pearson HA. Newborn screening coupled with comprehensive follow-up reduced early mortality of sickle cell disease in Connecticut. Conn Med. 2007;71:9–12. [PubMed] [Google Scholar]
- 26.Rahimy MC, Gangbo A, Ahouignan G, Adjou R, Deguenon C, Goussanou S, et al. Effect of a comprehensive clinical care program on disease course in severely ill children with sickle cell anemia in a sub-Saharan African setting. Blood. 2003;102:834–8. doi: 10.1182/blood-2002-05-1453. [DOI] [PubMed] [Google Scholar]