

Abstract
Background
Low-dose ketamine is used as analgesic for acute and chronic pain. It is metabolized in the liver to norketamine via cytochrome P450 enzymes. There are few human data on the involvement of CYP enzymes on the elimination of norketamine and its possible contribution to analgesic effect. The aim of this study was to investigate the effect of cytochrome P450 enzyme induction by rifampicin on the pharmacokinetics of S-ketamine and its major metabolite, S-norketamine, in healthy volunteers.
Methods
Twenty healthy male subjects received 20 mg/70kg/h (n = 10) or 40 mg/70kg/h (n = 10) intravenous S-ketamine for 2-h following either 5 days of oral rifampicin (once daily 600 mg) or placebo treatment. During and 3-h following drug infusion arterial plasma concentrations of S-ketamine and S-norketamine were obtained at regular intervals. The data were analyzed with a compartmental pharmacokinetic model consisting of three compartments for S-ketamine, three sequential metabolism compartments and two S-norketamine compartments using the statistical package NONMEM version VII.
Results
Rifampcin caused a 10% and 50% reduction in the area-under-the-curve of the plasma concentrations of S-ketamine and S-norketamine, respectively. The compartmental analysis indicated a 13% and 200% increase in S-ketamine and S-norketamine elimination from their respective central compartments by rifampicin.
Conclusions
A novel observation is the large effect of rifampicin on S-norketamine concentrations and indicates that rifampicin induces the elimination of S-ketamine’s metabolite, S-norketamine, probably via induction of the CYP3A4 and/or CYP2B6 enzymes.
Introduction
Ketamine is an arylcycloalkylamine structurally related to phencyclidine and first synthesized in 1963.1 At relatively high dose, ketamine produces anesthesia while at low, subanesthetic dose it is a potent analgesic. Ketamine analgesia is thought to result from non-competitive antagonism at the ionotropic glutamatergic N-methyl-D-aspartate (NMDA) receptor.2 NMDA receptors are excitatory and involved in enhanced nociceptive processing at spinal and supraspinal sites.3 By blocking the NMDA receptor, ketamine effectively reduces signal propagation in the pain circuitry from the spinal cord to the cortex and consequently produces analgesia. Ketamine-induced acute pain relief is driven by its pharmacokinetics, that is, upon termination of infusion (causing a brisk decrease in plasma concentration) acute pain relief ends rapidly,4,5 while in chronic pain patients the relief of spontaneous pain outlasts the treatment period by weeks to months.6-9
Ketamine undergoes extensive metabolism by hepatic cytochrome P450. In rat liver microsomes, norketamine, hydroxynorketamine, and hydroxyketamine account for 80%, 15% and 5%, respectively, of ketamine metabolism (fig. 1).10 In vivo, rabbits convert 68% of ketamine to norketamine.11 In humans, N-demethylation to norketamine is the major route of metabolism, which can undergo further metabolism via cyclohexanone ring hydroxylation to form 4-, 5,- and 6-hydroxynorketamine. Ketamine itself can be hydroxylated, forming 4-hydroxyketamine, although this is considered quantitatively insignificant.10,12 Other minor metabolites have recently been reported.13 Finally, norketamine and the hydroxy metabolic products are subsequently glucuronidated and eliminated via the kidney and bile.14 Just 15-10% of ketamine is eliminated unchanged. Like ketamine, norketamine is a non-competitive antagonist at the NMDA receptor.15,16 Nonhuman data indicate that norketamine passes the blood-brain barrier, has about one-fifth to one-third the potency of ketamine and is thought to contribute up to 30% of ketamine analgesia, and, albeit to a lesser extent, to the development of psychomimetic side effects.15,17,18 No pharmacological activity is attributed to the hydroxynorketamines.17
Figure 1.
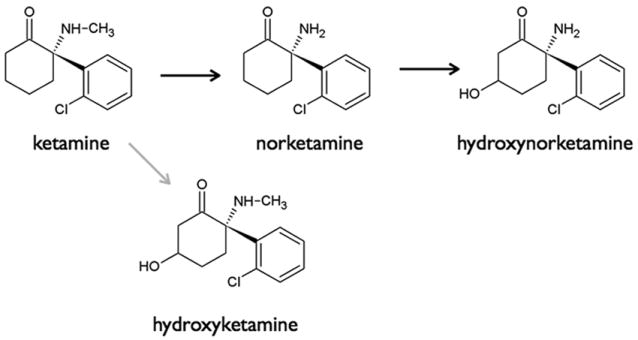
Metabolic pathways of ketamine. The black arrows indicate the major pathway; the grey arrow the minor pathway.
The major human hepatic cytochrome P450s catalyzing ketamine N-demethylation in vitro are CYP2B6 and CYP3A4, although there is ambiguity as to their comparative contributions to clinical ketamine metabolism.19,20 Rifampicin is an effective and non-selective inducer of multiple hepatic P450s, including those (CYPs 2B6 and 3A4) which catalyze ketamine N-demethylation.21
In the current study, we performed a compartmental pharmacokinetic analysis to quantify the effect of CYP induction on the elimination of S(+)-ketamine (S-ketamine) and formation and elimination of S(+)-norketamine (S-norketamine). We took this approach (i.e., CYP induction) as norketamine is unavailable for testing in humans. Next, using a sigmoid EMAX model with S-ketamine and S-norketamine contributions, we simulated the effect of rifampicin on analgesia to obtain an indication of the importance of variations in S-norketamine concentration on analgesic effect. We tested the effect of the S(+)-enantiomer of ketamine as it is the only registered ketamine product for human use in The Netherlands. S-ketamine has greater analgesic potency than either the R(-)-variant or the racemic mixture, with possibly fewer side effects than the racemic mixture.16,22
Materials and Methods
Subjects
Twenty healthy male volunteers (age 19-29 yr, body mass index < 30 kg/m2) were recruited to participate in the study after approval of the protocol by the local Human Ethics Committee (Commissie Medische Ethiek, Leiden University Medical Center, Leiden, The Netherlands). Written informed consent was obtained prior to inclusion in the study according to the Declaration of Helsinki. All subjects were instructed not to eat or drink for at least 8 h before the study. Alcohol, coffee, and chocolate were not allowed for 24 h and grapefruit or grapefruit juice was not allowed 6 days prior to the study day. The study was registered under number NTR1328.*
Study design: S-ketamine infusion and blood sampling
The study design was randomised single-blind, placebo-controlled and crossover. Subjects were treated with daily oral rifampicin 600 mg (Sandoz BV, Almere, The Netherlands) (Session I) or placebo (Session II; cellulose tablets produced by the local pharmacy) in the five days preceding the experimental testing (five 600 mg doses were given). Rifampicin/placebo was taken at bedtime. The time interval between the last dose and the ketamine administration was 10-12 h. The order of the sessions was random with at least 3 weeks between sessions. On the study day the subjects received an arterial line in the radial artery of the nondominant hand for blood sampling and a venous line in the contra-lateral arm for drug infusion. Total study duration was 5 h, of which S-ketamine (Ketanest S, Pfizer BV, Capelle aan de IJssel, The Netherlands) was administered during the first 2 h. The subjects were randomly allocated to receive a 20 mg/70 kg/h or 40 mg/70 kg/h S-ketamine infusion. The ketamine infusions were similar for Sessions I and II. Randomization was done by the pharmacy using computer-generated randomization lists.
Arterial blood sampling was performed at times t = 0 (predrug baseline), 5, 10, 15, 30, 45, 60, 75, 90, 105, 120, 125, 130, 135, 150, 180, 210, 240, 270, and 300 min after the start of the infusion. Five ml of blood was collected per sample. The analysis has been described previously.4 In brief: The samples were centrifuged at a speed of 3,000 rpm for 10 min. Two to three ml plasma was separated within 15-min of blood collection and stored at −25 °C until analysis. For the construction of S-ketamine and S-norketamine calibration lines, solid substances were obtained from Parke-Davis (Dallas, TX) and Tocris (St. Louis, MO), respectively. After extraction from the specific sample, S(+)-ketamine and S(+)-norketamine were determined by HPLC on a Gemini C18 column (Phenomenex, Utrecht, The Netherlands) at 40 °C. Monitoring of the eluent was performed at 195 nm with a photodiode-array-detector (PDA 100, Dionex, Amsterdam, The Netherlands). The lower limit of quantitation was 10 ng/ml, the lower limit of detection was 3 ng/ml for both drugs. All samples with concentrations > 3 ng/ml were included in the analysis. None of the samples had ketamine concentrations below 3 ng/ml while the initial 2 norketamine samples of all subjects were below the detection limit.
Pharmacokinetic Analysis
A t-test was performed to compare the maximum plasma concentration of S-ketamine and S-norketamine (CMAX), their time of occurrence (TMAX), and the area-under-the-plasma concentration curve (AUC) divided by the duration of the experiment (AUC0→300) of the placebo versus ketamine treatment. The analysis was performed in SPSS for Windows version 16.0 (SPSS Inc., Chicago, IL). P-values < 0.05 were considered significant. Values reported are mean ± standard deviation.
The compartmental model used for pharmacokinetic analysis was identical to a previously published model.4 The analysis was performed in two steps. Initially, only the ketamine data were analyzed. Subsequently, the combined ketamine norketamine data were analyzed. The model consisted of three ketamine compartments connected to two norketamine compartments (see fig. 2). Since we had found that norketamine’s formation and elimination (and inter-compartmental distribution) rates were not simultaneously estimable,4 the norketamine formation rate was set equal to the ketamine elimination rate. Volumes were scaled via WT/70 and clearances via (WT/70)0.75, where WT is body weight in kg.23 In order to test for a significant rifampicin effect on clearances, we introduced factor F as follows:
Figure 2.
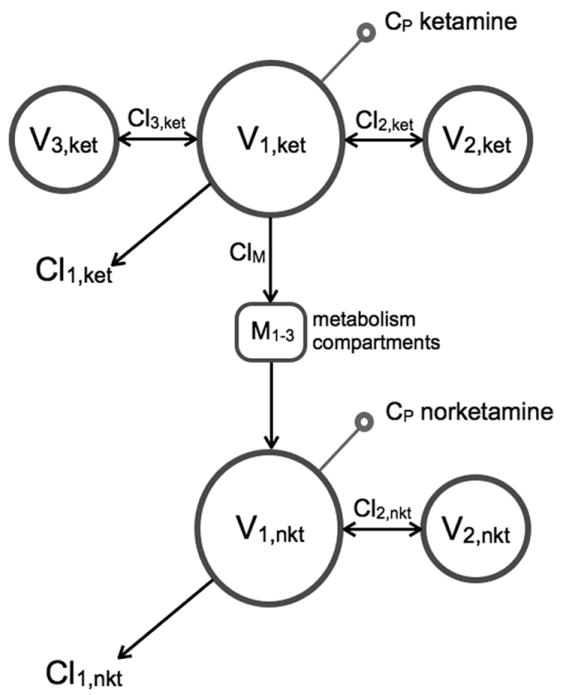
Schematic representation of the pharmacokinetic model used to analyze the combined S-ketamine and S-norketamine data. V1,ket, V2,ket and V3,ket are the three ketamine (ket) compartments, V1,nkt and V2,nkt the two norketamine (nket) compartments. M1-3 represents three sequential metabolism compartments. Cl1,ket is the clearance from compartment V1,ket, Cl2,ket the clearance from compartment V2,ket, Cl3,ket the clearance from compartment V3,ket, Cl1,nkt the clearance from compartment V1,nkt and Cl2,nkt is the clearance from compartment V2,nkt. ClM is the S-ketamine clearance responsible for the S-norketamine formation.
For example, an F value of 2 indicates that the parameter increased by 200% after rifampicin treatment. Significance of factors was tested using forward selection. Since experiments were performed on two separate days we estimated both interindividual (ω2) and interoccasion (ν2) variances. Exponential error models were used for inter- and intraindividual variability.
The compartmental analysis was performed with the statistical package NONMEM VII with first-order conditional estimation with interaction method (ICON Development Solutions, Ellicott City, MD).24 Data presented are median and 95% confidence intervals. A bootstrap analysis was used to check the final model and to obtain 95% confidence intervals for the model parameters (from 1,000 successful runs).
Side Effects
Drug high and sedation were scored using an 11-point numerical rating scale ranging from 0 (= no effect) to 10 (=maximum possible effect) at regular intervals during and following ketamine infusion.
Simulation Study
Simulation studies enable the systematic exploration of inferences made in our study with respect to anticipated norketamine effect size. Here we performed simulations to estimate the effect of the large change in norketamine concentration and relatively modest change in ketamine concentration that we observed after rifampicin treatment on pain relief. Two sets of simulation were performed, one set on pain relief induced by short-term ketamine infusion and another set on pain relief induced by chronic ketamine administration. To that end we made a priori assumptions with respect to the norketamine contributions to ketamine effect: Simulations with 0, 10, and 25% norketamine contribution were made. The difference in effect observed in the simulated pain relief with and without rifampicin treatment will give an indication of the norketamine contribution to effect.
Acute antinociception paradigm
Using the pharmacokinetic model parameters the effect of rifampicin treatment on acute antinociception was simulated for different norketamine contributions to effect. To that end, analgesic effect during and following a 2 h S-ketamine infusion of 40 mg/h was simulated using the current pharmacokinetic data set linked to pharmacodynamic data previously obtained and modeled in a similar subject population in our laboratory.4 Analgesia was simulated using a sigmoid EMAX model assuming an additive effect of S-ketamine and S-norketamine as follows:
where VAS is visual analogue score (ranging from 0 cm = no pain to 10 cm = severe pain), BLN is baseline (or predrug) VAS, γ a shape parameter, Cket and Cnkt the plasma concentrations of S-ketamine and S-norketamine, respectively; C50,ket the plasma concentration S-norketamine causing 50% effect, C50,nkt the concentration S-norketamine causing 50% effect. The following model parameters where used (Ref 4: BLN = 6.7 cm, γ = 2.5, and C50,ket = 375 ng/ml. The C50,nkt was varied in such a way that it contributed 0, 10 and 25% to total analgesic effect. We assumed no delay between blood concentration and acute antinociceptive effect (i.e., pain relief in response to an experimental heat pain stimulus) for both S-ketamine and S-norketamine.4,5
Chronic analgesia paradigm
Using pharmacokinetic model parameters (from refs. 6 and 7 on a study on the effect of ketamine on spontaneous chronic pain relief in complex regional pain syndrome type 1 patients), the effect of rifampicin treatment on chronic analgesia was simulated for different norketamine contributions to effect (0, 10 and 25%). To that end analgesic effect during and following a 4-day S-ketamine infusion of 5 mg/h on day 1, 10 hg/h on day 2, 15 mg/h on day 3 and 20 mg/h on day 4 was simulated.6,7 Before the simulations were performed the pharmacokinetic data obtained in the healthy volunteer population and chronic pain patients were compared. The differences were sufficiently small (data not shown) to allow the application of a rifampicin effect as observed in our current study to the kinetic data obtained in chronic pain patients. The pharmacodynamic model was as given above with model parameters: BLN = 7 cm, γ = 1.9, and C50,ket = 10 ng/ml. The C50,nkt was varied in such a way that it contributed 0, 10 and 25% to total analgesic effect. We previously observed that in our group of chronic pain patients, the effect of ketamine on spontaneous pain relief lasted beyond the duration of treatment.6,7 In a subgroup of these patients (i.e., responders to therapy) we estimated an effect half-life (t½k) of about 11 days and used this value in the chronic pain simulations.7
Results
Subjects
All subjects completed the protocol without unexpected side effects. One subject (in the 20 mg/h ketamine group) failed to return, for unknown reasons, for his second experimental session. The subjects age ranged from 20 to 29 yr (mean age 22.0 yr), their body mass index (BMI) from 19 to 28 (mean BMI 22.2). There were no differences in age or BMI between treatment groups.
Descriptive pharmacokinetic analysis
In figure 3, the mean S-ketamine and S-norketamine concentrations in plasma after rifampicin and placebo are plotted. Rifampicin decreased S-ketamine concentrations albeit the effects were modest in magnitude. The effect of rifampicin was most prominent in the lower S-ketamine infusion group: during 20 mg/h infusion rifampicin caused a 17% decrease in CMAX (P < 0.001), 14% decrease in AUC0→300 (P < 0.001). At double the ketamine infusion dose rifampicin decreased CMAX by 6% (ns) and AUC0→300 by 7% (P = 0.02; table 1). S-ketamine TMAX values remained unaffected by rifampicin (table 1). In contrast, the effects of rifampicin on S-ketamine’s metabolite S-norketamine concentrations in plasma were large with a 44% and 58% decrease in CMAX for the infusion of 20 mg/h (P = 0.001) and 40 mg/h (P < 0.001), and a 53% and 62% decrease in AUC0→300 for the 20 mg/h and 40 mg/h infusions (P < 0.001), respectively (table 1, fig. 3). S(+)-norketamine TMAX values remained unaffected by rifampicin (table 1). The metabolite-to-parent drug AUC0→300 ratios (AUCM/AUCP in table 1) were about 50% lowered by rifampicin compared to placebo irrespective of the S-ketamine infusion dose.
Figure 3.
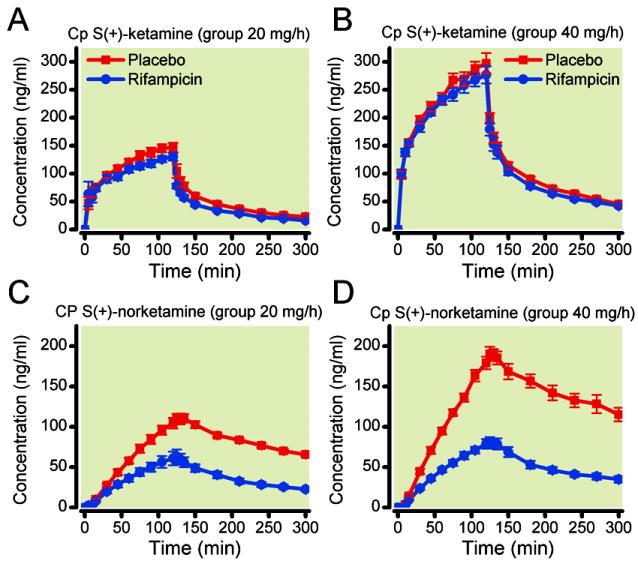
Effect of rifampicin treatment on plasma concentrations of S-ketamine (A and B) and S-norketamine (C and D) during and 3-h following a 2-h S(+)-ketamine infusion of 20 mg/h (A and C) and 40 mg/h (B and D). Values are mean ± SEM. The grey bars depict the 2-h infusion period.
Table 1.
Effect of Rifampicin on S-ketamine and Norketamine Concentrations: CMAX, TMAX and Area-Under-the-Curve (AUC) Values.
| Placebo | Rifampicin | P-value | |
|---|---|---|---|
|
|
|||
| [S-ketamine]20 | |||
| CMAX (ng/ml) | 150.4 ± 3.9 | 132.6 ± 7.1 | < 0.001 |
| TMAX (min) | 114 ± 3 | 113 ± 4 | 0.72 |
| AUC0→300 (ng/ml) | 71.5 ± 5.4 | 60.8 ± 5.1 | < 0.001 |
| [S-norketamine]20 | |||
| CMAX (ng/ml) | 113.8 ± 5.9 | 64.5 ± 8.7 | 0.001 |
| TMAX (min) | 129 ± 2 | 125 ± 1 | 0.06 |
| AUC0→300 (ng/ml) | 73.3 ± 8.4 | 35.5 ± 11.7 | < 0.001 |
| AUCM/AUCP | 1.03 ± 0.19 | 0.58 ± 0.19 | < 0.001 |
| [S-ketamine]40 | |||
| CMAX (ng/ml) | 304.7 ± 17.5 | 285.6 ± 14 | 0.27 |
| TMAX (min) | 110 ± 5 | 113 ± 3 | 0.51 |
| AUC0→300 (ng/ml) | 142 ± 15.8 | 132.0 ± 17.4 | 0.02 |
| [S-norketamine]40 | |||
| CMAX (ng/ml) | 198.3 ± 8.1 | 83.6 ± 6.2 | < 0.001 |
| TMAX (min) | 132 ± 2 | 128 ± 2 | 0.17 |
| AUC0→300 (ng/ml) | 124.8 ± 18.5 | 47.9 ± 11.6 | < 0.001 |
| AUCM/AUCP | 0.90 ± 0.12 | 0.36 ± 0.10 | < 0.001 |
AUC0→300 = the area-under-the-curve determined over the 300 min study period divided by 300 min; CMAX is the peak concentration; TMAX is the time of occurrence of CMAX.
20 and 40 denote data obtained during and after the 2-h infusion of 20 mg/h and 40 mg/h S-ketamine infusion (both per 70 kg), respectively.
AUCP = the parent AUC0→300 (S-ketamine); AUCM = the metabolite AUC0→300 (S-norketamine).
Values are mean ± SD.
Compartmental pharmacokinetic analysis
The pharmacokinetic model is given in figure 2. In agreement with an earlier observation,4 extending the three compartmental model (in which just the ketamine data were modeled) with a part describing the norketamine data did not cause any change in S-ketamine pharmacokinetic parameter values (data not shown). This indicates that adding the metabolism compartments (M1-3 in fig. 2) and the two norketamine compartments did not affect the analysis of ketamine. As judged by the eye, the model adequately described the data. Best, median and worst data fits as determined by the coefficient of determination (R2) for the combined S-ketamine/S-norketamine data fits are given in figure 4; goodness of fit plots are given in figure 5 (individual predicted versus measured concentration). Model parameter estimates together with their 95% confidence intervals are given in table 2. The pharmacokinetic model parameters were in agreement with previous findings.4,25-27 Rifampicin affected S-ketamine elimination (and consequently S-norketamine formation) modestly: clearance from the central compartment (V1,ket) increased by 13% (F Cl1,ket = 0.13 in fable 2). In contrast, rifampicin had a much greater effect on S-norketamine elimination: S-norketamine clearance form its central compartment (V1,nkt) increased by about 200% (F Cl1,nkt = 2.02 in table 2).
Figure 4.
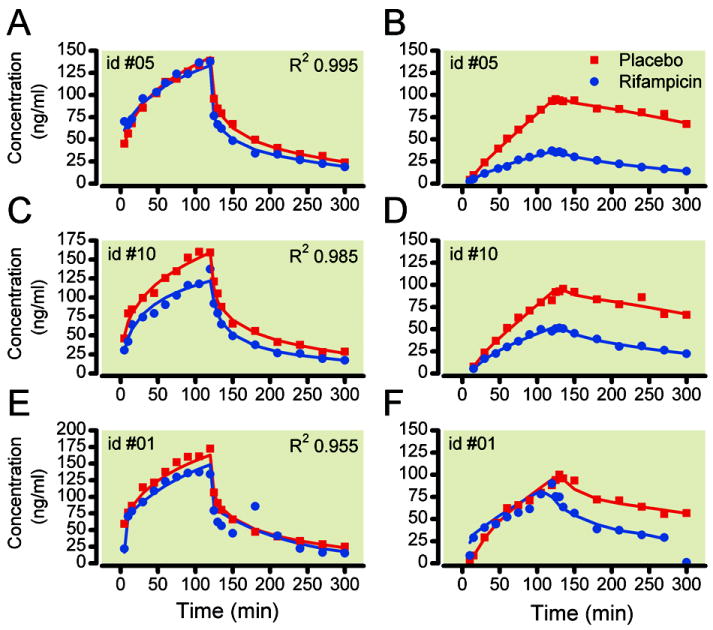
Pharmacokinetic data fits of the combined S-ketamine – S-norketamine model. A and B. Best S-ketamine and S-norketamine fits of subject #05; C and D. Median S-ketamine and S-norketamine data fits of subject #10; E and F. Worst S-ketamine and S-norketamine data fits of subject #01. Closed circles and continuous lines are the placebo data; Open squares and dashed lines are the rifampicin data. Goodness of fit was determined by the coefficient of determination (R2).
Figure 5.
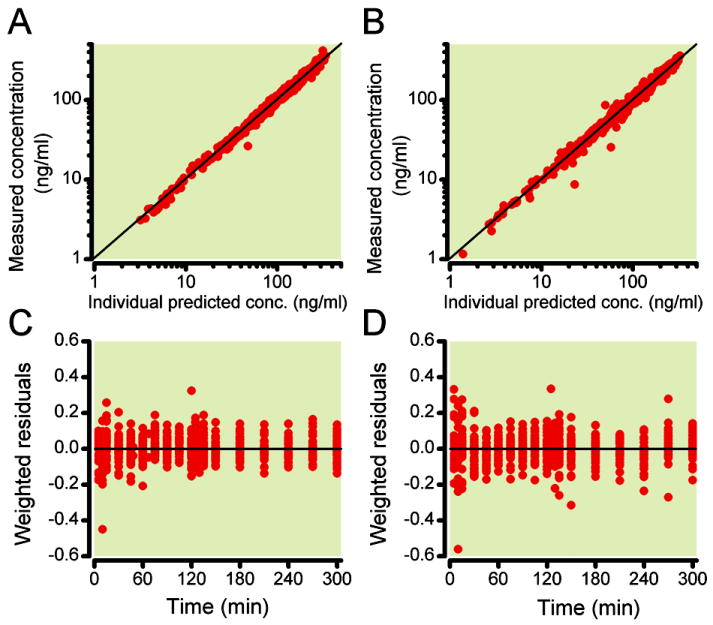
Goodness of fits plots: Individual predicted PK data versus measured data for S-ketamine (A) and S-norketamine (B). C and D: Weighted residuals, (Ymeasured – Ypredicted)/Ypredicted, versus time for S-ketamine concentrations (C) and S-norketamine concentrations (D).
Table 2.
Pharmacokinetic Model Parameters of the Combined Ketamine-Norketamine Model.
| Estimate | SE | ω2 | SE | ν2 | SE | ||
|---|---|---|---|---|---|---|---|
|
|
|||||||
| S(+)-ketamine | |||||||
| V1,ket (L) | 17.0 | 1.91 | - | - | 0.148 | 0.048 | |
| 95% ci | 13.6 – 21.0 | 0.06 – 0.25 | |||||
|
| |||||||
| V2.ket (L) | 28.3 | 2.97 | - | - | - | - | |
| 95% ci | 23.3 – 34.1 | ||||||
|
| |||||||
| V3,ket (L) | 147 | 10.2 | 0.065 | 0.019 | 0.029 | 0.020 | |
| 95% ci | 129 – 172 | 0.03 – 0.10 | 0.00001 – 0.08 | ||||
|
| |||||||
| Cl1,ket (L/h) | 93.5 | 2.70 | 0.007 | 0.004 | 0.006 | 0.002 | |
| 95% ci | 87.6 – 98.8 | 0.001 – 0.02 | 0.002 – 0.01 | ||||
| F Cl1,ket* | 0.13 | 0.03 | |||||
| 95% ci | 0.07 – 0.20 | ||||||
|
| |||||||
| Cl2,ket (L/h) | 127 | 12.3 | - | - | - | - | |
| 95% ci | 107 – 148 | - | - | ||||
|
| |||||||
| Cl3,ket (L/h) | 91.9 | 5.10 | 0.02 | 0.006 | - | - | |
| 95% ci | 82 - 101 | 0.01 – 0.03 | - | ||||
|
| |||||||
| σ2 | 0.008 | 0.002 | |||||
|
| |||||||
| S(+)-norketamine | |||||||
| MTT (h) | 0.25 | 0.02 | 0.05* | 0.03* | 0.02 | 0.01 | |
| 95% ci | 0.21 – 0.28 | 0.01 – 0.14 | 0.004 – 0.05 | ||||
|
| |||||||
| V2.nkt (L) | 193 | 10.6 | - | - | 0.14 | 0.03 | |
| 95% ci | 173 – 217 | - | 0.09 – 0.19 | ||||
|
| |||||||
| Cl1,nkt (L/h) | 64.9 | 2.81 | 0.03 | 0.01 | 0.03 | 0.01 | |
| 95% ci | 58.5 – 70.8 | 0.01 – 0.06 | 0.01 – 0.05 | ||||
| F Cl1,nkt* | 2.02 | 0.19 | |||||
| 95% ci | 1.67 – 2.41 | ||||||
|
| |||||||
| Cl2,nkt (L/h) | 334 | 30.5 | 0.05* | 0.03* | 0.14 | 0.04 | |
| 95% ci | 285 - 411 | 0.01 – 0.14 | 0.07 – 0.22 | ||||
|
| |||||||
| σ2 | 0.007 | 0.001 | |||||
|
| |||||||
* one parameter was sufficient for the interindividual error of Cl2,nket and MTT;
Units of the volumes are L/kg at 70 kg; units of the clearances are L/h at 70 kg;
Cl1,ket is the clearance from compartment 1,ket;
Cl2,ket is the clearance from compartment 2,ket;
Cl3,ket is the clearance from compartment 3,ket;
Cl1,nket is the clearance from compartment 1,nkt;
Cl2,nket is the clearance from compartment 2,nkt.
Factor F denotes the significant effect of rifampicin treatment on model parameters Cl1,nkt and Cl2,nkt (significance level < 0.01);
MTT is mean transit time;
ν2 is the interoccasion variability (in the log-domain);
σ2 is the within-subject variability (in the log domain);
SE is standard error;
V1,ket is the volume of compartment 1,ket;
V2,ket is the volume of compartment 2,ket;
V3,ket is the volume of compartment 3,ket;
V2,nkt is the volume of compartment 2,nkt.
ω2 is the inter-subject variability (in the log-domain).
P < 0.01.
Side Effects
Side effects were present during ketamine infusion and resolved promptly upon termination of infusion. The highest scores measured during ketamine infusion all occurred at the end of the infusion-period and were for drug high: 6.5 ± 2.3 (placebo, low-dose ketamine) versus 5.5 ± 3.0 (rifampicin, low-dose ketamine; P > 0.05), 8.1 ± 2.0 (placebo, high-dose ketamine) versus 7.8 ± 2.6 (rifampicin, high-dose ketamine; P > 0.05); and for sedation 3.8 ± 3.6 (placebo, low-dose ketamine) versus 3.8 ± 2.6 (rifampicin, low-dose ketamine; P > 0.05), and 5.1 ± 3.6 (placebo, high-dose ketamine) versus 4.5 ± 3.8 (rifampicin, high-dose ketamine; P > 0.05).
Simulation study
Acute analgesia paradigm
The simulation study was based on the current pharmacokinetic model parameters (table 2) linked to a pharmacodynamic model in which both S-ketamine and S-norketamine contribute to effect. The results of the simulations are shown in figure 6. At increasing norketamine contributions to analgesic effect, peak VAS is reduced by a maximum of 0.7 cm at 25% contribution. At 0, 10 and 25% norketamine contribution rifampicin treatment causes an increase in peak VAS of 0.3 cm (= 5% of peak VAS), 0.4 cm (10%) and 0.7 cm (21%), respectively (fig 6).
Figure 6.
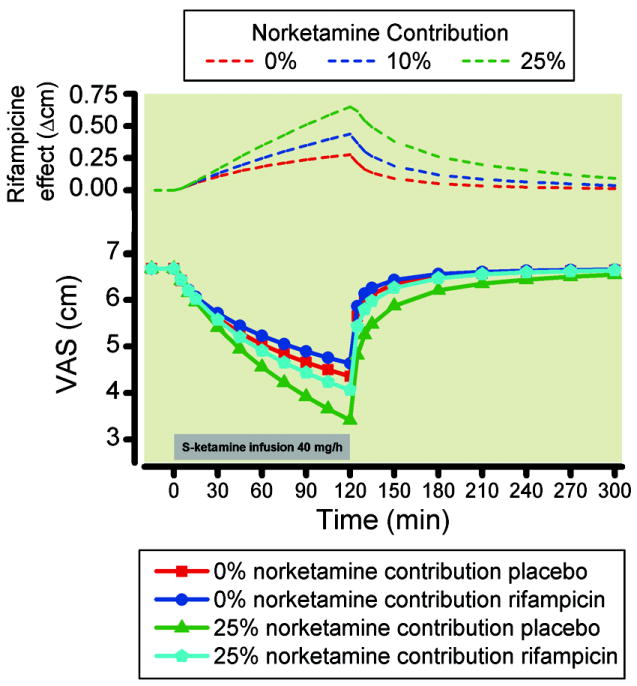
Simulation study for an acute pain paradigm. Simulation studies enable the systematic exploration of inferences made in our study with respect to anticipated norketamine effect size. We performed simulations to estimate the effect of the large change in norketamine concentration and relatively modest change in ketamine concentration that we observed after rifampicin treatment on acute pain relief induced by 2-h 40 mg/h ketamine infusion (grey bar). We made a priori assumptions with respect to the norketamine contributions to ketamine effect: Simulations with 0, 10 and 25% norketamine contribution were made. The difference in effect observed in the simulated pain relief with and without rifampicin treatment will give an indication of the norketamine contribution to effect. The top diagram shows the difference in VAS between placebo and rifampicin data for the three norketamine contribution to effect: 0, 10 and 25%. VAS = visual analogue score.
Chronic pain paradigm
The study was based on the application of the rifampicin effect to pharmacokinetic and pharmacodynamic data obtained in chronic pain patients. Hence, the effect of S-ketamine infusion mimics the effect observed in chronic pain patients (fig. 7).19 The effect of rifampicin was relatively small with maximum increases in VAS of 0.4 cm (0% norketamine contribution), 0.8 cm (10% contribution) and 1.3 cm (25% contribution, fig. 7). A biphasic effect of rifampicin on analgesia was observed with an initial peak during infusion (peak on day 2 of the infusion) and a second peak between days 20 and 30, irrespective of the magnitude of the norketamine contribution.
Figure 7.
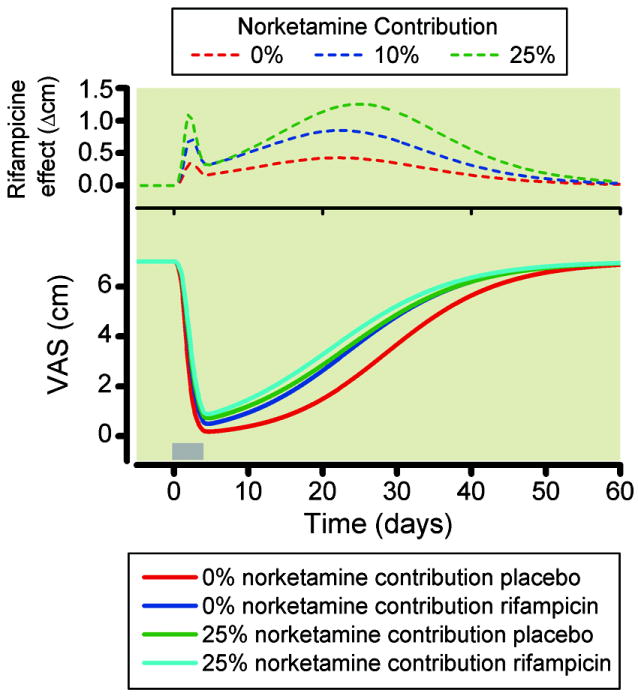
Simulation study for a chronic pain paradigm. We performed simulations to estimate the effect of the large change in norketamine concentration and relatively modest change in ketamine concentration that we observed after rifampicin treatment on chronic pain relief. Induced by a 4-day intravenous ketamine infusion (grey bar: day 1 = 5 mg/h, day 2 = 10 mg/h, day 3 = 15 mg/h and day 4 = 20 mg/h). Simulations with 0, 10, and 25% norketamine contribution were made. The difference in effect observed in the simulated pain relief with and without rifampicin treatment will give an estimate of the norketamine contribution to effect. The top diagram shows the difference in VAS between placebo and rifampicin data for the three norketamine contribution to effect: 0, 10, and 25%. VAS is visual analogue score.
Discussion
Rifampicin caused a 10% reduction in S-ketamine plasma concentrations coupled to a 50% reduction in plasma concentrations of ketamine’s metabolite S-norketamine. We relate these changes to the induction of S-ketamine and S-norketamine elimination. The compartmental pharmacokinetic analysis indicated a 13% increase in S-ketamine elimination, and a 200% increase in S-norketamine elimination.
The significant reduction in norketamine plasma concentrations in rifampin-treated subjects was an unexpected finding. Both norketamine CMAX and ratio AUCnorketamine/AUCketamine were diminished. Several explanations, not necessarily mutually exclusive, are possible. The first is a diminished rate of norketamine formation. This seems unlikely. CYP2B6 and CYP3A4 are the major human hepatic cytochrome P450s catalyzing ketamine N-demethylation in vitro, although their relative contributions in vivo remain unknown.19,20 At therapeutic substrate concentrations, both the rate and intrinsic clearance of N-demethylation to norketamine by CYP2B6 were found to be more than 10-fold greater than by CYP3A4 in two different studies.19,20 Based on this observation, using individual S(+) and R(-) ketamine enantiomers, Yanagihara et al.19 concluded that CYP2B6 mainly mediates ketamine N-demethylation. In contrast, Hijazi et al.20 concluded that CYP3A4 is the predominant CYP responsible for ketamine N-demethylation, which is based in part on the greater hepatic expression of CYP3A4 than CYP2B6. At clinical ketamine concentrations (< 5 μM) there is no evidence for other important CYPs involved. At supraclinical ketamine concentrations (50 μM) other CYP involvement has been described,28 although also then CYP2B6 and 3A4 are the enzymes with the highest activity. The clinical involvement of CYP3A4 was supported by the effect of the CYP3A4 inhibitor clarithromycin, which caused a 3.6-fold increase in S-ketamine CMAX while norketamine CMAX was reduced by 54%.29 Regardless of the relative contributions of CYPs 2B6 and 3A4 to ketamine N-demethylation, since rifampicin (600 mg daily for 5 days) induces the hepatic expression and catalytic activity of CYP2B6 and CYP3A4 several-fold, as well as the clinical metabolism of numerous CYP2B6 and CYP3A4 substrates,21 induction of ketamine metabolism by rifampicin is expected. Patients treated with chronic barbiturates (another inducer of hepatic CYP enzymes) have lower plasma ketamine concentrations.30 Together these findings do not suggest that rifampicin inhibits ketamine N-demethylation in vivo.
Another possible explanation for reduced norketamine concentrations is increased ketamine metabolism via an alternate pathway, and metabolic switching. Human liver microsomes form 4- and 5-hydroxyketamine.10 Although ketamine hydroxylation is considered minor compared with N-demethylation, the enzymes responsible for ketamine hydroxylation and the effect of rifampin induction are unknown.
A third potential explanation for rifampicin reduction of plasma norketamine concentrations is increased secondary metabolism of norketamine. The major secondary metabolite is 6-hydroxynorketamine, with lesser formation of 4- and 5- hydroxynorketamine. At clinical ketamine concentrations the enzymes responsible for norketamine hydroxylation and the effect of rifampin induction are unknown. A recent study performed at supra clinical concentrations (50 μM norketamine) indicates involvement of CYP2B6 and 2A6.28 Our compartmental analysis indicates that this possible mechanism is most likely. While norketamine formation may have increased by 13%, its elimination was increased by more than 200% (table 2). Assuming that also at clinical concentrations norketamine metabolism is predominantly regulated via CYP enzymes 2B6 and 2A6, it is highly probable that rifampicin caused induction of these enzymes and consequently increased the elimination of S-norketamine and caused the 50% lower S-norketamine plasma concentrations that we observed (figs. 3 and 4).
Finally, another potential explanation is an effect of hepatic and/or renal drug transporters on ketamine and/or norketamine disposition. In addition to induction of several CYP isoforms, rifampicin is an effective inducer of several drug transporters, including the efflux transporters P-glycoprotein, breast cancer resistance protein, and multidrug resistance proteins 1 and 2, and an inhibitor of the hepatic uptake transporter organic anion–transporting polypeptide.21,31 Inhibition of hepatic ketamine uptake or induction of efflux might reduce intracellular concentrations and hence the extent of N-demethylation, despite an accelerated rate. No information is available on ketamine or norketamine and hepatic drug transport.
Previously we modeled the contribution of norketamine to ketamine effect in a study on the effect of a ketamine infusion on acute antinociception using an experimental heat pain model.4 In that study we were unable to estimate a contribution of norketamine to ketamine antinociception, suggesting that, in humans, norketamine is not analgesic. However, since norketamine concentrations were not manipulated there was considerable uncertainty in the estimation of norketamine contribution to effect. In order to get an indication of the pharmacodynamic effect of the variations in S-norketamine concentration, we performed a simulation study with acute analgesia as end-point. Animal data show that S-norketamine is a noncompetitive NMDA receptor antagonist in the spinal cord and cortex.15 Its affinity to the NMDA receptor is weaker than that of S-ketamine, but there seems consensus that, at least in animals, norketamine does contribute significantly to the antinociceptive properties of ketamine (up to 30%), irrespective whether the S(+)-isomer or the racemic mixture is tested.14,15,17,18 Extrapolating the animal data to our simulations we assumed that S-norketamine contributed to overall analgesic effect in a range of 0 to 25%. Our simulation indicates that rifampicin caused a reduction in acute pain responses with a maximum in effect in VAS increase of less than 1 cm (fig. 6). Part of this effect is attributed to changes in S-ketamine concentration (7%) while the remainder is related to the changes in the concentration of S-norketamine (up to 16%). In actual practice, such an effect is small and may not be clinically detectable or important. This then suggests that the dose of ketamine for treatment of acute pain in patients on rifampicin needs little or no adjustment.
We infused S-ketamine for 2 h and observed a ratio AUCM/AUCP of about 1 after placebo treatment (table 1). Similar ratios are observed after long-term intravenous S-ketamine infusion in patients with chronic pain.6 Our simulation study revealed only a relatively small effect of variations in norketamine concentration on analgesia in chronic pain patients (maximum increase in VAS = 1.3 cm, fig. 7). However, a prolonged exposure to norketamine may cause a greater passage of the drug into the central nervous system than occurred during our short-term infusion and this may then increase its contribution to ketamine’s pharmacodynamics.18 We therefore may have underestimated the norketamine effect in our estimation of analgesia from S-ketamine in chronic pain patients. Also the route of administration may play a role in norketamine’s contribution to effect. For example, after oral and sublingual ketamine administration the concentration S-norketamine exceeds that of S-ketamine due to the first pass-effect in the liver and enterocyte.27 These higher concentrations may increase norketamine’s bioavailabilty to the brain compartment with a corresponding increase in contribution to ketamine’s analgesic effect. Further studies are needed to assess the effect of the induction of cytochrome P450 enzymes on norketamine’s contribution to ketamine-induced analgesia.
The two simulations studies that we performed used different values for effect-half life (no delay for acute pain and a t½k of 11 days for chronic pain relief). The absence of a delay for acute pain is derived from various studies on the acute effects of ketamine (measuring ketamine-induced electroencephalographic changes, arousal and recall during anesthesia and acute pain relief),4,5,32,33 and suggests a rapid passage of ketamine across the blood-brain-barrier. The equilibration half-life (t½k) of 11 days used in the chronic study is not a blood-effect-site equilibration constant but rather a disease modulatory factor. Various studies indicate that ketamine produces long-term analgesic effects beyond the treatment period with a half-life of many days.5-9 We recently estimate a value for t½k of about 11 days in patients with chronic pain from complex regional pain syndrome type 1.7 Interestingly, side effects in chronic pain patients treated with ketamine still show a rapid onset and offset suggesting that the drug rapidly crosses the blood-brain barrier and that the persistent analgesia observed is unrelated to the brain ketamine concentration.5,6 We recently argued that ketamine initiate a cascade of events (of which NMDA receptor antagonism is a first step) resulting in persistent analgesia.34
In conclusion, using rifampicin as inducer of specific cytochrome P450 enzymes, we observed that rifampicin induces the elimination of S-ketamine’s metabolite, S-norketamine, probably via induction of the CYP3A4, CYP2B6, and/or CYP2A6 enzymes.
Acknowledgments
Support: This study is supported in part by TREND (Trauma RElated Neuronal Dysfunction), a nonprofit consortium of academic hospitals, technical research groups and companies focused on the study of Complex Regional Pain Syndrome type 1 (The Hague, The Netherlands); also supported by National Institutes of Health grant K24DA00417 (Bethesda Maryland).
Footnotes
See www.trialregister.nl. Last accessed January 21, 2011.
Attribution: This work is attributed to Leiden University Medical Center.
Contributor Information
Ingeborg Noppers, Department of Anesthesiology, Leiden University Medical Center, Leiden, The Netherlands.
Erik Olofsen, Department of Anesthesiology, Leiden University Medical Center, Leiden, The Netherlands.
Marieke Niesters, Department of Anesthesiology, Leiden University Medical Center, Leiden, The Netherlands.
Leon Aarts, Department of Anesthesiology, Leiden University Medical Center, Leiden, The Netherlands.
René Mooren, Department of Anesthesiology, Leiden University Medical Center, Leiden, The Netherlands.
Albert Dahan, Department of Anesthesiology, Leiden University Medical Center, Leiden, The Netherlands.
Evan Kharasch, Department of Anesthesiology, Washington University in St. Louis, St. Louis, Missouri.
Elise Sarton, Department of Anesthesiology, Leiden University Medical Center, Leiden, The Netherlands.
References
- 1.Corssen G. Historiical aspects of ketamine – First clinical experience. In: Dominio EF, editor. Status of Ketamine in Anesthesiology. NPP Books; Ann Arbous: 1990. pp. 1–5. [Google Scholar]
- 2.Chizh BA. Low dose ketamine: A therapeutic and research tool to explore N-methyl-D-aspartate (NMDA) receptor-mediated plasticity in pain pathways. J Psychopharmacol. 2007;21:259–71. doi: 10.1177/0269881105062484. [DOI] [PubMed] [Google Scholar]
- 3.Petrenko AB, Yamakura T, Baba H, Shimoji K. The role of N-methyl-D-aspartate (NMDA) receptors in pain: A review. Anesth Analg. 2003;97:1108–16. doi: 10.1213/01.ANE.0000081061.12235.55. [DOI] [PubMed] [Google Scholar]
- 4.Sigtermans M, Dahan A, Mooren R, Bauer M, Kest B, Sarton E, Olofsen E. S(+)-ketamine effect on experimental pain and cardiac output: A population pharmacokinetic-pharmacodynamic modeling study in healthy volunteers. Anesthesiology. 2009;111:892–903. doi: 10.1097/ALN.0b013e3181b437b1. [DOI] [PubMed] [Google Scholar]
- 5.Sigtermans M, Noppers I, Sarton E, Bauer M, Mooren R, Olofsen E, Dahan A. An observational study on the effect of S(+)-ketamine on chronic pain versus experimental acute pain in Complex Regional Pain Syndrome type 1 patients. Eur J Pain. 2010;14:302–7. doi: 10.1016/j.ejpain.2009.05.012. [DOI] [PubMed] [Google Scholar]
- 6.Sigtermans M, van Hilten JJ, Bauer NCR, Arbus MS, Marinus J, Sarton EY, Dahan A. Ketamine produces effective and long-term pain relief in patients with Complex Regional Pain Syndrome Type 1. Pain. 2009;145:304–11. doi: 10.1016/j.pain.2009.06.023. [DOI] [PubMed] [Google Scholar]
- 7.Dahan A, Olofsen E, Sigtermans M, Noppers I, Niesters M, Aarts L, Bauer M, Sarton E. Population pharmacokinetic-pharmacodynamic modeling of ketamine-induced pain relief of chronic pain. Eur J Pain. 2010 doi: 10.1016/j.ejpain.2010.06.016. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 8.Amr YM. Multi-day low dose ketamine infusion as adjuvant to oral gabapentin in spinal cord injury related chronic pain: A prospective, randomized, double blind trial. Pain Physician. 2010;13:245–9. [PubMed] [Google Scholar]
- 9.Schwartzman RJ, Alexander GM, Grothusen JR, Paylor T, Reichenberger E, Perreault M. Outpatient intravenous ketamine for the treatment of complex regional pain syndrome: A double-blind placebo controlled study. Pain. 2009;147:107–15. doi: 10.1016/j.pain.2009.08.015. [DOI] [PubMed] [Google Scholar]
- 10.Adams JD, Baillie TA, Trevor AJ, Castagnoli N. Studies on the biotransformation of ketamine. 1. Identification of metabolites produced in vitro from rat liver microsomal preparations. Biomed Mass Spectrom. 1981;8:527–38. doi: 10.1002/bms.1200081103. [DOI] [PubMed] [Google Scholar]
- 11.Pedraz JL, Lanao JM, Dominguez-Gil A. Kinetics of ketamine and its metabolites in rabbits with normal and impaired renal function. Eur J Drug Metab Pharmacokinet. 1985;10:33–9. doi: 10.1007/BF03189695. [DOI] [PubMed] [Google Scholar]
- 12.Woolf TF, Adams JD. Biotransformation of ketamine, (Z)-6-hydroxyketamine, and (E)-6-hydroxyketamine by rat, rabbit, and human liver microsomal preparations. Xenobiotica. 1987;17:839–47. doi: 10.3109/00498258709043993. [DOI] [PubMed] [Google Scholar]
- 13.Turfus SC, Parkin MC, Cowan DA, Halket JM, Smith NW, Braithwaite RA, Elliot SP, Steventon GB, Kicman AT. Use of human microsomes and deuterated substrates: An alternative approach for the identification of novel metabolites of ketamine by mass spectrometry. Drug Metab Dispos. 2009;37:1769–78. doi: 10.1124/dmd.108.026328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hocking G, Visser EJ, Schug SA, Cousins MJ. Ketamine, does life begin at 40? Pain Clinical Updates. 2007;XV(3) [Google Scholar]
- 15.Ebert B, Mikkelsen S, Thorkildsen C, Borgbjerg FM. Norketamine, the main metabolite of ketamine, is a non-competitive NMDA receptor antagonist in the rat cortex and spinal cord. Eur J Pharmacol. 1997;333:99–104. doi: 10.1016/s0014-2999(97)01116-3. [DOI] [PubMed] [Google Scholar]
- 16.Holtman JR, Jr, Crooks PA, Johnson-Hardy JK, Hojomat M, Kleven M, Wala EP. Effects of norketamine enantiomers in rodent models of persistent pain. Pharmacol Biochem Behav. 2008;90:675–85. doi: 10.1016/j.pbb.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 17.Leung LY, Baillie TA. Comparative pharmacology in the rat of ketamine and its two principal metabolites, norketamine and (Z)-6-hydroxynorketamine. J Med Chem. 1986;29:2396–9. doi: 10.1021/jm00161a043. [DOI] [PubMed] [Google Scholar]
- 18.Shimoyama M, Shimoyama N, Gorman AL, Elliott KJ, Inturrisi CE. Oral ketamine is antinociceptive in the rat formalin test: Role of the metabolite, norketamine. Pain. 1999;81:85–93. doi: 10.1016/s0304-3959(98)00269-3. [DOI] [PubMed] [Google Scholar]
- 19.Yanagihara Y, Kariya S, Ohtani M, Uchino K, Aoyama T, Yamamura Y, Iga T. Involvement of CYP2B6 in N-demethylation of ketamine in human liver microsomes. Drug Metab Dis. 2001;29:887–90. [PubMed] [Google Scholar]
- 20.Hijazi Y, Boulieu R. Contribution of CYP3A4, CP2B6 and CYP2C9 isoforms to N-demethylation of ketamine in human liver microsomes. Drug Metab Dispos. 2002;30:853–8. doi: 10.1124/dmd.30.7.853. [DOI] [PubMed] [Google Scholar]
- 21.Niemi M, Backman JT, Fromm MF, Neuvonen PJ, Kivistö KT. Pharmacokinetic interactions with rifampicin. Clin Pharmacokinet. 2003;42:819–50. doi: 10.2165/00003088-200342090-00003. [DOI] [PubMed] [Google Scholar]
- 22.Pfenniger EG, Durieux ME, Himmelseher S. Cognitive impairment after small-dose ketamine isomers in comparison to equianalgesic racemic ketamine in human volunteers. Anesthesiology. 2002;96:357–66. doi: 10.1097/00000542-200202000-00022. [DOI] [PubMed] [Google Scholar]
- 23.Holford NHG. A size standard for pharmacokinetics. Clin Pharmacokin. 1996;30:329–32. doi: 10.2165/00003088-199630050-00001. [DOI] [PubMed] [Google Scholar]
- 24.Beal BL, Sheiner LB, Boeckman AJ, Bauer RJ. NONMEM User’s Guide. Icon development Solutions; Ellicott City, Maryland: 1989-2009. [Google Scholar]
- 25.Ihmsen H, Geisslinger G, Schüttler J. Stereoselective pharmacokinetics of ketamine: R(–)-ketamine inhibits the elimination of S(+)-ketamine. Clin Pharmacol Ther. 2001;70:431–8. doi: 10.1067/mcp.2001.119722. [DOI] [PubMed] [Google Scholar]
- 26.Persson J, Hasselström J, Maurset A, Öye I, Svensson J-O, Almqvist A, Scheinin H, Gustafsson LL, Almqvist O. Pharmacokinetics and non-analgesic effect of S- and R-ketamines in healthy volunteers with normal and reduced metabolic capacity. Eur J Clin Pharmacol. 2002;57:869–75. doi: 10.1007/s002280100353. [DOI] [PubMed] [Google Scholar]
- 27.Yanagihara Y, Ohtani M, Kariya S, Uchino K, Hiraishi T, Ashizawa N, Aoyama T, Yamamura Y, Yamada Y, Iga T. Plasma concentration profiles of ketamine and norketamine after administration of various ketamine preparations to healthy Japanese volunteers. Biopharm Drug Dis. 2003;24:37–43. doi: 10.1002/bdd.336. [DOI] [PubMed] [Google Scholar]
- 28.Portmann S, Kwan HY, Theurillat R, Schmitz A, Mevissen M, Thormann W. Enantioselective capillary electrophoresis for identification and characterization of human cytochrome P450 enzymes which metabolize ketamine and norketamine in vitro. J Chromatogr A. 2010;1217:7942–8. doi: 10.1016/j.chroma.2010.06.028. [DOI] [PubMed] [Google Scholar]
- 29.Hagelberg NM, Peltoniemi MA, Saari TI, Kurkinen KJ, Laine K, Neuvonen PJ, Olkkola KT. Clarithromycin, a potent inhibitor of CYP3A, greatly increases exposure to oral S-ketamine. Eur J Pain. 2009;14:625–9. doi: 10.1016/j.ejpain.2009.10.003. [DOI] [PubMed] [Google Scholar]
- 30.Koppel C, Arndt I, Ibe K. Effects of enzyme induction, renal and cardiac function on ketamine plasma kinetics in patients with ketamine long-term analgosedation. Eur J Drug Metab Pharmacokinet. 1990;15:259–63. doi: 10.1007/BF03190213. [DOI] [PubMed] [Google Scholar]
- 31.Vavricka SR, van Montfoort J, Ha HR, Meier PJ, Fattinger K. Interactions of rifamycin SV and rifampicin with organic anion uptake systems of human liver. Hepatology. 2002;36:164–72. doi: 10.1053/jhep.2002.34133. [DOI] [PubMed] [Google Scholar]
- 32.Schüttler J, Stanski DR, White PF, Trevor AJ, Horai Y, Verotta D, Sheiner LB. Pharmacodynamic modeling of the EEG effects of ketamine and its enantiomers in man. J Pharmacokinet Biopharm. 1987;15:241–53. doi: 10.1007/BF01066320. [DOI] [PubMed] [Google Scholar]
- 33.Herd DW, Anderson BJ, Keene NA, Holford NHG. Investigating the pharmacodynamics of ketamine in children. Ped Anesth. 2008;18:36–42. doi: 10.1111/j.1460-9592.2007.02384.x. [DOI] [PubMed] [Google Scholar]
- 34.Dahan A, Bauer M, Sarton E, Sigtermans M, van Hilten B, Marinus H. Response to drs. Kapural and Stanton-Hicks. Pain. 2010;149:410–1. [Google Scholar]