

Abstract
Several types of myeloid suppressor cell are currently being developed as cell-based immunosuppressive agents. Despite detailed knowledge about the molecular and cellular functions of these cell types, expert opinions differ on how to best implement such therapies in solid organ transplantation. Efforts in our laboratory to develop a cell-based medicinal product for promoting tolerance in renal transplant patients have focused on a type of suppressor macrophage, which we call the regulatory macrophage (M reg). Our favoured clinical strategy is to administer donor-derived M regs to recipients one week prior to transplantation. In contrast, many groups working with tolerogenic dendritic cells (DCs) advocate post-transplant administration of recipient-derived cells. A third alternative, using myeloid-derived suppressor cells, presumably demands that cells are given around the time of transplantation, so that they can infiltrate the graft to create a suppressive environment. On present evidence, it is not possible to say which cell type and treatment strategy might be clinically superior. This review seeks to position our basic scientific and early-stage clinical studies of human regulatory macrophages within the broader context of myeloid suppressor cell therapy in transplantation.
Keywords: Regulatory macrophage, M reg, Cell-based medicinal product, The ONE Study
Introduction
The existence of anti-inflammatory T cell-suppressive cells of the myeloid lineage has long been recognised and the ability of such cells to induce tolerance to auto- and allo-antigens after adoptive transfer has been studied extensively. Although often mooted, progress towards clinical applications of myeloid suppressor cell therapy was limited until recently, when several independent groups began trials in transplantation [1-3], rheumatoid arthritis [4] and diabetes [5]. Results from these early-phase clinical studies have been promising, at least in terms of demonstrating the feasibility and risk profile of such approaches, but evidence of efficacy in patients is still lacking. In this regard, the inception of The ONE Study, a clinical trial of cell therapy as an adjunct immunosuppressive treatment in renal transplantation, is a critical step forward [6].
Currently, the concept of promoting immunological regulation in transplant recipients by treating with myeloid suppressor cells of various types is being pursued by a number of groups [7-9]. Depending upon their exact nature and whether they are of donor, recipient or third-party origin, different myeloid suppressor cells exert their therapeutic effects through very different mechanisms [10]. In turn, the immunological actions of particular myeloid suppressor cells determine how those cells might be optimally delivered to patients, especially with respect to timing and route of administration, and immunosuppressive co-treatments. This review examines three radically alternative approaches to myeloid suppressor cell therapy in transplantation, each with its own clinical and immunological merits.
Broadly speaking, myeloid suppressor cells are characterised either by an arrested state of immaturity, when they are known as myeloid-derived suppressor cells (MDSCs) or tolerogenic DCs, or by a more mature phenotype, reflecting the ability of myeloid antigen-presenting cells to switch into a suppressive mode under certain conditions ( 1). Diverse anti-inflammatory treatments prevent DC maturation in vitro, including generation in the presence of IL-10 (DC-10) [11] or rapamycin (Rapa-DC) [12], culture in low concentrations of GM-CSF (Tol-DC)[13] or exposure to dexamethasone and vitamin D [4]. Paradoxically, various pro-inflammatory factors can also drive macrophages and DCs to a suppressive state, including IFN-γ, prostaglandin E2 (PGE2) and repetitive Toll-like Receptor (TLR) stimulation. As we describe below, the regulatory macrophage (M reg) is an important example of an activation-induced myeloid suppressor cell.
Figure 1.

The spectrum of monocyte-derived suppressor APCs. Suppressor macrophages and DCs can be generated from monocytes using M-CSF or GM-CSF, with or without IL-4. Development of immature DCs into mature, activating DCs can be blocked by various substances, including rapamycin or dexamethasone and vitamin D. DCs can also be rendered tolerogenic by culture in low-dose GM-CSF or by addition of suppressive cytokines, such as IL-10 or TGF-β1. Mesenchymal stem cells (MSC) can induce a suppressor phenotype in co-cultured macrophages. Myeloid-derived suppressor cells (MDSC) can be generated by exposing monocytes/macrophages to tumour-secreted factors, most notably PGE2.
It is not known whether the distinction between myeloid suppressor cells in a state of arrested immaturity and those in an activation-induced suppressor state is biologically meaningful. Certainly, many of the same molecular mechanisms account for the suppressive activities of both immature and activation-induced myeloid suppressor cells (Table 1). It is also unclear whether the various types of tolerogenic DCs and MDSCs described in the literature represent unique cell subtypes, or whether they are functionally interchangeable cells with only superficial phenotypic differences. A workshop recently convened by The ONE Study consortium in Regensburg, Germany, should provide some insight into these unresolved issues (see accompanying editorial [6]).
Table 1.
Main characteristics of different myeloid suppressor APCs
| Human phenotype | Function | Mechanism | Trafficking | Ref. | |
|---|---|---|---|---|---|
| M reg |
CD14-/low, CD16-, CD80-/low, CD40-/low, HLA-DR+, TLR2-, CD83-, CD163-/low |
T cell elimination, suppression of T cell proliferation |
IDO |
Lung, blood, liver, spleen |
[3,14-16] |
| Tol-DC |
HLA-DR+, low costimulation, CD14+, CD11b+, resistant to maturation |
Allo-Ag capture and presentation, suppression of T cell proliferation |
HO-1, EBl3, iNOS |
Spleen |
[8,17-20] |
| DC-10 |
CD14+, CD16+, CD83+, CD1a-, ILT4+, HLA-G+, IL-12- |
Induction T cell anergy, Tr-1 cell induction |
IL-10, ILT-4-HLA-G interactions |
Blood and secondary lymphoid organs |
[21] |
| Rapa-DC |
HLA-DR+, low costimulation, resistant to maturation, IL-12+ |
T cell hyporesponsiveness and apoptosis, T reg induction |
Low costimulation |
LN, spleen |
[22-26] |
| MDSC | HLA-DR-/low, CD11b++, CD14+, CD33+, CD34+ | Suppression of T cell proliferation, cytokine production, apoptosis in T cells | iNOS, Arg-1, ROS, PGE2, HO-1, TGF-β, cys depletion | Blood, graft, spleen, LN | [27-32] |
Regulatory macrophages
Human regulatory macrophages
Efforts in our laboratory to develop a cell-based medicinal product for use in promoting transplant tolerance in renal transplant patients have focussed on M regs. The human M reg reflects a unique state of macrophage differentiation, distinguished from macrophages in other activation states by its particular mode of derivation, robust phenotype and potent T cell suppressor function. These cells arise from CD14+ peripheral blood monocytes during a seven-day culture period during which the cells are exposed to M-CSF, 10% human serum and a final 24-hour pulse of IFN-γ [14]. M regs derived in this manner adopt a characteristic morphology and are homogeneously CD14-/low HLA-DR+ CD80-/low CD86+ CD16- CD64+ TLR2- TLR4- and CD163-/low. M regs do not stimulate allogeneic T cell proliferation in vitro and, when co-cultured with polyclonally stimulated T cells, are potently suppressive of proliferation. The suppressive capacity of M regs has been attributed to IFN-γ-induced indoleamine 2,3-dioxygenase (IDO) activity, as well as contact-dependent deletion of activated T cells [3]. Critically, human M regs are relatively resistant to maturation upon stimulation with lipopolysaccharide (LPS), possibly as a consequence of TLR down-regulation.
In order to assess their pattern of trafficking after central venous infusion, allogeneic M regs labelled with 111Indium-oxine were administered to a single patient, MM, whose case is described below [3]. Subsequently, the anatomical distribution of the M regs was tracked over 30 hours in serial whole-body Single Photon Emission Computed Tomography (SPECT) studies. Initially, M regs were only detected in the lungs, but within 2.5 hours were found circulating in blood. By 30 hours post-infusion, most M regs had emigrated from the lungs to the spleen, liver and haematopoietically-active bone marrow. M regs did not accumulate in lymph nodes. We can be confident that the majority of infused M regs survived for the duration of the follow-up, because tracer was not observed in the urinary tract or blood.
Mouse regulatory macrophages
Mouse CD11b+ Ly6C+ bone marrow monocytes cultured under conditions analogous to those used in the generation of human M regs give rise to a population of suppressive macrophages which are highly similar to human M regs in morphology, cell-surface phenotype and in vitro function [16]. Mouse M regs express a selection of typical macrophage markers, including CD11b, CD11c, CD68, F4/80 and CD14, and exhibit a partially-matured phenotype with intermediate levels of MHC Class II and CD80, and no expression of CD40 or CD86. Mouse M regs express sialoadhesin (CD169), macrophage scavenger receptor (CD204) and Dectin-1, but lack other markers of notable tissue macrophage subsets, such as Dectin-2, MARCO, CD4, CD206 and CD209. M regs do not express Ly6C or Ly6G, which together constitute the Gr-1 antigen that is expressed by all mouse MDSCs. CD11c is homogenously expressed by mouse M regs, but they do not express other DC subset-defining markers, including 33D1, OX40L (CD252), CD103, CD205 and CD207. Importantly, like human M regs, mouse M regs do not express TLR2 or TLR4. Given its mode of derivation, morphology and cell-surface phenotype, it seems most appropriate to classify the M reg as a macrophage; however, mouse M regs do not express markers typical of either M1-polarised macrophages (eg. TNF-α, IL-6 and IL-12b) or M2-polarised macrophages (eg. CD206, Ym1 or Fizz1). To better understand the phenotypic relationship between mouse M regs and previously-described macrophage polarisation states, we performed whole-genome gene expression studies to compare M regs to monocytes, monocyte-derived DCs, resting macrophages, IFN-γ-stimulated macrophages and M1-, M2a-, M2b- and M2c-polarised macrophages: These experiments showed that mouse M regs represent a novel and unique state of macrophage activation. Mouse M regs inhibit T cell responses in vitro by several mechanisms. Mitogen-stimulated T cell proliferation is non-specifically inhibited in co-cultures with M regs through the action of inducible nitric oxide synthase (iNOS). M regs delete co-cultured allogeneic T cells (but not isogeneic T cells) through an unknown mechanism that ultimately leads to phagocytosis of the T cells. Any T cells which survive direct co-culture with M regs are impaired in their ability to secrete IL-2 and IFN-γ upon specific and non-specific restimulation [16].
Preclinical experiments using a heterotopic mouse heart transplant model demonstrate the potential of mouse M regs to prolong allograft survival [16]. A single intravenous administration of 5×106 donor-strain M regs at 8 days prior to transplantation significantly prolongs allograft survival in unconditioned, non-immunosuppressed recipients using both the stringent C3H-to-BALB/c (32.6 ± 4.5 vs. 8.7 ± 0.2 days) and B6-to-BALB/c (31.1 ± 12 vs 9.7 ± 0.4 days) strain combinations. This graft-protective effect is specific to donor cells, as recipient cells do not prolong graft survival compared to untreated controls and third party-derived M regs provide only a marginal benefit (11.0 ± 0.6 days). Improved graft survival is observed irrespective of whether M regs are administered 8 or 35 days prior to transplantation. Co-treatment with M regs and 1 mg/kg/day rapamycin for 10 days post-transplant enhances the graft-protective effect of M regs (64.1 ± 8.6 days) compared to treatment with M regs alone or rapamycin alone, and some recipients co-treated with M regs and rapamycin accept their allografts indefinitely. The mechanism of M reg-mediated allograft protection is iNOS-dependent because M regs derived from Nos2-deficient mice only marginally prolong graft survival (12.0 ± 1.8 days). Very importantly, the iNOS-dependence of M reg treatment in vivo proves that the graft-protective effect of M regs is not simply due to alloantigen exposure, but must be mediated by living, metabolically-competent cells.
Several mutually redundant mechanisms might be invoked to explain the effects of M regs in vivo and, in our opinion, more than one mechanism is likely to be in operation (Figure 2). It is well-known that pre-transplant exposure to donor alloantigen promotes allograft acceptance [33] and that delivery of alloantigen as apototic cell debris enhances this effect [34,35]. Both CD8α+ DCs [36] and F4/80+ PD-L1+ IL-10-producing macrophages [37] of the splenic marginal zone appear to be important for the tolerogenic effects of complement-opsonised apoptotic antigens [38]. After intravenous injection into mice, isogeneic and allogeneic mouse M regs are initially trapped in the pulmonary vasculature, then rapidly redistribute to other peripheral organs, especially the liver and spleen, but not to lymph nodes [16]. Isogeneic and allogeneic M regs are relatively short-lived after transfer into immunocompetent mice, both being detectable at 2 weeks post-infusion, but not 4 weeks. These experiments suggest that M regs have an inherently limited lifespan after transfer; therefore, M regs probably serve as a source of apoptotic donor alloantigen-expressing material. Such a mechanism operates in tolerance induction protocols using donor-specific transfusion (DST) and αCD154 treatment, in which indirect presentation of donor alloantigen results in a predominantly deletional tolerance [39]. And yet, the graft-protective effect of donor alloantigen exposure in the absence of costimulatory blockade or lymphodepletive conditioning [40] is rarely as profound as that achieved with M reg treatment, even in less stringent transplant models. Moreover, it is difficult to reconcile the requirement for iNOS expression by transferred M regs with the suggestion that M regs act merely as a passive source of alloantigen. It is possible that mouse M regs directly suppress T cell responses in vivo through iNOS activity, as they do in vitro, but the critical action of iNOS might equally be mediated by recipient APCs [41,42]. Accordingly, one important fate of M regs might be to migrate into tissues, induce a suppressive condition in recipient APCs, before dying in a suitably self-conditioned environment.
Figure 2.

Proposed mode of M reg action. (1) When administered prior to transplantation, donor M regs migrate to spleen, where they present donor antigen through the direct pathway to alloreactive T cells and either delete or anergise them, or induce expansion of regulatory T cells. Human M regs have been shown to delete activated T cells through a contact-dependent mechanism and to suppress T cell proliferation through IDO; however, other suppressor mechanisms may also contribute to M reg function, such as IL-10 and TGF-β secretion, or iNOS activity. (2) It is likely that M regs serve as a source of donor alloantigen, which is captured and presented by immature recipient DCs to alloreactive T cells via the indirect and semi-direct pathways of alloantigen recognition. In consequence, responding T cells may be deleted or anergised, and antigen-specific T regs may be induced. (3) Through these mechanisms, the recipient T cell pool is enriched for T regs and depleted of donor-reactive T cells. (4) After transplantation, recipient T regs could induce tolerogenic DCs in secondary lymphoid organs. (5) Recipient tolerogenic DCs could then suppress activation of T cells. (6) In consequence, an immunological environment conducive to allograft acceptance is established.
MDSCs and tolerogenic DCs
Naturally-occuring myeloid suppressor cells fall into three ‘classes’: those present in non-inflamed tissues; those arising in inflamed tissues; and dedicated myeloid suppressor cell populations which are recruited to both inflamed and non-inflamed tissues. The ‘default’ condition of immature DCs and macrophages in non-inflamed tissues is suppressive; such cells play crucial roles in the maintenance of tissue homeostasis and self-tolerance, as well as resisting the otherwise perpetual activation of inflammatory responses [43]. Suppressive macrophages and DCs can also arise in inflamed tissues through conversion from activated, mature APCs; such cells limit the extent of inflammatory responses and promote tissue-reparative processes and the re-establishment of tissue homeostasis [44]. Committed myeloid suppressor cell populations, collectively known as MDSCs, are present in blood and can infiltrate both inflamed and non-inflamed tissues, including allografts and tumours [45]. These three general classes of naturally-occurring myeloid suppressor cell are reflected by the types of myeloid suppressor cell that can be generated in vitro.
Immature macrophages and DCs are normal stromal components of almost all peripheral tissues, where they serve many vital functions, including the removal of dead cells, microbial products and other tissue debris by phagocytosis, and the maintenance of a non-inflammatory environment through secretion of suppressive mediators. The immature state of tissue-resident macrophages and DCs is stabilised by anti-inflammatory factors present in non-inflamed tissues, such as IL-10 and glucocorticoid. Colonic macrophages exemplify the role of immature, tissue-resident APCs in preventing aberrant inflammatory responses; these cells respond to IL-10 by secreting IL-10, which suppresses the continual inflammation that would otherwise be caused by gut commensals and their products [46]. Besides their local effects in tissues, immature DCs deliver self-antigens to secondary lymphoid organs, where they are presented to T cells in a poor co-stimulatory context to propagate self-tolerance. Many in vitro-derived tolerogenic DCs fit this description of a phagocytic, maturation-resistant cell with the ability to present antigen in the absence of strong co-stimulation, including DC-10, Rapa-DC and Tol-DC [10]. A wide range of unrelated stimuli drive DCs to suppressive states, so there is no unique phenotype or mechanism of action associated with tolerogenic DC populations (Table 1). Generically, human tolerogenic DCs express cell-surface markers typical of immature monocyte-derived DCs, but there are markers of specific subtypes: Tol-DC are CD11c+ CD11b+ EBI3+ cells; DC-10 express high levels of HLA-G, ILT4 and secrete IL-10; Rapa-DC lack expression of CD80 and CD86, express only low-levels of HLA-DR, but secrete significant amounts of IL-12. These three tolerogenic DC subtypes exert their suppressive effects through alternative mechanisms, specifically: Tol-DC inhibit T cell responses through haemoxygenase (HO)-1 activity and T reg expansion [17]; DC-10 suppress T cell proliferation and induce antigen-specific Tr-1 cells through IL-10 production [21]; Rapa-DC induce effector T cell anergy and promote FoxP3+ T reg generation by presenting antigen in the absence of costimulation. Tolerogenic DC populations also differ in their migratory capacity in vivo. Studies in animals have shown that Rapa-DCs traffic to lymph nodes [25,26], but that Tol-DCs migrate preferentially to spleen [8,20].
In response to tissue injury or detection of pathogens, tissue-resident macrophages and DCs initiate a localised inflammatory response, involving the recruitment of other immunological effector cells, resulting in secondary tissue damage. However, this destructive behaviour is usually only transient, because activated macrophages and DCs switch to an anti-inflammatory and tissue-reparative mode [44]. The ability of macrophages to both exacerbate and attenuate inflammatory reactions is neatly illustrated by their contribution to ischaemia-reperfusion injury and its resolution: Experimental depletion of macrophages prior to renal ischaemia-reperfusion injury reduces the number of infiltrating leucocytes and helps to preserve short-term kidney function [47]; however, the cost of preventing early macrophage-mediated injury is a worse tissue repair response and impaired long-term function [48]. We regard human M regs, which are activated during cell culture by adherence to plastic, serum components and IFN-γ, as belonging to the class of activation-induced (or ‘deactivated’) myeloid suppressor cells.
MDSCs represent a third class of myeloid suppressor cell, which is characterised by the expression of markers associated with myeloid progenitors and commitment to a suppressor phenotype prior to entering tissues from blood. Although MDSC populations in mice and humans are highly heterogenous, any standard definition of mouse MDSCs includes expression of CD11b and Gr-1, whereas human MDSCs universally express CD11b, CD33, CD34 and VEGFR1 [45]. It is well-established that MDSCs exert a local immunosuppressive effect within solid tumours [49,50] and it appears that they have a similar function in transplanted organs, because induction of tolerance to kidney, skin and cardiac allografts is associated with infiltration of grafts by MDSCs [28,51].
Alternative clinical approaches
In principle, myeloid suppressor cells could favour allograft acceptance in several ways. Firstly, myeloid suppressor cells could exert transient, general immunosuppressive effects by secreting anti-inflammatory mediators or releasing of apoptotic debris. Secondly, myeloid suppressor cells could suppress inflammation and promote tissue repair processes within allografts during the immediate postoperative period. Thirdly, myeloid suppressor cells could anergize or delete recipient effector T cells, or induce alloantigen-specific regulatory T cells. Intuitively, the relative contribution of these allograft-protective mechanisms to transplant survival will depend on the route and timing of therapeutic cell administration, and the type of myeloid suppressor cell being used.
Pre-transplant versus post-transplant cell administration
Arguably, the state of the immune system prior to transplantation is more conducive to the establishment of tolerance than afterwards. Under steady-state, physiological conditions, immature DCs capture and present innocuous antigens leading to the anergy or deletion of antigen-reactive effector T cells and the expansion of specific T regs [9]. Organ transplantation disrupts this homeostatic condition by causing massive inflammation and the abrupt activation of vast numbers of alloreactive recipient T cells. Hence, the rationale for using myeloid suppressor cells prior to transplantation is that enrichment of alloantigen-specific T regs and deletion of effector T cells should be easier in an immunological environment naturally predisposed to the maintenance of tolerance. In addition, conventional immunosuppressive therapy might antagonise the regulatory action of myeloid suppressor cells given after transplantation.
Whilst exploiting the pro-tolerogenic condition of the pre-transplant immune system seems a sensible therapeutic strategy, a few arguments have been levelled against this approach. Firstly, and most importantly, in order to induce antigen-specific immunological regulation prior to transplantation, it is necessary to deliberately expose the recipient to donor alloantigen, which carries an attendant risk of allo-sensitisation. Secondly, patients with end-stage organ failure awaiting transplantation may not be in a immunologically quiescent state, either because of their underlying disease or concurrent subclinical infections. Thirdly, pre-treatment with donor-derived myeloid suppressor cells is not possible in the case of transplantation from deceased donors.
Myeloid suppressor cells of donor versus recipient origin
From an immunological standpoint, the principal reason for using myeloid suppressor cells of donor origin is to expose the recipient to intact donor alloantigen via the direct and semi-direct pathways. Alloantigen released by donor-derived cells could also be captured and presented in a pro-tolerogenic context by immature recipient DCs via the indirect pathway [52]. Consequently, donor-derived myeloid suppressor cells find their main application in pre-transplant conditioning therapies [3,53]. Donor-derived cells have the advantage that they can be reliably obtained from healthy, living donors. Recipient-derived myeloid suppressor cells are less likely to be eliminated by recipient T cells and NK cells, so have a greater capacity to migrate and engraft, and are less likely to sensitise the recipient against donor alloantigen. Production of recipient-derived myeloid suppressor cells prior to deceased-donor transplantation is feasible; however, since recipient-derived myeloid suppressor cells must capture and present graft-derived alloantigen in the indirect pathway, postoperative administration is the generally favoured approach of groups working with recipient-derived cells. Recipient-derived myeloid suppressor cells can be loaded with donor-alloantigen prior to infusion and this approach has proven to be a very effective therapy in animal models. Notably, antigen-pulsed tolerogenic DC were found to induce transplantation tolerance by expanding T regs which recognised alloantigen in the indirect pathway [25]. It has also been suggested that using third-party myeloid suppressor cells could eliminate some of the risks inherent to using donor or recipients cells, although it is difficult to envisage how third-party cells could induce allo-specific regulation, especially considering that the use of third-party tolerogenic DCs shows little or no effect in many animal models [20,25,54].
Route of administration
Selecting a route of administration for a tolerogenic cell therapy involves a trade-off between the most efficient means of delivering cells to their site of action and issues of clinical practicality and safety. Most animal experiments with myeloid suppressor cells have evaluated their therapeutic potential after intravenous administration [16,20,25], although some groups have investigated subcutaneous [55] and intramuscular injection [26]. The intravenous route seems quite suitable for M regs, since they normally traffic to liver, spleen and bone marrow. Tol-DCs and Rapa-DCs prolong allograft survival after intravenous injection; however, because recipient-derived tolerogenic DCs must capture graft antigens and suppress T cell activation in graft-draining lymph nodes, direct application of cells into lymph nodes may prove a superior route of administration. Since MDSCs exert important immunosuppressive actions within allografts, the possibility of injecting them directly into the arterial supply of the transplanted organ deserves further investigation.
Clinical applications of M reg therapy
As we have seen, when applying myeloid suppressor cell therapy in solid organ transplantation, the choice of myeloid suppressor cell type, whether it is given pre- or post-transplantation, whether it is of donor or recipient origin, and its route of administration are interdependent considerations (Table 2).
Table 2.
Clinical translation of myeloid suppressor cell therapy in solid organ transplantation
| Origin | Time | Pre-clinical | Clinical | Ref. | |
|---|---|---|---|---|---|
| M reg |
Donor |
Pre- |
Mouse, pig, dog |
TAIC- I, TAIC-II, The ONE Study |
[1-3,6,16,56-58] |
| Tol-DC |
Recipient |
Peri-/post- |
Rat, NHP |
The ONE Study |
[6,18,20,53,54] |
| DC-10 |
Recipient/Donor DC10 + recipient Tr-1 |
Peri-/post- |
Rat |
- |
[59] |
| Rapa-DC |
Donor-pulsed recipient |
Pre-Post |
Mouse |
- |
[25] |
| MDSC | Recipient | Peri-/Post- | Mouse | - | [60] |
On the present evidence, we cannot say which cell type or clinical approach represents an optimal therapy; however, based on our preclinical animal studies and the outcomes of the TAIC-I and TAIC-II clinical trials, our research group favours the preoperative administration of donor-derived M regs. A handful of clinical trials have been conducted (or are presently underway) using tolerogenic DCs in the treatment of Type I diabetes [5] and rheumatoid arthritis [4]; however, these cells have not yet been applied in solid organ transplantation. As we describe below, M reg-containing cell preparations have now been trialled in a total of 21 renal transplant recipients (Table 3).
Table 3.
Clinical studies with regulatory macrophages
| Study | n | Source | Time | Total cell number | Cells/kg body weight | Ref. |
|---|---|---|---|---|---|---|
| KW |
1 |
Donor spleen |
+5 |
|
1.1 × 106 |
[56] |
| TAIC-I |
12 |
Donor spleen |
+5 |
0.9-5 × 108 |
1-7.5 × 106 |
[1] |
| FR |
1 |
Donor blood |
−17 |
4.8 × 109 |
6.9 × 107 |
[57] |
| TAIC-II |
5 |
Donor blood |
−5 |
1.4-5.9 × 109 |
1.7-10.4 × 107 |
[2] |
| M reg: MM, CA | 2 | Donor blood | −6/7 | 4.3-7.5 × 108 | 7-8 × 106 | [3] |
The TAIC-I clinical trial
The TAIC-I trial was a single-centre, open-label, single-arm study with the objective of obtaining information on the safety and tolerability of administering M reg-containing cell preparations to renal transplant recipients (http://www.clinicaltrials.gov, NCT00223093) [1]. A total of 12 patients receiving their first transplant from a deceased donor were enrolled in the study. Initially immunosuppression comprised tacrolimus, sirolimus and corticosteroids. From week 4 posttransplantation, patients were aggressively weaned from immunosuppressive therapy with the intention of achieving tacrolimus monotherapy (8–10 ng/ml trough level) by week 12 and further reduction to ≤4 ng/ml within 24 weeks. Patients were treated with 0.9–5.0 × 108 donor-derived cells by central venous infusion at 5 days after transplantation (Figure 3). No acute complications or later adverse reactions relating to the cell infusion were observed. Thus, the TAIC-I trial demonstrated the clinical feasibility of producing and administering M reg-containing cell preparations to kidney transplant recipients.
Figure 3.

Overview of the TAIC-I trial. Patients enrolled in the TAIC-I Study each received a kidney transplant from a deceased donor. The mean age of the patients was 46.3 years and 9/12 patients were male. The median HLA-A,-B and –DR mismatch was 5/6. Initially, patients were treated with a combination of tacrolimus (trough levels of 10–15 ng/ml), sirolimus (trough levels of 4–8 ng/ml) and corticosteroids. Cells were infused on day 5 post-transplant. Steroids were tapered in weeks 5 and 6. Sirolimus was withdrawn in weeks 7 and 8. If graft function remained stable, tacrolimus treatment was first minimised to trough tacrolimus levels of 8–10 ng/ml by week 12 and then to levels of 5–8 ng/ml by week 24. Further reductions in tacrolimus therapy were undertaken in patients with stable graft function and no histological evidence of rejection. Figure reproduced with permission from Hutchinson, JA. et al. Transplant International (2008) 21:728–741.
The TAIC-II clinical trial
The TAIC-II study was a phase I/II clinical trial (http://www.clinicaltrials.gov, NCT00223067) designed to assess the safety and efficacy of administering donor-derived M reg-containing cell preparations to recipients of living-donor renal transplants [2]. Five days prior to surgery, five living-related kidney transplant recipients were treated with 1.4-5.9 × 108 cells (Figure 4). No clinical complications of the cell infusion were observed. All patients received induction therapy with anti-thymocyte globulin (ATG) on days 0, 1 and 2. From the time of transplantation onwards, patients received a dual immunosuppressive regime comprising conventional steroid treatment and tacrolimus therapy, aiming for trough levels of 8–12 ng/ml. Steroids were weaned by week 8 post-transplantation and tacrolimus was reduced to 5–8 ng/ml over several weeks. Four patients were successfully minimised to low-dose tacrolimus monotherapy. No rejection occurred in two of five patients. Following the reduction of tacrolimus treatment to <2 ng/ml for 6 weeks, one patient underwent a rejection episode at 36 weeks. The two remaining patients experienced acute rejection episodes only after complete cessation of immunosuppression for 2 and 34 weeks. All patients in TAIC-II were monitored for indices of graft acceptance and rejection through the RISET network. None of the patients developed anti-donor HLA antibodies as a consequence of M reg administration and no accelerated graft loss occurred. Anti-donor T cell reactivity was serially assayed by MLR and was found to be consistently reduced.
Figure 4.

Overview of the TAIC-II trial. Patients enrolled in the TAIC-II Study each received a kidney transplant from a living donor. The mean age of the patients was 35.4 years and 4/5 patients were male. The median HLA-A,-B and –DR mismatch was 3/6. Cells were infused 5 days prior to transplantation. All patients received ATG induction therapy on days 0, 1 and 2. Initial maintenance immunosuppression comprised glucocorticoids and tacrolimus (8–12 ng/ml trough levels). Steroid therapy was withdrawn by week 10. Tacrolimus dosing was then adjusted into a target range of 5 – 8 ng/ml trough levels. From week 24 onwards, further reductions were made in tacrolimus monotherapy, leading to complete drug withdrawal in two patients.
Patients MM and CA
Since the TAIC-I and TAIC-II clinical trials, we have arrived at a detailed understanding of the derivation, phenotype and T cell-suppressive functions of in vitro-derived human regulatory macrophages. This knowledge has inspired methodological advances in regulatory macrophage manufacture, leading to a far purer and more homogeneous cell product, which has now been applied to two further living-donor renal transplant recipients with encouraging results [3].
The first of these patients, MM, a 23 year-old female with renal failure owing to IgA nephropathy, received a living-donor kidney transplant from her 58 year-old mother. Mother and daughter had only single HLA-B and- DR mismatches (Figure 5). Six days prior to transplantation, patient MM received 8.0 × 106 donor-derived M regs/kg by slow central venous infusion under cover of 2 mg/kg/d azathioprine. Conventional treatment with steroids and tacrolimus was started at the time of transplantation. Azathioprine was stopped at 8 weeks post-transplant and steroids were weaned by 14 weeks. Thereafter, MM was maintained on tacrolimus monotherapy with trough levels of less than 6 ng/ml. Protocol biopsies at 8 and 24 weeks showed no signs of rejection. At 3 years, MM was in a stable clinical condition, receiving tacrolimus 2 mg BD with trough levels of 4–5 ng/ml as her sole maintenance immunosuppression.
Figure 5.
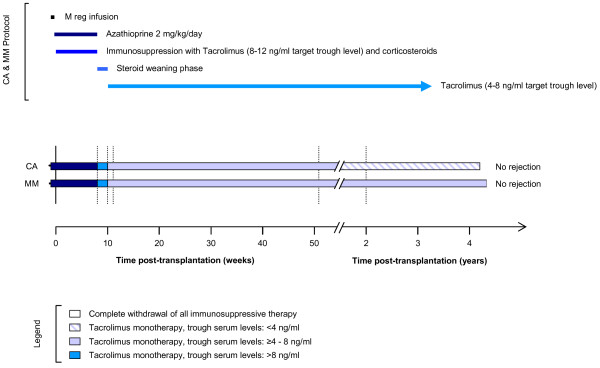
Overview of the treatment of patients MM and CA. Both patients received a living-donor kidney transplant. M regs were infused 6 (MM) or 7 (CA) days prior to transplantation under cover of 2 mg/kg/day azathioprine. Initial maintenance immunosuppression comprised glucocorticoids and tacrolimus (>8 ng/ml trough levels). Steroid therapy was withdrawn by week 10. Tacrolimus dosing was then adjusted into a target range of 4 – 8 ng/ml trough levels.
The second patient, CA, a 47 year-old man, received a fully-mismatched kidney from a 40 year-old living, unrelated male donor. CA was treated with 7.1 × 106 donor-derived M regs/kg seven days prior to transplantation under cover of 2 mg/kg/d azathioprine. Treatment with tacrolimus and steroids began the time of transplantation. Protocol biopsies at 8, 24 and 52 weeks showed no signs of rejection. At 3 years post-transplantation, CA had stable renal function and was being maintained with sustained-release tacrolimus 5 mg OD with a trough tacrolimus level of 2.7 ng/ml.
Minimising the maintenance immunosuppression of renal transplant recipients to tacrolimus monotherapy is not recommended by the Kidney Disease Improving Global Outcomes (KDIGO) guidelines, which advise the combination of a calcineurin inhibitor and an antiproliferative agent, with or without corticosteroids [61]. Nevertheless, in practice, patients receiving tacrolimus monotherapy are not uncommon and use of Alemtuzumab induction as a means of establishing patients on maintenance tacrolimus monotherapy has stimulated much recent interest. So, should we be surprised by the clinical outcomes of MM and CA? Although, we must be cautious in our interpretation of these two case studies, there are intriguing aspects of MM and CA’s histories that deserve particular mention: Both patients were minimised to low-dose tacrolimus monotherapy; this reduction in tacrolimus dosing was made relatively early after transplantation; neither MM nor CA received conventional induction therapy; and, both MM and CA developed a peripheral blood biomarker profile converging upon the IOT-RISET signature of tolerance [62].
What constitutes low-dose tacrolimus therapy? Definitions of low-dose and standard-dose tacrolimus treatment are largely a matter of convention. The Symphony Study, which assessed whether a mycophenolate mofetil (MMF)-based regimen allows minimisation of adjunct immunosuppression, incorporated a low-dose tacrolimus arm in which 75% of patients had trough tacrolimus levels of 4.3-10.0 ng/mL [63]. By this standard, MM and CA were treated with very low-dose tacrolimus, since both patients registered drug levels in the lowest 12.5 percentile of this range [64]. Unlike subjects in the Symphony Study, patients MM and CA were not given Dacluzimab induction or maintenance MMF therapy. Viewed in this context, the fact that both MM and CA remain rejection-free and with stable graft function at >4 years post-transplantation is an encouraging outcome.
Is low-dose tacrolimus monotherapy difficult to achieve in renal transplant recipients? Shapiro’s 2003 study remains a benchmark trial of minimised tacrolimus monotherapy in renal transplant recipients [65]. 150 patients were treated with 5 mg/kg ATG and bolus prednisone as an induction therapy, and were subsequently maintained tacrolimus monotherapy, which was minimised in a step-wise fashion over many months (Figure 6). Under this regimen, 37% of patients underwent acute rejection prior to minimisation of tacrolimus dosing. 113 patients were then selected to undergo tacrolimus weaning: These patients were followed-up for a mean of 11 ± 5.4 months, during which time 23% of patients underwent acute rejection. Other studies with the aim of establishing renal transplant patients on tacrolimus monotherapy after ATG induction achieved similar outcomes [66]. More recently, Alemtuzumab induction with tacrolimus monotherapy has been used with some success [67]. Margreiter et al. reported a 20% 1-year biopsy-proven acute rejection rate in patients undergoing Alemtuzumab induction, followed by tacrolimus monotherapy with trough drug levels of 8–12 ng/ml for 6 months, reduced to 5–8 ng/ml thereafter [68]. More impressively, Chan and colleagues observed an 89.9% 2-year rejection-free renal allograft survival rate using Alemtuzumab and tacrolimus monotherapy with a target-range of 5–8 ng/ml [69]. Clearly, tacrolimus monotherapy can be achieved in renal transplant patients treated with a powerful induction agent, such as Alemtuzumab; nevertheless, it is still surprising that patients MM and CA, who were not treated with T cell-depleting monoclonal antibodies, were able to tolerate an early, fairly abrupt minimisation of immunosuppression.
Figure 6.
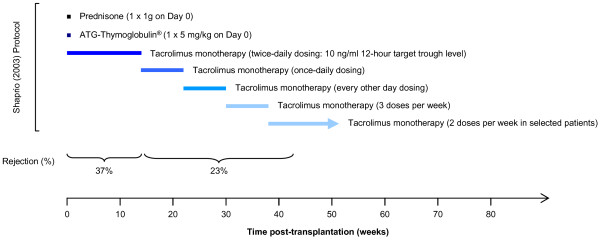
Summary of a tacrolimus-minimisation study conducted by Shapiro et al.[[65]]. This clinical trial remains a benchmark study of minimised tacrolimus monotherapy in renal transplant recipients. 150 patients were treated with ATG induction therapy and bolus prednisone before being maintained on tacrolimus monotherapy. Over several months, tacrolimus was weaned in a stepwise fashion from 113 patients. The acute rejection rate prior to weaning was 37% and the acute rejection rate during weaning was 23%.
A pattern of peripheral blood gene expression associated with a drug-free, tolerant state in renal transplant recipients has been defined by the IOT-RISET consortium [62]. To assess the immunological consequences of M reg therapy on patients MM and CA, expression of the ten most discriminatory gene markers of tolerance identified by the IOT-RISET group was quantified in serial blood samples taken from both patients [3]. Over the first year post-transplant, the pattern of expression of these markers gradually converged upon the IOT-RISET gene signature, suggesting that MM and CA might have achieved a degree of immunological regulation against their donors. To put this result into context, we have to consider the frequency of renal transplant recipients on CNI monotherapy whose gene expression profiles converge on the IOT-RISET signature: Of the 30 patients on CNI monotherapy included in the IOT-RISET cohort, all of whom were 6 to 9 years post-transplantation, only 5 displayed such a gene expression profile. Because the IOT-RISET study purposefully selected long-term, clinically stable patients, it is likely that 16% is an over-estimation of the frequency of CNI monotherapy patients in the general transplant population with the tolerance signature.
Conclusions
Studies in animal models have shown the potential of immature DCs, MDSCs and regulatory macrophages to suppress T cell responses against alloantigen and to prolong allograft survival. This review has presented three very different strategies for myeloid cell therapy in solid organ transplantation. The particular cellular and molecular functions of different myeloid suppressor APCs might make each cell type more suitable for different clinical applications: Donor-derived M regs are to be used pre-operatively to induce donor-specific regulation, whereas recipient-derived suppressor cells and MDSCs have to be used peri- or post-operatively. Translation of myeloid suppressor APC therapy to the clinic is already underway. Several patients have now been treated with M regs in early-phase clinical trials and tolerogenic DCs are currently being tested in rheumatoid arthritis and diabetes studies. The ONE Study consortium is undertaking a clinical trial which should allow a side-by-side comparison of M regs and Tol-DC as adjunct immunosuppressive therapies in renal transplantation. The results of this study are awaited with great excitement.
Abbreviations
Ag: Antigen; APC: Antigen presenting cell; CNI: Calcineurin inhibitor; IL: Interleukin; M-CSF: Monocyte colony stimulating factor; GM-CSF: Granulocyte monocyte colony stimulating factor; LN: lymph node; T reg: Regulatory T cell; MLR: Mixed lymphocyte reaction; NHP: Non-human primate.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
PR wrote the manuscript with assistance from EKG and JAH. All authors read and approved the final manuscript.
Contributor Information
Paloma Riquelme, Email: paloma.riquelme@ukr.de.
Edward K Geissler, Email: edward.geissler@ukr.de.
James A Hutchinson, Email: james.hutchinson@ukr.de.
Acknowledgements
The authors wish to thank Mr. Ben James for proof-reading this article.
References
- Hutchinson JA, Riquelme P, Brem-Exner BG, Schulze M, Matthai M, Renders L, Kunzendorf U, Geissler EK, Fandrich F. Transplant acceptance-inducing cells as an immune-conditioning therapy in renal transplantation. Transpl Int. 2008;21:728–741. doi: 10.1111/j.1432-2277.2008.00680.x. [DOI] [PubMed] [Google Scholar]
- Hutchinson JA, Brem-Exner BG, Riquelme P, Roelen D, Schulze M, Ivens K, Grabensee B, Witzke O, Philipp T, Renders L. et al. A cell-based approach to the minimization of immunosuppression in renal transplantation. Transpl Int. 2008;21:742–754. doi: 10.1111/j.1432-2277.2008.00692.x. [DOI] [PubMed] [Google Scholar]
- Hutchinson JA, Riquelme P, Sawitzki B, Tomiuk S, Miqueu P, Zuhayra M, Oberg HH, Pascher A, Lutzen U, Janssen U. et al. Cutting Edge: immunological consequences and trafficking of human regulatory macrophages administered to renal transplant recipients. J Immunol. 2011;187:2072–2078. doi: 10.4049/jimmunol.1100762. [DOI] [PubMed] [Google Scholar]
- Harry RA, Anderson AE, Isaacs JD, Hilkens CM. Generation and characterisation of therapeutic tolerogenic dendritic cells for rheumatoid arthritis. Ann Rheum Dis. 2010;69:2042–2050. doi: 10.1136/ard.2009.126383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannoukakis N, Phillips B, Finegold D, Harnaha J, Trucco M. Phase I (safety) study of autologous tolerogenic dendritic cells in type 1 diabetic patients. Diabetes Care. 2011;34:2026–2032. doi: 10.2337/dc11-0472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler EK. The ONE Study compares cell therapy products in organ transplantation: introduction to a review series on suppressive monocyte-derived cells. Transplantation Research. 2012;1:11. doi: 10.1186/2047-1440-1-11. doi:10.1186/2047-1440-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson JA, Riquelme P, Geissler EK. Human regulatory macrophages as a cell-based medicinal product. Curr Opin Organ Transplant. 2012;17:48–54. doi: 10.1097/MOT.0b013e32834ee64a. [DOI] [PubMed] [Google Scholar]
- Beriou G, Moreau A, Cuturi MC. Tolerogenic dendritic cells: applications for solid organ transplantation. Curr Opin Organ Transplant. 2012;17:42–47. doi: 10.1097/MOT.0b013e32834ee662. [DOI] [PubMed] [Google Scholar]
- Thomson AW. Tolerogenic dendritic cells: all present and correct? Am J Transplant. 2010;10:214–219. doi: 10.1111/j.1600-6143.2009.02955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli AE, Thomson AW. Tolerogenic dendritic cells and the quest for transplant tolerance. Nat Rev Immunol. 2007;7:610–621. doi: 10.1038/nri2132. [DOI] [PubMed] [Google Scholar]
- Amodio G. Human tolerogenic DC-10: perspectives for clinical applications. Transplantation Research. 2012;1:14. doi: 10.1186/2047-1440-1-14. doi:10.1186/2047-1440-1-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macedo C. Immunoregulatory properties of rapamycin-conditioned monocyte-derived dendritic cells and their role in transplantation. Transplantation Research. 2012;1:16. doi: 10.1186/2047-1440-1-16. doi:10.1186/2047-1440-1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau A. Cell therapy using tolerogenic dendritic cells in transplantation. Transplantation Research. p. 13. in press. [DOI] [PMC free article] [PubMed]
- Hutchinson JA, Riquelme P, Geissler EK, Fandrich F. Human regulatory macrophages. Methods Mol Biol. 2011;677:181–192. doi: 10.1007/978-1-60761-869-0_13. [DOI] [PubMed] [Google Scholar]
- Riquelme P, Govert F, Geissler EK, Fandrich F, Hutchinson JA. Human transplant acceptance-inducing cells suppress mitogen-stimulated T cell proliferation. Transpl Immunol. 2009;21:162–165. doi: 10.1016/j.trim.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Riquelme P, Tomiuk S, Kammler A, Fandrich F, Schlitt HJ. Geissler EK. Hutchinson JA: IFN-gamma-induced iNOS Expression in Mouse Regulatory Macrophages Prolongs Allograft Survival in Fully Immunocompetent Recipients. Mol Ther; 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauveau C, Remy S, Royer PJ, Hill M, Tanguy-Royer S, Hubert FX, Tesson L, Brion R, Beriou G, Gregoire M. et al. Heme oxygenase-1 expression inhibits dendritic cell maturation and proinflammatory function but conserves IL-10 expression. Blood. 2005;106:1694–1702. doi: 10.1182/blood-2005-02-0494. [DOI] [PubMed] [Google Scholar]
- Moreau A, Hill M, Thebault P, Deschamps JY, Chiffoleau E, Chauveau C, Moullier P, Anegon I, liot-Licht B, Cuturi MC. Tolerogenic dendritic cells actively inhibit T cells through heme oxygenase-1 in rodents and in nonhuman primates. FASEB J. 2009;23:3070–3077. doi: 10.1096/fj.08-128173. [DOI] [PubMed] [Google Scholar]
- Hill M, Thebault P, Segovia M, Louvet C, Beriou G, Tilly G, Merieau E, Anegon I, Chiffoleau E, Cuturi MC. Cell therapy with autologous tolerogenic dendritic cells induces allograft tolerance through interferon-gamma and epstein-barr virus-induced gene 3. Am J Transplant. 2011;11:2036–2045. doi: 10.1111/j.1600-6143.2011.03651.x. [DOI] [PubMed] [Google Scholar]
- Peche H, Trinite B, Martinet B, Cuturi MC. Prolongation of heart allograft survival by immature dendritic cells generated from recipient type bone marrow progenitors. Am J Transplant. 2005;5:255–267. doi: 10.1111/j.1600-6143.2004.00683.x. [DOI] [PubMed] [Google Scholar]
- Gregori S, Tomasoni D, Pacciani V, Scirpoli M, Battaglia M, Magnani CF, Hauben E, Roncarolo MG. Differentiation of type 1T regulatory cells (Tr1) by tolerogenic DC-10 requires the IL-10-dependent ILT4/HLA-G pathway. Blood. 2010;116:935–944. doi: 10.1182/blood-2009-07-234872. [DOI] [PubMed] [Google Scholar]
- Turnquist HR, Cardinal J, Macedo C, Rosborough BR, Sumpter TL, Geller DA, Metes D, Thomson AW. mTOR and GSK-3 shape the CD4+ T-cell stimulatory and differentiation capacity of myeloid DCs after exposure to LPS. Blood. 2010;115:4758–4769. doi: 10.1182/blood-2009-10-251488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naranjo-Gomez M, Raich-Regue D, Onate C, Grau-Lopez L, Ramo-Tello C, Pujol-Borrell R, Martinez-Caceres E, Borras FE. Comparative study of clinical grade human tolerogenic dendritic cells. J Transl Med. 2011;9:89. doi: 10.1186/1479-5876-9-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnquist HR, Fischer RT, Thomson AW. Pharmacological modification of dendritic cells to promote their tolerogenicity in transplantation. Methods Mol Biol. 2010;595:135–148. doi: 10.1007/978-1-60761-421-0_8. [DOI] [PubMed] [Google Scholar]
- Taner T, Hackstein H, Wang Z, Morelli AE, Thomson AW. Rapamycin-treated, alloantigen-pulsed host dendritic cells induce ag-specific T cell regulation and prolong graft survival. Am J Transplant. 2005;5:228–236. doi: 10.1046/j.1600-6143.2004.00673.x. [DOI] [PubMed] [Google Scholar]
- Reichardt W, Durr C, Von ED, Juttner E, Gerlach UV, Yamada M, Smith B, Negrin RS, Zeiser R. Impact of mammalian target of rapamycin inhibition on lymphoid homing and tolerogenic function of nanoparticle-labeled dendritic cells following allogeneic hematopoietic cell transplantation. J Immunol. 2008;181:4770–4779. doi: 10.4049/jimmunol.181.7.4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten TF, Manns MP, Korangy F. Myeloid derived suppressor cells in human diseases. Int Immunopharmacol. 2011;11:802–807. doi: 10.1016/j.intimp.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugast AS, Haudebourg T, Coulon F, Heslan M, Haspot F, Poirier N, de Vuillefroy SR, Usal C, Smit H, Martinet B. et al. Myeloid-derived suppressor cells accumulate in kidney allograft tolerance and specifically suppress effector T cell expansion. J Immunol. 2008;180:7898–7906. doi: 10.4049/jimmunol.180.12.7898. [DOI] [PubMed] [Google Scholar]
- De Wilde V, Van RN, Hill M, Lebrun JF, Lemaitre P, Lhomme F, Kubjak C, Vokaer B, Oldenhove G, Charbonnier LM. et al. Endotoxin-induced myeloid-derived suppressor cells inhibit alloimmune responses via heme oxygenase-1. Am J Transplant. 2009;9:2034–2047. doi: 10.1111/j.1600-6143.2009.02757.x. [DOI] [PubMed] [Google Scholar]
- Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood. 2011;118:5498–5505. doi: 10.1182/blood-2011-07-365825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Pang Y, Gara SK, Achyut BR, Heger C, Goldsmith PK, Lonning S, Yang L. Gr-1+CD11b+cells are responsible for tumor promoting effect of TGF-beta in breast cancer progression. Int J Cancer. 2012;131:2584–2595. doi: 10.1002/ijc.27572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010;70:68–77. doi: 10.1158/0008-5472.CAN-09-2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushell A, Karim M, Kingsley CI, Wood KJ. Pretransplant blood transfusion without additional immunotherapy generates CD25 + CD4+ regulatory T cells: a potential explanation for the blood-transfusion effect. Transplantation. 2003;76:449–455. doi: 10.1097/01.TP.0000083043.84630.99. [DOI] [PubMed] [Google Scholar]
- Morelli AE, Larregina AT. Apoptotic cell-based therapies against transplant rejection: role of recipient’s dendritic cells. Apoptosis. 2010;15:1083–1097. doi: 10.1007/s10495-010-0469-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman RM, Turley S, Mellman I, Inaba K. The induction of tolerance by dendritic cells that have captured apoptotic cells. J Exp Med. 2000;191:411–416. doi: 10.1084/jem.191.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu CH, Miyake Y, Kaise H, Kitamura H, Ohara O, Tanaka M. Novel subset of CD8{alpha} + dendritic cells localized in the marginal zone is responsible for tolerance to cell-associated antigens. J Immunol. 2009;182:4127–4136. doi: 10.4049/jimmunol.0803364. [DOI] [PubMed] [Google Scholar]
- Getts DR, Turley DM, Smith CE, Harp CT, McCarthy D, Feeney EM, Getts MT, Martin AJ, Luo X, Terry RL. et al. Tolerance induced by apoptotic antigen-coupled leukocytes is induced by PD-L1+ and IL-10-producing splenic macrophages and maintained by T regulatory cells. J Immunol. 2011;187:2405–2417. doi: 10.4049/jimmunol.1004175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli AE, Larregina AT, Shufesky WJ, Zahorchak AF, Logar AJ, Papworth GD, Wang Z, Watkins SC, Falo LD Jr, Thomson AW. Internalization of circulating apoptotic cells by splenic marginal zone dendritic cells: dependence on complement receptors and effect on cytokine production. Blood. 2003;101:611–620. doi: 10.1182/blood-2002-06-1769. [DOI] [PubMed] [Google Scholar]
- Quezada SA, Fuller B, Jarvinen LZ, Gonzalez M, Blazar BR, Rudensky AY, Strom TB, Noelle RJ. Mechanisms of donor-specific transfusion tolerance: preemptive induction of clonal T-cell exhaustion via indirect presentation. Blood. 2003;102:1920–1926. doi: 10.1182/blood-2003-02-0586. [DOI] [PubMed] [Google Scholar]
- Pearson TC, Madsen JC, Larsen CP, Morris PJ, Wood KJ. Induction of transplantation tolerance in adults using donor antigen and anti-CD4 monoclonal antibody. Transplantation. 1992;54:475–483. doi: 10.1097/00007890-199209000-00018. [DOI] [PubMed] [Google Scholar]
- Ren G, Su J, Zhao X, Zhang L, Zhang J, Roberts AI, Zhang H, Das G, Shi Y. Apoptotic cells induce immunosuppression through dendritic cells: critical roles of IFN-gamma and nitric oxide. J Immunol. 2008;181:3277–3284. doi: 10.4049/jimmunol.181.5.3277. [DOI] [PubMed] [Google Scholar]
- Lu L, Bonham CA, Chambers FG, Watkins SC, Hoffman RA, Simmons RL, Thomson AW. Induction of nitric oxide synthase in mouse dendritic cells by IFN-gamma, endotoxin, and interaction with allogeneic T cells: nitric oxide production is associated with dendritic cell apoptosis. J Immunol. 1996;157:3577–3586. [PubMed] [Google Scholar]
- Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broichhausen C, Riquelme P, Geissler EK, Hutchinson JA. Regulatory macrophages as therapeutic targets and therapeutic agents in solid organ transplantation. Curr Opin Organ Transplant. 2012;17:332–342. doi: 10.1097/MOT.0b013e328355a979. [DOI] [PubMed] [Google Scholar]
- Boros P, Ochando JC, Chen SH, Bromberg JS. Myeloid-derived suppressor cells: natural regulators for transplant tolerance. Hum Immunol. 2010;71:1061–1066. doi: 10.1016/j.humimm.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes MJ, Powrie F. Regulatory T cells reinforce intestinal homeostasis. Immunity. 2009;31:401–411. doi: 10.1016/j.immuni.2009.08.011. [DOI] [PubMed] [Google Scholar]
- Day YJ, Huang L, Ye H, Linden J, Okusa MD. Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: role of macrophages. Am J Physiol Renal Physiol. 2005;288:F722–F731. doi: 10.1152/ajprenal.00378.2004. [DOI] [PubMed] [Google Scholar]
- Jang HS, Kim J, Park YK, Park KM. Infiltrated macrophages contribute to recovery after ischemic injury but not to ischemic preconditioning in kidneys. Transplantation. 2008;85:447–455. doi: 10.1097/TP.0b013e318160f0d1. [DOI] [PubMed] [Google Scholar]
- Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–174. doi: 10.1038/nri2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermajer N. Generation of myeloid-derived suppressor cells using prostaglandin E2. Transplantation Research. 2012. in press. [DOI] [PMC free article] [PubMed]
- Garcia MR, Ledgerwood L, Yang Y, Xu J, Lal G, Burrell B, Ma G, Hashimoto D, Li Y, Boros P. et al. Monocytic suppressive cells mediate cardiovascular transplantation tolerance in mice. J Clin Invest. 2010;120:2486–2496. doi: 10.1172/JCI41628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divito SJ, Wang Z, Shufesky WJ, Liu Q, Tkacheva OA, Montecalvo A, Erdos G, Larregina AT, Morelli AE. Endogenous dendritic cells mediate the effects of intravenously injected therapeutic immunosuppressive dendritic cells in transplantation. Blood. 2010;116:2694–2705. doi: 10.1182/blood-2009-10-251058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan YY, Wang Z, Raimondi G, Wu W, Colvin BL, De CA, Thomson AW. “Alternatively activated” dendritic cells preferentially secrete IL-10, expand Foxp3 + CD4+ T cells, and induce long-term organ allograft survival in combination with CTLA4-Ig. J Immunol. 2006;177:5868–5877. doi: 10.4049/jimmunol.177.9.5868. [DOI] [PubMed] [Google Scholar]
- Beriou G, Peche H, Guillonneau C, Merieau E, Cuturi MC. Donor-specific allograft tolerance by administration of recipient-derived immature dendritic cells and suboptimal immunosuppression. Transplantation. 2005;79:969–972. doi: 10.1097/01.TP.0000158277.50073.35. [DOI] [PubMed] [Google Scholar]
- Dhodapkar MV, Steinman RM, Krasovsky J, Munz C, Bhardwaj N. Antigen-specific inhibition of effector T cell function in humans after injection of immature dendritic cells. J Exp Med. 2001;193:233–238. doi: 10.1084/jem.193.2.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson JA, Govert F, Riquelme P, Brasen JH, Brem-Exner BG, Matthai M, Schulze M, Renders L, Kunzendorf U, Geissler EK. et al. Administration of donor-derived transplant acceptance-inducing cells to the recipients of renal transplants from deceased donors is technically feasible. Clin Transplant. 2009;23:140–145. doi: 10.1111/j.1399-0012.2008.00953.x. [DOI] [PubMed] [Google Scholar]
- Hutchinson JA, Roelen D, Riquelme P, Brem-Exner BG, Witzke O, Philipp T, Matthai M, Govert F, Claas FH, Westphal E. et al. Preoperative treatment of a presensitized kidney transplant recipient with donor-derived transplant acceptance-inducing cells. Transpl Int. 2008;21:808–813. doi: 10.1111/j.1432-2277.2008.00712.x. [DOI] [PubMed] [Google Scholar]
- Warnecke G, Hutchinson JA, Riquelme P, Kruse B, Thissen S, Avsar M, Zehle G, Steinkamp T, Peters C, Baumann R. et al. Postoperative intravenous infusion of donor-derived transplant acceptance-inducing cells as an adjunct immunosuppressive therapy in a porcine pulmonary allograft model. Transpl Int. 2009;22:332–341. doi: 10.1111/j.1432-2277.2008.00778.x. [DOI] [PubMed] [Google Scholar]
- Tiurbe G, Matuschek A, Kammerer U, Schneider M, Thiede A, Ulrichs K, Otto C. Inhibitory effects of rat bone marrow-derived dendritic cells on naive and alloantigen-specific CD4+ T cells: a comparison between dendritic cells generated with GM-CSF plus IL-4 and dendritic cells generated with GM-CSF plus IL-10. BMC Res Notes. 2009;2:12. doi: 10.1186/1756-0500-2-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou HS, Hsieh CC, Charles R, Wang L, Wagner T, Fung JJ, Qian S, Lu LL. Myeloid-derived suppressor cells protect islet transplants by B7-H1 mediated enhancement of T regulatory cells. Transplantation. 2012;93:272–282. doi: 10.1097/TP.0b013e31823ffd39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidney D. Improving Global Outcomes (KDIGO) Transplant Work Group. KDIGO clinical practice guideline for the care of kidney transplant recipients. Am J Transplant. 2009;9(Suppl 3):S1–S155. doi: 10.1111/j.1600-6143.2009.02834.x. [DOI] [PubMed] [Google Scholar]
- Sagoo P, Perucha E, Sawitzki B, Tomiuk S, Stephens DA, Miqueu P, Chapman S, Craciun L, Sergeant R, Brouard S. et al. Development of a cross-platform biomarker signature to detect renal transplant tolerance in humans. J Clin Invest. 2010;120:1848–1861. doi: 10.1172/JCI39922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekberg H, Tedesco-Silva H, Demirbas A, Vitko S, Nashan B, Gurkan A, Margreiter R, Hugo C, Grinyo JM, Frei U. et al. Reduced exposure to calcineurin inhibitors in renal transplantation. N Engl J Med. 2007;357:2562–2575. doi: 10.1056/NEJMoa067411. [DOI] [PubMed] [Google Scholar]
- Ekberg H, Mamelok RD, Pearson TC, Vincenti F, Tedesco-Silva H, Daloze P. The challenge of achieving target drug concentrations in clinical trials: experience from the Symphony study. Transplantation. 2009;87:1360–1366. doi: 10.1097/TP.0b013e3181a23cb2. [DOI] [PubMed] [Google Scholar]
- Shapiro R, Jordan ML, Basu A, Scantlebury V, Potdar S, Tan HP, Gray EA, Randhawa PS, Murase N, Zeevi A. et al. Kidney transplantation under a tolerogenic regimen of recipient pretreatment and low-dose postoperative immunosuppression with subsequent weaning. Ann Surg. 2003;238:520–525. doi: 10.1097/01.sla.0000089853.11184.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan HP, Kaczorowski D, Basu A, McCauley J, Marcos A, Donaldson J, Unruh M, Randhawa P, Zeevi A, Shapiro R. Steroid-free tacrolimus monotherapy after pretransplantation thymoglobulin or Campath and laparoscopy in living donor renal transplantation. Transplant Proc. 2005;37:4235–4240. doi: 10.1016/j.transproceed.2005.10.020. [DOI] [PubMed] [Google Scholar]
- Tan HP, Donaldson J, Basu A, Unruh M, Randhawa P, Sharma V, Morgan C, McCauley J, Wu C, Shah N. et al. Two hundred living donor kidney transplantations under alemtuzumab induction and tacrolimus monotherapy: 3-year follow-up. Am J Transplant. 2009;9:355–366. doi: 10.1111/j.1600-6143.2008.02492.x. [DOI] [PubMed] [Google Scholar]
- Margreiter R, Klempnauer J, Neuhaus P, Muehlbacher F, Boesmueller C, Calne RY. Alemtuzumab (Campath-1 H) and tacrolimus monotherapy after renal transplantation: results of a prospective randomized trial. Am J Transplant. 2008;8:1480–1485. doi: 10.1111/j.1600-6143.2008.02273.x. [DOI] [PubMed] [Google Scholar]
- Chan K, Taube D, Roufosse C, Cook T, Brookes P, Goodall D, Galliford J, Cairns T, Dorling A, Duncan N. et al. Kidney transplantation with minimized maintenance: alemtuzumab induction with tacrolimus monotherapy-an open label, randomized trial. Transplantation. 2011;92:774–780. doi: 10.1097/TP.0b013e31822ca7ca. [DOI] [PubMed] [Google Scholar]