

Abstract
Cytogenetic studies in clonal plasma cell disorders have mainly been done in whole bone marrow or CD138+ microbead-enriched plasma cells and suggest that recurrent immunoglobulin heavy chain translocations - e.g. t(4;14) -are primary oncogenetic events. The aim of this study was to determine cytogenetic patterns of highly purified aberrant plasma cells (median purity ≥98%) in different clonal plasma cell disorders. We analyzed aberrant plasma cells from 208 patients with multiple myeloma (n=148) and monoclonal gammopathy of undetermined significance (n=60) for the presence of del(13q14), del(17p13) and t(14q32) using multicolor interphase fluorescence in situ hybridization. Additionally, immunoglobulin heavy chain gene arrangements were analyzed and complementarity determining region 3 was sequenced in a subset of patients and combined multicolor interphase fluorescence in situ hybridization/immunofluorescent protein staining analyses were performed in selected cases to confirm clonality and cytogenetic findings. At diagnosis, 96% of cases with multiple myeloma versus 77% of monoclonal gammopathy of undetermined significance cases showed at least one cytogenetic alteration and/or hyperdiploidy. The cytogenetic heterogeneity of individual cases reflected coexistence of cytogenetically-defined aberrant plasma cell clones, and led to the assumption that karyotypic alterations were acquired stepwise. Cases of multiple myeloma and monoclonal gammopathy of undetermined significance frequently showed different but related cytogenetic profiles when other cytogenetic alterations such as deletions/gains of the immunoglobulin heavy chain or the fibroblast growth factor receptor 3 were additionally considered. Interestingly, in 24% of multiple myeloma versus 62% of monoclonal gammopathy of undetermined significance patients with an immunoglobulin heavy chain translocation, aberrant plasma cells with and without t(14q32) coexisted in the same patient. Our data suggest that recurrent immunoglobulin heavy chain translocations might be absent in the primordial plasma cell clone in a significant proportion of patients with clonal plasma cell disorders carrying these cytogenetic alterations.
Introduction
Acquisition of del(13q14), immunoglobulin heavy chain (IGH) translocations at chromosome 14q32 - t(14q32) - and numerical gains of different chromosomes (e.g., trisomies of chromosomes 3, 5, 7 and 9) are considered to be early cytogenetic events in clonal plasma cell disorders, whereas other molecular changes (e.g, MYC dysregulation and RAS mutation) are characteristic of malignant transformation and advanced stages of the disease.1-3 Despite this, limited and controversial data are currently available about the patterns and the chronological sequence of acquisition of cytogenetic alterations at the intratumoral cell level in multiple myeloma (MM) and monoclonal gammopathy of undetermined significance (MGUS).4-9 This is related to the fact that the majority of studies reported so far have relied on conventional cytogenetics with relatively few metaphases analyzed and/or single color/single chromosome interphase fluorescence in situ hybridization (iFISH) analyses of whole bone marrow samples or plasma cells (PC) enriched by CD138+ microbeads.4,10,11
Although significantly more karyotypic aberrations can be identified in MM and MGUS when CD138+ PC are enriched rather than when unsorted whole bone marrow is studied,11 the PC enrichment technique is associated with several pitfalls, particularly in MGUS. CD138 is commonly expressed by normal PC (N-PC), reactive PC and aberrant PC (aPC).12 Normal PC are typically absent or infrequent in MM, but they account for >5% of all bone marrow PC in nearly 90% of MGUS patients, with a mean percentage of <2% of N-PC in MM and of around 65% in MGUS.13-16 Moreover, previous studies suggest that CD138 could be rapidly lost by apoptotic PC17 and a subpopulation of CD138- PC frequently coexists with the major fraction of CD138+ PC in clonal plasma cell disorders.18 Finally, the 85%-95% median purity of magnetically enriched CD138+ PC limits detailed evaluation of clonal profiles in cases carrying one or multiple cytogenetic changes, particularly as regards identification of a fraction of non-altered aPC,19 as also described for other B-cell neoplasias.20 Simultaneous usage of the FISH technique and May-Grünwald-Giemsa staining or immunopheno-typing (e.g., staining for CD138 or cytoplasmic light chains) through the so-called FICTION technique has occasionally been described.21,22 However, both approaches are limited by the fact that only a low number of PC are analyzed in a cytospin preparation, a bone marrow smear or a formalin-fixed, paraffin-embedded section. Conversely, flow cytometry allows reliable differentiation and large scale, high purity (median ≥98%) sorting of N-PC and aPC.23 To the best of our knowledge, only one cytogenetic study using flow cytometry-sorted aPC has been published so far, but the analysis was focused on numerical chromosomal alterations and did not include the prognostically informative IGH gene rearrangements.24 In the present study we investigated the intratumoral patterns of cytogenetic alterations in a large series of MM and MGUS patients, using multicolor iFISH analysis of highly purified flow cytometry-sorted aPC. Further analysis of IGH gene arrangements and complementarity determining region 3 (CDR3) sequencing was carried out in a subset of patients and the simultaneous application of iFISH and immunofluorescent protein staining (FICTION or immunoFISH technique) was performed in selected cases to confirm clonality and cytogenetic findings. Overall, the cytogenetic heterogeneity of aPC from individual patients reflected coexistence of cytogenetically-defined aPC clones; this suggests that acquisition of cyto-genetic alterations occurs stepwise within the bone marrow aPC of individual patients with plasma cell disorders. It is noteworthy that different but related cytogenetic patterns were frequently found in MM versus MGUS. Interestingly, in individual MGUS cases, and to a lesser extent also in MM, a significant proportion of aPC carrying an IGH translocation - including recurrent cytogenetic alterations such as t(11;14) - coexists with aPC lacking these alterations.
Design and Methods
Patients
Patients with newly diagnosed MM (n=148) and MGUS (n=60) were included in this study; three MM patients underwent assessment of cytogenetic alterations during follow-up at different disease stages. Written informed consent was given by each subject. In order to avoid borderline cases we excluded from the analysis those MGUS patients with ≤5% N-PC (since they are at high risk of progression to MM15) as well as those MM patients with the opposite pattern (>5% N-PC) who have a more indolent disease course.13,15,16 Patients with IgM MGUS were also specifically excluded from this series. The study was approved by the local ethics committee and conducted in accordance with the declaration of Helsinki.
Multiparameter flow cytometry and sorting of aberrant plasma cells
Immunophenotyping of N-PC and aPC was performed on EDTA-anticoagulated, erythrocyte-lysed bone marrow samples with a conventional direct immunofluorescence stain-and-then-lyse technique, as previously described.16,25 A standard FACSCanto II flow cytometer equipped with the FACSDiVa software [Becton Dickinson Biosciences (BD), San José, CA, USA] was used for data acquisition and Infinicyt software (Cytognos SL, Salamanca, Spain) was employed for data analysis. Positivity for a particular antigen was recorded if ≥20% of the aPC stained for this antigen above the autofluorescence levels of unstained aPC.
Normal PC and aPC were identified in bone marrow samples from all patients using either four-color (127 MM and 55 MGUS patients) or eight-color (21 MM and 5 MGUS patients) immunostaining panels of fluorochrome-conjugated monoclonal antibodies based on the following combinations of reagents: fluorescein isothiocyanate (FITC)/phycoerythrin (PE)/peridinin chlorophyll protein-cyanin 5.5 (PerCP Cy5.5)/allophycocyanin (APC) and FITC/PE/PerCP-Cy5.5/PE-Cyanin 7 (PE Cy7)/APC/APCH7/Pacific blue (PacB)/Pacific orange (PacO) -: CD38/CD56/CD19/CD45, CD38/CD27/CD45/CD28, β-2 microglobulin/CD81/CD38/CD117 and CD38/CD56/β -2 microglobulin/CD19/cytoplasmic immunoglobulin kappa light chain (cyIgκ)/cyIg lambda light chain (cyIgλ)/CD45/CD138, CD38/CD28/CD27/CD19/CD117/CD81/CD45/CD138.
Plasma cells were identified based on their unique light scatter profile and their pattern of expression of CD38, after excluding cell doublets and debris. Aberrant PC were distinguished from N-PC based on their aberrant patterns of expression of two or more of the following antigens previously described in detail:23,26 CD19-, CD27-/lo, CD28+, CD38+/lo, CD45-, CD56+, CD81-/lo, CD117+ and/or monotypic cyIg light chain expression. The most frequent immunophenotype of aPC - considering only some of the antigens analyzed - included the following patterns: CD19- CD45- CD56+ (101/205 patients, 49%), CD19- CD45- CD56- (32/205 patients, 16%), CD19- CD45+CD56+ (30/205 cases, 15%), CD19+CD45-CD56+ (16/205 cases, 8%); other aberrant profiles were found more rarely (P>0.05 for MM versus MGUS).
Aberrant PC were sorted with a FACSAria II cell sorter (BD). The median purity of aPC was ≥98% (range: 90%-100%).
Interphase fluorescence in situ hybridization studies
Interphase FISH was done on flow cytometry-sorted aPC (from all MM and MGUS cases) and N-PC (from 8 MGUS and 2 MM patients containing chromosomal alterations in their aPC by iFISH), as previously reported.25,27 Briefly, translocations involving the IGH gene at chromosome 14q32 and either del(14q32) or +14q32 in the absence of t(14q32) were identified with a panel of standard LSI IGH dual-color break-apart rearrangement probes purchased from Vysis (Downers Grove, IL, USA). Samples showing an IGH gene rearrangement or del(14q32) potentially associated with an IGH gene rearrangement (one fusion and one split signal) were further analyzed using specific fusion probes for the most frequent t(14q32) chromosomal translocations - e.g, t(4;14), t(11;14), and t(14;16) - (Vysis).28 Deletions and gains of FGFR3, CCND1 and MAF were also systematically recorded in these patients if not involved in the IGH gene rearrangement. The presence of del(13q14) and del(17p13) or +17p13 was simultaneously analyzed with the LSI RB1 (13q14) and LSI P53 (17p13.1) probes (Vysis), respectively. Near tetraploid cases (flow cytometry DNA index ≥1.8) were considered to carry del(13q14) if two signals were detected in a tetraploid cell. Thresholds of 8% and 15% were applied for the detection of deletions/gains and t(14q32), respectively. In turn, cases with ≥10% of iFISH signals compatible with t(14q32) using the IGH break-apart probe and ≥10% positive signals for specific t(14q32) - or three IGH copies in the case of ‘other t(14q32)’ - using fusion probes were considered to have IGH gene rearrangements. All thresholds were based on the results obtained through the analysis of control (flow cytometry sorted) bone marrow cells for the same probes (mean + 3 standard deviations of the values obtained).
Cases with an IGH gene rearrangement without t(4;14), t(11;14) or t(14;16) were classified as ‘other t(14q32)’, while cases with a deletion potentially associated or not with an IGH gene rearrangement but lacking t(4;14), t(11;14) or t(14;16) were considered to carry del(14q32) in the absence of t(14q32). The calculation of the percentage of aPC carrying either t(4;14), t(11;14) or t(14;16) was based on the mean of the percentages of aPC showing a positive signal for the IGH break-apart probe and aPC identified with a specific t(14q32), respectively. In a single case which showed two different types of IGH translocation - t(4;14) and t(14;16) - in distinct populations of aPC, percentages refer to the respective t(14q32) specific probes. Percentages of aPC with a distinct cytogenetic alteration were systematically adjusted to the purity of aPC using the following formula: ‘% of cells with a distinct cytogenetic alteration x 100/% purity of aPC’. The karyotypic patterns of cytogenetic alterations were assessed separately in every case in order to define individual aPC clones based on the percentage of aPC with a distinct cytogenetic alteration using a 15%-threshold as specified below. Only those clones in which >15% of the aPC differed were considered to be cytogenetically different. This criterion [>15% aPC without a specific cytogenetic alteration, e.g. t(14q32)] was also used for cytogenetically altered cases to define the presence of an aPC clone lacking the tested cytogenetic alteration. In those few cases with two different cytogenetically altered aPC populations differing by ≤15% aPC but only one of them comprising ≥85% of all aPC, the two cell populations were considered to belong to the same (i.e. initial) clone (because of the estimated maximum contamination with N-PC in the bone marrow and the variability associated with iFISH spot counting). Karyotypic alterations found in the majority of aPC (≥85%) were considered to be early oncogenetic events, whereas those found only in a fraction of aPC were considered to occur later during disease evolution. Confirmation of parent-progeny relationships and coexistence of multiple clones was achieved by simultaneous hybridization with all involved probes in addition to the hybridizations performed with the double stainings documented above, as illustrated in Figure 1. Interphase FISH slides from cases with t(14q32) were blindly evaluated by two independent expert observers with a high reproducibility (Online Supplementary Table S1). Of note, none of the N-PC analyzed showed chromosomal alterations for all iFISH probes assayed.
Figure 1.

Illustrating examples of clonal profiles of MM patients found by iFISH analysis of highly purified aPC, as defined in Table 2. (A and C): karyotypic patterns of group I; (B): karyotypic patterns of group II. Each number in the panels corresponds to a cell and identifies its sequence from the ancestral clone (1) to additional subsequent clones (2 and 3) (x1000 original magnification). BA: break-apart; DC: dual color; DF: dual fusion; SG: spectrum green; SO: spectrum orange. →: sequential acquisition. &: simultaneous acquisition.
Analysis of DNA ploidy of aberrant plasma cells
Evaluation of flow cytometry DNA ploidy was based on a double staining for nuclear DNA (propidium iodide) and surface PC antigens (anti-CD38 and anti-CD138 monoclonal antibodies), as previously reported in detail.27,29 The DNA index was defined as the ratio between the modal channel of the G0-G1 peak of PC and the G0-G1 peak of the remaining residual normal cell populations. Hypodiploidy was defined when the DNA index was ≤0.9, hyperdiploidy when it was ≥1.08; all other cases with a DNA index >0.9 and <1.08 were classified as being DNA diploid.
FICTION analyses
Briefly for FICTION analyses (n=4 MM cases) flow cytometry-sorted aPC cells were sequentially washed with phosphate-buffered saline (PBS) containing 1% bovine serum albumin (BSA), permeabilized (PBS with 1% BSA + 0.1% TritonX), and washed again (PBS with 1% BSA). The cells were then stained in parallel with anti-CyIgκ-APC (DAKO, Glostrup, Denmark) and anti-CyIgλ -APCH7 (BD) antibodies, followed by the iFISH protocol as described above (with slight modifications). Stained samples were analyzed using a Confocal Laser Scanning Microscope (TCS SP2, LEICA, Heidelberg, Germany).
Analysis of IGH gene rearrangements and CDR3 sequencing
The technique used for analysis of IGH gene rearrangements and CDR3 sequencing performed in a subgroup of nine patients is detailed in the Online Supplementary Design and Methods. Briefly, two different multiplex polymerase chain reactions were tested for the IGH gene regions (VDJ-FR2 and DJ) using highly purified aPC. Clonal IGH gene rearrangements were identified by fragment analysis in an ABI PRISM 3130 Avant sequencer, using GEN-EMAPPER 3.1 software (Applied Biosystems, Melbourne, Australia). A clonal population was defined by the presence of either a single peak or a predominant peak over a polyclonal background. Clonal polymerase chain reaction products were then purified with ExoSap (USB Corp., Cleveland, OH, USA) and directly sequenced in both directions in an ABI 3130 DNA sequence analyzer using the Big-Dye 3.1 Terminator cycle sequencing chemistry (Applied Biosystems).
Statistical methods
Comparisons of frequencies in the different groups were analyzed with the χ2 test or Fisher's exact test. For continuous measurements the Mann-Whitney U test was used. P values <0.05 (two-sided) are considered statistically significant. The commercially available PASW software (Version 18.0, Chicago, IL, USA) was used for the statistical analyses.
Results
Frequencies and patterns of cytogenetic alterations
Frequencies of different cytogenetic alterations in MM versus MGUS are shown in Table 1. At least one of the cytogenetic alterations investigated (including DNA hyperdiploidy) was observed in 96% of MM and 77% of MGUS patients (P<0.001). Among these cases, a large proportion had cytogenetic alterations of chromosomes 13q14, 17p13 and 14q32 detected by iFISH (84% of MM and 55% of MGUS cases, P<0.001), while the other altered cases only had DNA hyperdiploidy (12% of MM and 22% of MGUS cases, P=0.08). Patients were further classified into distinct cytogenetic groups according to the ‘karyotypic pattern’ based on the presence or absence of del(13q14) and del(17p13) with or without t(14q32) (Table 2).
Table 1.
Frequency of individual cytogenetic alterations in MM and MGUS.
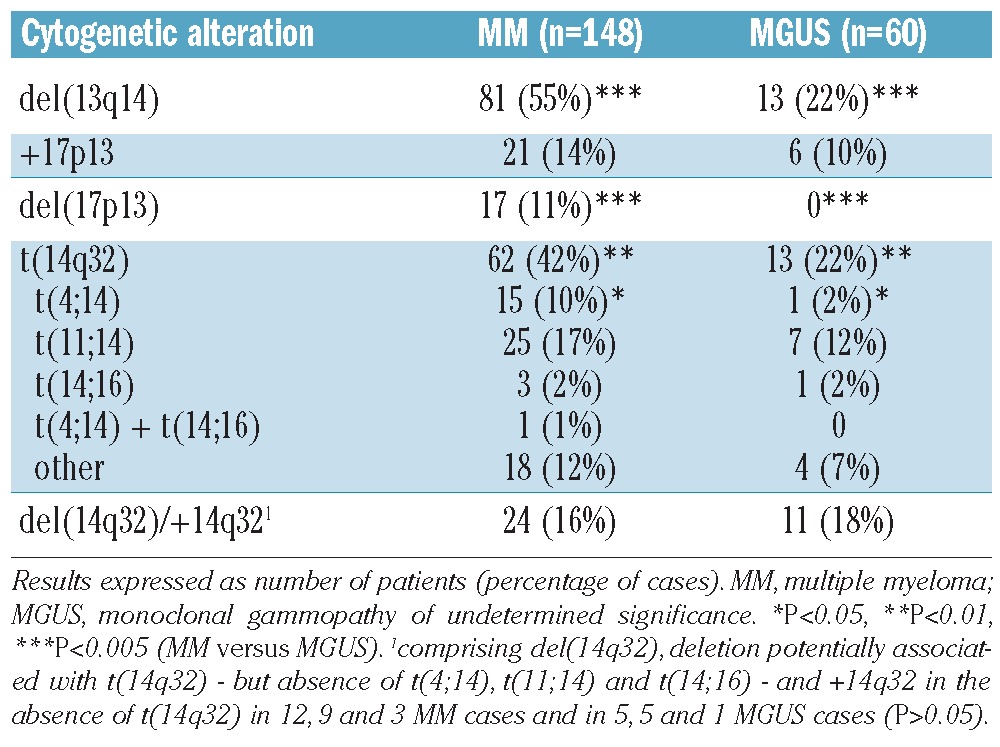
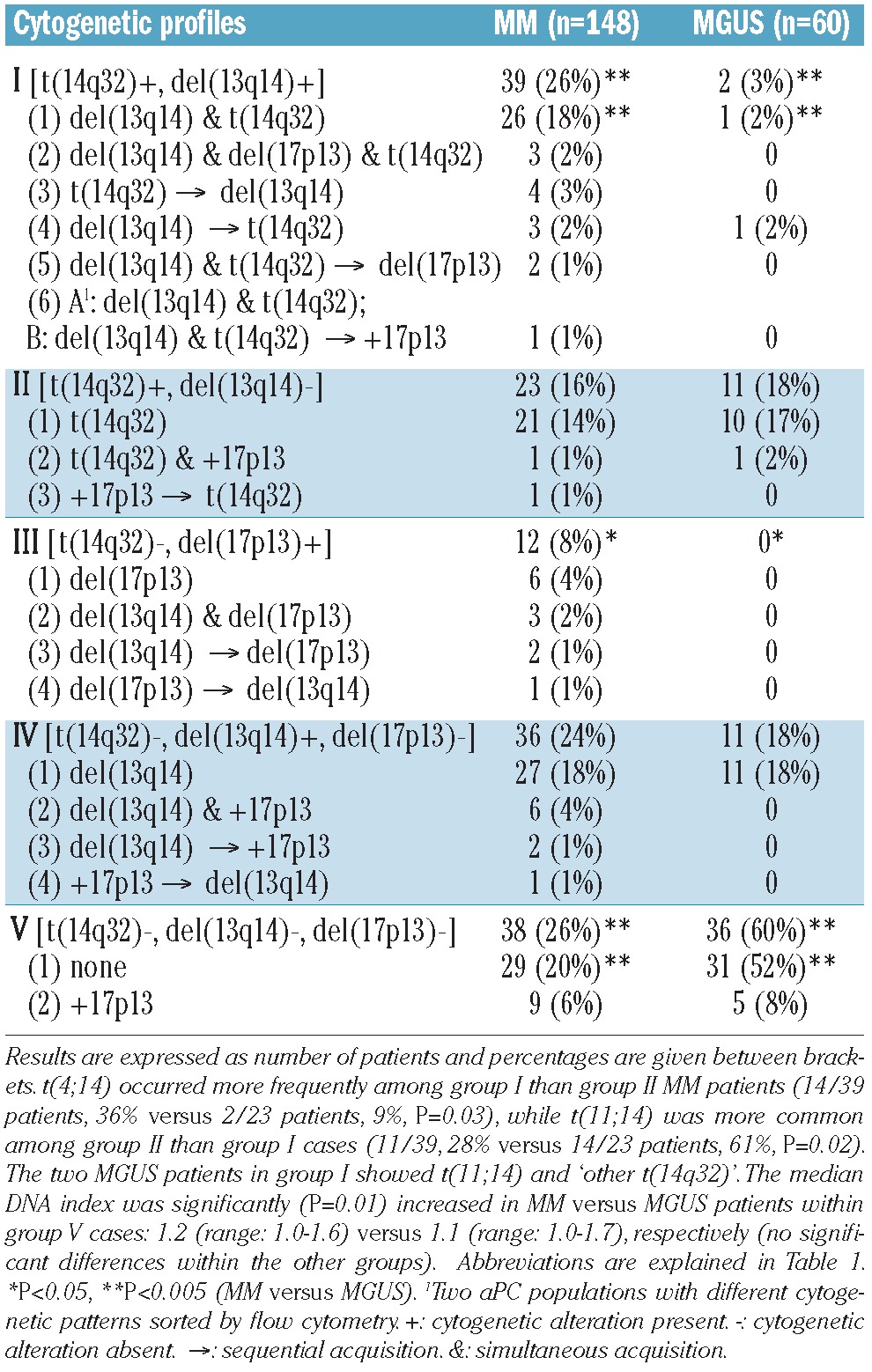
Table 2.
Cytogenetic patterns defined by the simultaneous assessment of del(13q14), del(17p13), +17p13 and t(14q32) in different cytogenetic subgroups of patients with MM or MGUS.
The median percentage of aPC carrying individual cytogenetic alterations, including different types of t(14q32), did not differ significantly between MM and MGUS, although the percentage of aPC with t(14q32) was slightly higher (P=0.08) in IGH-translocated MM than in IGH-translocated MGUS cases (Figure 2; Online Supplementary Figure S1). Focusing on MM patients, the median percentages of aPC with del(13q14) and t(14q32) were higher (P=0.02 and P<0.001) than the median percentage with del(17p13) (Figure 2); these findings support the assumption that the former two cytogenetic alterations reflect mainly primary cytogenetic events, whereas del(17p13) typically corresponds to a secondary genetic change. The percentage of aPC with del(13q14) was also significantly (P=0.03) higher than the percentage with +17p13, both alterations being found in MM at lower percentages (P=0.01 and P<0.001) than t(14q32). Finally, the percentage of aPC with t(14q32) and del(14q32) or +14q32 in the absence of t(14q32) differed significantly (P=0.01) in MM patients. By contrast, the median percentages of aPC with different cytogenetic alterations [e.g. del(13q14), del(17p13)] did not differ significantly in the cohort of MGUS patients (Figure 2). Likewise, the percentage of aPC with different types of t(14q32) did not differ significantly among IGH-translocated cases, either in MM or in MGUS (Online Supplementary Figure S1).
Figure 2.
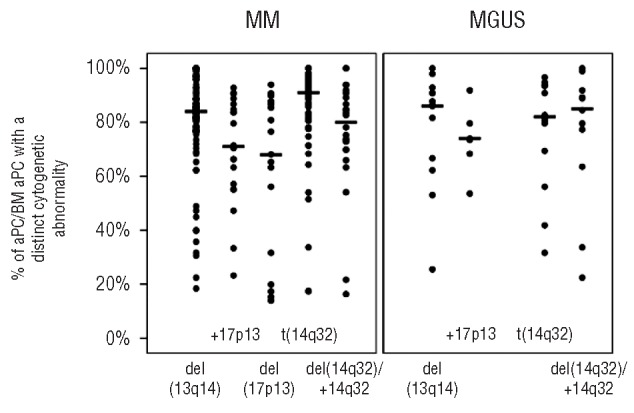
Percentage of highly purified aPC with del(13q14), +17p13, del(17p13), t(14q32) and del(14q32)/+14q32 in the absence of t(14q32) from all bone marrow aPC in individual MM versus MGUS patients. Circles indicate individual patients and bars correspond to the median values of altered cells. Statistical comparisons are given in the text.
Specific evaluation by iFISH for the presence of PC positive for IGH gene rearrangements was also performed in purified residual N-PC fractions from a subgroup of ten patients who showed coexisting residual N-PC and aPC in their bone marrow at diagnosis, and carried a t(14q32) in a high percentage of aPC (>82%); in all these cases normal iFISH patterns were systematically found within the purified N-PC fraction, with <1% nuclei having a t(14q32).
Intratumoral patterns of cytogenetic alterations
The presence of two or more cytogenetically different clones in aPC from the same patient and sample was more commonly observed in MM (95/148 cases, 64%) than in MGUS (22/60 cases, 37%) (P<0.001) (Table 3). In order to determine the specific potential clonal profiles in MM versus MGUS we investigated whether there were shared profiles between the two groups of patients, based on both the presence versus absence of del(13q14), del(17p13) and/or t(14q32) and the presumed chronological sequence of acquisition of these cytogenetic alterations (Table 2). Only MM and MGUS patients with cytogenetic alterations by iFISH for the respective chromosomal regions were included in this part of the analysis. Considering the karyotypic patterns defined by the presence of del(13q14), del(17p13) and t(14q32), 98/110 (89%) patients with MM showed patterns that were also found in MGUS and 24/24 (100%) MGUS cases had patterns also found in MM (P>0.05). Despite this, when the presumed chronological sequence of acquisition of these alterations together with +17p13 was considered, the frequency of shared patterns decreased to 87/119 (73%) in MM but remained 100% among MGUS cases (29/29) (P<0.001; Table 2). Additionally, when further cytogenetic variables were considered, such as DNA ploidy, type of t(14q32) and deletions/gains of IGH, FGFR3, CCND1 and MAF without t(14q32) in their presumed chronological sequences, the frequencies of overlapping karyotypic patterns were 32/125 (26%) for MM and 18/33 (55%) for MGUS (P=0.003; Online Supplementary Table S2). Altogether, these findings could suggest that progression of MGUS to MM frequently requires acquisition of specific cytogenetic alterations in individual aPC which are uniquely associated with the advanced form of the disease.
Table 3.
Number of cytogenetically different clones in the distinct cytogenetic groups of MM versus MGUS patients.
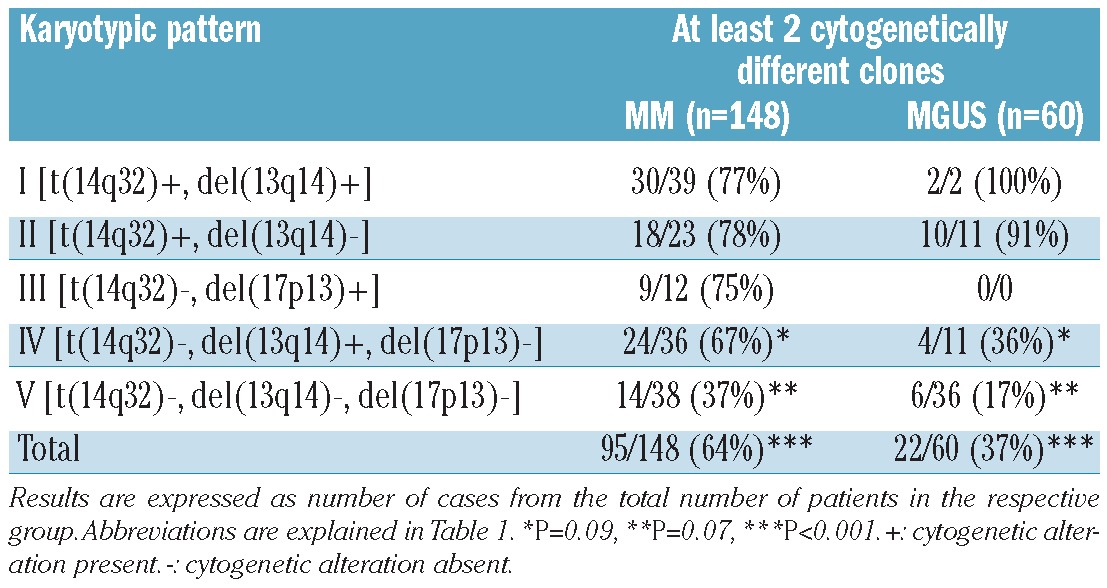
Interestingly, 73/148 (49%) MM and 44/60 (73%) MGUS cases (P=0.002) showed a primary aPC clone (primordial or ancestral tumor cell clone) representing >15% aPC without any cytogenetic alteration for all probes tested (Online Supplementary Table S3). Among cases with t(14q32), this alteration was absent in the primordial aPC clone in a smaller proportion of cases: 15/62 (24%) MM: t(4;14), t(11;14) and ‘other t(14q32)’ in three, five and seven cases, respectively versus 8/13 (62%) MGUS patients: t(11;14), t(14;16) and ‘other t(14q32)’ in four, one and three cases, respectively; P=0.02) (Figure 3; Online Supplementary Table S2). In order to confirm these apparently unexpected findings, we further analyzed some exemplarily cases with a recurrent IGH translocation, e.g. t(4;14) or t(11;14), by FICTION studies. These studies showed that aPC with and without the translocation can coexist in the same patient/sample, whereas all investigated cells expressed only one single light chain type (cyIgκ or cyIgλ) confirming the high purity of flow cytometry-sorted aPC (Figure 4).
Figure 3.

Sequence of acquisition of cytogenetic alterations - del(13q14), del(17p13) and t(14q32) -and frequency of clonal profiles in MM (A) and MGUS (B). Results are expressed as number of patients and percentages are given in brackets. Evolution of the primary clone differed significantly between hyperdiploid and non-hyperdiploid MM cases (P=0.005) and between non-hyperdiploid MM and non-hyperdiploid MGUS patients (P<0.001). -: cytogenetic alteration absent; *presence of at least one cytogenetic alteration.
Figure 4.
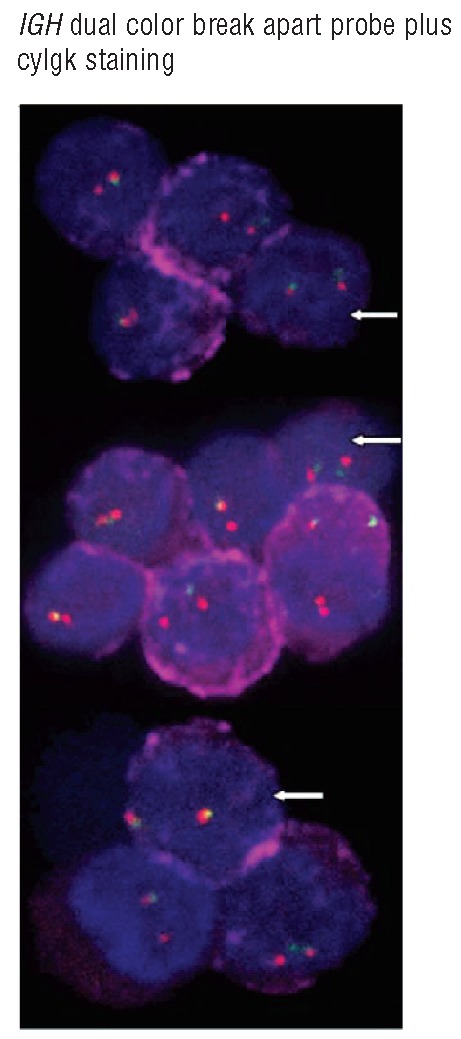
Simultaneous analysis of cyIgκ light chain expression (diffuse red staining) and iFISH spots for the IGH dual color break apart probe in purified aPC from a representative MM patient carrying t(4;14) in only part of all aPC. IGH-rearranged aPC show a fused red/green signal plus an isolated red signal together with positive cyIgκ whereas non-rearranged aPC show two red/green spots together with similar cyIgκ red staining (white arrows).
Interestingly, in some of the MM cases which were exemplarily evaluated during follow-up at different disease stages, the non-altered aPC clone persisted during disease evolution, even in cases carrying a recurrent IGH translocation such as t(4;14) (Online Supplementary Table S4).
DNA ploidy status and immunophenotype of neoplastic plasma cells
The frequency of DNA hyperdiploidy was similar in patients with MM or MGUS (47% versus 45%; Figure 3). As expected, the t(14q32) prevailed in non-hyperdiploid versus hyperdiploid cases in both MM (57% versus 25%; P<0.001) and MGUS (30% versus 11%; P>0.05). Del(13q14) was likewise more frequent among non-hyperdiploid than hyperdiploid cases in MM (63% versus 45%; P=0.03). In contrast, del(17p13) was found at similar frequencies in hyperdiploid and non-hyperdiploid MM cases (14% versus 9%; P>0.05). +17p13 was more frequent among hyperdiploid MM (25%) and MGUS (19%) than in non-hyperdiploid cases (5% and 3%; P<0.001 and P=0.08, respectively), while del(14q32)/+14q32 in the absence of t(14q32) showed no significant association with DNA ploidy either in MM or in MGUS (data not shown).
Analysis of IGH gene rearrangements and CDR3 sequencing of aberrant plasma cells
IGH gene rearrangements and CDR3 sequences of highly purified aPC were analyzed in nine exemplary (2 MGUS and 7 MM) cases; two of them had none of the tested cytogenetic alterations and the other seven showed at least one cytogenetic alteration (Online Supplementary Table S5). In two out of these latter seven MM cases a fraction of the aPC purified had a recurrent IGH translocation - t(4;14) in one case and t(11;14) in the other - and coexisted with another aPC fraction without the translocation (36% and 23% of all aPC in cases n. 93759 and 85864; Online Supplementary Table S5). Only one unique CDR3 sequence was identified for each sample analyzed, indicating a clonal relationship between the IGH-translocated and non-translocated cases and absence of a significance proportion of contaminating polyclonal N-PC.
Discussion
In the present study virtually all patients with MM and the great majority of those with MGUS showed hyperdiploidy and/or cytogenetic alterations involving chromosomes 13q14, 17p13 and 14q32. Overall, del(13q14), del(17p13) and t(14q32) were identified at significantly higher frequencies among MM patients than among MGUS cases; in contrast, the incidence of +17p13 and del(14q32)/+14q32 in the absence of t(14q32) did not differ significantly between the two groups of patients. The frequencies found here for these cytogenetic alterations in MM are concordant with previous data,27,30,31 but those observed in MGUS are considerably lower than described by others.3,8 Different inclusion criteria used here for MGUS - only cases with >5% N-PC - and MM - only cases with ≤5% N-PC - might contribute to these discrepancies since the usage of this 5% threshold for N-PC excludes a group of MGUS patients (15-20% of all cases) with a higher risk of progression into MM, at the same time as avoiding inclusion of some cases of smoldering MM.13,15,16
Interestingly, unique karyotypic patterns were found in MM versus MGUS cases. For example, the simultaneous presence of del(13q14) and t(14q32) occurred frequently in MM but not in MGUS. As mentioned above, at least one cytogenetic alteration and/or hyperdiploidy occurred in virtually all MM patients, confirming the previously suggested high frequency of genetic alterations in this disease,2 but only in 77% of MGUS cases. This observation and the fact that two or more cytogenetically different clones were found significantly more frequently in MM than in MGUS supports the previous assumption that evolution of MM from MGUS is associated with a clonal expansion of genetically abnormal PC5 and potentially also with the acquisition of additional chromosomal/molecular alterations in individual aPC clones uniquely associated with malignant transformation of the disease.
Despite the limitations of a mainly non-longitudinal study - a large-scale longitudinal study to detect cytogenetic alterations in primordial aPC prior to diagnosis of a plasma cell disorder seems to be virtually impossible - further remarkable differences were found when the intratumoral patterns of cytogenetic changes were analyzed in individual patients at the single aPC level. In MM, the median percentage of aPC with different cytogenetic alterations differed significantly indicating that during disease evolution some alterations appear at an earlier stage than others. Thus, we observed a significantly higher incidence of aPC carrying t(14q32) and del(13q14) than aPC with del(17p13); this is in line with the general belief that t(14q32) and del(13q14) are mainly primary lesions while del(17p13) commonly occurs later as a secondary cytogenetic change.2,3,32 Most interestingly, however, in about one quarter of MM cases with t(14q32) we identified a significant proportion of aPC that did not carry the respective cytogenetic alteration, indicating that this alteration - i.e. t(14q32) - might be absent in the primordial (ancestral) PC clone and be also acquired during disease evolution. It is noteworthy that these cases also comprised different types of t(14q32), including t(4;14) and t(11;14). These findings support a previous hypothesis that t(14q32) are relatively early cytogenetic events;2,3 however, they also argue against the assumption that they almost always correspond to primary oncogenic events except for the rare, possibly random, t(14q32) other than t(4;14), t(11;14) or t(14;16).2,3 In this context our results strongly support recently published single cell level FISH data questioning the initiating role of recurrent IGH translocations.33 In addition, our results are also similar to those reported by Fonseca et al.7 in a series of 23 MGUS patients who had t(14q32), most of whom [15/23 (65%)] had a significant proportion (>15%) of clonal Ig-light chain-restricted PC without t(14q32) coexisting with the t(14q32)-positive cells.
It should be mentioned further that normal iFISH patterns were systematically found within the purified residual N-PC fractions in ten cases carrying a coexisting aPC population with an IGH translocation in the present analysis. In addition, analysis of IGH gene rearrangements and CDR3 sequencing revealed that aPC populations with distinct cytogenetic alterations coexisting in the same patient are clonally related. Additionally, the detection of only one unique CDR3 sequence in the absence of an oligo-/polyclonal background in all tested samples (including those cases with a recurrent IGH translocation present only in an aPC subclone) argues against a significant proportion of contaminating polyclonal N-PC. Moreover simultaneous immunostaining for cyIgκ and cyIgλ plus iFISH (FICTION assay) was applied in some exemplary cases, further supporting the notion that aPC and N-PC might be clearly distinguished by the flow-cytometry based approach used here, as suggested previously.13,23 Thus it is unlikely that our findings are due to contaminating N-PC. Finally, our observation that an aPC clone without any of the tested cytogenetic alterations can persist - and even increase - at different disease stages during follow-up, even in cases carrying a recurrent IGH translocation such as t(4;14), further supports our hypothesis that these karyotypic alterations might not always correspond to primary events during aPC expansion.
Taken together, our results suggest that also plasma cell disorders may evolve from low numbers of aPC, which lack cytogenetic alterations commonly associated with MM and MGUS and that they may occur in a significant percentage of otherwise healthy individuals, similarly to what has been described for healthy subjects who show circulating monoclonal chronic lymphocytic leukemia-like B cells among whom cytogenetic alterations, such as trisomy 12 or del(13q), are typically found within those cases with higher percentages of clonal chronic lymphocytic leukemia-like B cells.34,35 In any case, the potential existence of other underlying primary genetic lesions versus increased aPC survival mechanisms (e.g. cell senescence) as early events triggering the expansion of aPC in MGUS requires further investigation.
As in MM, cytogenetic heterogeneity and different sizes of cytogenetically defined aPC clones with partially overlapping chromosomal alterations suggest that such alterations were probably acquired stepwise also in MGUS; since the median percentage of aPC with distinct cytogenetic alterations did not differ significantly between MM and MGUS, it could be hypothesized that comparable chronological sequences of acquisition of individual cytogenetic alterations might in principle be found in both diseases. The presence of contaminating N-PC (e.g., due to PC sorting by CD138+ microbeads) might explain the discrepant results found in other studies, which suggested that del(13q14) typically occurs only in a subpopulation of aPC in MGUS but in the majority of aPC in MM.9,30
Interestingly, our results showed that among cases with IGH translocation the presence of a relatively high proportion of aPC without the translocation was significantly more frequent in MGUS than in MM patients, supporting the notion that IGH translocations could, more frequently than expected, correspond to a secondary event, particularly in MGUS.3,32
Detailed analysis of distinct cytogenetic profiles of individual cases showed only partially overlapping intratumoral pathways of clonal evolution in MM versus MGUS when additional cytogenetic variables such as DNA ploidy, lack of cytogenetic alterations in the primordial clone, type of t(14q32) and deletion or gain of IGH, FGFR3, CCND1 and MAF in the absence of t(14q32) were considered together with del(13q14), del(17p13) and +17p13. Of note, these overlapping cytogenetic profiles were found in about half of MGUS patients and one quarter of MM cases. Altogether, these findings suggest that at the single aPC level, clinically stable MGUS (cases with >5% N-PC/all bone marrow PC13-16) frequently display different cytogenetic profiles from those of symptomatic MM (MM cases with typically ≥95% aPC/all bone marrow PC13-16) and that progression from MGUS to MM may frequently require acquisition of additional cytogenetic alterations at the MGUS stage, which lead to the cytogenetic profiles typically found in MM. However, the disappearance and emergence of specific cytogenetic clones observed in a subset of MM cases analyzed here after therapy suggest that marked genetic instability and intratumoral cytogenetic heterogeneity, presumably also affected by clonal selection, frequently occurs in the course of MM and not only at transformation from MGUS to MM.
In summary, the cytogenetic heterogeneity found here in both MM and MGUS patients led us to assume that acquisition of cytogenetic alterations occurs stepwise within the aPC compartment of patients with plasma cell disorders, but with only partially overlapping pathways between the two entities. Interestingly, both in patients with MGUS and in those with MM, significant percentages of aPC coexist with and without t(14q32) at the intratumoral aPC level indicating that also recurrent IGH translocations might frequently correspond to secondary events, which are not common to all aPC within individual patients.
Acknowledgments
The authors would like to thank the Cooperative Research Thematic Network (RTICs; RTICC RD06/0020/0035, RD06/0020/0006 and G03/136), MM Jevitt, SL firm, Instituto de Salud Carlos III/Subdirección General de Investigación Sanitaria (FIS: PI060339; 02/0905; 01/0089/01-02; PS09/01897) and Gerencia Regional de Salud de Castilla y León; Ayuda de Excelencia de Castilla y León, Consejeria de Educación (EDU/894/2009, GR37), and Consejería de Sanidad (557/A/10), Junta de Castilla y León, Valladolid, Spain for supporting this study. JMS is supported by a grant (CP05/00321) from the ISCIII, Ministerio de Ciencia e Innovacion, Madrid, Spain.
Funding:This work was partially supported by grants from the Fundacion Memoria de Don Samuel Solorzano Barruso, Salamanca, Spain (FS/4-2010). The authors would also like to thank the Dr. Werner Jackstädt Foundation (Wuppertal, Germany) for grant supporting the work of Martin Schmidt-Hieber.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures: Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Chng W-J, Huang G-F, Chung T-H, Ng S-B, Gonzalez-Paz N, Troska-Price T, et al. Clinical and biological implications of MYC activation: a common difference between MGUS and newly diagnosed multiple myeloma. Leukemia. 2011;25(6):1026-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fonseca R, Bergsagel P-L, Drach J, Shaughnessy J, Gutierrez N, Stewart A-K, et al. ; International Myeloma Working Group. International Myeloma Working Group molecular classification of multiple myeloma: spotlight review. Leukemia. 2009;23(12):2210-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fonseca R, Barlogie B, Bataille R, Bastard C, Bergsagel P-L, Chesi M, et al. Genetics and cytogenetics of multiple myeloma: a workshop report. Cancer Res. 2004;64(4):1546-58 [DOI] [PubMed] [Google Scholar]
- 4.Pérez-Simón J-A, García-Sanz R, Tabernero M-D, Almeida J, González M, Fernández-Calvo J, et al. Prognostic value of numerical chromosome aberrations in multiple myeloma: a FISH analysis of 15 different chromosomes. Blood. 1998;91(9):3366-71 [PubMed] [Google Scholar]
- 5.López-Corral L, Gutiérrez N-C, Vidriales M-B, Mateos M-V, Rasillo A, García-Sanz R, et al. The progression from MGUS to smoldering myeloma and eventually to multiple myeloma involves a clonal expansion of genetically abnormal plasma cells. Clin Cancer Res. 2011;17(7):1692-700 [DOI] [PubMed] [Google Scholar]
- 6.Tabernero D, San Miguel J-F, Garcia-Sanz M, Nájera L, García-Isidoro M, Peréz-Simon J-A, et al. Incidence of chromosome numerical changes in multiple myeloma: fluorescence in situ hybridization analysis using 15 chromosome-specific probes. Am J Pathol. 1996;149(1):153-61 [PMC free article] [PubMed] [Google Scholar]
- 7.Fonseca R, Bailey R-J, Ahmann G-J, Rajkumar S-V, Hoyer J-D, Lust J-A, et al. Genomic abnormalities in monoclonal gammopathy of undetermined significance. Blood. 2002;100(4):1417-24 [PubMed] [Google Scholar]
- 8.Bochtler T, Hegenbart U, Cremer F-W, Heiss C, Benner A, Hose D, et al. Evaluation of the cytogenetic aberration pattern in amyloid light chain amyloidosis as compared with monoclonal gammopathy of undetermined significance reveals common pathways of karyotypic instability. Blood. 2008;111(9):4700-5 [DOI] [PubMed] [Google Scholar]
- 9.Chiecchio L, Dagrada G-P, Ibrahim A-H, Dachs Cabanas E, Protheroe R-K, Stockley D-M, et al. ; UK Myeloma Forum. Timing of acquisition of deletion 13 in plasma cell dyscrasias is dependent on genetic context. Haematologica. 2009;94(12):1708-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Avet-Loiseau H, Attal M, Moreau P, Charbonnel C, Garban F, Hulin C, et al. Genetic abnormalities and survival in multiple myeloma: the experience of the Intergroupe Francophone du Myélome. Blood. 2007;109(8):3489-95 [DOI] [PubMed] [Google Scholar]
- 11.Put N, Lemmens H, Wlodarska I, Konings P, Moreau Y, Hagemeijer A, et al. Interphase fluorescence in situ hybridization on selected plasma cells is superior in the detection of cytogenetic aberrations in plasma cell dyscrasia. Genes Chromosomes Cancer. 2010;49(11):991-7 [DOI] [PubMed] [Google Scholar]
- 12.Bataille R, Jégo G, Robillard N, Barillé-Nion S, Harousseau J-L, Moreau P, et al. The phenotype of normal, reactive and malignant plasma cells. Identification of “many and multiple myelomas” and of new targets for myeloma therapy. Haematologica. 2006; 91(9):1234-40 [PubMed] [Google Scholar]
- 13.Ocqueteau M, Orfao A, Almeida J, Bladé J, González M, García-Sanz R, et al. Immunophenotypic characterization of plasma cells from monoclonal gammopathy of undetermined significance patients. Implications for the differential diagnosis between MGUS and multiple myeloma. Am J Pathol. 1998;152(6):1655-65 [PMC free article] [PubMed] [Google Scholar]
- 14.Lima M, Teixeira Mdos A, Fonseca S, Gonçalves C, Guerra M, Queirós M-L, et al. Immunophenotypic aberrations, DNA content, and cell cycle analysis of plasma cells in patients with myeloma and monoclonal gammopathies. Blood Cells Mol Dis. 2000;26(6):634-45 [DOI] [PubMed] [Google Scholar]
- 15.Pérez-Persona E, Vidriales M-B, Mateo G, García-Sanz R, Mateos M-V, de Coca A-G, et al. New criteria to identify risk of progression in monoclonal gammopathy of uncertain significance and smoldering multiple myeloma based on multiparameter flow cytometry analysis of bone marrow plasma cells. Blood. 2007;110(7):2586-92 [DOI] [PubMed] [Google Scholar]
- 16.Paiva B, Vidriales M-B, Mateo G, Pérez J-J, Montalbán M-A, Sureda A, et al. ; GEM (Grupo Español de MM)/PETHEMA (Programa para el Estudio de la Terapéutica en Hemopatías Malignas) Cooperative Study Groups. The persistence of immunophenotypically normal residual bone marrow plasma cells at diagnosis identifies a good prognostic subgroup of symptomatic multiple myeloma patients. Blood. 2009;114(20):4369-72 [DOI] [PubMed] [Google Scholar]
- 17.Jourdan M, Ferlin M, Legouffe E, Horvathova M, Liautard J, Rossi J-F, et al. The myeloma cell antigen syndecan-1 is lost by apoptotic myeloma cells. Br J Haematol. 1998;100(4):637-46 [DOI] [PubMed] [Google Scholar]
- 18.Reid S, Yang S, Brown R, Kabani K, Aklilu E, Ho P-J, et al. Characterisation and relevance of CD138-negative plasma cells in plasma cell myeloma. Int J Lab Hematol. 2010;32(6Pt1):e190-6 [DOI] [PubMed] [Google Scholar]
- 19.Cumova J, Kovarova L, Potacova A, Buresova I, Kryukov F, Penka M, et al. Optimization of immunomagnetic selection of myeloma cells from bone marrow using magnetic activated cell sorting. Int J Hematol. 2010;92(2):314-9 [DOI] [PubMed] [Google Scholar]
- 20.Quijano S, López A, Rasillo A, Barrena S, Luz Sánchez M, Flores J, et al. Association between the proliferative rate of neoplastic B cells, their maturation stage, and underlying cytogenetic abnormalities in B-cell chronic lymphoproliferative disorders: analysis of a series of 432 patients. Blood. 2008;111(10):5130-41 [DOI] [PubMed] [Google Scholar]
- 21.Cook J-R, Hartke M, Pettay J, Tubbs R-R. Fluorescence in situ hybridization analysis of immunoglobulin heavy chain translocations in plasma cell myeloma using intact paraffin sections and simultaneous CD138 immunofluorescence. J Mol Diagn. 2006;8(4):459-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mercer B-R, Rayeroux K-C. Detection of chromosome abnormalities using cytoplasmic immunoglobulin staining and FISH in myeloma. Methods Mol Biol. 2011;730: 159-71 [DOI] [PubMed] [Google Scholar]
- 23.Rawstron A-C, Orfao A, Beksac M, Bezdickova L, Brooimans R-A, Bumbea H, et al. ; European Myeloma Network. Report of the European Myeloma Network on multiparametric flow cytometry in multiple myeloma and related disorders. Haematologica. 2008;93(3):431-8 [DOI] [PubMed] [Google Scholar]
- 24.Rasillo A, Tabernero M-D, Sánchez M-L, Pérez-Andrés M, Martín Ayuso M, Hernández J, et al. Fluorescence in situ hybridization analysis of aneuploidization patterns in monoclonal gammopathy of undetermined significance versus multiple myeloma and plasma cell leukemia. Cancer. 2003;97(3):601-9 [DOI] [PubMed] [Google Scholar]
- 25.Schmidt-Hieber M, Pérez-Andrés M, Paiva B, Flores-Montero J, Perez J-J, Gutiérrez N-C, et al. CD117 expression in gammopathies is associated with an altered maturation of the myeloid and lymphoid hematopoietic cell compartments and favorable disease features. Haematologica. 2011;96(2):328-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Paiva B, Almeida J, Pérez-Andrés M, Mateo G, López A, Rasillo A, et al. Utility of flow cytometry immunophenotyping in multiple myeloma and other clonal plasma cell-related disorders. Cytometry B Clin Cytom. 2010;78(4):239-52 [DOI] [PubMed] [Google Scholar]
- 27.Mateo G, Castellanos M, Rasillo A, Gutiérrez N-C, Montalbán M-A, Martín M-L, et al. Genetic abnormalities and patterns of antigenic expression in multiple myeloma. Clin Cancer Res. 2005;11(10):3661-7 [DOI] [PubMed] [Google Scholar]
- 28.Trakhtenbrot L, Hardan I, Koren-Michowitz M, Oren S, Yshoev G, Rechavi G, et al. Correlation between losses of IGH or its segments and deletions of 13q14 in t(11;14) (q13;q32) multiple myeloma. Genes Chromosomes Cancer. 2010;49(1): 17-27 [DOI] [PubMed] [Google Scholar]
- 29.San Miguel J-F, Gutiérrez N-C, Mateo G, Orfao A. Conventional diagnostics in multiple myeloma. Eur J Cancer. 2006;42(11): 1510-9 [DOI] [PubMed] [Google Scholar]
- 30.Avet-Loiseau H, Facon T, Grosbois B, Magrangeas F, Rapp M-J, Harousseau J-L, et al. ; Intergroupe Francophone du Myélome. Oncogenesis of multiple myeloma: 14q32 and 13q chromosomal abnormalities are not randomly distributed, but correlate with natural history, immunological features, and clinical presentation. Blood. 2002;99(6):2185-91 [DOI] [PubMed] [Google Scholar]
- 31.Neben K, Jauch A, Bertsch U, Heiss C, Hielscher T, Seckinger A, et al. Combining information regarding chromosomal aberrations t(4;14) and del(17p13) with the International Staging System classification allows stratification of myeloma patients undergoing autologous stem cell transplantation. Haematologica. 2010;95(7):1150-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chng W-J, Glebov O, Bergsagel P-L, Kuehl W-M. Genetic events in the pathogenesis of multiple myeloma. Best Pract Res Clin Haematol. 2007;20(4):571-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagy Z, Kajtár B, Jáksó P, Dávid M, Kosztolányi S, Hermesz J, et al. Evolutionary sequence of cytogenetic aberrations during the oncogenesis of plasma cell disorders. Direct evidence at single cell level. Leuk Res. 2011;35(8):1114-6 [DOI] [PubMed] [Google Scholar]
- 34.Nieto W-G, Almeida J, Romero A, Teodosio C, López A, Henriques A-F, et al. Primary Health Care Group of Salamanca for the Study of MBL Increased frequency (12%) of circulating chronic lymphocytic leukemia-like B-cell clones in healthy subjects using a highly sensitive multicolor flow cytometry approach. Blood. 2009;114(1):33-7 [DOI] [PubMed] [Google Scholar]
- 35.Almeida J, Nieto W-G, Teodosio C, Pedreira C-E, López A, Fernández-Navarro P, et al. CLL-like B-lymphocytes are systematically present at very low numbers in peripheral blood of healthy adults. Leukemia. 2011;25(4):718-22 [DOI] [PubMed] [Google Scholar]