

Abstract
BCL11A was the focus of recent studies on its inhibiting effect when bound onto the β-globin cluster in the mechanism of hemoglobin switching and HbF downregulation. We examined a cohort of 10 patients displaying different HbF levels and short deletions within the γβ-δ intergenic region to find a possible correlation with the BCL11A binding site located 5' to the δ-globin gene. Precise characterization of deletions was achieved using a custom DNA-array chip and breakpoint sequencing. The α-globin cluster and major SNP associated with HbF expression were genotyped. Our results show that the loss of the BCL11A binding domain located 5' to the δ-globin gene is correlated with a strong HbF difference (mean+2.7 g/dL, ratio 2.81). This result provides evidence for the use of BCL11A level down-regulation or this domain blockage for new therapies in sickle cell disease and β-thalassemia major patients.
Introduction
A growing number of studies describe BCL11A as a major repressor of fetal hemoglobin (HbF) expression. These include the description of several single nucleotide polymorphisms (SNPs) located in the BCL11A gene and linked to HbF level, and thus, the severity of sickle cell disease and beta-thalassemia (β-thal)1-4 and the demonstration of the direct binding of this transcription factor onto several locations in the β-globin cluster, in particular, to a region located 5' to the δ-globin gene.5 All these studies allow us to draw a network of co-operating transacting factors (KLF1, SOX6, GATA1 and BCL11A) involved in the switch between fetal and adult hemoglobin (Hb). A recent study6 using the analysis of large deletions has defined a critical area located 5' to the δ-globin gene that appears to exert the main repressor activity of BCL11A.
However, if the role of the BCL11A binding region is now well documented, no studies have evaluated the consequence of the deletion of that region in terms of HbF output. We report the precise analysis of the correlation between phenotype and genotype of patients displaying a short deletion (under 25 kb) differing at the 5' δ-globin gene breakpoint. The short length of the deletions found in our patients makes these events not comparable with hereditary persistence of fetal Hb (HPFH) deletions whose mechanism is likely to bring the 3'HS (3' hypersensitive site) activator site close to the γ-globin genes, and thus enhances HbF expression.7
Design and Methods
A cohort of 10 patients was selected from our DNA bank of patients referred to our laboratory for the molecular analysis of a β-thal phenotype. Prior to sampling, patients received genetic counseling and gave their written, informed consent to the study. The selected patients were heterozygous carriers of a short deletion featuring the 5' breakpoint within the γβ-δ intergenic region. The phenotypic analysis of patients included red blood cell (RBC) count carried out on an automated cell counter, and Hb study using cation exchange high performance liquid chromatography (HPLC) (VARIANT II™; Bio-Rad Laboratories, Hercules, CA, USA).8 The results are shown in Table 1. Relevant hematologic data were obtained to verify that no patient had received a blood transfusion which could compromise data interpretation. This retrospective study was approved by the Institutional Review Board of the Henri Mondor Hospital, Créteil, France.
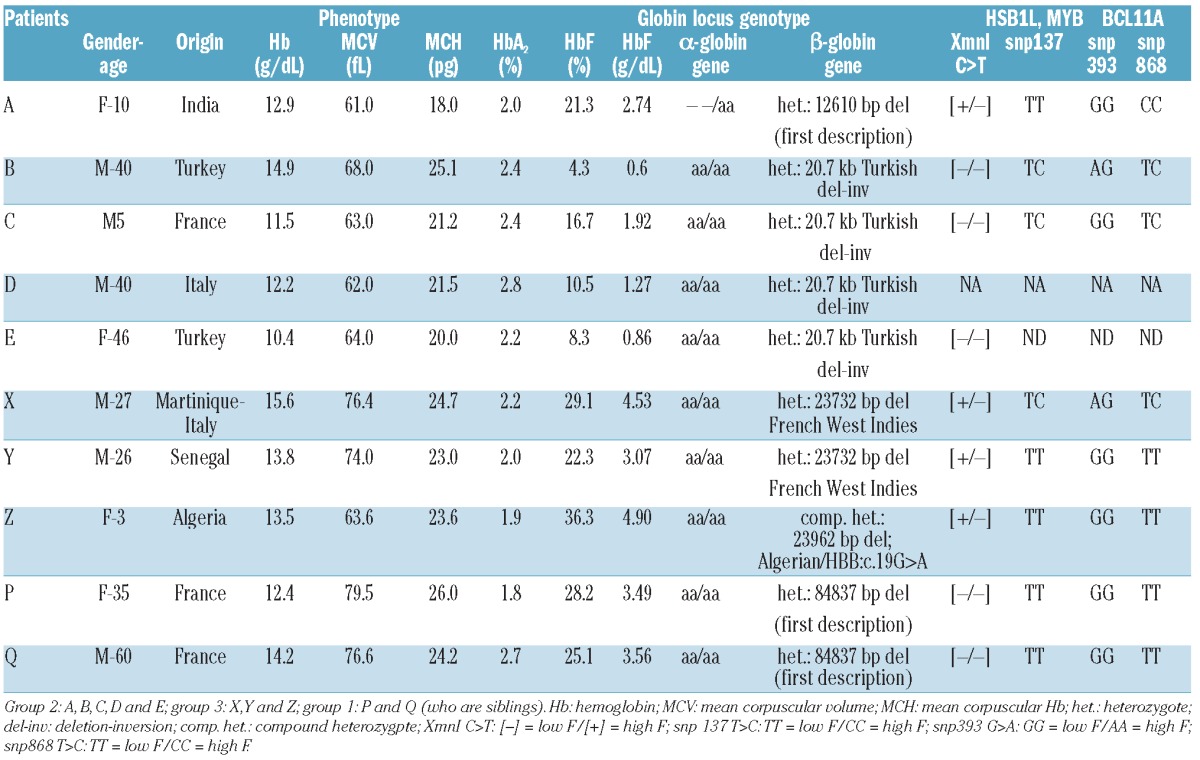
Table 1.
Phenotypic and genotypic data of the studied patients.
The molecular analysis of the β-globin cluster was achieved including deletion detection using the quantitative multiplex polymerase chain reaction of short fragment (QMPSF) procedure.9 Precise localization of unknown breakpoints was determined using a custom CGH-array chip with high probe density on the β- and α-globin clusters,10 and achieved by amplification then sequencing, using primers extrapolated from probes flanking the deletions (Online Supplementary Figures S1-S6).
In order to evaluate any correlation as precisely as possible, genotyping of the α-globin cluster and of the major SNPs associated with high HbF was carried out (i.e. BCL11A rs4671393 and rs11886868; HSB1L-MYB rs9399137; HB rs7482144-XMN1 Gγ polymorphic site) (Online Supplementary Figure S6).
We also ensured that neither a mutation/deletion in the BCL11A gene nor a sequence modification within the three BCL11A binding sites described in the β-globin cluster 5 was present (Online Supplementary Tables S1-S4).
Statistical analyses were carried out using the Mann-Whitney U test (non-parametric statistical analysis).
Results and Discussion
Results of phenotype analysis and molecular studies are reported in Table 1. The studied patients can be divided into three groups.
The first group to be considered includes Patients P and Q. They were identified as carriers of a new 84.8 kb deletion (84837 bp del, NC_000011.9:g.5175177_5260013del84837) not described before nor registered in HbVar (globin.cse.psu.edu/hbvar), and the breakpoints have not been found in GenBank.
This deletion overlaps with Black HPFH-1 (NC_000011.9:g.5174451_5259368del84918) and thus the HbF rate observed in these patients is most likely caused by the same molecular configuration as that of HPFH-1, i.e. the proximity of the 3'HS activatory region close to the γ-globin genes.7 Even if it removed the BCL11A binding domain located 5' to the δ-globin gene, it would not be possible to differentiate between the mechanism involved in deletional HPFH and the effect of the BCL11A domain loss.
The second group includes Patients A to E. They carry deletions that do not remove the 5' δ-globin BCL11A binding domain (Figure 1). Patient A carries a 12.6 kb deletion, described for the first time (12610 bp del, NC_000011.9:g.5241853_5254462del12610), extending from the δ-globin gene IVS-II to 4 kb downstream of the β-globin gene (Figure 1). This patient was also found to be heterozygous for a large α0 deletion (- -/αα). Patients B, C, D and E are heterozygous for the rare 20.7 kb Turkish deletion/inversion, described only once before in an Anatolian family.10 The phenotype of these patients was not precisely reported. Patients A to E have an average HbF expression of 1.48 g/dL (SD 0.85; n=5). Patient A displayed the highest level of HbF (2.74 g/dL). The analysis of the reasons that could explain this difference includes the complex network of factors involved in HbF expression and some features of the patient which are in favor of a higher HbF level: Patient A is heterozygous for the 5' Gγ XmnI SNP which other patients in this group do not carry, and has a higher number of RBCs (7.16×109/L vs. an average 5.64×1 09/L; SD 0.47, n=4) than the other patients.
Figure 1.
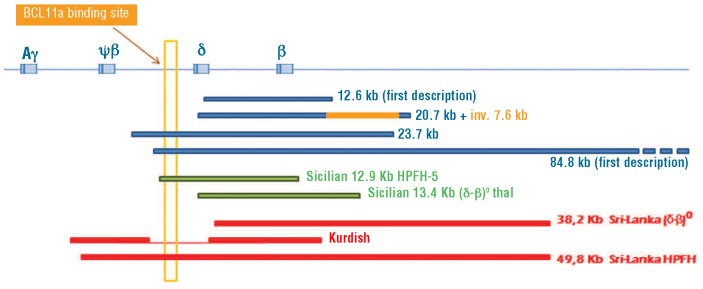
Comparison of the extent of the four deletions found in our study along with the three deletions shown in Sankaran et al.,6 the Sicilian (δβ)0 13.4 kb deletion and the Sicilian HPFH-5 12 kb deletion. The BCL11A binding-site laying 1.5 kb 5' δ-globin gene (yellow vertical rectangle) is removed for 23.7 kb deletion as in the 49.8 kb Sri Lanka deletion6 and the Sicilian HPFH-5 deletion, while it is maintained for 12.6 kb, 20.7 kb Turkish deletion-inversion, Sri Lanka (δβ)0 and Sicilian (δβ)0 deletions.
The third group includes Patients X to Z. They carry recently described deletions removing the the 5' δ-globin BCL11A binding domain (Figure 1): the French West-Indies deletion or the Algerian deletion.10 The average HbF level found in these patients was 4.16 g/dL (SD 0.96, n=3) and the highest values of HbF (5.24 and 4.10 g/dL) previously reported for these deletions11 is in agreement with our findings.
Thus, these two groups allowed us to estimate an evaluation of the repressive action of BCL11A when bound onto the 5' δ-binding site. Like in other similar cases, the Sicilian (δβ)0 deletion of 13.4 kb, which is a short deletion frequently found with breakpoints close to that of the deletions reported here but not removing the BCL11A domain, is also associated with low HbF levels (ES Ghedira et al., personal data, 2012: average HbF 1.21 g/dL; SD=0.47, n=6).
On the other hand, the Sicilian HPFH-5 deletion, which is also a short range (δβ)0 deletion (12 kb) but removing the BCL11A 5' δ-domain, is associated with high HbF. These data give further evidence of the direct functional importance of this domain.
The genotyping of HbF-modifying SNPs in all our patients did not show any evidence of a haplotype supposed to be clearly in favor and correlated with a high HbF expression. Thus, we could hypothesize that the main difference between the Patient A-E group (1.48 g/dL; n=5) and the Patient X-Z group (4.16 g/dL; n=3) is only the removal of the 5' δ-globin gene BCL11A binding domain, inducing a significant mean HbF difference of 2.7 g/dL (Mann-Whitney U test; P<0.05).
Our results are consistent with a recent study6 which reported three deletions within the β-globin locus that demonstrates the functional role of the 5' δ-globin gene BCL11A binding site in silencing the γ-globin. In our study, the extent of the shorter deletions found, and the absence of several studied HbF modifiers, make the hypothesis of the major importance of this domain stronger. Furthermore, the literature to date cites that most of the deletions described as HPFH feature this domain loss at the 5' breakpoint, whatever its 3' end (i.e. the 12.9 kb Sicilian HPFH-5).
The HbF expression rate is one of the main modulating factors of several frequent red cell diseases and decades of research into the elements involved in HbF-silencing have retrieved numerous regions and transacting sequences supposed to play a role in this phenomenon.12 The discovery of BCL11A focused the research on that transinhibitor factor. Our study, together with recent publications, demonstrate that its binding domain, located 5' to the δ-gene, is a major player in the complex network of factors involved in HbF expression. We found that the loss of this region is associated with an increase of 2.7 g/dL of HbF, which would be sufficient to compensate the Hb lack in β-thal major patients13 and would be sufficient to prevent sickle cell anemia complications. These findings would help to focus efforts towards defining new therapeutic strategies through BCL11A silencing or preventing its binding for HbF reactivation.
Acknowledgments
The authors would like to thank Professor Michel Goossens, Chief, Molecular Genetics Department, Henri Mondor Hospital APHP, Créteil, France; Kinda Atassi (Pharm. D.), from the pharmacy department Henri Mondor Hospital APHP, Créteil, France, for technical support ; Alain Zider, Ph.D., from the Genetics Department at the Paris Diderot Univesity, Paris, France; and Mrs. Marianne F.H. Carver, Evans, GA, USA, for editing the manuscript.
Footnotes
The online version of this article has a Supplementary Appendix.
Authorship and Disclosures: Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Menzel S, Garner C, Gut I, Matsuda F, Yamaguchi M, Heath S, Foglio M, et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat Genet. 2007; 39:1197-9 [DOI] [PubMed] [Google Scholar]
- 2.Uda M, Galanello R, Sanna S, Lettre G, Sankaran VG, Chen W, et al. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of β-thalassemia. Proc Natl Acad Sci USA. 2008; 105:1620-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lettre G, Sankaran VG, Bezerra MA, Araújo AS, Uda M, Sanna S, et al. DNA polymorphisms at the BCL11A, HBS1L-MYB, and δ-dglobin loci associate with fetal hemoglobin levels and pain crises in sickle cell disease. Proc Natl Acad Sci USA. 2008;105:11869-74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galarneau G, Palmer CD, Sankaran VG, Orkin SH, Hirschhorn JN, Lettre G. Fine-mapping at three loci known to affect fetal hemoglobin levels explains additional genetic variation. Nat Genet. 2010;42:1049-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sankaran VG, Menne TF, Xu J, Akie TE, Lettre G, Van Handel B, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322:1839-42 [DOI] [PubMed] [Google Scholar]
- 6.Sankaran VG, Xu J, Byron R, Greisman HA, Fisher C, Weatherall DJ, et al. A functional element necessary for fetal hemoglobin silencing. N Engl J Med. 2011;365:807-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feingold EA, Forget BG. The breakpoint of a large deletion causing hereditary persistence of fetal hemoglobin occurs within an ery-throid DNA domain remote from the β-glo-bin gene cluster. Blood. 1989;74:2178-86 [PubMed] [Google Scholar]
- 8.Wilson JB, Huisman THJ. Detection and quantitation of normal and abnormal hemoglobins in adults and newborn by HPLC. In: Huisman THJ, ed. The Hemoglobinopathies. Methods in Hematology, Vol. 15. Edinburgh: Churchill Livingstone; 1986:32-46 [Google Scholar]
- 9.Casilli F., Di Rocco ZC., Gad S., Tournier I., Stoppa-Lyonnet D., Frebourg T, et al. , rapid detection of novel BRCA1rearrangements in high-risk breast-ovarian cancer families using short fluorescent fragments. Human Mutation. 2002;20:218-26 [DOI] [PubMed] [Google Scholar]
- 10.Kulozik AE, Bellan-Koch A, Kohne E, Kleihauer E. A deletion/inversion rearrangement of the β-globin gene cluster in a Turkish family with δβ0-thalassemia intermedia. Blood. 1992;79:2455-9 [PubMed] [Google Scholar]
- 11.Joly P, Lacan P, Garcia C, Couprie N, Francina A. Identification and molecular characterization of four new large deletions in the β-glo-bin gene cluster. Blood Cells Mol Dis 2009;43:53-7 [DOI] [PubMed] [Google Scholar]
- 12.Badens C, Joly P, Agouti I, Thuret I, Gonnet K, Fattoum S, et al. Variants in genetic modifiers of β-thalassemia can help to predict the major or intermedia type of the disease. Haematologica. 2011;96:1712-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilber A, Nienhuis AW, Persons DA. Transcriptional regulation of fetal to adult hemoglobin switching: new therapeutic opportunities. Blood. 2011;117:3945-53 [DOI] [PMC free article] [PubMed] [Google Scholar]