

Abstract
The heart's pumping capacity results from highly regulated interactions of actomyosin molecular motors. Mutations in the gene for a potential regulator of these motors, cardiac myosin binding protein-C (cMyBP-C), cause hypertrophic cardiomyopathy. However, cMyBP-C's ability to modulate cardiac contractility is not well understood. Using single particle fluorescence imaging techniques, transgenic protein expression, proteomics, and modeling, we found that cMyBP-C slowed actomyosin motion generation in native cardiac thick filaments. This mechanical effect was localized to where cMyBP-C resides within the thick filament (i.e. the C-zones) and was modulated by phosphorylation and site-specific proteolytic degradation. These results provide molecular insight into why cMyBP-C should be considered a member of a tripartite complex with actin and myosin that allows fine tuning of cardiac muscle contraction.
Main Text
Cardiac muscle's pumping capacity is produced by the sarcomere (Fig. 1A), a parallel array of proteins assembled into thick filaments, composed of myosin molecular motors that cyclically interact with actin-containing thin filaments, generating force that propels the thin filaments past the thick filaments. These actomyosin interactions can be modulated on a beat to beat basis (Fig. 1B) by cardiac myosin binding protein-C (cMyBP-C) (Fig. 1C), a 140 kDa immunoglobulin (Ig) protein superfamily member that is confined to two distinct regions (i.e. C-zones) of the thick filament (1, 2) (Fig. 1A). Mutations in the MYBPC3 gene are a leading cause of familial hypertrophic cardiomyopathy (FHC) (1, 2). Proposed mechanisms of cMyBP-C's function assume that several Ig-like domains and their linkers (C0–C2, Fig. 1C) extend away from the thick filament backbone (Fig. 1B) (3) and reversibly bind to myosin's motor domains and/or actin filaments (1, 2), with this binding tunable by phosphorylation of four serines (S273, S282, S302, and S307) in the motif linker between domains C1 and C2 (Fig. 1C) (4, 5). Insight into cMyBP-C's function and regulation by phosphorylation has benefited from intact heart and muscle fiber studies, but these complex preparations make molecular level interpretations difficult. Isolated protein studies, although simpler, lack the sarcomere's spatial relationship between the thin and thick filaments. Here, we developed an in vitro sarcomere model system in which single actin filaments could be visualized moving over native cardiac thick filaments with and without cMyBP-C.
Fig. 1.
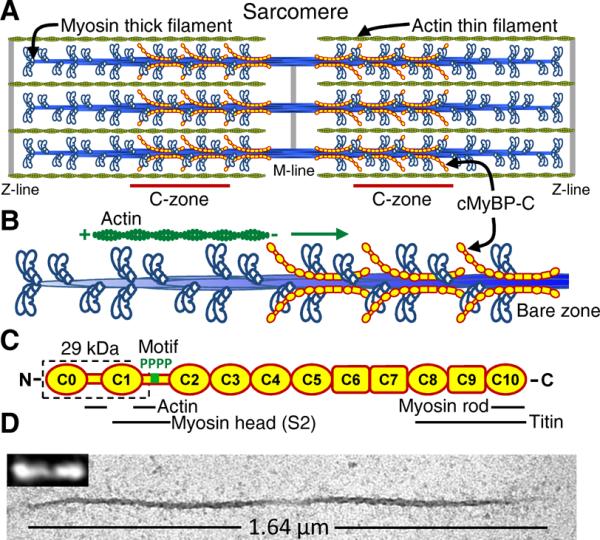
Native cardiac thick filaments and cMyBP-C. (A) Cardiac muscle sarcomere with interdigitating thick and thin filaments with cMyBP-C localized to thick filament C-zones. (B) Illustration of one half of thick filament with an actin filament traveling towards bare zone as in experiments. (C) Schematic diagram of cMyBPC's Ig-like (oval) and fibronectin (rectangle) domains, with 4 phosphorylation sites (P) in motif linker, and 29kDa fragment (dashed box). Sarcomeric protein domain interactions identified. (D) Native cardiac thick filament imaged by electron microscopy and TIRFM (inset) by effectively labeling the myosin heads with fluorescent ATP (6).
Cardiac thick filaments that retained their native length (~1.6 μm), bipolar structure, and central bare zone devoid of myosin heads (Fig. 1D) were isolated from mouse hearts by fine tissue dissection and limited enzyme-induced protein degradation (0.2 U/μl μ-calpain) (6).
Quantitative liquid chromatography-mass spectrometry (LC-MS) (6) showed that filaments contained the normal complement of cMyBP-C (Fig. S1). Because cMyBP-C is a target for calpain-mediated protein degradation (8), protein immunoblotting with domain specific antibodies was used to quantify that 79 ± 4% (SD, N = 3) of the cMyBP-C molecules were intact (Fig. S2), and in combination with LC-MS analyses we determined that the remainder of the molecules had 29 kDa of their N-terminus removed (i.e. C0–C1 plus 17 amino acids of the motif (Fig. 1C); C0C1f) by cleavage between amino acids R266 and R271 (Fig. S3).
To assess cMyBP-C's mechanical impact on actin filament sliding, native cardiac thick filaments were adhered to a microscope coverslip. Fluorescently-labeled actin filaments were then introduced onto the coverslip (25 mM KCl, 100 μM ATP, 22 °C) and their sliding along the thick filaments tracked (6) with high time (8.3 ms) and spatial (30 nm) resolution (Fig. 2A). The use of short (250 ± 9 nm (SEM)) actin filaments (Fig. S4) prevented these filaments from spanning the bare zone (Fig. S5), allowing us to probe one half of the thick filament where the actomyosin interactions were oriented as in the sarcomere (Fig. 1B). To ensure that a given actin filament traversed regions of the thick filament with and without cMyBP-C, we only analyzed trajectories greater than the C-zone length (i.e. ~350 nm (8)), averaging 658 ± 8.7 nm (SEM) before the actin filament diffused away from the thick filament. Displacement versus time traces for these trajectories were characterized by two different modes of travel. The majority (73%) displayed an initial fast velocity followed by a 45% slower velocity (Fig. 2B and 2D; Table 1), whereas the remaining 27% of trajectories (Fig. 2C and 2E; Table 1) had a constant velocity for the entire encounter, no different than the faster velocity observed for actin filaments that displayed two velocity phases. The distance traveled during the slow velocity phase (286 ± 57 nm; SD, N = 58) being similar to the C-zone length suggested that cMyBP-C had its effect restricted to the C-zone. To confirm this, we isolated thick filaments from cMyBP-C null mice (9), which displayed only a single fast velocity (Table 1). Thus, cMyBP-C slowed actomyosin motion generation only within the C-zone, providing a molecular basis for sudden reductions in unloaded velocity observed in skeletal muscle fibers (10).
Fig. 2.

Effect of cMyBP-C on actin motility. (A) TIRFM image series of actin shard moving along a native thick filament. (B) Displacement-time plots for 5 actin filaments on wild-type thick filaments demonstrated two velocity phases (fast, blue; slow, red). Inset for 20 filaments with distance traveled during slow velocity phase identified. (C) As in B except actin filaments exhibited constant velocities. (D) Frequency-velocity histograms and Gaussian fits for actin trajectories as in B (N = 58; fast phase, blue; slow phase, red). (E) Frequency-velocity histogram and Gaussian fit for actin trajectories as in C with constant velocities (N = 21). (F) Spatial relations for an analytic model where actin filaments (green) moved over a thick filament with myosin crowns at the same azimuthal position separated by 43 nm and cMyBP-C localized in the C-zone (red highlighted crowns 3–11; (8)). Actin detached upon reaching the bare zone (crown 0). (G) Model-generated displacement-time plots for a 250 nm actin filament on a thick filament with 78% intact cMyBP-C, as in B. Fast phase = 1.98 ± 0.03 μm/s (SEM), slow phase = 1.15 ± 0.02 μm/s (SEM), N = 5 (inset, N = 20, slow phase travel distance identified). (H) As in G but for a theoretical thick filament with 10% intact cMyBP-C that showed only a constant velocity (2.06 ± 0.01 μm/s (SEM), N = 5; inset, 20 runs). (I) Inhibition of actin sliding velocity by C0C1f and C0C3 fragments (mean ± SD) in motility assay. (J) Effect of motif phosphorylation on actin filament velocity inhibition by C0C3 in motility assay or by cMyBP-C in native thick filaments. Percent phosphorylation was defined as the average percent phosphorylation at S273, S282, S302, S307. Percent inhibition of motility assay data was normalized to thick filament inhibition data, where inhibition is the percent velocity reduction compared to control without cMyBP-C or fragment.
Table 1.
Velocities measured on native cardiac thick filaments.
| Runs with slowing | Runs without slowing | |||||
|---|---|---|---|---|---|---|
| Thick filament source | Calpain (U/μl) | Initial velocity (μm/s ± SE) | C-zone velocity (μm/s ± SE) | N | Velocity (μm/s ± SE) | N |
| Wild-type | 0.2 | 1.89 ± 0.06 | 1.04 ± 0.05 | 58 | 1.81 ± 0.09 | 21 |
| cMyBP-C null | 0.2 | 0 | 1.72 ± 0.05 | 55 | ||
| Wild-type | 1.2 | 2.02 ± 0.08 | 1.27 ± 0.07* | 22 | 1.94 ± 0.05 | 87 |
| Wild-type: PKA-treated | 0.2 | 1.87 ± 0.06 | 0.92 ± 0.04 | 65 | 1.78 ± 0.09 | 31 |
| All P+ | 0.2 | 1.91 ± 0.07 | 1.05 ± 0.04 | 58 | 1.76 ± 0.07 | 27 |
| Wild-type: λ phosphatase-treated | 0.2 | 1.77 ± 0.05 | 0.69 ± 0.05* | 61 | 1.80 ± 0.09 | 20 |
Significant difference in C-zone velocity when compared to WT, p < 0.01, Mann-Whitney-Wilcoxon.
To explain the various modes of travel on wild-type thick filaments, we implemented an analytical model to predict the velocity generated by two spatially distinct populations of myosin molecules (i.e. within the C-zone and outside of it) that mechanically interacted to propel a 250 nm actin filament (Fig. 2F). The predicted trajectories showed a 50% reduction in velocity for 334 ± 50 nm (SD; N = 20) (Fig. 2G), nearly equal to that of the C-zone length and that observed experimentally (Fig. 2B). The model also predicted that given our spatial resolution, trajectories would be described by a single velocity once the abundance of intact cMyBP-C within the thick filament fell between 10–20% (Fig. 2H, Fig. S6). Assuming that removal of cMyBP-C's N-terminal 29 kDa fragment effectively eliminated cMyBP-C's mechanical impact, then the 27% of actin trajectories over wild-type thick filaments with a single velocity (Fig. 2C and 2E) most likely originated from thick filaments with a higher complement of truncated cMyBP-C. If so, then increasing the content of N-terminally cleaved cMyBP-C should increase the percentage of single velocity trajectories, which was the case.
Following increased enzymatic degradation (1.2 U/μl μ-calpain), 70 ± 10% (SD, N = 3) of the cMyBP-C had its N-terminus removed (Fig. S2), correspondingly 80% of actin trajectories were described by a single velocity (Table 1). For the remaining 20% of actin trajectories that displayed two velocity phases, the faster velocity was unaffected by the increased N-terminal degradation, whereas the reduction in velocity within the C-zone was not as pronounced (i.e. 37%, Table 1), due to at least 50% of the cMyBP-C molecules being cleaved (based on model predictions; Fig. S6). Thus, the 29 kDa C0C1f domain probably mediated the slowing of velocity to a large extent. Indeed, bacterially-expressed C0C1f was able to inhibit actin filament movement over a surface of monomeric mouse cardiac myosin in an in vitro motility assay (Fig. 2I). Given that C0C1f stereospecifically and reversibly binds actin (11, 12), this portion of the protein may tether the actin filament to the motility surface and act as a viscous load (6) to slow myosin's motion generation (12). Alternately, C0C1f could slow actin movement by directly binding to myosin, altering myosin's kinetics of motion generation (10, 13, 14).
β-adrenergic stimulated motif phosphorylation and its effect on cMyBP-C is believed to be a major contributor to enhanced cardiac contractility (15–17). We used LC-MS to quantify the degree of phosphorylation at each of the motif's phosphorylated serines (6), and varied phosphorylation by either kinase/phosphatase treating wild-type thick filaments or isolating filaments from transgenic mice (AllP+; 18) that expressed mutant cMyBP-C in which S273, S282, and S302 were replaced by phosphomimetic aspartic acids with S307 phosphorylated endogenously. The high phosphorylation levels observed in the wild-type thick filaments (Table 2), agreed with that found in healthy hearts from mice and humans (19, 20). Despite a modest increase in phosphorylation (<20%) observed when thick filaments were treated with protein kinase A (PKA) or isolated from AllP+ transgenic mice (Table 2), actin filament trajectories were indistinguishable from non-treated wild-type thick filaments (Table 1). In contrast, substantial dephosphorylation following lambda phosphatase treatment (Table 2) resulted in actin velocities within the C-zone that were 61% slower than the fast phase (Table 1), and suggested that dephosphorylated cMyBP-C was a more potent inhibitor of actomyosin motion generation.
Table 2.
Degree of cMyBP-C phosphorylation quantified by LC-MS.
| Amino acid | Phosphopeptide observed | Wild-type | Wild-type: PKA-treated | All P+ | Wild-type: λ phosphatase-treated |
|---|---|---|---|---|---|
| % phos ± SD (N = 4) | % phos ± SD (N = 4) | % phos ± SD (N = 3) | % phos ± SD (N = 2) | ||
| S273 | 271RTSpLAGAGR279 | 73 ± 8 | 66 ± 5 | 100* | 3 |
| S282 | 280RTSpDSHEDAGTLDFSSLLK298 | 90 ± 3 | 85 ± 2 | 100* | 5 |
| S302 | 299KRDSpFR304 | 37 ± 18 | 73 ± 16 | 100* | 47 |
| S307 | 305RDSpKLEAPAEEDVWEILR322 | 57 ± 6 | 82 ± 5 | 29 ± 6 | 32 |
| Average | 64 ± 20 | 76 ± 16 | 82 ± 6 | 22 |
Phos = phosphorylation. Average = (S273+S282+S302+S307) / 400 ± error propagation.
Amino acid was replaced with aspartic acid to mimic phosphorylation.
To better define the relationship between cMyBP-C phosphorylation and inhibition of actomyosin motility, we bacterially-expressed a longer N-terminal cMyBP-C fragment (C0C3) that contained the entire motif with its four phosphorylatable serines, and was as inhibitory as C0C1f in the motility assay (Fig. 2I). Mutant C0C3 with the four serines replaced with combinations of alanines and aspartic acids (as phosphomimetics) were expressed, which provided 25%, 50%, 75%, and 100% phosphorylation levels (6). Increased levels of phosphomimetic substitution resulted in a proportional reduction in C0C3's inhibitory effect, similar to that observed in the limited thick filament data set (Fig. 2J). A potential mechanism for this effect is that phosphorylation alters the motif's intrinsically disordered structure (21) and being directly connected to the 29 kDa domain, may limit the C0C1f's spatial freedom to bind actin and/or myosin. Thus, the phosphorylation-dependent increase in velocity within the thick filament C-zone should contribute to both the increased unloaded shortening velocity (15) and faster tension recovery following stretch-activation (16, 17) observed in mouse myocardial preparations after PKA-treatment.
Here we found a mechanical role for cMyBP-C in modulating cardiac contractility even when restricted to the C-zone. Despite being spatially localized within the sarcomere, cMyBP-C's effective load should be transmitted through the 1 μm long thin filaments to all attached myosins (Fig. 1A). Normal cardiac structure and function may rely on cMyBP-C's internal load to act as a governor, lowering power output and energy utilization because sustained power elevation in cMyBP-C null mice leads to cardiac hypertrophy (21, 22). Thus, cardiac hypertrophy in FHC patients with cMyBP-C haploinsufficiency (2) may be a secondary response to the reduced content of cMyBP-C, which could lead to a hypercontractile heart. In contrast, excessive dephosphorylation of cMyBP-C, which is associated with cardiac ischemia (7, 23) and failure (20), would lead to reduced power output based on data presented here. Because dephosphorylated cMyBP-C is highly susceptible to proteolytic cleavage (7, 23), the increased presence of the 29 kDa fragment in the plasma of patients and animal models with heart failure (7), points to N-terminal cleavage and its effective reduction in the number of functional cMyBP-C being a compensatory mechanism to restore cardiac power to more normal levels. Thus, cMyBP-C provides a measure of contractile tunability to a fully active muscle.
Supplementary Material
Acknowledgements
National Institutes of Health funds supported M.P. (HL07647); J.G., J.R., and D.W. (HL059408) and the Vermont Genetics Network for the LC-MS instrumentation (8P20GM103449). We thank B. Palmer and Y. Wang for mouse colony management, S. Tremble for technical assistance, M. Jennings for LC-MS expertise, M. Von Turkovich and the UVM Microscopy Imaging Center for EM assistance, G. Kennedy, from the Instrumentation and Modeling Facility, for imaging expertise. Data described in the paper are presented in the Supporting Online Material.
Footnotes
This manuscript has been accepted for publication in Science. This version has not undergone final editing. Please refer to the complete version of record at http://www.sciencemag.org/. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of AAAS.
References and Notes
- 1.Winegrad S. Cardiac myosin binding protein C. Circ Res. 1999;84:1117. doi: 10.1161/01.res.84.10.1117. [DOI] [PubMed] [Google Scholar]
- 2.Harris SP, Lyons RG, Bezold KL. In the thick of it: HCM-causing mutations in myosin binding proteins of the thick filament. Circ Res. 1999;108:751. doi: 10.1161/CIRCRESAHA.110.231670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luther PK, et al. Direct visualization of myosin-binding protein C bridging myosin and actin filaments in intact muscle. Proc Natl Acad Sci U S A. 2011;108:11423. doi: 10.1073/pnas.1103216108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gautel M, Zuffardi O, Freiburg A, Labeit S. Phosphorylation switches specific for the cardiac isoform of myosin binding protein-C: a modulator of cardiac contraction? EMBO J. 1995;14:1952. doi: 10.1002/j.1460-2075.1995.tb07187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jia W, Shaffer JF, Harris SP, Leary JA. Identification of novel protein kinase A phosphorylation sites in the M-domain of human and murine cardiac myosin binding protein-C using mass spectrometry analysis. J Proteome Res. 2010;9:1843. doi: 10.1021/pr901006h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Materials and methods are available as supplementary materials on Science Online.
- 7.Govindan S, et al. Cardiac myosin binding protein-C is a potential diagnostic biomarker for myocardial infarction. J Mol Cell Cardiol. 2012;52:154. doi: 10.1016/j.yjmcc.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luther PK, et al. Understanding the organisation and role of myosin binding protein C in normal striated muscle by comparison with MyBP-C knockout cardiac muscle. J Mol Biol. 2008;384:60. doi: 10.1016/j.jmb.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zoghbi ME, Woodhead JL, Moss RL, Craig R. Three-dimensional structure of vertebrate cardiac muscle myosin filaments. Proc Natl Acad Sci U S A. 2008;105:2386. doi: 10.1073/pnas.0708912105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hofmann PA, Greaser ML, Moss RL. C-protein limits shortening velocity of rabbit skeletal muscle fibres at low levels of Ca2+ activation. J Physiol. 1991;439:701. doi: 10.1113/jphysiol.1991.sp018689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mun JY, et al. Electron microscopy and 3D reconstruction of F-actin decorated with cardiac myosin-binding protein C (cMyBP-C) J Mol Biol. 2011;410:214. doi: 10.1016/j.jmb.2011.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weith A, et al. Unique single molecule binding of cardiac myosin binding protein-C to actin and phosphorylation-dependent inhibition of actomyosin motility requires 17 amino acids of the motif domain. J Mol Cell Cardiol. 2012;52:219. doi: 10.1016/j.yjmcc.2011.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Razumova MV, et al. Effects of the N-terminal domains of myosin binding protein-C in an in vitro motility assay: Evidence for long-lived cross-bridges. J Biol Chem. 2006;281:35846. doi: 10.1074/jbc.M606949200. [DOI] [PubMed] [Google Scholar]
- 14.Ratti J, Rostkova E, Gautel M, Pfuhl M. Structure and interactions of myosin-binding protein C domain C0: cardiac-specific regulation of myosin at its neck? J Biol Chem. 2011;286:12650. doi: 10.1074/jbc.M110.156646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sadayappan S, et al. Cardiac myosin binding protein-C phosphorylation in a β-myosin heavy chain background. Circulation. 2009;119:1253. doi: 10.1161/CIRCULATIONAHA.108.798983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stelzer JE, Patel JR, Moss RL. Protein kinase A-mediated acceleration of the stretch activation response in murine skinned myocardium is eliminated by ablation of cMyBPC. Circ Res. 2006;99:884. doi: 10.1161/01.RES.0000245191.34690.66. [DOI] [PubMed] [Google Scholar]
- 17.Tong CW, Stelzer JE, Greaser ML, Powers PA, Moss RL. Acceleration of crossbridge kinetics by protein kinase A phosphorylation of cardiac myosin binding protein C modulates cardiac function. Circ Res. 2008;103:974. doi: 10.1161/CIRCRESAHA.108.177683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sadayappan S, et al. Cardiac myosin binding protein C phosphorylation is cardioprotective. Proc Natl Acad Sci U S A. 2006;103:16918. doi: 10.1073/pnas.0607069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sadayappan S. Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circ Res. 2005;97:1156. doi: 10.1161/01.RES.0000190605.79013.4d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jacques AM, et al. Myosin binding protein C phosphorylation in normal, hypertrophic and failing human heart muscle. J Mol Cell Cardiol. 2008;45:209. doi: 10.1016/j.yjmcc.2008.05.020. [DOI] [PubMed] [Google Scholar]
- 21.Karsai A, Kellermayer MS, Harris SP. Mechanical unfolding of cardiac myosin binding protein-C by atomic force microscopy. Biophys J. 2011;101:1968. doi: 10.1016/j.bpj.2011.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Korte FS, McDonald KS, Harris SP, Moss RL. Loaded shortening, power output, and rate of force redevelopment are increased with knockout of cardiac myosin binding protein-C. Circ Res. 2003;93:752. doi: 10.1161/01.RES.0000096363.85588.9A. [DOI] [PubMed] [Google Scholar]
- 23.Decker RS, et al. Myosin-binding protein C phosphorylation, myofibril structure, and contractile function during low-flow ischemia. Circulation. 2005;111:906. doi: 10.1161/01.CIR.0000155609.95618.75. [DOI] [PubMed] [Google Scholar]
- 24.Palmer BM, et al. Roles for cardiac MyBP-C in maintaining myofilament lattice rigidity and prolonging myosin cross-bridge lifetime. Biophys J. 2011;101:1661. doi: 10.1016/j.bpj.2011.08.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Debold EP, et al. Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse alpha-cardiac myosin in the laser trap assay. Am J Physiol Heart Circ Physiol. 2007;293:H284. doi: 10.1152/ajpheart.00128.2007. [DOI] [PubMed] [Google Scholar]
- 26.Silva JC, Gorenstein MV, Li GZ, Vissers JP, Geromanos SJ. Absolute quantification of proteins by LCMSE: a virtue of parallel MS acquisition. Mol Cell Proteomics. 2006;5:144. doi: 10.1074/mcp.M500230-MCP200. [DOI] [PubMed] [Google Scholar]
- 27.Harada Y, Sakurada K, Aoki T, Thomas DD, Yanagida T. Mechanochemical coupling in actomyosin energy transduction studied by in vitro movement assay. J Mol Biol. 1990;216:49. doi: 10.1016/S0022-2836(05)80060-9. [DOI] [PubMed] [Google Scholar]
- 28.Sage D, Neumann FR, Hediger F, Gasser SM, Unser M. IEEE Trans Image Process. 2005;14:1372. doi: 10.1109/tip.2005.852787. [DOI] [PubMed] [Google Scholar]
- 29.Harris DE, Work SS, Wright RK, Alpert NR, Warshaw DM. Smooth, cardiac and skeletal muscle myosin force and motion generation assessed by cross-bridge mechanical interactions in vitro. J Muscle Res Cell Motil. 1994;15:11. doi: 10.1007/BF00123828. [DOI] [PubMed] [Google Scholar]
- 30.Kaya M, Higuchi H. Nonlinear elasticity and an 8-nm working stroke of single myosin molecules in myofilaments. Science. 2010;329:686. doi: 10.1126/science.1191484. [DOI] [PubMed] [Google Scholar]
- 31.Kulikovskaya I, McClellan G, Levine R, Winegrad S. Effect of MyBP-C binding to actin on contractility in heart muscle. Am J Physiol Heart Circ Physiol. 2003;285:H857. doi: 10.1152/ajpheart.00841.2002. [DOI] [PubMed] [Google Scholar]
- 32.Greenberg MJ, Moore JR. The molecular basis of frictional loads in the in vitro motility assay with applications to the study of the loaded mechanochemistry of molecular motors. Cytoskeleton (Hoboken) 2010;67:273. doi: 10.1002/cm.20441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sellers JR, Kachar B. Polarity and velocity of sliding filaments: control of direction by actin and of speed by myosin. Science. 1990;249:406. doi: 10.1126/science.2377894. [DOI] [PubMed] [Google Scholar]
- 34.Scholz T, Brenner B. Actin sliding on reconstituted myosin filaments containing only one myosin heavy chain isoform. J Muscle Res Cell Motil. 2003;24:77. doi: 10.1023/a:1024871825135. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.