

Abstract
Brown Norway rats (BN, BN/NHsdMcwi) are profoundly resistant to developing acute kidney injury (AKI) following ischemia reperfusion. To help define the genetic basis for this resistance, we used consomic rats, in which individual chromosomes from BN rats were placed into the genetic background of Dahl SS rats (SS, SS/JrHsdMcwi) to determine which chromosomes contain alleles contributing to protection from AKI. The parental strains had dramatically different sensitivity to ischemia reperfusion with plasma creatinine levels following 45 minutes of ischemia and 24 hours reperfusion of 4.1 and 1.3 mg/dl in SS and in BN, respectively. No consomic strain showed protection similar to the parental BN strain. Nine consomic strains (SS-7BN, SS-XBN, SS-8BN, SS-4BN, SS-15BN, SS-3BN, SS-10BN, SS-6BN, and SS-5BN) showed partial protection (plasma creatinine about 2.5-3.0 mg/dl), suggesting that multiple alleles contribute to the severity of AKI. In silico analysis was performed using disease ontology database terms and renal function quantitative trait loci from the rat genome database on the BN chromosomes giving partial protection from AKI. This tactic identified at least 36 candidate genes, with several previously linked to the pathophysiology of AKI. Thus, natural variants of these alleles or yet to be identified alleles on these chromosomes provide protection against AKI. These alleles may be potential modulators of AKI in susceptible patient populations.
Keywords: acute kidney injury, genetic susceptibility
Introduction
Physicians who care for acutely ill people have long observed that individual patients, with otherwise similar co-morbidities, have differential susceptibility to develop acute renal failure (ARF) or acute kidney injury (AKI) (1). This well-recognized phenomenon raises the question of whether an underlying genetic background may significantly affect an individual’s susceptibility or resistance to developing AKI brought on by a specific insult.
Ischemia reperfusion (I/R) injury is a frequent cause of clinical AKI and therefore has been commonly used in rats and mice to model the pathophysiology of acute kidney injury. A complex interaction of pathophysiological pathways influences the severity of I/R induced AKI, such as pathways that affect cellular oxidant stress, metabolism, inflammation, immune cell activation, cell death and vascular tone (2-6). In addition, the activation of cell stress response factors (e.g. Hmox1, Hif1a and heat shock proteins) may protect against ischemic injury and are thought to mediate, in part, ischemic preconditioning (7-16). Clearly, diverse biochemical and physiological pathways integrate to generate the final profile of renal injury in response to I/R.
Differences in the sensitivity to injury between rodent strains provide the potential for a powerful experimental system to study allelic contributions to kidney injury. Previously, our group identified the Brown Norway rat (BN/NHsdMcwi) as strongly resistant to renal damage induced by I/R (17). Independently, another group has shown that BN rats have reduced inflammatory responses and accelerated healing following I/R relative to Sprague Dawley rats (18). In our initial study, BN rat kidneys had higher basal levels of inducible heat shock proteins (HSP) compared with the unprotected strain (17). However, it is unlikely that the modestly increased HSP expression is the sole contributing factor that conveys protection in the BN kidney. Indeed, Nilikantan et al., demonstrated a favorable profile of antioxidant proteins in BN rat kidneys (19), and Saenz-Morales et al., demonstrated attenuated pro-inflammatory gene expression in the kidney and in circulating cells following I/R in BN vs SD rats (18). Taken together, multiple pathways affected by the unique genetic background of the BN likely work in concert to afford the profound resistance to injury observed in this strain.
One way to determine the genetic contribution to disease severity has been to make use of a chromosomal substitution paradigm in which one chromosome from an inbred disease resistant strain is introgressed into the background of an alternate inbred, disease prone strain (20). The feasibility of this approach was verified when a panel of consomic rats was developed by introgressing individual chromosomes from the normotensive BN/Mcwi rat strain into the genetic background of the Dahl SS strain (SS/JrHsdMcwi), which exhibits salt sensitive hypertension. These consomic strains were initially developed to validate linkage studies identifying QTLs in a chromosomal region. This strategy provides a strain to test the functional role of two sets of alleles within a narrowed genomic region, an entire chromosome in this case. Although the use of consomics fails to validate all QTLs, the ability to run groups of genetically identical animals may be advantageous when measuring traits with some physiological variability.
While it is unlikely that a complex trait is attributable to a single allelic difference, differences in complex traits can be observed by substitution of single chromosomes using this strategy (20). For example, substitution of individual chromosomes of the Dahl SS rat can attenuate the severity of hypertension and proteinuria (21). Once so defined, informative consomic strains can be used to generate congenic strains to narrow the region of interest on a chromosome and facilitate positional cloning of candidate genes. Recently this strategy was used to define regions on chromosome 13 which can attenuate the development of hypertension in Dahl S rats (22).
Our hypothesis, then, was that the dramatic resistance to ischemic renal injury observed in BN rats is the result of contributions from multiple BN alleles present on separate chromosomes. The goal of this study was to identify protective BN alleles using the two allele system available through the SSxBN consomic panel and the model of ischemia induced renal injury. We found differential susceptibility to I/R injury in several distinct consomic SS-BN lines, suggesting multiple alleles on separate chromosomes influence the susceptibility to injury in the SS rat, and the full profound resistance to AKI in the BN rat.
Results
Dahl SS/Mcwi salt-sensitive rats (SS) were utilized as the background strain for the generation of a panel of consomic rats, by introgressing individual chromosomes from the AKI-resistant BN/Mcwi (BN) (20). We studied this consomic panel to define which specific BN chromosomes carried genes that afford protection against renal I/R injury. When subjected to 45 minutes of renal ischemia then allowed to recover for 24 hours, SS rats develop significant AKI as illustrated by the marked rise in the level of serum creatinine to ~ 4.1 ± 0.2 mg · dl-1. This level of serum creatinine is similar to that which we previously reported using this model of I/R injury for the commonly studied, outbred Sprague-Dawley rat (17). Important to highlight, these results derive from multiple cohorts spread out over a 5-year period as consomic strains became available. The individual mean of each surgical cohort of the parental SS strain was always between 3.5 mg · dl-1 and 4.5 mg · dl-1 and remained consistent over the study period. By comparison, and as we previously reported, the rise in serum creatinine in the BN rat subjected to the same ischemic insult was significantly less profound at 24 hours of reperfusion (1.3 ± 0.1 mg · dl-1, Figure 1). This resistance to injury in BN rats also remained stable across all surgical cohorts over the 5 year study period, with no cohort manifesting a mean 24 hr creatinine > 1.6 mg · dl-1.
Figure 1.
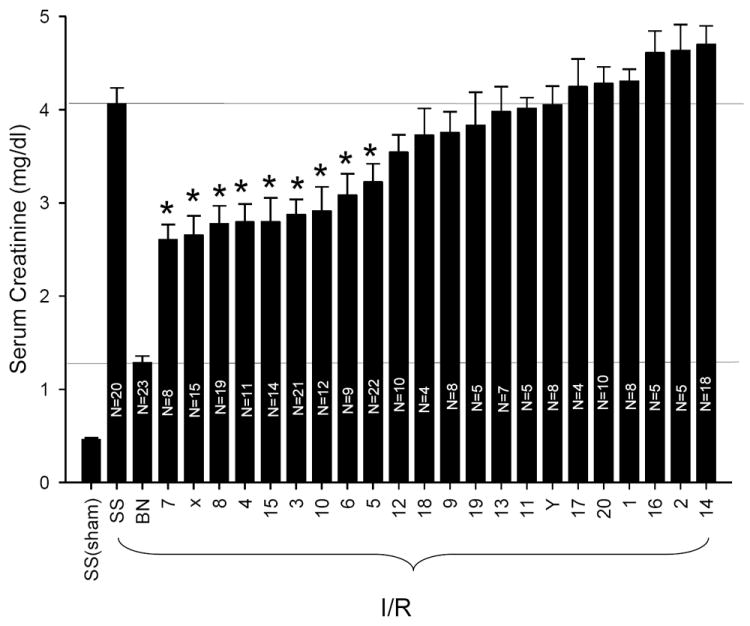
Resistance to I/R induced AKI in BN and SS-BN consomic rats. Except for SS sham, rats were subjected to 45 min bilateral renal ischemia with 24 hrs reflow. Data are mean ± 1 S.D. of the serum creatinine values at 24 hrs. The N for each group is indicated in individual bars; * indicates P < 0.001 vs. SS rats post I/R. Numbers on x-axis are the BN chromosome introgressed into the SS background (e.g. 7 is SS-7BN).
All consomic strains showed sensitivity to I/R induced injury that was significantly greater than the BN- parental strain (P < 0.05, by ANOVA). Most of the consomic strains had equivalent or worse AKI, as measured by serum Cr, as the parental SS strain. However, several consomic strains manifested renal insufficiency following I/R that was intermediate between the ischemia susceptible SS and the resistant BN parental strains. Consomic animals from nine different BN chromosome substitutions showed a statistically significant reduction in serum creatinine level at 24 hours reperfusion relative to the parental SS strain subjected to the same ischemic insult. Nine consomic strains (SS-7BN, SS-XBN, SS-8BN, SS-4BN, SS-15BN, SS-3BN, SS-10BN, SS-6BN, and SS-5BN) had mean serum creatinine values less than 3.0 mg · dl-1 at 24 hours recovery, but no strain had serum creatinine values less than 2.5 mg · dl-1. Therefore, all of the protected consomic strains showed only partial protection against ischemia induced AKI when compared with the dramatic protection observed in the parental BN strain. To ensure the phenotypic stability of the protected consomic strains, animals from each of these strains were evaluated at intervals ranging between 18 months and 5 years, with most of the consomic strains studied over 4-5 years; in each instance, the partial protection remained stable through the course of the study (not shown).
Randomly selected samples of kidney tissue from both protected and non-protected groups were processed for analysis of renal histology after I/R. Figure 2 illustrates representative periodic acid Schiff stained tissues from the susceptible parental strain SS and the ischemia resistant parental strain BN. SS rats show evidence of significant necrotic debris in the lumen of corticomedullary tubules (Figure 2A, indicated by *). A large percentage of severely damaged tubules was prominent in the outer medulla (89 ± 2%), as well as the renal cortex (36 ± 5%)(Figure 2G). By contrast, the BN rat kidney was largely resistant to damage; following I/R, BN rat kidneys demonstrated largely intact brush border staining (Figure 2B, white arrow) and a significantly lower percentage of tubules with severe damage (< 5%, Figure 2G).
Figure 2.
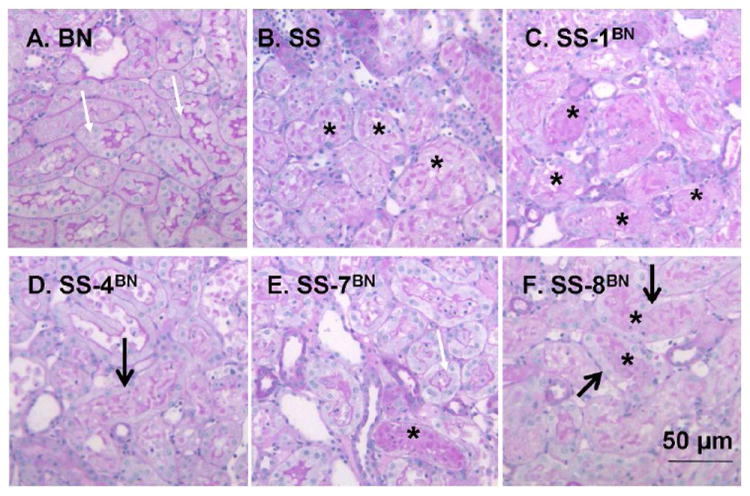
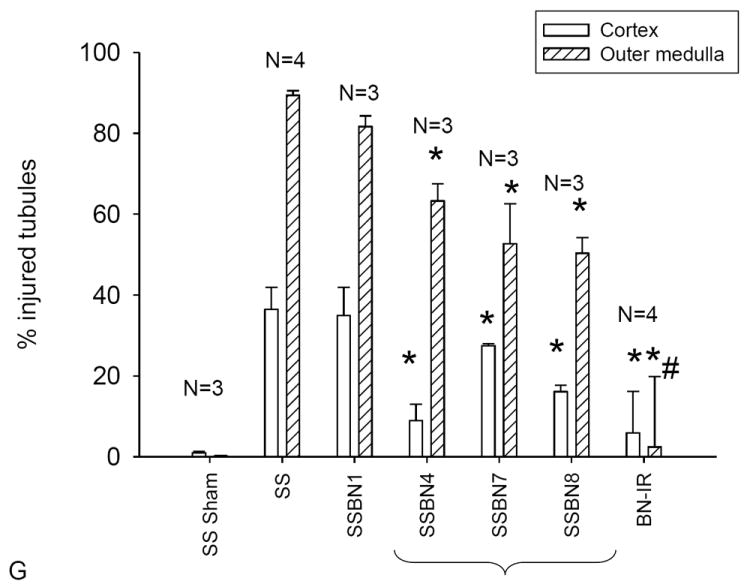
Representative renal histology of SS, BN and selected consomic rat strains 24 hours following I/R injury. Photomicrographs are shown in the renal outer medulla for the BN (A), SS (B), SS-1BN (C), SS-4BN (D), SS-7BN (E) and SS-8BN (F). White arrows indicate PAS-positive brush border staining, while area of necrotic cellular debris are indicated by *. In some tubules, although cellular debris is present, dedifferentiated and viable cells are clearly seen (thick arrow). Panel G is quantitative analysis of tubular injury scores for renal cortex (open) or outer medulla (hashed bar). Data are expressed as percent affected tubules and are mean ± SE. The N for each group is shown; * P < 0.05 indicates significantly reduced vs SS rats; # indicates injury in BN is less than the consomic protected strains.
We also examined three consomic strains with a high resistance to injury (SS-7BN, SS-8BN, SS-4BN; Figure 2D, E, F) and one consomic strain that was susceptible to injury (SS-1BN; Figure 2C). The partially protected consomic strains showed an intermediate level of damage relative to the parental strains. In these protected consomic animals, there is found intact but dedifferentiated cells in tubules containing cellular debris (see thick black arrows in figure 2, D and F) but also the appearance of intact brush border staining remaining in adjacent tubules (white arrows). Although tubular damage and necrotic debris was present in the protected strains (indicated by * in Figure 2), the percent of damaged tubules was significantly less than parental SS rats (Figure 2G). In contrast, the tubule damage associated with the injury prone SS-1BN consomic strain was highly similar to the parental SS strain, with little evidence of intact brush border staining (Figure 2, B and C), and a similar percentage of damaged tubules (Figure 2G).
In order to identify potential alleles that may affect significantly the course of ischemia-reperfusion induced AKI, the identified chromosomes were subjected to further in silico analysis using available gene and quantitative trait loci (QTL) annotations and ontology enrichment tools (see methods section). Figure 3 summarizes the process used to narrow the list of candidate genes contained on the BN rat chromosomes found to be protective. The first step (Fig. 3A) limited the genes on each protective chromosome (X, 3, 4, 5, 6, 7, 8, 10, 15) for three disease ontology terms (reperfusion injury, ischemia, kidney disease). Chromosomes 4 and 10 contained the largest number of genes (104 and 99, respectively) with DO annotations for the three terms. The second step (Fig. 3B) identified renal function QTL for each protective chromosome. Chromosome 6 contains the largest number of renal function QTL, a total of 18, although all protective chromosomes had at least one renal function QTL mapped. The third step (Fig. 3C) used the intersection of genes that were contained within the renal function QTL and also had a disease annotation for reperfusion injury, ischemia, and kidney disease which reduced the number of candidate genes to 36 (listed in Fig. 3D). The full gene names corresponding to the abbreviations in Figure 3D is provided in Table 1. These candidate genes are shown in full genome view with overlapping QTL in Figure 4. Further prioritization of the candidate genes is possible through review of the renal function QTL that overlap with the intersection of genes annotated with reperfusion injury, ischemia, and kidney disease since these QTLs were mapped using a variety of inbred strains and under different experimental conditions (e.g. control conditions, high salt diet, L-NAME administration, unilateral nephrectomy).
Figure 3.
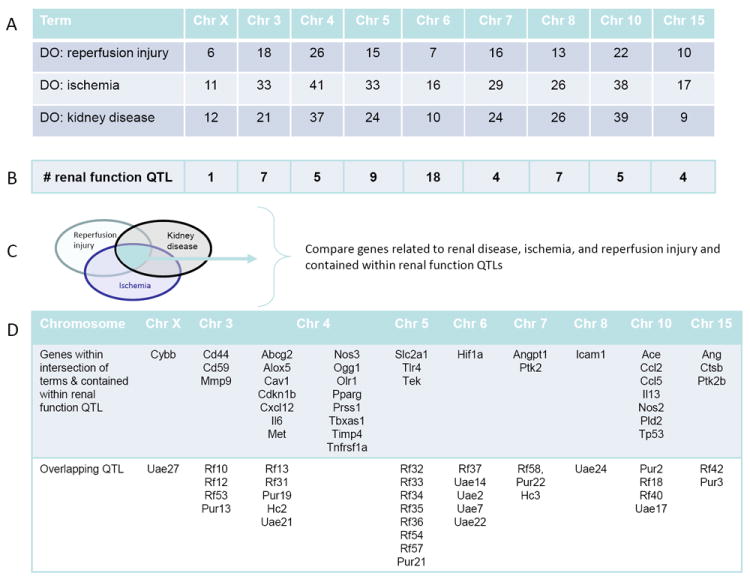
In silico analysis to identify potential candidate genes that confer protection in AKI. To generate a list of potential candidate genes that play a role in the resistance to ischemic injury found in the BN strain, an in silico analysis was done to identify candidate genes involved in the resistance to I/R induced AKI. A: Number of genes with annotations to three disease ontology (DO) terms (reperfusion injury, ischemia, kidney disease) for the resistant chromosomes (consomics with a resistant phenotype). B: Number of renal function quantitative trait loci (QTL) located on chromosomes that protect against I/R injury. C: Potential candidate genes were identified if gene has DO annotations for reperfusion injury, ischemia, and kidney disease and were found within a renal function QTL. D: Summary of candidate genes and the QTLs that overlap with the candidate genes.
Table 1.
Full names for genes identified as potential candidates listed in Figure 3.
| Abbreviation | Gene Name |
|---|---|
| Abcg2 | ATP-binding cassette, sub-family G (WHITE), member 21 |
| Ace | angiotensin I converting enzyme (peptidyl-dipeptidase A) 1 |
| Alox5 | arachidonate 5-lipoxygenase |
| Ang | angiogenin, ribonuclease, RNase A family, 5 |
| Angpt1 | angiopoietin 1 |
| Cav1 | caveolin 1, caveolae protein, 22kDa |
| Ccl2 | chemokine (C-C motif) ligand 2 |
| Ccl5 | chemokine (C-C motif) ligand 5 |
| Cd44 | CD44 molecule |
| Cd59 | CD59 molecule, complement regulatory protein |
| Cdkn1b | cyclin-dependent kinase inhibitor 1B (p27, Kip1) |
| Ctsb | cathepsin B |
| Cxcl12 | chemokine (C-X-C motif) ligand 12 |
| Cybb | cytochrome b-245, beta polypeptide |
| Hif1a | hypoxia inducible factor 1, alpha subunit (basic helix-loop-helix transcription factor) |
| Icam1 | intercellular adhesion molecule 1 |
| Il13 | interleukin 13 |
| Il6 | interleukin 6 (interferon, beta 2) |
| Met | met proto-oncogene (hepatocyte growth factor receptor) |
| Mmp9 | matrix metallopeptidase 9 (gelatinase B, 92kDa gelatinase, 92kDa type IV collagenase |
| Nos2 | nitric oxide synthase 2, inducible |
| Nos3 | nitric oxide synthase 3 (endothelial cell) |
| Ogg1 | 8-oxoguanine DNA glycosylase |
| Olr1 | oxidized low density lipoprotein (lectin-like) receptor 1 |
| Pld2 | phospholipase D1, phosphatidylcholine-specific |
| Pparg | peroxisome proliferator-activated receptor gamma |
| Prss1 | protease, serine, 1 (trypsin 1) |
| Ptk2 | PTK2 protein tyrosine kinase 2 |
| Ptk2b | PTK2B protein tyrosine kinase 2 beta |
| Slc2a1 | solute carrier family 2 (facilitated glucose transporter), member 1 |
| Tbxas1 | thromboxane A synthase 1 (platelet) |
| Tek | TEK tyrosine kinase, endothelial |
| Timp4 | TIMP metallopeptidase inhibitor 4 |
| Tlr4 | toll-like receptor 4 |
| Tnfrsf1a | tumor necrosis factor receptor superfamily, member 1A |
| Tp53 | tumor protein p53 |
Figure 4.
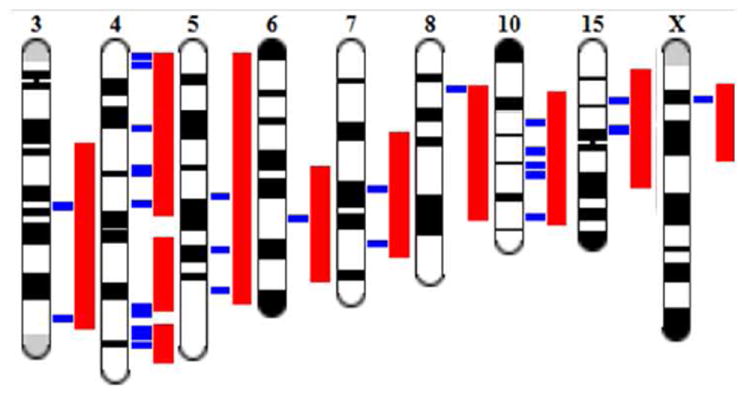
Illustrative map of renal QTLs and candidate genes associated with protection from AKI in BN consomic strains. Genome Viewer (GViewer) output from the Rat Genome Database provides a complete genome view of the prioritized candidate genes (indicated in blue) and QTL (indicated in red). These overlapping renal function QTLs were included in the final step of the in silico analysis, and candidate alleles identified using disease ontology terms to narrow the candidate gene list.
Discussion
Because of the significant clinical impact of AKI, research efforts have focused on defining individual and specific pathways that either promote or prevent renal injury, which might then be exploited to impose resistance to AKI. Studies of the injury process using in vivo and in vitro models have provided a number of potential pathways to target, including those that affect vasoconstriction, inflammation, cell metabolism, and pathways to necrotic or apoptotic cell death (2-6). Experimental models designed to investigate the role of specific genes typically use transgenic over-expression or null-mutation strategies. Another approach has been to use models of ischemic, chemical, or heat preconditioning to identify candidate genes thought to afford resistance to injury.
We chose an alternate method to identify pathways potentially protective against ischemia induced AKI, by taking advantage of the natural differences in susceptibility found in different inbred rat strains. Baker et al., demonstrated that isolated hearts from different rat strains had differential sensitivity to ischemia. That hearts from BN rats were more resistant to injury than other strains suggested a genetic component of resistance to ischemia (23). Similarly, we discovered that BN rats have profound intrinsic resistance to renal I/R injury when compared with the commonly studied Sprague-Dawley rat (17). In that study, we also found that BN rat kidneys have higher basal levels of HSP25 and HSP72, stress proteins implicated in the cytoprotection seen in preconditioning models (24-28), but this is not likely to be the sole reason that the BN rat has such profound resistance to renal injury. Rather, we suspected that several factors regulated by the unique BN genome work together to mediate resistance to injury.
In the current study, we screened a consomic rat panel as described by Cowley et al., (20) so as to define where alleles that contribute to the intrinsic resistance against renal I/R injury could be found within the BN genome. The availability of this unique resource, which was originally generated to define the genetic differences contributing to renal function and blood pressure regulation (20), allowed us to investigate sensitivity to renal I/R using a similar approach. A critical feature that enables this approach is that the inbred parental strains used to generate the consomic panel display a wide and consistent difference in the phenotype being investigated. In this case, the background SS strain used in the current study shows a strong susceptibility to renal I/R that is similar to the commonly studied outbred Sprague Dawley rat, while BN rats display remarkable resistance to injury [Figure 1 and reference (17)].
In these studies, we attempted to minimize the influence of blood pressure by using Dahl S rats maintained on low salt diet. Although these animals do not elicit the dramatic levels of hypertension relative to those on high salt (mean arterial pressure ~170 mm Hg), Dahl S rats on low salt do have higher blood pressures than BN rats (MAP ~ 124 mm Hg vs. ~ 110 mm Hg in BN rats) (21). Whether these differences might contribute to the sensitivity to I/R injury is not known. Nevertheless, the susceptibility of SS rats to AKI does not appear to be explained by blood pressure and salt-sensitivity for the following reasons. First, the level of injury in SS rats is essentially the same as that found in the normotensive SD outbred strain of rat that is routinely used in these I/R studies. Second, the SS-13BN consomic rat, which displays MAP levels nearly equivalent to BN rats and does not show salt sensitive hypertension (20), is completely sensitive to I/R, with a level of AKI exactly the same as the parental SS (see figure 1).
Were a single allele responsible for resistance to injury in BN rats, a lone consomic strain would likely have shown resistance to I/R similar to the parental BN strain. However, no single consomic strain displayed the full resistance to renal injury seen in the parental BN strain, while several BN consomic lines manifested partial protection. Even the most protected consomic strains still developed significant injury (e.g. strains SS.BN7, SS.BN8, SS.BN4), suggesting that no single genetic element is responsible for the strong protection against renal injury observed in BN rats. Rather, the resistance to injury appears to be mediated by the contribution of multiple different alleles, on separate chromosomes. The functional differences conveyed by these discrete alleles likely work in concert to provide the full protection observed in the parental BN strain. In context of the view that AKI is a multifactorial disease, where multiple processes mediate the pathogenesis, our findings, therefore, are not surprising.
An important feature of the present study is that it begins the process of mapping the genomic location of rat alleles that contribute either resistance or susceptibility to AKI following an ischemic insult. Since both the rat and human genome are fully sequenced, further defining pertinent alleles in the rat model of AKI has several potential future benefits. First, identifying genes that affect outcome following I/R injury, which heretofore were not known to be involved in this disease process, could provide new potential therapeutic targets. Second, defining more precisely the complex of genes that affect outcome could eventually lead to studies seeking to identify susceptible patients prior to a procedure that puts them at risk for AKI. Third, knowing a particular genetic susceptibility for an individual patient might then allow a targeted therapy to prevent or ameliorate AKI in that individual.
To date, several studies have already suggested that genes associated with discrete biochemical pathways may contribute to the severity of AKI in different patient populations. For example, in the setting of kidney transplantation, donor kidneys with a TLR4 loss-of-function allele contained less TNFα, less MCP-1, more heme oxygenase 1 (Hmox1) and exhibited a higher rate of immediate graft function (29). Interestingly, in the current study, TLR4 was identified as a potential candidate on chromosome 5. In another study, variation in a catalase allele and serum catalase activity was associated with a difference in outcome of patients in the ICU setting (30), suggesting that the intrinsic antioxidant defense pathways of individuals influence AKI. Finally, another study showed that genetic polymorphisms in HSP-72, specifically, the Hsp72 1267-GG allele, may increase the risk of acute kidney injury in low birth weight neonates (31). Studies of these genes in humans were driven by prior knowledge of specific pathophysiology of the disease process defined in basic science studies. We propose that bioinformatic analytical approaches combined with a genetic model of resistance to ischemic renal injury may identify new candidate genes that can then be explored in patients as potential predictors of risk to develop AKI.
With those considerations, further effort to define allelic variations that significantly affect the development and outcome of AKI is warranted. Several approaches to achieve this goal could be productive. One approach is to use breeding strategies, with protected consomic lines identified in the current study, to narrow genomic regions of interest with the goal to identify candidate genes involved in the disease process. This allows development of congenic strains with narrowed regions of interest on a specific chromosome, which can then be phenotyped in a manner similar to the consomic panel that we studied (20, 22). While potentially quite informative, this method involves an extended effort of breeding and genotyping to narrow the regions of interest, followed by phenotyping to determine whether there is effect on this complex disease process. Since our current results implicate several chromosomes containing genes of interest, developing congenic and congenic substrains to define the pertinent genes of interest would be challenging, due to the number of lines needed to cover nearly half of the genome.
For this reason, we sought here to begin narrowing the range of possible genes of interest using in silico analysis, from which the information gleaned may provide reasonable targets for future additional analysis. Although more than 15,000 genes are on the nine chromosomes that confer protection from ischemic injury in the BN strain, information about many of these genes can be obtained using a variety of genomic data sets. The current strategy was to use curated data and studies investigating similar disease mechanisms, which significantly limits the genomic regions of interest. One potentially significant drawback to this strategy is that the results are restricted to published and curated data, thus limiting the power of the results if no data has been published related to the gene or the role of the gene in certain diseases. Finally, our analysis required that genes lie within a renal function QTL, substantially limiting the number of genes identified.
Thus, the strategy employed here was conservative and could miss genes that might well be very pertinent to the disease process. For example, rat chromosome 7 that we found to be the most protected consomic strain, contains the gene for the transcription factor Hsf1, which regulates induction of all stress inducible heat shock proteins including HSP 72 and 25. Both of these stress proteins, regulated by Hsf1, have been extensively studied in this model of renal injury and have been implicated in some studies as affording protection against ischemia (24-28). Furthermore, the relationship of HSF-1 activation to cellular processes triggered by renal ischemia has also been described in several reports (1, 32, 33). Despite this, Hsf1 was not identified in the in silico analysis as a potential gene of interest. This highlights the potential limitation of this analytical approach, and indicates that results should not be considered a comprehensive list of candidate genes. However, if candidate genes such as Hsf1 have been identified through other studies, the gene or surrounding genomic region can be further investigated using other approaches. For example, single nucleotide variation between the SS and BN strains can be obtained at the Rat Genome Database (http://rgd.mcw.edu) for any region of the genome. For Hsf1, 58 variants exist between the SS and BN, although none of these variants have predicted functional changes. This fundamental search strategy can be modified to include other ontology terms related to inflammation, cell death, mitochondrial function and oxidative stress to yield different, but informative results.
Nevertheless, the current strategy identified 36 alleles with the potential to modify the renal response to I/R injury, with many of these genes having obvious prior implications in the I/R process. Specifically, genes related to vasoreactivity (ACE, Nos3), inflammation (Nos2, ICAM, Il6, Cxcl12, Ccl2, Ccl5), tissue injury (MMP9, Timp4) and cytoresistance (Hif1a) were all identified using this screening approach. Such a list generated by this or alternative in silico analysis, not only emphasizes the contributory role of these genes in the pathogenesis of AKI, but also imply that naturally occurring variants may modulate the severity of AKI in patients.
In summary, we have shown that the BN genome contains discrete elements that ameliorate the degree of kidney injury following I/R. No single genetic element appears to be responsible for the profound resistance to injury observed in BN rats. Full protection against ischemic injury in the BN rat likely is from multiple beneficial alleles working in concert, and we have identified the chromosomes on which those alleles appear to reside. This unique genetic model of profound resistance to injury, combined with currently available genetic database tools, can identify candidate genes with the potential to mediate heritable protection against AKI. This information should be exploited further to investigate multi-genetic factors modulating development of AKI in animal models, and then to translate this knowledge into clinical studies evaluating the genetic basis both of risk to develop AKI and of the severity of AKI once established.
Methods
Animals
Care of the rats before and during the experimental procedures was conducted in accordance with the policies of the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All protocols had received prior approval by the Institutional Animal Care and Use Committees at the Medical College of Wisconsin
Experiments were performed on inbred lines of SS/JrHsdMcwi (Dahl salt-sensitive rats), BN/NHsdMcwi (BN), and a panel of consomic rats. The consomic rat lines were derived from inbred BN and SS rats as previously described (34, 35). The consomic rat panel consisted of individual rat strains in which each of the 20 autosomes as well as the X and Y chromosomes from the BN rat were transferred onto the SS genetic background. The consomic strains are formally designated as SS-Chr nBN/Mcwi (SS-nBN throughout the text), where n designates the substituted chromosome (e.g., SS-1BN/Mcwi, SS-2BN/Mcwi, etc). In one of the strains, SS-8BN/Mcwi, the entire chromosome was not transferred from the BN to the SS, resulting in a congenic strain rather than a consomic strain. In this strain, greater than 70% of the BN chromosome 8 was transferred to the SS/Mcwi genetic background, specifically in the region indicated by the markers D8rat163-D8rat81. Therefore, this strain is referred to properly as SS.BN-(D8rat163-D8rat81)/Mcwi.
The breeders of each strain were maintained on chow containing 0.4% NaCl obtained from Harlan Teklad (3075S, Madison, WI) and the weaned animals were maintained on standard rat chow containing 0.4% NaCl prior to surgery. Surgery was performed on male rats, age 8-12 weeks, weighing 200 - 250 g between the time of 0900 and 1200.
Surgical procedures
Acute kidney injury was induced by bilateral ischemia reperfusion injury to the kidneys by clamping the renal pedicle for 45 minutes using a surgical approach that has been described previously under anesthesia induced ketamine (100 mg · kg -1) and pentobarbital (25-50 mg · kg -1) (17). To minimize experimental variation, only two individuals contributed to the surgeries comprising this study group. A total of 268 rats were used for these studies. Of these 43 were of the parental strains (20 for SS, and 23 for BN), and the remaining 225 rats were of the various 22 different consomic strains. Based on the availability of these strains as they were being generated, the surgeries to induce AKI were spread out over a 5-year period of time. Repeated measurements of both parental strains were routinely included to ensure that the level of injury remained consistent over time. Both parental strains showed the same level of susceptibility (SS strain) and resistance (BN strain) to injury at the beginning and the end of this 5-year window. Typically, during any surgical session, up to 2-4 rats from different consomic strains would be evaluated in parallel. The final N, then, was between 4-22 animals per strain. Typically, the higher N for a particular consomic strain is due to repeated surgical sessions to strengthen statistical power, when the initial results indicated that a particular strain displayed some level of protection against AKI. Thus, results for the most promising strains derive from a larger N and more than one surgical session that showed consistent results. The higher N for the parental SS and BN strains resulted from routine inclusion of each strain throughout the course of the study to confirm ongoing and unchanging ischemia susceptible and resistant parental phenotypes, respectively. This approach ensured that observed differences in susceptibility to I/R injury between consomic strains were not due to variations in surgical technique over the course of repetitive sessions. For the final statistical analysis, serum creatinine values for each animal were analyzed together with all other individuals from a particular strain, representing the cumulative activity of multiple surgical sessions.
Assessment of renal function and histology
Blood samples were collected into heparinized tubes at 24 hrs of reperfusion. Plasma creatinine was measured with an assay based on the Jaffe reaction by an autoanalyzer (ACE, Alfa Wasserman, Fairfield, NJ) to determine the extent of renal injury. Following the conclusion of the study, kidneys were removed from deeply anaesthetized rats, bisected and fixed by immersion into 10% buffered formalin and stored at room temperature. Formalin fixed tissues were embedded in paraffin and stained using periodic acid Schiff (PAS) for standard histological assessment. Images were acquired using a Nikon Optiphot-2 upright microscope equipped with a Spot digital camera and image acquisition software (version 3.4.5., Diagnostic Instruments). A minimum of 3 cortical and 3 outer medullary field images were obtained using a 20X objective generating a field-of-view of ~.24 mm2. Scoring by a blinded reviewer was based on the method described by Kelly et al. to assess the severity of necrotic damage (36). However, rather than assigning an injury index, the percentage of damaged tubules were quantified using the following classifications: Normal tubules showed no evidence of injury, “moderate” damage was indicated by loss of brush border staining and simplification but no evident necrosis, “severe” damage was indicated by tubules with apparent necrosis, or celluar debris in the lumen. For each kidney, approximately 500 tubules were scored. The percent of damaged tubules was calculated by (number of severe injured tubules) / (total number tubules) X (100).
Data analysis for genomic data from selected chromosomes or narrowed genomic regions
After identifying several consomic strains as manifesting protection again I/R induced AKI, steps were taken to prioritize candidate genes or genomic regions for additional study. First, the genes on protective chromosomes were analyzed for enrichment of either disease-ontology or gene-ontology (GO) annotations. No specific disease or GO enrichment was found for any of the protective chromosomes using the enrichment analysis available through RatMine (http://ratmine.mcw.edu). RatMine, a data warehouse that is powered by InterMine, houses biological data including large-scale functional genomic and proteomic datasets for a wide range of model organisms (fly, rat, yeast, mouse, zebrafish, and worm). Ontology enrichment analysis can be done through template queries (e.g Chromosome – all genes, “find all genes on a chromosome”) or by uploading a list of identifiers (e.g. genes)(37). List-detail pages are generated and provide genomic distribution and enrichment for several ontologies (Gene Ontology, Disease Ontology, Pathway Ontology, and Mammalian Phenotype Ontology) with several options for post-hoc analyses.
The second step in the analysis involved using specific disease ontology (DO maintained by the Comparative Toxicogenomics Database) terms to identify genes on the protective chromosomes. The DO terms for this analysis were “reperfusion injury”, “ischemia”, and “kidney disease”. The protective chromosomes were analyzed for renal function quantitative trait loci (QTL) using the Rat Genome Database (http://rgd.mcw.edu). Finally, for each protective chromosome, the genes related to renal disease, ischemia, reperfusion injury, which were also contained within a renal function QTL, were prioritized as candidate genes for follow-up experiments.
Statistical Analysis
For analysis of serum creatinine, a one-way ANOVA was carried out with the aid of SigmaPlot software (Systat Software, San Jose, CA) after verifying that the dataset was normal and of equal variance. A Holm-Sidak post hoc test was used to evaluate differences between the parental SS/Mcwi and each of the other strains. P < 0.05 was considered significant. For statistical analysis of morphometric scoring, because the scores were similar among the protected consomic strains, those protected consomic strains were grouped for ANOVA and compared with the unprotected SS parental strain, the unprotected consomic strains, as well as with the protected BN parental strain.
Acknowledgments
SVW was funded by a Pilot and Feasibility grant for P50 DK079306 (PI Ellis Avner) and RS was funded by KO8-DK075470. Additional funding was provided by the Children’s Research Institute and the Department of Pediatrics, Medical College of Wisconsin. DB was supported by RO1 DK 063114
Footnotes
Disclosure:
None:
References
- 1.Sreedharan R, Devarajan P, Van Why S. Pathogenesis of acute renal failure. In: Avner E, Harmon W, Niaudet P, Yoshikawa N, editors. Pediatric Nephrology. 6. Springer-Verlag; Heidleberg: 2009. pp. 1579–1602. [Google Scholar]
- 2.Padanilam BJ. Cell death induced by acute renal injury: a perspective on the contributions of apoptosis and necrosis. American Journal of Physiology - Renal Fluid & Electrolyte Physiology. 2003;284:F608–627. doi: 10.1152/ajprenal.00284.2002. [DOI] [PubMed] [Google Scholar]
- 3.Nath K, Norby S. Reactive oxygen species and acute renal failure. The American journal of medicine. 2000;109:665–678. doi: 10.1016/s0002-9343(00)00612-4. [DOI] [PubMed] [Google Scholar]
- 4.Rabb H. Immune Modulation of Acute Kidney Injury. Journal of the American Society of Nephrology. 2006;17:604–606. doi: 10.1681/ASN.2006010060. [DOI] [PubMed] [Google Scholar]
- 5.Bonventre JV, Zuk A. Ischemic acute renal failure: An inflammatory disease? Kidney Int. 2004;66:480–485. doi: 10.1111/j.1523-1755.2004.761_2.x. [DOI] [PubMed] [Google Scholar]
- 6.Basile DP. The endothelial cell in ischemic acute kidney injury: implications for acute and chronic function. Kidney Int. 2007;72:151–156. doi: 10.1038/sj.ki.5002312. [DOI] [PubMed] [Google Scholar]
- 7.Nath KA. Heme oxygenase-1: A provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int. 2006;70:432–443. doi: 10.1038/sj.ki.5001565. [DOI] [PubMed] [Google Scholar]
- 8.Yang C-C, Lin L-C, Wu M-S, Chien C-T, et al. Repetitive Hypoxic Preconditioning Attenuates Renal Ischemia/Reperfusion Induced Oxidative Injury via Upregulating HIF-1[alpha]-Dependent bcl-2 Signaling. Transplantation. 2009;88:1251–1260. doi: 10.1097/TP.0b013e3181bb4a07. 1210.1097/TP.1250b1013e3181bb1254a1207. [DOI] [PubMed] [Google Scholar]
- 9.Weidemann A, Bernhardt WM, Klanke B, Daniel C, et al. HIF Activation Protects From Acute Kidney Injury. Journal of the American Society of Nephrology. 2008;19:486–494. doi: 10.1681/ASN.2007040419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rosenberger C, Heyman SN, Rosen S, Shina A, et al. Up-regulation of HIF in experimental acute renal failure: Evidence for a protective transcriptional response to hypoxia. Kidney Int. 2005;67:531–542. doi: 10.1111/j.1523-1755.2005.67110.x. [DOI] [PubMed] [Google Scholar]
- 11.Kim M, Park SW, Kim M, Chen SWC, et al. Selective renal overexpression of human heat shock protein 27 reduces renal ischemia-reperfusion injury in mice. American Journal of Physiology - Renal Physiology. 2010;299:F347–F358. doi: 10.1152/ajprenal.00194.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo S, Wharton W, Moseley P, Shi H. Heat shock protein 70 regulates cellular redox status by modulating glutathione-related enzyme activities. Cell Stress and Chaperones. 2007;12:245–254. doi: 10.1379/CSC-265.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park KM, Cho HJ, Bonventre JV. Orchiectomy reduces susceptibility to renal ischemic injury: a role for heat shock proteins. Biochemical & Biophysical Research Communications. 2005;328:312–317. doi: 10.1016/j.bbrc.2004.12.177. [DOI] [PubMed] [Google Scholar]
- 14.Kumar Y, Tatu U. Stress protein flux during recovery from simulated ischemia: induced heat shock protein 70 confers cytoprotection by suppressing JNK activation and inhibiting apoptotic cell death. Proteomics. 2003;3:513–526. doi: 10.1002/pmic.200390065. [DOI] [PubMed] [Google Scholar]
- 15.Van Why SK, Siegel NJ. Heat shock proteins: Role in prevention and recovery from acute renal failure. In: Molitoris BA, Finn WF, editors. Acute renal failure: A companion to Brenner and Rectors The Kidney. WB Sanders; Philadelphia: 2001. pp. 143–156. [Google Scholar]
- 16.Gomes M, Cancherini V, Reboucas MA, Reboucas NA. Ischemic Preconditioning of Renal Tissue: Identification of Early Up-Regulated Genes. Nephron Experimental Nephrology. 2003;93:e107–e116. doi: 10.1159/000069553. [DOI] [PubMed] [Google Scholar]
- 17.Basile DP, Donohoe DL, Cao X, Van Why S. Resistance to ischemic acute renal failure in the Brown Norway rat: A new model to study cytoprotection. Kidney Int. 2004;65:2201–2211. doi: 10.1111/j.1523-1755.2004.00637.x. [DOI] [PubMed] [Google Scholar]
- 18.Saenz-Morales D, Conde E, Blanco-Sanchez I, Ponte B, et al. Differential resolution of inflammation and recovery after renal ischemia-reperfusion injury in Brown Norway compared with Sprague Dawley rats. Kidney Int. 2010;77:781–793. doi: 10.1038/ki.2010.10. [DOI] [PubMed] [Google Scholar]
- 19.Nilakantan V, Hilton G, Maenpaa C, Van Why S, et al. Favorable balance of anti-oxidant/pro-oxidant systems and ablated oxidative stress in Brown Norway rats in renal ischemia-reperfusion injury. Molecular and Cellular Biochemistry. 2007;304:1–11. doi: 10.1007/s11010-007-9480-z. [DOI] [PubMed] [Google Scholar]
- 20.Cowley AW, Jr, Liang M, Roman RJ, Greene AS, et al. Consomic rat model systems for physiological genomics. Acta Physiologica Scandinavica. 2004 Aug;181:585–592. doi: 10.1111/j.1365-201X.2004.01334.x. [DOI] [PubMed] [Google Scholar]
- 21.Mattson DL, Dwinell MR, Greene AS, Kwitek AE, et al. Chromosome substitution reveals the genetic basis of Dahl salt-sensitive hypertension and renal disease. American Journal of Physiology - Renal Physiology. 2008;295:F837–F842. doi: 10.1152/ajprenal.90341.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moreno C, Williams J, Lu L, Liang M, et al. Narrowing a region on rat chromosome 13 that protects against hypertension in Dahls SS-13BN congenic strains. Am J Physiol Heart Circ Physiol. 2011;300 doi: 10.1152/ajpheart.01026.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baker JE, Konorev EA, Gross GJ, Chilian WM, et al. Resistance to myocardial ischemia in five rat strains: is there a genetic component of cardioprotection? American Journal of Physiology - Heart & Circulatory Physiology. 2000;278:H1395–1400. doi: 10.1152/ajpheart.2000.278.4.H1395. [DOI] [PubMed] [Google Scholar]
- 24.Aufricht C, Ardito T, Thulin G, Kashgarian M, et al. Heat-shock protein 25 induction and redistribution during actin reorganization after renal ischemia. American Journal of Physiology. 1998;274:F215–222. doi: 10.1152/ajprenal.1998.274.1.F215. [DOI] [PubMed] [Google Scholar]
- 25.Aufricht C, Lu E, Thulin G, Kashgarian M, et al. ATP releases HSP-72 from protein aggregates after renal ischemia. American Journal of Physiology. 1998;274:F268–274. doi: 10.1152/ajprenal.1998.274.2.F268. [DOI] [PubMed] [Google Scholar]
- 26.Emami A, Schwartz JH, Borkan SC. Transient ischemia or heat stress induces a cytoprotectant protein in rat kidney. Am J Physiol. 1991;260:F479–F485. doi: 10.1152/ajprenal.1991.260.4.F479. [DOI] [PubMed] [Google Scholar]
- 27.Havasi A, Wang Z, Gall JM, Spaderna M, et al. Hsp27 inhibits sublethal, Src-mediated renal epithelial cell injury. American Journal of Physiology - Renal Physiology. 2009;297:F760–F768. doi: 10.1152/ajprenal.00052.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park KM, Chen A, Bonventre JV. Prevention of kidney ischemia/reperfusion-induced functional injury and JNK, p38, and MAPK kinase activation by remote ischemic pretreatment. 2001:11870–11876. doi: 10.1074/jbc.M007518200. [DOI] [PubMed] [Google Scholar]
- 29.Kruger B, Krick S, Dhillon N, Lerner SM, et al. Donor Toll-like receptor 4 contributes to ischemia and reperfusion injury following human kidney transplantation. Proceedings of the National Academy of Sciences. 2009;106:3390–3395. doi: 10.1073/pnas.0810169106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Perianayagam MC, Liangos O, Kolyada AY, Wald R, et al. NADPH Oxidase p22phox and Catalase Gene Variants Are Associated with Biomarkers of Oxidative Stress and Adverse Outcomes in Acute Renal Failure. Journal of the American Society of Nephrology. 2007;18:255–263. doi: 10.1681/ASN.2006070806. [DOI] [PubMed] [Google Scholar]
- 31.Vásárhelyi B, Tóth-Heyn P, Treszl A, Tulassay T. Genetic polymorphisms and risk for acute renal failure in preterm neonates. Pediatric Nephrology. 2005;20:132–135. doi: 10.1007/s00467-004-1711-x. [DOI] [PubMed] [Google Scholar]
- 32.Van Why SK, Mann AS, Thulin G, Zhu XH, et al. Activation of heat-shock transcription factor by graded reductions in renal ATP, in vivo, in the rat. Journal of Clinical Investigation. 1994;94:1518–1523. doi: 10.1172/JCI117492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Van Why SK, Kim S, Geibel J, Seebach FA, et al. Thresholds for cellular disruption and activation of the stress response in renal epithelia. American Journal of Physiology. 1999;277:F227–234. doi: 10.1152/ajprenal.1999.277.2.F227. [DOI] [PubMed] [Google Scholar]
- 34.Cowley AW, Roman RJ, Kaldunski ML, Dumas P, et al. Brown Norway Chromosome 13 Confers Protection From High Salt to Consomic Dahl S Rat. Hypertension. 2001;37:456–461. doi: 10.1161/01.hyp.37.2.456. [DOI] [PubMed] [Google Scholar]
- 35.PhysGen: Program for genomic applications: physiogenomics of stressors in derived consomic rats. Milwaukee, WI: Medical College of Wisconsin; Online. [Google Scholar]
- 36.Kelly KJ, Williams WW, Jr, Colvin RB, Meehan SM, et al. Intercellular adhesion molecule-1-deficient mice are protected against ischemic renal injury. Journal of Clinical Investigation. 1996;97:1056–1063. doi: 10.1172/JCI118498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lyne R, Smith R, Rutherford K, Wakeling M, et al. FlyMine: an integrated database for Drosophila and Anopheles genomics. Genome Biology. 2007;8:R129. doi: 10.1186/gb-2007-8-7-r129. [DOI] [PMC free article] [PubMed] [Google Scholar]