

Background: Ceramide is a critical lipid mediator of cellular stress responses relating to inflammation and insulin resistance.
Results: Palmitate and LPS synergistically induce macrophage de novo ceramide biosynthesis.
Conclusion: TLR4 signaling via TRIF and MyD88 in combination with palmitate up-regulates ceramide synthesis through a non-transcriptional mechanism.
Significance: Understanding how lipid excess intersects with inflammatory signaling may provide insights into the pathogenesis of metabolic disease.
Keywords: Ceramide, Cytokine, Lipopolysaccharide (LPS), Lipotoxicity, Macrophages
Abstract
Macrophages play a key role in host defense and in tissue repair after injury. Emerging evidence suggests that macrophage dysfunction in states of lipid excess can contribute to the development of insulin resistance and may underlie inflammatory complications of diabetes. Ceramides are sphingolipids that modulate a variety of cellular responses including cell death, autophagy, insulin signaling, and inflammation. In this study we investigated the intersection between TLR4-mediated inflammatory signaling and saturated fatty acids with regard to ceramide generation. Primary macrophages treated with lipopolysaccharide (LPS) did not produce C16 ceramide, whereas palmitate exposure led to a modest increase in this sphingolipid. Strikingly, the combination of LPS and palmitate led to a synergistic increase in C16 ceramide. This response occurred via cross-talk at the level of de novo ceramide synthesis in the ER. The synergistic response required TLR4 signaling via MyD88 and TIR-domain-containing adaptor-inducing interferon beta (TRIF), whereas palmitate-induced ceramide production occurred independent of these inflammatory molecules. This ceramide response augmented IL-1β and TNFα release, a process that may contribute to the enhanced inflammatory response in metabolic diseases characterized by dyslipidemia.
Introduction
Obesity is increasing at an epidemic rate worldwide and is accompanied by growing rates of chronic metabolic diseases such as diabetes. Current evidence suggests that elevated circulating and tissue fatty acids, particularly saturated fatty acids (SFAs), play a pathologic role in the development of insulin resistance and end-organ dysfunction in these conditions (2–4). Obesity and diabetes are also associated with immune cell dysfunction, which is thought to contribute to increased rates of infection and poor wound healing after tissue injury (i.e., myocardial infarction or surgery) in affected individuals (5–8). Because macrophages play a central role in the response to infectious pathogens and tissue damage, macrophage dysfunction may be relevant to the pathogenesis of diabetic complications (9, 10). For these reasons, investigating the impact of lipotoxic stimuli on inflammatory signaling pathways in macrophages is highly relevant.
Toll-like receptor 4 (TLR4)2 is activated on macrophages in response to bacterial infection and tissue damage. The Gram-negative bacterial outer membrane lipid lipopolysaccharide (LPS) is the prototypical TLR4 ligand (11). However, damage-associated endogenous molecules or alarmins that are released from injured cells can also signal through this receptor (12, 13). TLR4 signaling utilizes two distinct adaptor proteins known as MyD88 and TIR-domain-containing adaptor-inducing interferon beta (TRIF). The MyD88-dependent pathway culminates in the activation of MAP kinases and NF-κB, whereas TRIF mediates IRF3 activation/IFNβ production as well as delayed NF-κB and MAP kinase activation (14). In addition to cytokine production, another consequence of TLR4-mediated macrophage activation is the remodeling of cellular lipids. Recent lipidomic data has revealed that TLR4 activation of RAW 264.7 cells increases total cellular sphingolipids, including ceramides (15, 16). These findings are of particular interest because ceramides have been implicated in a wide variety of cell stress responses including cell death, proliferation, inflammation, autophagy, and insulin resistance (17, 18).
The production of ceramides can occur via de novo synthesis, sphingomyelinase cleavage of membrane lipids, or recycling of more complicated glycosylated ceramides (19). De novo synthesis takes place in the endoplasmic reticulum (ER) where serine and palmitoyl-CoA are condensed to form 3-ketosphinganine. This reaction is the rate-limiting step of de novo synthesis and is catalyzed by the enzyme serine palmitoyltransferase (SPT). LPS stimulation of TLR4 in RAW cells generates ceramide by multiple mechanisms, including the de novo synthetic pathway (15). The SFA palmitate also increases de novo ceramide synthesis in a variety of cell types through an SPT1-dependent mechanism (20). Although lipotoxic concentrations of palmitate can increase availability of palmitoyl-CoA substrate for SPT, recent data demonstrate that palmitate-induced ceramide production in skeletal muscle cells requires TLR4 and its adaptor MyD88 and involves IKKβ- and NF-κB-mediated transcriptional up-regulation of genes involved in ceramide biosynthesis (21). Consistent with these findings, there is a binding site for NF-κB in the promoter of the SPT1 gene, and overexpression of the NF-κB p65 subunit can increase total cellular ceramide levels (22). However, whether transcriptional regulation of SPT1 is the predominant mechanism accounting for increased ceramide synthesis in TLR4-activated cells is not known.
As a model to assess macrophage function in the altered metabolic milieu of nutrient excess, we stimulated primary peritoneal macrophages with LPS in the presence of media containing pathophysiological concentrations of the SFA palmitate and analyzed sphingolipid remodeling by LC-MS/MS. Our findings revealed that the combination of TLR4 ligation and palmitate supplementation synergistically augmented macrophage ceramide production via enhanced de novo synthesis. The synergistic response required TLR4, MyD88, and TRIF but occurred independent of NF-κB and MAP kinase activation. Furthermore, TLR4-mediated IL-1β and TNFα secretion was augmented under lipotoxic conditions through a mechanism involving this synergistic stimulation of de novo ceramide synthesis.
EXPERIMENTAL PROCEDURES
Reagents
SB203580, PD98059, SB600125, and SN50 were from EMD-Millipore. Fumonisin B and myriocin were from Cayman Chemicals. Z-YVAD was from Enzo Life Sciences. Ultrapure Escherichia coli LPS was from Invivogen. Thioglycollate was from Difco. Fatty acids were from Nu-Chek Prep. Ultrapure-bovine serum albumin (BSA) was from Seracare and was tested for TLR ligand contamination before use. All sphingolipids, serine, homoserine, palmitoyl-CoA, heptadecanoyl-CoA, and dansyl chloride were purchased from commercial sources (Matreya Inc,; AVANTI Polar Lipids; Sigma; Alfa Aesar).
Cell Culture
Peritoneal macrophages were isolated from C57BL/6 or the indicated knock-out mice 4 days after intraperitoneal injection of 3.85% thioglycollate and plated at a density of 5 × 105 cells/ml in DMEM containing 10% inactivated fetal serum, 50 units/ml penicillin G sodium, and 50 units/ml streptomycin sulfate. Stimulations were performed on the day after harvest. Growth medium was supplemented with palmitate complexed to BSA at a 2:1 molar ratio as described previously (20), and BSA-supplemented media was used as control. For cell stimulations, PBS or LPS (50 ng/ml) were added to BSA or palmitate media.
Mice
Wild type (WT) and TLR4 knock-out (KO) C57BL/6 mice were obtained from Oriental Bioscience. TRIF KO, MyD88 KO, and TRIF/MyD88 DBL KO mice were from Marco Colonna (Washington University); IKKβ floxed mice crossed with Lys-M Cre were from Michael Karin (University of California San Diego). All lines were in the C57BL/6 background. Mice were maintained in a pathogen-free facility on a standard chow diet ad libitum (6% fat). All animal experiments were conducted in accordance with National Institutes of Health guidelines for humane treatment of animals and were reviewed by the Animal Studies Committee of Washington University School of Medicine.
RNA Isolation and Quantitative RT-PCR
Total cellular RNA was isolated using Qiagen RNeasy columns and reverse transcribed using a high capacity cDNA reverse transcription kit (Applied Biosystems). Real-time quantitative PCR was performed using SYBR Green reagent (Applied Biosystems) on an ABI 7500 fast thermocycler. Relative gene expression was determined using the ΔΔCT method normalized to 36B4 expression. Mouse primers sequences were as follows (all 5′-3′): 36B4 (forward, ATC CCT GAC GCA CCG CCG TGA; reverse, TGC ATC TGC TTG GAG CCC ACG TT); TNFα (forward, CAT CTT CTC AAA ATT CGA GTG ACA A; reverse, TGG GAG TAG ACA CAA GGT ACA ACC C); SPT1 (forward, AGT GGT GGG AGA GTC CCT TT; reverse, CAG TGA CCA CAA CCC TGA TG); SPT2, forward, GGA TAC ATC GGA GGC AAG AA; reverse, ACC TGG TGT TCT CAG CCA AC).
IL-1β and TNFα ELISA
Supernatants were harvested from macrophages 20 h after the indicated stimulations. IL-1β and TNFα was quantified using a DuoSet ELISA kit (R&D Systems) according to the manufacturer's instructions.
Cell Lysates for Lipidomics
Macrophages (5 × 105 cells) were grown in 24-well tissue culture plates. After stimulation, the cells were washed 3 times with PBS, and lysates were prepared by scraping cells in 100 μl of ice-cold PBS followed by homogenization by 10 passes through a 26-gauge syringe. The resulting homogenate (50 μl) was mixed 1:1 with methanol for protein precipitation. The supernatant was spiked with internal standards and was adjusted to 1 ml with 1:1 (v/v) methanol/water solution for sphingolipid, ceramide, and palmitoyl-CoA analyses.
Modified SPT Activity Assay
Macrophages (2.5 × 105 cells) were cultured with the indicated stimuli for 2 h in 24-well plates. Cell lysis and SPT assay were performed as described (23) with quantification of 3-ketosphinganine after a 1-h reaction at 37 ºC.
Instrumentation
An online column trapping HPLC/MS/MS system was established using an API-4000 mass spectrometer (Applied Biosystems) connected with a Valco electro-actuated valve (Valco Instruments Co), two LC systems (Shimadzu 20AR: Shimadzu Scientific Instruments; Agilent 1100: Agilent Technologies), and a Leap HTC PAL autosampler (Leap Inc.). Thermo Betasil C18 trapping columns (2.1 × 10 and 4.6 × 10 mm, 5 μm particle size (Thermo Scientific) and four analytical HPLC columns (Thermo Betasil C18, 2 × 100 mm, 5 μm; Agilent Eclipse XDR-C18, 3 × 100 mm, 3.5 μm; Agilent Zorbax Eclipse C18, 3 × 50 mm, 3.5 μm; Vydac C18, 2 × 150 mm, 5 μm) were used for sphingolipid, serine, and palmitoyl-CoA analysis. Mobile phases of 1.5% formic acid (FA) in water and acetonitrile were used for trapping. Mobile phases for analytical HPLC are described in the quantification of sphingolipids section. Multiple reaction monitoring with Q1 and Q3 ions using positive ion electrospray (electrospray ionization) mode was applied, and Analyst 1.51v software (Applied Biosystems) was used to quantify each of the sphingolipids, serine, and palmitoyl-CoA with calibration curves from analyte standard solutions containing internal standards.
Quantification of Sphingolipid Metabolites, Seine, and Palmitoyl-CoA
Macrophage lysates (50 μl) containing sphingoid base internal standards (C17-sphingosine and C17-sphingosine 1-phosphate) were analyzed using an Agilent Eclipse XRD-C18 column with a solvent gradient of 70% B (2% FA in 1:1 methanol/acetonitrile) and 30% A (2% FA in water) to 100% B in 5 min. Positive Q1 and Q3 ions for all four analytes (sphingosine, 3-ketosphinganine, sphinganine, and sphingosine 1-phosphate) including the internal standards were determined by analyte infusion. For C16 ceramide 1-phosphate, 50 μl of the lysates containing C17 sphingosine 1-phosphate as the internal standard was directly injected onto an Agilent Eclipse XRD-C18 column with a solvent gradient of 70% B (10 mm NH4OAc in 1:1 methanol/isopropyl alcohol) and 30% A (10 mm NH4OAc in 3:7 acetonitrile/water) to 100% B in 4 min. Positive Q1 and Q3 ions for C16 ceramide 1-phosphate were also determined by analyte infusion. To determine ceramide, dihydroceramide, and sphingomyelin concentrations, 10 μl of macrophage lysates spiked with 100 ng of internal standards (C17, C22 d4, C24 d4-ceramide, and C17 sphingomyelin) were directly injected into a Thermo Electron Betasil C18 analytical column. The solvent gradient was 90% B (10 mm NH4OAc in 1:1 methanol/isopropyl alcohol) and 10% A (10 mm NH4OAc in 3:7 acetonitrile/water) to 100% B in 5 min. Calibration standards for sphingomyelin were not used so that peak area comparison of each analyte with the internal standard was applied for sphingomyelin quantification. Published positive Q1 and Q3 ions of these analytes were used for these analyses (23). For serine quantification, 20 μl of macrophage lysate from a dansyl chloride-derivatized sample and calibration solution were injected into a Vydac C18 peptide column, and the solvent gradient was 50% B (1% formic acid in acetonitrile) and 50% A (1% formic acid in water) to 100% B in 4 min. Positive Q1 and Q3 ions including the internal standard (l-homoserine) were determined by analyte infusion. For palmitoyl-CoA, 20 μl of macrophage lysate was spiked with 50 ng of heptadecanoyl-CoA (C17-CoA). The samples were analyzed using an Agilent Zobax C18 column with a solvent gradient of 0% B (0.1% FA in 3:7 water/acetonitrile) and 100% A (15 mm ammonium hydroxide in 9:1 water acetonitrile) to 10% B in 2 min, 50% B in 5 min, and 100% B in 6 min.
RESULTS
LPS and Palmitate Lead to Synergistic C16 Ceramide Production in Macrophages
To determine the impact of LPS stimulation on sphingolipid content of primary macrophages cultured for 16 h in the presence of 500 μm palmitate, we used LC-MS/MS to examine metabolites in this pathway (24). As seen in Table 1 and Fig. 1, the most dramatic change was in the production of C16-ceramide, which was synergistically induced by the combination of LPS and palmitate. Interestingly, in these primary cells and in contrast to RAW cells (15), LPS alone did not increase the abundance of this ceramide species. The combined stimulus of LPS and palmitate also increased the abundance of other long-chain ceramides but to a lesser extent. C16-ceramide phosphate was reduced by LPS and increased by palmitate, and the synergy response for this ceramide species was blunted in comparison to that seen for C16-ceramide. The observed synergy for C16-ceramide was dependent on the dose of palmitate and was apparent at free fatty acid concentrations as low as 100 μm (Fig. 1A). Increased ceramide was observed as early as 4 h after stimulation and was markedly increased by 16 h (Fig. 1B). This synergy required co-treatment with palmitate and LPS, as the effects were not observed with sequential exposures (not shown).
TABLE 1.
Characterization of sphingolipids in macrophages stimulated with 500 μm palmitate and 50 ng/ml LPS for 16 h
| Analyte (units) | BSA |
Palm |
||
|---|---|---|---|---|
| PBS | LPS | PBS | LPS | |
| C16:0 ceramide (ng/μg DNA) | 1.7 ± 0.1 | 1.94 ± 0.06 | 6.7 ± 0.09a | 17.5 ± 0.32a,b |
| C20:0 ceramide (pg/μg DNA) | 103 ± 3.5 | 65 ± 1.9b | 114 ± 1.0a | 210 ± 20.9a,b |
| C22:0 ceramide (pg/μg DNA) | 319 ± 17.9 | 212 ± 16.8b | 894 ± 22.5a | 1,932 ± 343a,b |
| C24-ceramide (pg/μg DNA) | 894 ± 89.1 | 735 ± 62.1b | 2143 ± 142a | 2787 ± 490a,b |
| C16:0 ceramide phosphate (pg/μg DNA) | 240 ± 41.7 | 140 ± 16b | 540 ± 45.9a | 1190 ± 126a,b |
| C16-sphingomyelin (ng/μg DNA) | 93 ± 3.6 | 81 ± 5.3 | 89 ± 5.6 | 85±.5.3 |
| Sphingosine (pg/μg DNA) | 79 ± 2.7 | 38 ± 4.4b | 116 ± 8.1a | 65 ± 11.9a,b |
| Sphingosine 1-phosphate (pg/μg DNA) | 2.1 ±.26 | 2.7±.48 | 56 ± 4.4a | 166 ± 49a,b |
a p < 0.05 BSA vs. palm.
b p < 0.05 PBS vs. LPS.
FIGURE 1.
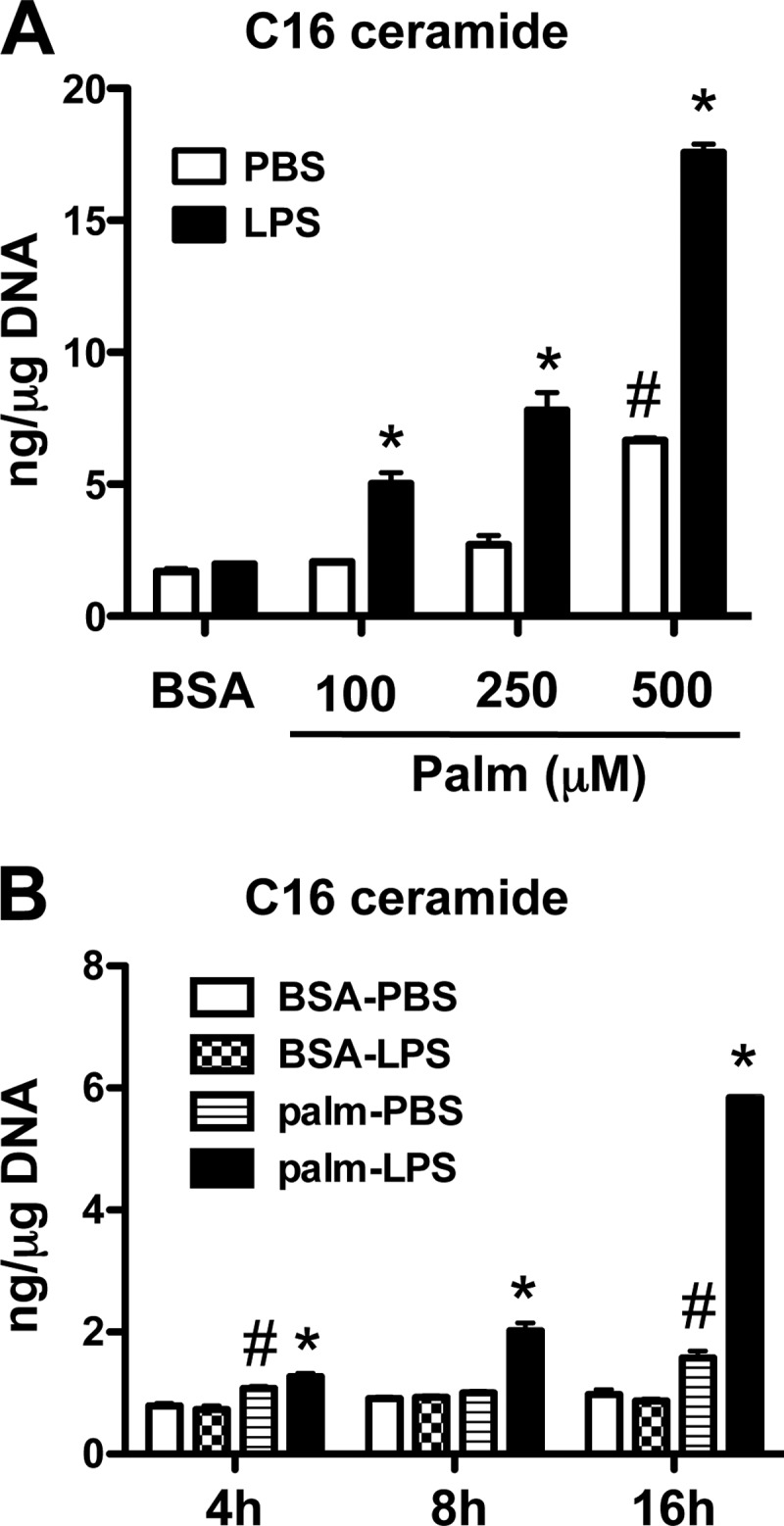
LPS synergizes with palmitate to increase ceramide in macrophages. A, peritoneal macrophages (pMACs) were incubated for 16 h with the indicated concentrations of palmitate (Palm) (or with BSA as control) alone or in combination with 50 ng/ml of LPS (or with PBS as control). C16 ceramide concentrations were determined by LC-MS/MS and normalized per μg DNA. B, pMACs were stimulated with 500 μm palm ± LPS for the indicated time points, and C16 ceramide was quantified by LC-MS/MS. Bar graphs report the means ± S.E. (S.E.) for a minimum of three experiments, each performed in triplicate. *, p < 0.05 for LPS versus PBS; #, p < 0.05 for palm versus BSA. n.s., not significant.
Synergistic Ceramide Production Occurs via the de Novo Synthetic Pathway
Ceramide can be produced by de novo synthesis, sphingomyelin cleavage via sphingomyelinases, and/or recycling pathways (19). To gain insight into which pathway(s) was likely responsible for our ceramide phenotype, we determined the levels of sphingomyelin, sphingosine, and de novo synthesis intermediates in macrophages after palmitate-LPS stimulation (Fig. 2A). Although C16 sphingomyelin levels remained constant and sphingosine levels were reduced under conditions of LPS treatment, C16-dihydroceramide was synergistically induced by LPS and palmitate, suggesting a role for de novo synthesis (Table 1, Fig. 2B). To further investigate the induction of this pathway, we performed an early time course analysis of the first two intermediates in this pathway, 3-ketosphinganine and sphinganine (Fig. 2A). Interestingly, as early as 2 h after stimulation the synergistic generation of these early lipid products was observed in macrophages (Fig. 2, C and D). To determine the contribution of de novo synthesis in palmitate-LPS induced ceramide generation, we employed two chemical inhibitors of this pathway. Myriocin (myr) inhibits SPT and blocks the first step in sphingolipid biosynthesis, and fumonisin B (FB) is a ceramide synthase inhibitor that prevents the conversion of sphinganine to dihydroceramides (Fig. 2A). Both of these inhibitors almost completely blocked the induction of C16-ceramide by palmitate-LPS (Fig. 2E). To confirm the specificity of the inhibitors, we also determined sphinganine concentrations in the samples. Sphinganine levels in LPS-, palm-, and palm-LPS-treated cells were profoundly suppressed by myriocin, consistent with SPT inhibition (Fig. 2F, left panel). The lack of effect in PBS-BSA-treated cells suggests that under these control conditions pathways such as sphinganine 1-phosphate cycling or dihydroceramide ceramidase cycling may contribute substantially to cellular sphinganine levels. In contrast, FB-treated cells had markedly increased sphinganine levels, consistent with a block at the level of sphinganine conversion to dihydroceramide (Fig. 2F, right panel).
FIGURE 2.

Palmitate and LPS synergize to induce de novo ceramide biosynthesis. A, schematic shows the de novo ceramide synthesis pathway and the site of action of chemical inhibitors myriocin (myr) and fumonisin B1 (FB). B, C16:0 dihydroceramide levels were determined by LC-MS/MS in pMACs stimulated with 500 μm palm ± LPS for 16 h and normalized per μg of DNA. C and D, the de novo ceramide synthesis intermediates 3-ketosphinganine (C) and sphinganine (D) were quantified using LC-MS/MS at 2 and 4 h after treatment of pMACs with 500 μm palm ± LPS. E and F, pMACs were pretreated with vehicle (veh; white bars), myr (1 μm, hatched bars), or FB (5 μm, black bars) for 30 min before co-treatment with the indicated stimuli. C16 ceramide (E) and sphinganine (F) were determined at 16 h by LC-MS/MS. G and H, macrophages were treated with the indicated fatty acids ± LPS, and C16:0 ceramide (G) or 3-ketosphinganine (H) levels were determined at 16 h. Bar graphs report the mean ± S.E. for a minimum of 3 experiments, each performed in triplicate. *, p < 0.05 for LPS versus PBS; #, p < 0.05 for palm or other FA versus BSA; **, p < 0.05 for inhibitor versus vehicle.
To determine if the synergistic induction of de novo ceramide biosynthesis with LPS was specific to palmitate (C16:0), we co-treated macrophages with LPS and myristic (C14:0), stearic (C18:0), or oleic acid (C18:1). Palmitate and stearate treatment both increased macrophage C16 ceramide content; however, the augmentation by LPS was more pronounced in palmitate-treated cells (Fig. 2G). Neither myristate nor oleate increased C16 ceramide in the absence or presence of LPS. Although stearate alone and in combination with LPS promoted C16 ceramide production, the longer SFA had only modest effects on 3-ketosphinganine, suggesting a distinct mechanism(s) of ceramide generation compared with palmitate treatment (Fig. 2H). Consistent with this notion, stearate was significantly more toxic to macrophages than palmitate (DNA content was reduced by 65% in stearate-treated cells), which may have triggered other stress-related pathways for ceramide production.
Synergistic de Novo Synthesis of Ceramide Occurs via a TLR4-, MyD88-, and TRIF-dependent Mechanism
The above data indicated that LPS acts to augment de novo ceramide synthesis in the presence of palmitate. To investigate the LPS signaling module responsible for up-regulating this pathway, we determined C16-ceramide levels in TLR4 KO, MyD88 KO, TRIF KO, and MyD88-TRIF (DBL) KO macrophages. As expected, macrophages lacking the LPS receptor, TLR4, had a complete loss of synergistic ceramide production (Fig. 3A). Interestingly, palmitate-induced ceramide production still occurred in the absence of TLR4. Macrophages lacking either MyD88 or TRIF both had a significant reduction in C16-ceramide production after palmitate-LPS, with TRIF-deficient cells having a more pronounced phenotype (Fig. 3A). Similar to the TLR4 KO cells, DBL KO macrophages had a complete loss of synergistic ceramide production (Fig. 3B). As seen in the TLR4 KO cells, the increase of C16 ceramide after palmitate treatment was not impacted by the loss of MyD88 and TRIF. Thus, TLR4 signaling via both MyD88 and TRIF is required for the full synergistic de novo ceramide synthesis but is dispensable for palmitate-induced ceramide production.
FIGURE 3.
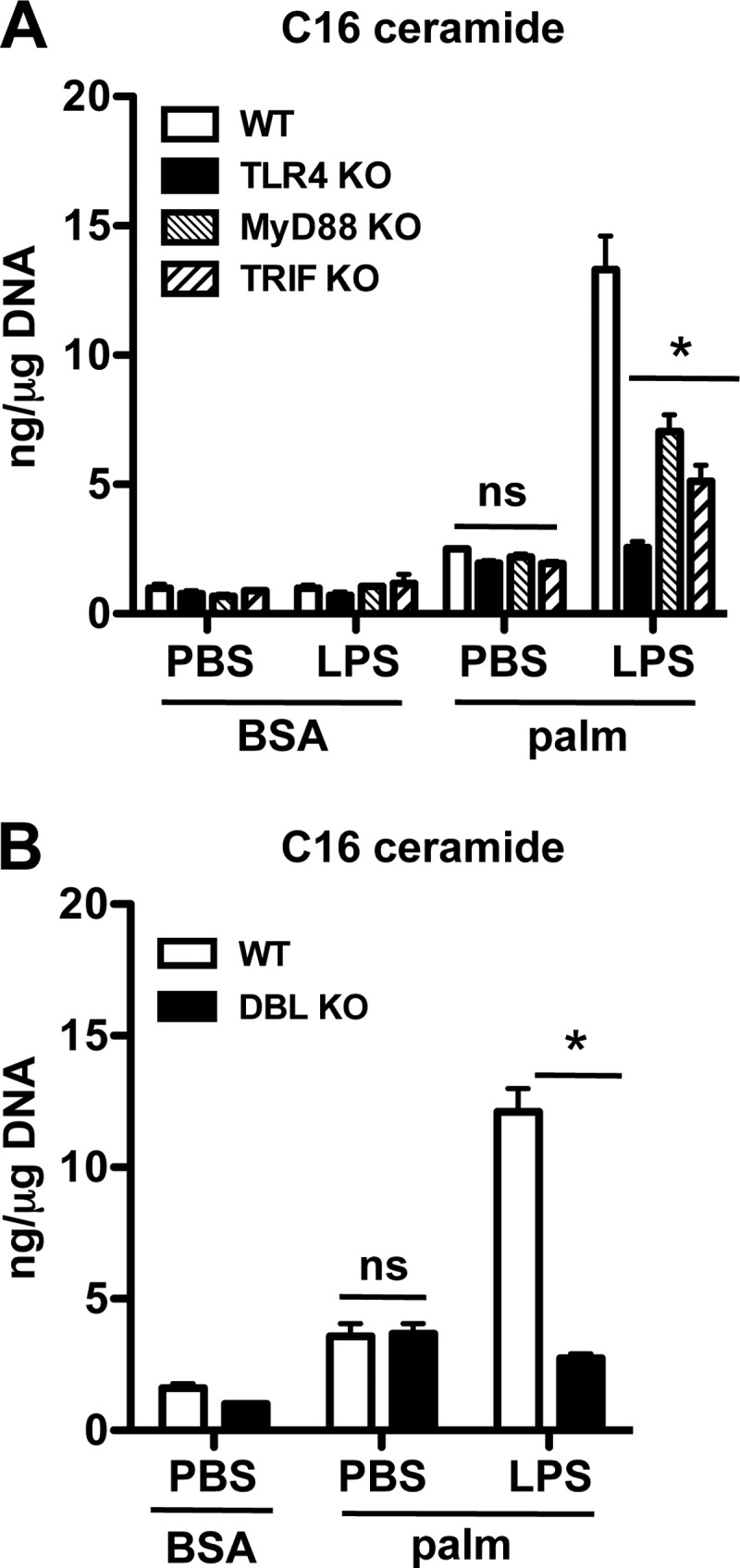
Synergistic ceramide production requires signaling via TLR4, MyD88, and TRIF. A, macrophages from WT, TLR4 KO, MyD88 KO, or TRIF KO mice were treated as indicated, and C16 ceramide was quantified at 16 h. B, pMACs from WT or MyD88/TRIF double KO mice (DBL KO) were treated with palm ± LPS, and C16 ceramide levels were quantified at 16 h. Bar graphs report the mean ± S.E. for a minimum of three experiments, each performed in triplicate. *, p < 0.05 for KO versus WT; ns, not significant.
Ceramide Synthesis after Palmitate-LPS Treatment Occurs Independent of NF-κB and MAP Kinases
TLR4 signaling culminates in the activation of the transcription factor NF-κB and the MAP kinases p38, ERK, and JNK. Full activation of these pathways requires both MyD88 and TRIF. To determine whether NF-κB was required for synergistic ceramide production, we employed pharmacologic and genetic approaches. SN50 is a peptide that inhibits NF-κB nuclear translocation. Consistent with its ability to prevent NF-κB-mediated gene expression, we observed a 65% decrease in LPS-induced TNFα release in SN50-treated cells (Fig. 4A). Despite the anti-inflammatory properties of this peptide, SN50 did not affect C16-ceramide production in palmitate-LPS-treated macrophages (Fig. 4B). IKKβ is upstream of NK-κB activation and has been implicated in the up-regulation of genes involved in ceramide biosynthesis (21). To test a role for IKKβ in the ceramide response to palmitate and LPS, we used macrophages from LysM-Cre mice crossed to IKKβ-floxed animals (25). Similar to SN50, there was no difference in ceramide production in IKKβ KO cells even though in this conditional knock-out system IKKβ mRNA expression in peritoneal macrophages was reduced by ∼60% (Fig. 4, C and D). To assess the contribution of MAP kinases to synergistic de novo ceramide biosynthesis, we used well characterized inhibitors of p38, ERK, and JNK before palmitate-LPS stimulation. There were no differences in C16 ceramide levels observed with any of these inhibitors despite their ability to modulate macrophage TNFα secretion (Fig. 4, E and F). Thus, the synergistic ceramide response to LPS and palmitate in primary macrophages occurs independent of NF-κB and is likely independent of signaling through classical MAP kinases.
FIGURE 4.

The induction of ceramide in response to palmitate and LPS occurs through a NF-κB and MAP kinase-independent mechanism. A and B, macrophages were pretreated with the NF-κB translocation inhibitor SN50 (25 μm) and then with palmitate ± LPS and SN50 for 16 h. TNFα secretion (A) and C16 ceramide levels (B) were quantified by ELISA and LC-MS/MS, respectively. C and D, mRNA expression of IKKβ relative to 36B4 was quantified by quantitative real-time-PCR (C), and ceramide levels were quantified after 16 h of treatment as indicated (D) in macrophages from IKKβ flox mice (FLOX) and from IKKβ flox mice crossed with LysM-Cre mice (KO). E and F, MAP kinase inhibitors for p38 (p38i SB203580, 20 μm), JNK (JNKi SP600125, 20 μm), and ERK (ERKi PD98059,10 μm) were preincubated with pMACs before stimulation with palm and LPS. TNFα secretion (E) and C16 ceramide levels (F) were determined 16 h after treatment. Bar graphs report the mean ± S.E. for a minimum of three experiments, each performed in triplicate. *p < 0.05 for KO/TG versus WT and for inhibitor versus vehicle. ns, not significant.
LPS and Palmitate Do Not Modulate SPT Expression or Activity
SPT catalyzes the rate-limiting step in de novo ceramide synthesis. Given previous reports that palmitate or LPS can induce mRNA expression of SPT, we determined the expression levels of SPT1 and SPT2 using quantitative real-time-PCR. At 2 h after stimulation, when de novo synthesis is already markedly induced, SPT1 mRNA was slightly decreased by palmitate and not affected by LPS, whereas SPT2 showed a very modest increase in expression with both stimuli together (Fig. 5A). These data combined with the rapid kinetics of 3-ketosphinganine production after stimulation with LPS and palmitate, make it unlikely that transcriptional induction of SPT explains the synergistic ceramide response. To determine whether changes in SPT activity could account for enhanced de novo ceramide synthesis, we performed an SPT activity assay on lysates from cells pretreated with all combinations of PBS or LPS with or without palmitate for 2h (26). At this time point 3-ketosphinganine levels are significantly increased in intact cells (Fig. 2C). SPT1 activity, as reflected by 3-ketosphinganine production, was robust in macrophage lysates exposed to palmitoyl CoA and serine, but surprisingly there were no differences between the treatment groups (Fig. 5B). As a control, macrophage lysates were incubated without substrate or in the presence of the SPT inhibitor myriocin, both of which prevented the generation of 3-ketosphinganine (Fig. 5B). This phenotype was also verified at several different concentrations of palmitoyl-CoA in the reaction mixture (Fig. 5C). Thus, it appears that de novo ceramide synthesis is increased via a mechanism that does not involve changes in the expression of SPT or its activity in cell extracts.
FIGURE 5.

SPT expression and activity in cell extracts is unchanged after LPS and palmitate treatment of macrophages. A, pMACs were treated PBS or LPS (50 ng/ml) in the presence of BSA (white bars) or 500 μm palm (black bars), and mRNA was harvested 2 h after stimulation. Gene expression levels for the 2 subunits of SPT (SPT1, SPT2) and TNFα were determined by quantitative real-time-PCR and normalized to 36B4 expression. B, cell lysates were prepared from pMACs treated for 2 h with the indicated stimuli and assayed in vitro for SPT activity by quantification of 3-ketosphinganine production using LC-MS/MS. Control reactions were performed without the addition of substrate or in the presence of myr. ns, not significant. C, palmitoyl CoA dose-response curves for SPT activity are shown for lysates prepared from cells treated with BSA-PBS or palm-LPS. D and E, pMACs were treated with the indicated stimuli for 2 h after which intracellular serine (D) or palmitoyl-CoA (E) concentrations were determined by LC-MS/MS. As a control for serine detection, intracellular serine concentrations were determined in pMACs after incubation with media containing (+) or not containing (−) 10 mm serine (SER) for 2 h (D, inset). All experiments were performed in triplicate a minimum of three times. *, p < 0.05 for LPS versus PBS; #, p < 0.05 for palm versus BSA.
Another potential mechanism to explain the disconnect between 3-ketosphinganine production and SPT activity would be a change in substrate presentation. Therefore, we determined the concentrations of serine and palmitoyl-CoA in treated macrophages. Serine levels remained constant for all combinations of palmitate and LPS (Fig. 5D). To validate our serine detection method, we incubated macrophages in standard media or media supplemented with 10 mm serine for 2 h. Cells exposed to serine-enriched media had a marked increase in intracellular serine levels using an LC-MS/MS-based approach (Fig. 5D, inset). In contrast, palmitoyl-CoA levels increased with palmitate exposure; however, there were no differences in relative amount between palmitate- and palmitate-LPS-treated macrophages (Fig. 5E). Thus, the synergistic induction of de novo ceramide synthesis seen with palmitate-LPS compared with LPS alone occurs without significant changes in SPT substrate availability.
De Novo Ceramide Biosynthesis Leads to Augmented IL-1β and TNFα Release from LPS-treated Lipid-overloaded Macrophages
Tight regulation of macrophage cytokine release is essential to generate an effective inflammatory response. Both IL-1β and TNFα are potent proinflammatory cytokines that have been implicated in tissue pathology associated with diabetes and lipid overload (27–31). IL-1β production is dependent on inflammasome activation and caspase 1-mediated cleavage of pro-IL-1β (32). Consistent with previous data (30), we observed that the combination of palmitate and LPS lead to robust release of IL-1β that was blocked by the caspase 1-specific inhibitor Z-YVAD (Fig. 6A). Notably, neither palmitate nor LPS treatment alone led to IL-1β release. To evaluate the role of de novo ceramide synthesis in this response, we quantified IL-1β release from palmitate-LPS-treated macrophages in the presence of the ceramide synthesis inhibitors myr and FB. Each compound significantly reduced the release of IL-1β (Fig. 6B). We also assessed the effects of de novo ceramide synthesis on macrophage TNFα secretion. LPS treatment led to robust TNFα release, and this response was further enhanced in the presence of palmitate (Fig. 6C). Importantly, palmitate did not trigger macrophage TNFα secretion by itself. Similar to IL-1β, myr and FB blunted the augmented TNFα production observed in palmitate-LPS-treated cells (Fig. 6, C and D). However, these inhibitors did not reduce LPS-induced TNFα secretion, consistent with our data that LPS alone does not increase ceramide levels. As shown in Fig. 6D, the augmented TNFα release induced by the combination of palmitate and LPS was reduced by ∼50% after treatment with the de novo ceramide synthesis inhibitors.
FIGURE 6.

Augmented de novo ceramide synthesis in macrophages exposed to palmitate and LPS leads to excessive IL-1β and TNFα secretion. A, pMACS were treated with BSA or palm (250 μm) ± LPS with or without 20 μm Z-YVAD or vehicle control, and IL-1β levels in the supernatant were determined at 24 h. B, macrophages were stimulated with LPS and palm in the presence of vehicle (white bars), myr (hatched bars), or FB (black bars), and IL-1β secretion was quantified by ELISA 24 h after treatment. C, TNFα secretion by pMACs was determined by ELISA 24 h after the indicated treatments in the presence of myr or FB. ns, not significant. D, augmented TNFα secretion, defined as the difference in TNFα release from palm-LPS versus LPS treated macrophages, was quantified in macrophages treated with veh, myr, or FB (vehicle-treated samples = 100%). All experiments were performed three times in triplicate. *, p < 0.05 for inhibitor versus veh; #, p < 0.05 for palm versus BSA. ns, not significant.
DISCUSSION
Obesity and diabetes are strongly associated with lipid abnormalities and increased inflammation (33). In addition, there is evidence that macrophage function is compromised in states of lipid excess, although the mechanisms are not well understood. To gain insight into the effects of lipid overload on macrophage inflammatory function, we explored changes in the sphingolipids of LPS activated peritoneal macrophages exposed to palmitate. Our experiments revealed marked alterations in sphingolipid and ceramide content in LPS-activated macrophages exposed to an environment rich in SFA. Although LPS alone did not change ceramide levels in these primary cells, it synergistically induced C16 ceramide when combined with palmitate. This response occurred via augmented flux through the de novo ceramide biosynthesis pathway. Signaling via the TLR4 adaptors MyD88 and TRIF was required for maximal ceramide biosynthesis in response to combined stimulation; however, neither was required for palmitate-induced ceramide production. Interestingly, palmitate enhanced the release of IL-1β and TNFα from LPS-stimulated macrophages. Whereas inhibition of de novo ceramide synthesis did not affect LPS-stimulated cytokine release, it modestly reduced this augmented release of inflammatory mediators observed with combined palmitate-LPS treatment.
Increased production of ceramides often occurs in response to stress and can modulate pathways related to cell death, proliferation, autophagy, and inflammation (34). The effects of lipids and inflammatory signaling on ceramide production have been studied in established cell lines, and our data show that these stimuli regulate ceramide production differently in primary macrophages. In cultured RAW 264.7 macrophages, TLR activation has been shown to increase sphingolipid and ceramide content (15, 16). As with other cell types, palmitate treatment of RAW cells has also been shown to increase total cellular ceramide (21). In contrast, our data show that LPS alone does not increase ceramide generation in primary macrophages, but the combination of palmitate and LPS triggers synergistic ceramide generation via enhanced de novo ceramide synthesis. A recent report investigating palmitate-induced ceramide production in C2C12 skeletal myoblasts demonstrated that increased de novo synthesis of ceramide occurred via TLR4-MyD88-IKKβ-dependent induction of SPT and ceramide synthase gene expression (21). In our study palmitate increased de novo ceramide synthesis in primary macrophages; however, macrophages from mice deficient in TLR4 did not have a defect in palmitate-induced ceramide production. These results support the notion that palmitate increases ceramide synthesis by providing more substrate for SPT independent of inflammatory pathway activation. Future studies to dissect the molecular basis of differences in this response among immortalized RAW cells, skeletal myoblasts, and primary macrophages may provide important new insights to its molecular underpinnings.
TLR4 signaling occurs via the adaptor proteins MyD88 and TRIF, both of which were required for the synergistic ceramide phenotype induced by palmitate and LPS. MyD88- and TRIF-dependent signaling pathways are distinct but overlap with the activation of NF-κB and MAP kinases. NF-κB has been implicated in the induction of genes involved in ceramide synthesis; however, inhibition of NF-κB or the MAP kinases in primary macrophages did not affect C16 ceramide production. Moreover, the upstream kinase IKKβ was also not required for the ceramide phenotype in palmitate- and LPS-treated cells. When considered with the rapid kinetics of 3-ketosphinganine production after stimulation, these data suggest that LPS modulates de novo synthesis through a non-transcriptional mechanism that is independent of canonical inflammatory signaling pathways.
The rate-limiting step of de novo synthesis is catalyzed by the enzyme SPT and results in the production of 3-ketosphinganine. Thus, de novo synthesis can be regulated by changes in SPT gene expression, SPT activity, or increased substrate availability. Our data point to mechanisms other than increased transcription of SPT. This is supported by our results with NF-κB inhibition, the very modest alterations in SPT2 mRNA, and the rapid kinetics of the de novo ceramide response. The synergistic response is also unlikely due to changes in substrate levels, as serine and palmitoyl-CoA concentrations in the cells were similar after treatment with palmitate and LPS as compared with palmitate alone. The observation that the palmitate-LPS response is accompanied by rapid generation of 3-ketosphinganine points to increases in SPT activity in intact cells; however, enzyme activity in lysates prepared from stimulated macrophages was not increased, suggesting that LPS must influence SPT function through a mechanism that requires integrity of the cell. Under lipotoxic conditions, remodeling of ER membrane lipids could alter the conformation of SPT, which is an integral membrane protein (35). Moreover, evidence is emerging that the functional activity of SPT can be modulated by other ER proteins such as Orm1/2. In yeast, the Orms have been shown to physically interact with SPT in the ER thereby reducing enzyme activity (36, 37). Phosphorylation of Orm1 disrupts its interaction with SPT1 and enhances the production of sphingolipids and ceramides (38). To date, studies of the regulation of SPT by ORMs have relied on transfection of epitope-tagged constructs into cells of interest. Alternative approaches will be required for study of these interactions in primary macrophages, which are not well suited for transient transfection approaches. Additionally, development of reagents to study endogenously expressed ORMs will facilitate future studies to probe whether their activity or interaction with SPT are affected in mouse macrophages under lipotoxic conditions.
Cytokine secretion is a particularly important mechanism through which activated macrophages control inflammatory responses. IL-1β and TNFα are proinflammatory cytokines that are critical for host defense and tissue repair (39, 40). Yet dysregulation of their expression and release has been linked to excessive tissue damage and the development of insulin resistance and diabetes (28, 30, 41). Although it has been suggested that fatty acids can directly activate TLRs, leading to the production of cytokines, this notion is still controversial given the presence of contaminants in many BSA preparations (3, 42, 43). Using ultrapure BSA we observed that palmitate-treated macrophages did not release IL-1β or TNFα even through the combination of palmitate and LPS significantly stimulated the release of these cytokines. IL-1β secretion in response to combined LPS and palmitate stimulation is known to be dependent on the NRLP3 inflammasome (30). Although it has previously been shown that exogenously added ceramides can trigger inflammasome activation (31), we demonstrate here that sphingolipids generated endogenously through the de novo synthetic pathway influence this pathway in palmitate-LPS treated macrophages. Although inhibition of de novo ceramide synthesis reduces the enhanced macrophage TNFα secretion after combined palmitate-LPS treatment, additional studies will be required to define the roles of ceramide, ceramide-phosphate, and other sphingolipid molecular species. Nonetheless, our findings argue that the de novo ceramide synthesis pathway can modulate the over-exuberant inflammatory responses in the setting of combined lipotoxic and inflammatory stimuli. This notion is consistent with another recent study suggesting that SFA can enhance LPS-induced IL-6 and IL-8 release in human monocytes via a ceramide-dependent mechanism (1).
Macrophages play a central role in the host response to infection and tissue injury, and evidence is accumulating that these cells are dysfunctional in patients with obesity and diabetes. In the present study we describe a cross-talk pathway between TLR4 and the fatty acid palmitate that leads to synergistic induction of ceramide production through enhanced de novo synthesis. This process occurs via a non-transcriptional mechanism that is independent of classically inflammatory signaling molecules such as NF-κB and MAP kinases. Our study provides evidence for a previously unappreciated interaction between lipid metabolic and inflammatory mediators that has the potential to influence the release of inflammatory signaling molecules from macrophages and that may be relevant to the pathogenesis of metabolic diseases.
Acknowledgment
Mass spectrometry analyses were performed in the Washington University Metabolomics Facility.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 DK064989 and P20 HL113444 (to J. E. S.), K08HL098373 (to J. D. S.), U24 DK076169, and P60 DK020579. This work was also supported by Burroughs Wellcome Foundation Grant 1005935 (to J. E. S.).
- TLR4
- Toll-like receptor 4
- LPS
- lipopolysaccharide
- TRIF
- TIR-domain-containing adaptor-inducing interferon beta
- SFA
- saturated fatty acid
- ER
- endoplasmic reticulum
- SPT
- serine palmitoyltransferase
- FA
- formic acid
- FB
- fumonisin B
- pMAC
- peritoneal macrophage
- MAP
- mitogen-activated protein
- palm
- palmitate
- IKK
- IκB kinase.
REFERENCES
- 1. Schwartz E. A., Zhang W. Y., Karnik S. K., Borwege S., Anand V. R., Laine P. S., Su Y., Reaven P. D. (2010) Nutrient modification of the innate immune response. A novel mechanism by which saturated fatty acids greatly amplify monocyte inflammation. Arterioscler. Thromb. Vasc. Biol. 30, 802–808 [DOI] [PubMed] [Google Scholar]
- 2. Cascio G., Schiera G., Di Liegro I. (2012) Dietary fatty acids in metabolic syndrome, diabetes, and cardiovascular diseases. Curr. Diabetes Rev. 8, 2–17 [DOI] [PubMed] [Google Scholar]
- 3. Shi H., Kokoeva M. V., Inouye K., Tzameli I., Yin H., Flier J. S. (2006) TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Invest. 116, 3015–3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Assimacopoulos-Jeannet F. (2004) Fat storage in pancreas and in insulin-sensitive tissues in pathogenesis of type 2 diabetes. Int. J. Obes. Relat. Metab. Disord. 28, S53–S57 [DOI] [PubMed] [Google Scholar]
- 5. Joshi N., Caputo G. M., Weitekamp M. R., Karchmer A. W. (1999) Infections in patients with diabetes mellitus. N. Engl. J. Med. 341, 1906–1912 [DOI] [PubMed] [Google Scholar]
- 6. Rosen D. A., Hung C. S., Kline K. A., Hultgren S. J. (2008) Streptozocin-induced diabetic mouse model of urinary tract infection. Infect. Immun. 76, 4290–4298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Greer J. J., Ware D. P., Lefer D. J. (2006) Myocardial infarction and heart failure in the db/db diabetic mouse. Am. J. Physiol. Heart Circ. Physiol. 290, H146–H153 [DOI] [PubMed] [Google Scholar]
- 8. Czyzk A., Królewski A. S., Szabłowska S., Alot A., Kopczyński J. (1980) Clinical course of myocardial infarction among diabetic patients. Diabetes Care 3, 526–529 [DOI] [PubMed] [Google Scholar]
- 9. Mirza R., DiPietro L. A., Koh T. J. (2009) Selective and specific macrophage ablation is detrimental to wound healing in mice. Am. J. Pathol. 175, 2454–2462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mirza R., Koh T. J. (2011) Dysregulation of monocyte/macrophage phenotype in wounds of diabetic mice. Cytokine 56, 256–264 [DOI] [PubMed] [Google Scholar]
- 11. Kaisho T., Akira S. (2000) Critical roles of Toll-like receptors in host defense. Crit. Rev. Immunol. 20, 393–405 [PubMed] [Google Scholar]
- 12. Timmers L., Sluijter J. P., van Keulen J. K., Hoefer I. E., Nederhoff M. G., Goumans M. J., Doevendans P. A., van Echteld C. J., Joles J. A., Quax P. H., Piek J. J., Pasterkamp G., de Kleijn D. P. (2008) Toll-like receptor 4 mediates maladaptive left ventricular remodeling and impairs cardiac function after myocardial infarction. Circ. Res. 102, 257–264 [DOI] [PubMed] [Google Scholar]
- 13. Andersson U., Tracey K. J. (2011) HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 29, 139–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Takeda K., Akira S. (2004) TLR signaling pathways. Semin. Immunol. 16, 3–9 [DOI] [PubMed] [Google Scholar]
- 15. Sims K., Haynes C. A., Kelly S., Allegood J. C., Wang E., Momin A., Leipelt M., Reichart D., Glass C. K., Sullards M. C., Merrill A. H., Jr. (2010) Kdo2-lipid A, a TLR4-specific agonist, induces de novo sphingolipid biosynthesis in RAW264.7 macrophages, which is essential for induction of autophagy. J. Biol. Chem. 285, 38568–38579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dennis E. A., Deems R. A., Harkewicz R., Quehenberger O., Brown H. A., Milne S. B., Myers D. S., Glass C. K., Hardiman G., Reichart D., Merrill A. H., Jr., Sullards M. C., Wang E., Murphy R. C., Raetz C. R., Garrett T. A., Guan Z., Ryan A. C., Russell D. W., McDonald J. G., Thompson B. M., Shaw W. A., Sud M., Zhao Y., Gupta S., Maurya M. R., Fahy E., Subramaniam S. (2010) A mouse macrophage lipidome. J. Biol. Chem. 285, 39976–39985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mullen T. D., Obeid L. M. (2012) Ceramide and apoptosis. Exploring the enigmatic connections between sphingolipid metabolism and programmed cell death. Anticancer Agents in Med. Chem. 12, 340–363 [DOI] [PubMed] [Google Scholar]
- 18. Grösch S., Schiffmann S., Geisslinger G. (2012) Chain length-specific properties of ceramides. Progress in Lipid Research 51, 50–62 [DOI] [PubMed] [Google Scholar]
- 19. Gault C. R., Obeid L. M., Hannun Y. A. (2010) An overview of sphingolipid metabolism. From synthesis to breakdown. Adv. Exp. Med. Biol. 688, 1–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Listenberger L. L., Ory D. S., Schaffer J. E. (2001) Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J. Biol. Chem. 276, 14890–14895 [DOI] [PubMed] [Google Scholar]
- 21. Holland W. L., Bikman B. T., Wang L. P., Yuguang G., Sargent K. M., Bulchand S., Knotts T. A., Shui G., Clegg D. J., Wenk M. R., Pagliassotti M. J., Scherer P. E., Summers S. A. (2011) Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J. Clin. Invest. 121, 1858–1870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chang Z. Q., Lee S. Y., Kim H. J., Kim J. R., Kim S. J., Hong I. K., Oh B. C., Choi C. S., Goldberg I. J., Park T. S. (2011) Endotoxin activates de novo sphingolipid biosynthesis via nuclear factor κB-mediated up-regulation of Sptlc2. Prostaglandins Other Lipid Mediat. 94, 44–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shaner R. L., Allegood J. C., Park H., Wang E., Kelly S., Haynes C. A., Sullards M. C., Merrill A. H., Jr. (2009) Quantitative analysis of sphingolipids for lipidomics using triple quadrupole and quadrupole linear ion trap mass spectrometers. J. Lipid Res. 50, 1692–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schnute M. E., McReynolds M. D., Kasten T., Yates M., Jerome G., Rains J. W., Hall T., Chrencik J., Kraus M., Cronin C. N., Saabye M., Highkin M. K., Broadus R., Ogawa S., Cukyne K., Zawadzke L. E., Peterkin V., Iyanar K., Scholten J. A., Wendling J., Fujiwara H., Nemirovskiy O., Wittwer A. J., Nagiec M. M. (2012) Modulation of cellular S1P levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem. J. 444, 79–88 [DOI] [PubMed] [Google Scholar]
- 25. Greten F. R., Arkan M. C., Bollrath J., Hsu L. C., Goode J., Miething C., Göktuna S. I., Neuenhahn M., Fierer J., Paxian S., Van Rooijen N., Xu Y., O'Cain T., Jaffee B. B., Busch D. H., Duyster J., Schmid R. M., Eckmann L., Karin M. (2007) NF-κB is a negative regulator of IL-1β secretion as revealed by genetic and pharmacological inhibition of IKKβ. Cell 130, 918–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rütti M. F., Richard S., Penno A., von Eckardstein A., Hornemann T. (2009) An improved method to determine serine palmitoyltransferase activity. J. Lipid Res. 50, 1237–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Olson N. C., Callas P. W., Hanley A. J., Festa A., Haffner S. M., Wagenknecht L. E., Tracy R. P. (2012) Circulating levels of TNF-α are associated with impaired glucose tolerance, increased insulin resistance, and ethnicity. The insulin resistance atherosclerosis study. J. Clin. Endocrinol. Metab. 97, 1032–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hotamisligil G. S., Shargill N. S., Spiegelman B. M. (1993) Adipose expression of tumor necrosis factor-α. Direct role in obesity-linked insulin resistance. Science 259, 87–91 [DOI] [PubMed] [Google Scholar]
- 29. Dandona P., Aljada A., Bandyopadhyay A. (2004) Inflammation. The link between insulin resistance, obesity, and diabetes. Trends Immunol. 25, 4–7 [DOI] [PubMed] [Google Scholar]
- 30. Wen H., Gris D., Lei Y., Jha S., Zhang L., Huang M. T., Brickey W. J., Ting J. P. (2011) Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 12, 408–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Vandanmagsar B., Youm Y. H., Ravussin A., Galgani J. E., Stadler K., Mynatt R. L., Ravussin E., Stephens J. M., Dixit V. D. (2011) The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 17, 179–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Henao-Mejia J., Elinav E., Strowig T., Flavell R. A. (2012) Inflammasomes. Far beyond inflammation. Nat. Immunol. 13, 321–324 [DOI] [PubMed] [Google Scholar]
- 33. Chawla A., Nguyen K. D., Goh Y. P. (2011) Macrophage-mediated inflammation in metabolic disease. Nat. Rev. Immunol. 11, 738–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nikolova-Karakashian M. N., Rozenova K. A. (2010) Ceramide in stress response. Adv. Exp. Med. Biol. 688, 86–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Borradaile N. M., Han X., Harp J. D., Gale S. E., Ory D. S., Schaffer J. E. (2006) Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J. Lipid Res. 47, 2726–2737 [DOI] [PubMed] [Google Scholar]
- 36. Han S., Lone M. A., Schneiter R., Chang A. (2010) Orm1 and Orm2 are conserved endoplasmic reticulum membrane proteins regulating lipid homeostasis and protein quality control. Proc. Natl. Acad. Sci. U.S.A. 107, 5851–5856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Breslow D. K., Collins S. R., Bodenmiller B., Aebersold R., Simons K., Shevchenko A., Ejsing C. S., Weissman J. S. (2010) Orm family proteins mediate sphingolipid homeostasis. Nature 463, 1048–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Roelants F. M., Breslow D. K., Muir A., Weissman J. S., Thorner J. (2011) Protein kinase Ypk1 phosphorylates regulatory proteins Orm1 and Orm2 to control sphingolipid homeostasis in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 108, 19222–19227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Silke J. (2011) The regulation of TNF signaling. What a tangled web we weave. Curr. Opin. Immunol. 23, 620–626 [DOI] [PubMed] [Google Scholar]
- 40. Strowig T., Henao-Mejia J., Elinav E., Flavell R. (2012) Inflammasomes in health and disease. Nature 481, 278–286 [DOI] [PubMed] [Google Scholar]
- 41. Hotamisligil G. S. (2006) Inflammation and metabolic disorders. Nature 444, 860–867 [DOI] [PubMed] [Google Scholar]
- 42. Weatherill A. R., Lee J. Y., Zhao L., Lemay D. G., Youn H. S., Hwang D. H. (2005) Saturated and polyunsaturated fatty acids reciprocally modulate dendritic cell functions mediated through TLR4. J. Immunol. 174, 5390–5397 [DOI] [PubMed] [Google Scholar]
- 43. Erridge C., Samani N. J. (2009) Saturated fatty acids do not directly stimulate Toll-like receptor signaling. Arterioscler. Thromb. Vasc. Biol. 29, 1944–1949 [DOI] [PubMed] [Google Scholar]