

Background: Interleukin-6 and interleukin-10 both activate the same signaling mediator, STAT3, yet generate nearly opposing responses.
Results: Interleukin-6 and interleukin-10 signaling lead to different durations of STAT3 activation and consequently distinct responses.
Conclusion: The duration of receptor and STAT3 activation determines the specific cytokine response.
Significance: This work reveals a signaling coding mechanism that relieves a cellular information bottleneck.
Keywords: Cell Biology, Computational Biology, Drug Discovery, Immunology, STAT3
Abstract
Paradoxically, the pro-inflammatory cytokine IL-6 and the anti-inflammatory cytokine IL-10 both activate STAT3, yet generate nearly opposing cellular responses. Here, we show that the temporal pattern of STAT3 activation codes for the specific cytokine response. A computational model of IL-6 and IL-10 signaling predicted that IL-6 stimulation results in transient activation of STAT3, with a rapid decline in phosphorylation and nuclear localization. In contrast, simulated IL-10 signaling resulted in sustained STAT3 activation. The predicted STAT3 patterns produced by each cytokine were confirmed experimentally in human dendritic cells. Time course microarray studies further showed that the dynamic genome-wide transcriptional responses were nearly identical at early time points following stimulation (when STAT3 is active in response to both IL-6 and IL-10) but divergent at later times (when STAT3 is active only in response to IL-10). Truncating STAT3 activation after IL-10 stimulation caused IL-10 to elicit an IL-6–like transcriptional and secretory response. That the duration of IL-10 receptor and STAT3 activation can direct distinct responses reveals a complex cellular information-coding mechanism that may be relevant to improving the prediction of the effects of drug candidates using this mechanism.
Introduction
Cytokines are small, secreted peptides that play a key role in regulating and directing the immune response. Individual cells receive many different cytokine stimuli that each yield specific cellular responses, yet there are relatively few signaling pathways available to process the myriad inputs and produce the appropriate output responses. Cases in which different cytokines regulate the same signaling molecule but differ in their effects illustrate the limitation of the current understanding of the mechanism of action of cytokines and other extracellular stimuli.
Interleukin-6 (IL-6) and interleukin-10 (IL-10) both regulate the same signaling molecule, STAT3, yet generate different cellular responses. IL-6 is primarily a pro-inflammatory cytokine, whereas IL-10 generates a strong anti-inflammatory response. IL-6 and IL-10 each bind to their cognate receptor, leading to STAT3 phosphorylation, nuclear localization, and a cytokine-specific gene activation pattern. Aberrant IL-6 and IL-10 signaling has been implicated in a wide range of inflammatory diseases (1–4), and STAT3 itself is known to be dysregulated in many malignancies (5, 6). Paralleling its discordant role in mediating IL-6 and IL-10 responses, STAT3 has been considered both an important pro-inflammatory regulator of the tumor microenvironment (7–9) and a key anti-inflammatory mediator of tumor immune evasion (10). Explaining how activation of the same signaling molecule can paradoxically be both inflammatory and anti-inflammatory is important for clarifying the mechanism of STAT3-related malignancies and immunological diseases and facilitating therapeutic drug discovery in this area.
EXPERIMENTAL PROCEDURES
Differentiation of Dendritic Cells
Monocyte-derived dendritic cells (DCs)2 were obtained from anonymized healthy human blood donors (New York Blood Center) using a standard Ficoll density centrifugation, immunomagnetic purification, and differentiation protocol described elsewhere (11).
IL-6, IL-10, and LPS Treatment
DCs were generated as described above and treated with 10 ng/ml hIL-6 (Peprotech), 10 ng/ml hIL-10 (Peprotech), and/or 100 ng/ml LPS from Salmonella minnesota (InvivoGen). Following treatment, cells were used for flow cytometry, imaging flow cytometry, quantitative RT-PCR, or microarray analysis. Supernatants were used for ELISA or Luminex analysis.
Antibody Blockage
An effective inhibitory concentration for anti-IL10Rα monoclonal antibodies (R&D Systems) was determined by treating cells with 10 ng/ml IL-10 and 0, 0.8, 1.6, 4, or 8 μg/ml anti-IL10Rα monoclonal antibodies and measuring STAT3 phosphorylation by flow cytometry. To truncate STAT3 activation following IL-10 stimulation, DCs were treated with 4–5 μg/ml anti-IL10Rα monoclonal antibodies (depending on the lot) 30 min after initiation of IL-10 stimulation. To control for nonspecific effects of antibody treatment, DCs were treated with an equivalent concentration of mouse IgG1 isotype control (R&D Systems) antibodies 30 min after initiation of IL-10 stimulation.
Flow Cytometry Analysis
STAT3 phosphorylation was measured by flow cytometry. Cells were fixed with 1.6% paraformaldehyde (Sigma-Aldrich), washed with FACSFlow sheath buffer (BD Biosciences), permeabilized with 100% cold methanol (Fisher Scientific), washed with FACS staining buffer (BD Biosciences), and stained with mAbs for Tyr(P)705-STAT3 conjugated to Alexa Fluor 488 (BD Biosciences).
For high resolution analysis of STAT3 phosphorylation dynamics (and for the untreated barcoding setup and control set), cells were fluorescently barcoded to increase throughput, as described previously (12). In brief, cells were fixed, washed, and then permeabilized in methanol containing different combinations of 0, 6.25, or 100 ng/ml Alexa Fluor 350-NHS and 0, 6.25, or 100 ng/ml Alexa Fluor 750-NHS (Invitrogen), washed with FACS staining buffer, and then stained with mAbs. Single-color fluorescence compensation control samples were obtained by staining DCs with Alexa Fluor 350, Alexa Fluor 750, or a mAb against CD45-conjugated Alexa Fluor 488 (BioLegend). Stained cells were assayed on a LSRII flow cytometer (BD Biosciences), and data were analyzed using FlowJo software (Tree Star).
Imaging Flow Cytometry Analysis
Imaging flow cytometry was used to measure the nuclear localization of STAT3 at a single cell level. DCs were fixed and permeabilized as described previously. DCs were then stained with a mAb for Tyr(P)705-STAT3 conjugated to Alexa Fluor 488 (BD Biosciences) and the DNA dye Draq5 (Biostatus Limited). Single cell images were acquired using an ImageStream 100 (Amnis), and nuclear localization was quantified using image analysis software (IDEAS 4; Amnis), as described elsewhere (13). In brief, the pixel-by-pixel Pearson correlation, ρ, of the intensity of pSTAT3 staining with the intensity of Draq5 was calculated for each cell, and then log-transformed to give the similarity score, where similarity = ln((1 + ρ)/(1 − ρ)).
RNA Isolation
Total RNA was isolated from DCs using a Qiagen Mini RNeasy kit or a Qiagen BioRobot Universal with RNeasy 96 kit, with on-column DNase treatment, according to the manufacturer's protocol. RNA was quantified using a ND-1000 spectrophotometer (NanoDrop).
Microarray Analysis
RNA was isolated from DCs from four anonymous healthy human donors, treated with either IL-6 or IL-10 for 45 min, 2 h, 4 h, 8 h, or 12 h. The RNA integrity of all samples was confirmed using an Agilent Bioanalyzer 2100, according to the manufacturer's protocol. Gene expression analysis was performed utilizing the Illumina human HT-12 v4 Expression BeadChip arrays. As an additional quality control measure, the STAT3 phosphorylation response to IL-6 and IL-10 were determined by flow cytometry. Microarrays were processed at the Yale Center for Genome Analysis. BeadChips were scanned using the Illumina iScan, and data were analyzed using Matlab (Mathworks).
Data were first quantile-normalized, and then for each donor the expression value of each gene was normalized by the median expression value of each gene in that donor. Low expression genes were filtered out, and the remaining expression data were log2-transformed. Differentially expressed genes (versus untreated cells) were selected using a two-tail unpaired t test with a false discovery rate-corrected threshold of 0.05 (14). Principal component analysis was performed using the Statistics Toolbox in Matlab (Mathworks) using the normalized, log2-transformed expression values for the differentially expressed genes. Hierarchical clustering was performed using the Bioinformatics Toolbox in Matlab (Mathworks). For each of the differentially expressed genes, its median -fold change was calculated (relative to untreated), and clustering was performed on this data using correlation as the distance metric.
Quantitative RT-PCR
RNA expression was quantified using quantitative RT-PCR. cDNA was synthesized from total RNA using AffinityScript MultiTemp RT (Agilent) with an oligo(dT)18 primer. Real-time PCR was performed using PlatinumTaq DNA polymerase (Invitrogen) and SYBR Green (Invitrogen) on an ABI7900HT thermal cycler (Applied Biosystems), as described previously (15). A robust global normalization algorithm, using expression levels of the housekeeping genes ribosomal protein S11 (rps11), β-actin (actb), and α-tubulin (tuba), was used for all experiments, as described elsewhere (15, 16). In brief, all crossing threshold values were first adjusted by median difference of all samples from actb. Each individual sample was then further corrected by the median crossing threshold value of the three corrected housekeeping control for that sample. Finally, nominal copy numbers were calculated by assuming 2500 molecules of actB mRNA per cell, and an amplification efficiency of 93%. PCR primer sequences can be found in the supplementary Materials.
ELISA for TNFα
DCs were left untreated or treated for 6 h with LPS, LPS, and IL-6, LPS and IL-10, or LPS and IL-10 with neutralizing antibody, and the supernatants were isolated. The TNFα concentration in the supernatants was measured by ELISA according to the manufacturer's protocol (BioLegend).
Multiplex ELISA for Interferon Gamma-induced Protein 10 (IP-10) and IL-10
DCs were left untreated or treated for 6 h with IL-6, IL-10, or IL-10 with neutralizing antibody, and the supernatants were isolated. The IP-10 and IL-10 concentrations in the supernatants were assayed by Luminex (Millipore) according to the manufacturer's protocol.
Computational Models of IL-6 and IL-10 Signaling
We developed stochastic and deterministic models of IL-6 and IL-10 signaling to investigate the dynamics of STAT3 activation. These models build on previously developed JAK/STAT models (17–19) and used parameter values from previous models of STAT3 signaling (17). The deterministic versions of these models used a system of coupled ordinary differential equations (ODEs) based on mass action kinetics (similar to the initial models). The stochastic versions of these models were implemented using Gillespie's method (20, 21). All model simulations were performed in Matlab. Details of the model can be found in the supplementary Materials.
The computer simulations were used to predict time evolution of signaling components following the addition of IL-6 or IL-10 ligand. The model includes ligand-induced receptor activation, ligand-induced STAT3 phosphorylation, STAT3 dephosphorylation, STAT3 nuclear import, STAT3 nuclear export, phosphorylated STAT3 binding to and unbinding from the socs3 promoter, socs3 mRNA production, mRNA nuclear export, mRNA degradation, mRNA translation SOCS3 protein production, SOCS3 protein degradation, and in the case of IL-6 signaling, SOCS3-mediated receptor degradation.
Robustness of Model Simulation Predictions
A metric was created to determine whether a simulation with a given parameter set predicted that IL-6 would generate a transient activation of STAT3 and IL-10 would generate a sustained activation. The mean STAT3 phosphorylation responses over early time points (0–30 min) and late time points (120–180 min) were compared for IL-6 and IL-10 signaling according to the following metric: D = (IL-60–30min/IL-6120–180min)/(IL-100–30min/IL-10120–180min). Each simulation with a value of D ≥ 2 was considered to qualitatively recapitulate the transient versus sustained STAT3 dynamics.
We tested the global robustness of the model predictions by simultaneously selecting each parameter randomly from a 1,000,000-fold range (logarithmically uniform sampling centered around the parameter values used for the original simulations) and measuring whether the same qualitative STAT3 activation patterns (transient versus sustained) were observed using the D metric, similar to a method described previously (22). We ran ∼100,000 simulations with randomly chosen parameter sets and calculated the percentage of simulations with D ≥ 2. We calculated the fraction of values, C, that for the average parameter in the 1,000,000-fold range are compatible with the previously predicted STAT3 activation patterns, using Cn = (fraction of simulations with D > 2), where n is the number of parameters in the model.
We measured the local robustness of the model predictions by systematically varying each parameter value individually (while keeping all others constant) over a range from 4096-fold decrease to a 4096-fold increase, simulating the IL-6 and IL-10 models, and calculating an associated D value for each simulation.
RESULTS
Computational Models Predict Distinct STAT3 Activation Dynamics in Response to IL-6 and IL-10
We constructed a compartmentalized mathematical model of IL-6 and IL-10 signaling and simulated the responses to each cytokine. IL-6 and IL-10 both signal through STAT3 but differ in the architectures of their signaling networks. The signaling of the IL-6 receptor is inhibited by the STAT3-induced protein SOCS3, which does not affect the IL-10 receptor (23). The model for IL-6 signaling included ligand-induced receptor activation, phosphorylation, and nuclear localization of STAT3, and negative feedback through SOCS3 (Fig. 1A). The IL-10 model was identical except for lacking SOCS3 negative feedback (Fig. 1B).
FIGURE 1.
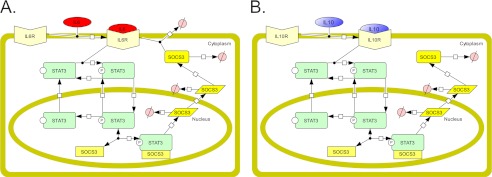
Schematic representations of IL-6 and IL-10 signaling models. A and B, schematics of molecular species and reactions included in the IL-6 model (A) and the IL-10 model (B). Reactions are depicted by black arrow lines, with the beginning of the arrow representing the reactants and the end of the arrow representing the products. The SOCS3 rectangle species in the nucleus represents socs3 DNA, the SOCS3 parallelogram species represents SOCS3 mRNA, and the SOCS3 rounded rectangle species represents SOCS3 protein. The degradation reactions are depicted by arrows ending in Ø.
Model simulations predicted that the temporal dynamics of STAT3 activation obtained with each cytokine were distinct (Fig. 2). IL-6 elicited transient STAT3 phosphorylation and nuclear translocation, with a decline in STAT3 activation corresponding to the effects of the induced feedback inhibitor SOCS3 (Fig. 2, A and B, and supplemental Fig. 1). In contrast, IL-10 signaling resulted in sustained STAT3 activation (Fig. 2, A and B, and supplemental Fig. 2). The robustness of the model behavior to changes in model parameters was studied by individual parameter sensitivity analysis (supplemental Fig. 3) and by global parameter variation (Fig. 2C). Model stability to global parameter variation was tested by randomly selecting values for each parameter over a 106 range and determining whether the simulation produced distinct STAT3 activation responses to IL-6 and IL-10. The response patterns were stable to large variations in model parameters. More than one third of the global parameter variation simulations (36.98%), representing on average 93.6% of the range of each randomly chosen parameter, displayed the same qualitatively distinct responses to IL-6 and IL-10 signaling.
FIGURE 2.
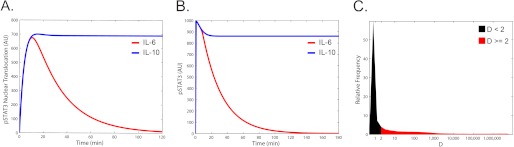
Simulation results of the IL-6 and IL-10 signaling models. A and B, simulated time course of the level of nuclear phosphorylated STAT3 (A) and total phosphorylated STAT3 (B) following IL-6 (red) or IL-10 (blue) stimulation. AU is arbitrary units. C, distribution of D values for ∼100,000 simulations with randomly chosen parameters, where D reflects the difference in STAT3 activity produced by each cytokine (see “Experimental Procedures”). All parameters were simultaneously, randomly selected from a 6 orders of magnitude range, simulations of IL-6 and IL-10 signaling were performed, and the D value was calculated. The distribution of D values from ∼100,000 simulations is shown. Simulations with D values that represent prolonged IL-10 versus IL-6 STAT3 duration (D ≥ 2) are shown in red, and simulations that were below this relative duration threshold are shown in black.
IL-6 and IL-10 Generate Different Temporal Patterns of STAT3 Activation
The differing temporal dynamics of STAT3 activation predicted by the simulations were tested experimentally in human monocyte-derived DCs. The dynamics of STAT3 phosphorylation in response to different concentrations of IL-6 (Fig. 3A) and IL-10 (Fig. 3B) were measured using high resolution imaging flow cytometry. For all concentrations tested, IL-6 produced a transient increase in STAT3 phosphorylation that returned to base line at about 100 min. IL-10, in contrast, produced a sustained STAT3 phosphorylation that reached peak levels after ∼20 min and remained well above base line for the entire 2-h period of study. These results indicate that, as predicted by the model, the duration of STAT3 activation elicited by IL-6 and IL-10 signaling has a distinct temporal dynamics for each cytokine that is conserved for different concentrations. For subsequent experiments, we chose a concentration of 10 ng/ml for IL-6 and for IL-10, levels that caused similar initial patterns of activation of STAT3 for both cytokines.
FIGURE 3.

Phosphorylation and of STAT3 in response to IL-6 or IL-10. A and B, time course of STAT3 tyrosine 705 phosphorylation in IL-6–treated (A) and IL-10–treated (B) DCs, measured by flow cytometry in response to cytokine concentrations of 1, 5, 10, and 20 ng/ml. MFI is median fluorescence intensity. Error bars are S.D. of MFI of three samples from the same blood donor.
Similar responses to IL-6 and IL-10 were obtained with multiple donors (supplemental Fig. 4). Because STAT1 has been reported to be activated by IL-10 (24, 25), the specificity of the patterns of JAK-STAT signaling was evaluated by measuring the levels of STAT1 activation. In dendritic cells, no significant activation of STAT1 by IL-6 or IL-10 was observed (supplemental Fig. 5).
Induction of socs3 mRNA Recapitulates the Different Dynamics of STAT3 Activation
Our signaling model identifies SOCS3 as the main factor responsible for the difference in signaling dynamics observed between IL-6 and IL-10 due to its ability to inhibit the IL-6 receptor and not the IL-10 receptor. As a result, model simulations further predicted that the dynamics of socs3 mRNA expression would mirror the temporal pattern of STAT3 activation, with IL-6 transiently inducing socs3 expression (Fig. 4A) and IL-10 producing a more sustained increase in socs3 mRNA (Fig. 4B). The dynamics of socs3 expression induced by IL-6 and IL-10 were tested experimentally using quantitative RT-PCR (Fig. 4C). IL-6 rapidly induced socs3 expression, and mRNA levels returned to base line after ∼90 min. IL-10 also rapidly induced socs3, but expression remained sustained above base line. In line with model predictions, the differing pattern of socs3 induction over time with each cytokine resembled the different dynamics of STAT3 activation.
FIGURE 4.

Expression of socs3 gene in response to IL-6 and IL-10. A and B, simulated time course of the level of socs3 gene induction in response to IL-6 (A) and IL-10 (B). AU is arbitrary units. C, time course of socs3 gene expression in IL-6–treated (red) and IL-10–treated (blue) DCs, measured by RT-PCR. Error bars are S.E. of three independent samples from the same blood donor.
Dynamic Genome-wide Transcriptional Responses Correlate with Duration of STAT3 Activation
Having established the difference in the dynamics of STAT3 activation elicited by IL-6 and IL-10, we explored whether these dynamics were responsible for the different gene responses associated with each of these two cytokines.
First, we proceeded to test whether the duration in STAT3 activation was a feature conserved following nuclear translocation of this transcription factor. A fine grain time course was obtained for phosphorylated STAT3 (Fig. 5A) and compared with STAT3 translocation to the nucleus (Fig. 5B) measured by high resolution imaging flow cytometry. As hypothesized by the model, IL-6 produced a transient increase in STAT3 nuclear translocation that attenuated after 60 min. IL-10 produced a sustained and stable increase in STAT3 nuclear translocation.
FIGURE 5.

Nuclear translocation of STAT3 and dynamic genome-wide transcriptional responses to IL-6 and IL-10. A, time course of STAT3 tyrosine 705 phosphorylation in IL-6–treated (red) and IL-10–treated (blue) DCs, measured by flow cytometry. MFI is median fluorescence intensity. Error bars are S.D. of MFI of three samples from the same blood donor. B, time course of the nuclear translocation of phosphorylated STAT3 (pSTAT3) in response to IL-6 (red) or IL-10 (blue). For each cell, the pixel-by-pixel correlation, ρ, between the pSTAT3 and nuclear fluorescence intensity images was determined, and similarity was calculated (see “Experimental Procedures”). The median similarity ± S.D. (error bars) of all cells in three samples are shown. C, hierarchical clustering by sample treatment/duration and by genes was performed using the median -fold change in expression relative to untreated cells. Rows represent differentially expressed genes, and columns represent different sample treatments and durations. D, principal component analysis of differentially expressed genes in response to IL-6 (red) or IL-10 (blue) stimulation. The three components plotted account for ∼90% of the total variance.
The levels of active STAT3 were nearly identical early after IL-6 and IL-10 stimulation and markedly different at later time points. We hypothesized that if the cellular responses result from STAT3 activation, the cellular programs activated by each cytokine should show a similar divergence over time. This prediction was tested on a genome-wide scale by determining the transcriptional programs induced by IL-6 and IL-10 over time by microarray analysis. Hierarchical clustering and principal component analysis performed on differentially expressed genes showed that IL-6 and IL-10 produced similar transcriptional responses at early times following stimulation and divergent responses at later times (Fig. 5, C and D). Together, these results suggest that the temporal pattern of STAT3 activation dictates the specific cellular response.
Truncating STAT3 Activity Transforms IL-10 Signal into an IL-6–like Response
We further studied the relationship of the temporal pattern of STAT3 activation and DC response by using a receptor antibody to abrogate IL-10 signaling. Blocking the receptor inhibited STAT3 phosphorylation by IL-10 in a concentration-dependent manner (data not shown). When the receptor-blocking antibody was added to cells 30 min after treatment with IL-10, STAT3 activation was truncated, such that IL-10 caused a transient increase in STAT3 phosphorylation that resembled the pattern obtained with IL-6 (Fig. 6A). The transcriptional and functional effects of truncating STAT3 activation following IL-10 stimulation was determined by measuring differentially regulated transcripts and secreted factors (Fig. 6, B–D) chosen based on our microarray study results and their established involvement with inflammatory and anti-inflammatory responses. Namely, the induction of gene mRNA transcripts of three chemokines ccl3, ccl4, and cxcl1 was used to characterize the inflammatory response elicited by IL-6. In contrast, the absence of these transcripts together with the induction of cepbd was chosen to characterize the anti-inflammatory response elicited by IL-10. These responses were consistent for different concentrations of IL-6 and IL-10 (see supplemental Fig. 6). Truncating the IL-10–induced activation of STAT3 caused IL-10 to induce an IL-6–like response. IL-10 alone caused induction of cebpd and reduction of ccl3 and ccl4 mRNAs. In contrast, IL-10 with STAT3 activation truncation caused induction of ccl3, ccl4, and cxcl1 transcripts, a response indistinguishable from that seen with IL-6 stimulation. Control experiments using isotype control antibodies showed no effects (supplemental Fig. 7). IL-10 suppresses the LPS-stimulated secretion of TNFα, a marker of its anti-inflammatory actions (24). IL-6 has no effect on TNFα induction and stimulates the inflammatory chemokine IP-10. When STAT3 activation was truncated following IL-10 stimulation, a functional response corresponding to the IL-6 response was observed (Fig. 6, C and D). Thus, although IL-6 and IL-10 both activate STAT3, they encode different temporal patterns of activation (transient versus sustained) that are associated with pro-inflammatory and anti-inflammatory cellular programs, respectively.
FIGURE 6.
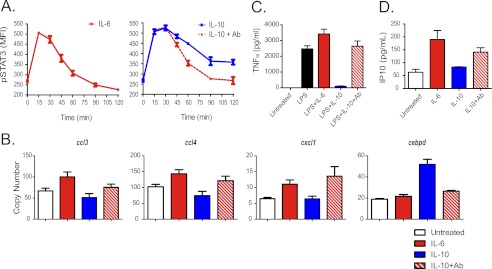
Truncation of IL-10–induced STAT3 activation leads to IL-6–like response. A, DCs were treated with IL-6 (red, left panel), IL-10 (blue, right panel), or IL-10 with anti-IL10Rα receptor-blocking antibodies added 30 min afterward (red dotted, right panel), and STAT3 phosphorylation (pSTAT3) was measured by flow cytometry. MFI is median fluorescence intensity. Error bars are S.D. of MFI of three samples from the same blood donor. B, truncation of IL-10–induced STAT3 activation leads to an IL-6–like transcriptional response. DCs were left untreated (white), or treated with IL-6 (red), IL-10 (blue), or IL-10 with anti-IL10Rα receptor-blocking antibodies added 30 min afterward (red and white), and the expression of four target genes was measured after 4 h by RT-PCR. Error bars are S.E. of six samples from the same blood donor. C, truncation of IL-10–induced STAT3 activation leads to loss of IL-10 anti-inflammatory activity. DCs were left untreated or were treated with LPS (black), LPS and IL-6 (red), LPS and IL-10 (blue), or LPS and IL-10 with anti-IL10Rα receptor-blocking antibodies added 30 min afterward (red and white), and secretion of TNFα into the media after 6 h was measured by ELISA. Error bars are S.E. of three samples from the same blood donor. D, truncation of IL-10–induced STAT3 activation leads to gain of IL-6 pro-inflammatory activity. DCs were left untreated or treated IL-6 (red), IL-10 (blue), or IL-10 with anti-IL10Rα receptor-blocking antibodies added 30 min afterward (red and white), and secretion of the inflammatory chemokine IP-10 into the media after 6 h was measured by Luminex essay. Error bars are S.E. of three samples from the same blood donor.
DISCUSSION
Simulations predicted and imaging flow cytometry experiments confirmed that the topology of the IL-6 and IL-10 signaling networks lead, respectively, to transient and sustained activation of STAT3. The steady divergence of the gene program induced by each cytokine over time suggested that the duration of STAT3 activation might dictate the specific cellular responses. SOCS3 expression is necessary for the pro-inflammatory effects of IL-6 (26). We showed that prolonged activation of STAT3 is necessary for the anti-inflammatory response to IL-10. Shortening the activation of STAT3 by IL-10 transforms the IL-10 cellular response into an IL-6–like response. These results support the hypothesis that the pro- and anti-inflammatory signals of IL-6 and IL-10 are encoded at the level of STAT3 by its transient and sustained activation.
The antibody blockade experiment shows that transient activation of the IL-10 receptor elicits an IL-6–like response. The absence of STAT1 activation eliminates this signal from contributing to the response specificity. However, our results cannot categorically exclude the contribution of some unmeasured or unknown signaling process to the divergent response elicited by IL-6 and IL-10. The evidence that the distinct responses elicited by activation of the two receptors result from the differences in the temporal dynamics of STAT3 activation includes (i) differing concentrations of IL-6 or IL-10 or of peak STAT3 activation levels do not influence the cytokine-specific pattern of the cellular responses observed. (ii) The microarray responses are similar at early time points, when the STAT3 activation patterns also correspond, and diverge at later time points. (iii) The STAT3 activation patterns correlate with the patterns of cell response observed in the antibody inhibition experiment and not with the cytokine used for stimulation.
Without accounting for the temporal pattern information encoding, it would be difficult to predict the cellular and physiological effects of STAT3 modulators. Temporal encoding may have contributed to the challenge of developing STAT3 modulators as therapeutic drugs and has implications for drug discovery in general. The current target-based drug discovery paradigm relies on selecting important therapeutic targets, such as STAT3, developing target modulators, and advancing them through preclinical characterization and clinical trials. The decreasing efficiency of this approach has been attributed to the so-called “valley of death,” the difficulty of bringing target modulators into clinical trials (27, 28). However, the decreasing success rate of drugs entering clinical trials (29) suggests that the pharmacology of target-based drug candidates predicts their therapeutic efficacy and toxicity poorly.
The mechanism of IL-6 and IL-10 information encoding we describe points to the need to refine the pharmacology underlying the current drug discovery paradigm. Temporal pattern encoding relieves the cellular information bottleneck, where many stimuli must signal through few intracellular pathways to generate diverse and specific cellular responses (30–32) but complicates the relationship between synthetic target modulators and their cellular and clinical effects. Several recent studies of drugs acting at cell surface receptor targets show that the activation mechanisms responsible for their cellular and systemic effects are more complex than accounted for by their classical pharmacological profiles (30–32). Elucidating the various mechanisms through which receptors and signaling mediators encode and transmit the information that determines cellular responses may improve the efficiency of drug discovery.
Acknowledgments
We thank Profs. Charles Peskin, Steven Kleinstein, Fernand Hayot, and Dr. Jeremy Seto for helpful discussions and/or comments on the manuscript and Nada Marjanovic for assistance with the RT-PCR experiments.
This study was supported, in whole or in part, by National Institutes of Health Contract HHSN272201000054C.

This article contains supplemental Materials and Figs. 1–7.
- DC
- dendritic cell
- IL10Rα
- IL-10 receptor α
- IP-10
- interferon gamma-induced protein 10.
REFERENCES
- 1. Ishihara K., Hirano T. (2002) IL-6 in autoimmune disease and chronic inflammatory proliferative disease. Cytokine Growth Factor Rev. 13, 357–368 [DOI] [PubMed] [Google Scholar]
- 2. Lio D., Licastro F., Scola L., Chiappelli M., Grimaldi L. M., Crivello A., Colonna-Romano G., Candore G., Franceschi C., Caruso C. (2003) Interleukin-10 promoter polymorphism in sporadic Alzheimer's disease. Genes Immun. 4, 234–238 [DOI] [PubMed] [Google Scholar]
- 3. Nishimoto N., Kishimoto T. (2004) Inhibition of IL-6 for the treatment of inflammatory diseases. Curr. Opin. Pharmacol. 4, 386–391 [DOI] [PubMed] [Google Scholar]
- 4. Tagore A., Gonsalkorale W. M., Pravica V., Hajeer A. H., McMahon R., Whorwell P. J., Sinnott P. J., Hutchinson I. V. (1999) Interleukin-10 (IL-10) genotypes in inflammatory bowel disease. Tissue Antigens 54, 386–390 [DOI] [PubMed] [Google Scholar]
- 5. Darnell J. E. (2005) Validating Stat3 in cancer therapy. Nat. Med. 11, 595–596 [DOI] [PubMed] [Google Scholar]
- 6. Levy D. E., Inghirami G. (2006) STAT3: a multifaceted oncogene. Proc. Natl. Acad. Sci. U.S.A. 103, 10151–10152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bollrath J., Phesse T. J., von Burstin V. A., Putoczki T., Bennecke M., Bateman T., Nebelsiek T., Lundgren-May T., Canli O., Schwitalla S., Matthews V., Schmid R. M., Kirchner T., Arkan M. C., Ernst M., Greten F. R. (2009) gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 15, 91–102 [DOI] [PubMed] [Google Scholar]
- 8. Grivennikov S., Karin E., Terzic J., Mucida D., Yu G. Y., Vallabhapurapu S., Scheller J., Rose-John S., Cheroutre H., Eckmann L., Karin M. (2009) IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 15, 103–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yu H., Pardoll D., Jove R. (2009) STATs in cancer inflammation and immunity: a leading role for STAT3. Nat. Rev. Cancer 9, 798–809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang T., Niu G., Kortylewski M., Burdelya L., Shain K., Zhang S., Bhattacharya R., Gabrilovich D., Heller R., Coppola D., Dalton W., Jove R., Pardoll D., Yu H. (2004) Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 10, 48–54 [DOI] [PubMed] [Google Scholar]
- 11. Fernandez-Sesma A., Marukian S., Ebersole B. J., Kaminski D., Park M. S., Yuen T., Sealfon S. C., García-Sastre A., Moran T. M. (2006) Influenza virus evades innate and adaptive immunity via the NS1 protein. J. Virol. 80, 6295–6304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Krutzik P. O., Nolan G. P. (2006) Fluorescent cell barcoding in flow cytometry allows high-throughput drug screening and signaling profiling. Nat. Methods 3, 361–368 [DOI] [PubMed] [Google Scholar]
- 13. George T. C., Fanning S. L., Fitzgeral-Bocarsly P., Medeiros R. B., Highfill S., Shimizu Y., Hall B. E., Frost K., Basiji D., Ortyn W. E., Morrissey P. J., Lynch D. H. (2006) Quantitative measurement of nuclear translocation events using similarity analysis of multispectral cellular images obtained in flow. J. Immunol. Methods 311, 117–129 [DOI] [PubMed] [Google Scholar]
- 14. Benjamini Y., Hochberg Y. (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. B Stat. Methods. 57, 289–300 [Google Scholar]
- 15. Bordería A. V., Hartmann B. M., Fernandez-Sesma A., Moran T. M., Sealfon S. C. (2008) Antiviral-activated dendritic cells: a paracrine-induced response state. J. Immunol. 181, 6872–6881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yuen T., Wurmbach E., Pfeffer R. L., Ebersole B. J., Sealfon S. C. (2002) Accuracy and calibration of commercial oligonucleotide and custom cDNA microarrays. Nucleic Acids Res. 30, e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mahdavi A., Davey R. E., Bhola P., Yin T., Zandstra P. W. (2007) Sensitivity analysis of intracellular signaling pathway kinetics predicts targets for stem cell fate control. PLoS Comput. Biol. 3, e130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qiao L., Phipps-Yonas H., Hartmann B., Moran T. M., Sealfon S. C., Hayot F. (2010) Immune response modeling of interferon β-pretreated influenza virus-infected human dendritic cells. Biophys. J. 98, 505–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zi Z., Cho K. H., Sung M. H., Xia X., Zheng J., Sun Z. (2005) In silico identification of the key components and steps in IFN-γ-induced JAK-STAT signaling pathway. FEBS Lett. 579, 1101–1108 [DOI] [PubMed] [Google Scholar]
- 20. Gillespie D. T. (1976) A general method for numerically simulating the stochastic time evolution of coupled chemical reactions. J. Comput. Physics 22, 403–434 [Google Scholar]
- 21. Gillespie D. T. (2007) Stochastic simulation of chemical kinetics. Annu. Rev. Phys. Chem. 58, 35–55 [DOI] [PubMed] [Google Scholar]
- 22. von Dassow G., Meir E., Munro E. M., Odell G. M. (2000) The segment polarity network is a robust developmental module. Nature 406, 188–192 [DOI] [PubMed] [Google Scholar]
- 23. Niemand C., Nimmesgern A., Haan S., Fischer P., Schaper F., Rossaint R., Heinrich P. C., Müller-Newen G. (2003) Activation of STAT3 by IL-6 and IL-10 in primary human macrophages is differentially modulated by suppressor of cytokine signaling 3. J. Immunol. 170, 3263–3272 [DOI] [PubMed] [Google Scholar]
- 24. Finbloom D. S., Winestock K. D. (1995) IL-10 induces the tyrosine phosphorylation of tyk2 and Jak1 and the differential assembly of STAT1 α and STAT3 complexes in human T cells and monocytes. J. Immunol. 155, 1079–1090 [PubMed] [Google Scholar]
- 25. Wehinger J., Gouilleux F., Groner B., Finke J., Mertelsmann R., Weber-Nordt R. M. (1996) IL-10 induces DNA binding activity of three STAT proteins (Stat1, Stat3, and Stat5) and their distinct combinatorial assembly in the promoters of selected genes. FEBS Lett. 394, 365–370 [DOI] [PubMed] [Google Scholar]
- 26. Yasukawa H., Ohishi M., Mori H., Murakami M., Chinen T., Aki D., Hanada T., Takeda K., Akira S., Hoshijima M., Hirano T., Chien K. R., Yoshimura A. (2003) IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat. Immunol. 4, 551–556 [DOI] [PubMed] [Google Scholar]
- 27. Butler D. (2008) Translational research: crossing the valley of death. Nature 453, 840–842 [DOI] [PubMed] [Google Scholar]
- 28. Coller B. S., Califf R. M. (2009) Traversing the valley of death: a guide to assessing prospects for translational success. Sci. Transl. Med. 1, 10cm19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Arrowsmith J. (2011) Trial watch: phase II failures, 2008–2010. Nat. Rev. Drug Discov. 10, 328–329 [DOI] [PubMed] [Google Scholar]
- 30. Fribourg M., Moreno J. L., Holloway T., Provasi D., Baki L., Mahajan R., Park G., Adney S. K., Hatcher C., Eltit J. M., Ruta J. D., Albizu L., Li Z., Umali A., Shim J., Fabiato A., MacKerell A. D., Jr., Brezina V., Sealfon S. C., Filizola M., González-Maeso J., Logothetis D. E. (2011) Decoding the signaling of a GPCR heteromeric complex reveals a unifying mechanism of action of antipsychotic drugs. Cell 147, 1011–1023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. González-Maeso J., Ang R. L., Yuen T., Chan P., Weisstaub N. V., López-Gimenez J. F., Zhou M., Okawa Y., Callado L. F., Milligan G., Gingrich J. A., Filizola M., Meana J. J., Sealfon S. C. (2008) Identification of a serotonin/glutamate receptor complex implicated in psychosis. Nature 452, 93–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. González-Maeso J., Weisstaub N. V., Zhou M., Chan P., Ivic L., Ang R., Lira A., Bradley-Moore M., Ge Y., Zhou Q., Sealfon S. C., Gingrich J. A. (2007) Hallucinogens recruit specific cortical 5-HT(2A) receptor-mediated signaling pathways to affect behavior. Neuron 53, 439–452 [DOI] [PubMed] [Google Scholar]