

Background: Osteopontin (OPN) is highly expressed in glioblastoma (GBM) and possesses inflammatory activity modulated by proteolytic cleavage.
Results: Cleaved OPN was increased in GBM and led to more adhesion of GBM cells. OPN conferred resistance to apoptosis in GBM cells.
Conclusion: Increased osteopontin proteolysis increased GBM cell resistance to apoptosis.
Significance: OPN cleavage links coagulation and inflammation providing a favorable niche for GBM development.
Keywords: Adhesion, Apoptosis, Biomarkers, Gene Expression, Glioblastoma, Integrins, Thrombin
Abstract
Osteopontin (OPN), which is highly expressed in malignant glioblastoma (GBM), possesses inflammatory activity modulated by proteolytic cleavage by thrombin and plasma carboxypeptidase B2 (CPB2) at a highly conserved cleavage site. Full-length OPN (OPN-FL) was elevated in cerebrospinal fluid (CSF) samples from all cancer patients compared with noncancer patients. However, thrombin-cleaved OPN (OPN-R) and thrombin/CPB2-double-cleaved OPN (OPN-L) levels were markedly increased in GBM and non-GBM gliomas compared with systemic cancer and noncancer patients. Cleaved OPN constituted ∼23 and ∼31% of the total OPN in the GBM and non-GBM CSF samples, respectively. OPN-R was also elevated in GBM tissues. Thrombin-antithrombin levels were highly correlated with cleaved OPN, but not OPN-FL, suggesting that the cleaved OPN fragments resulted from increased thrombin and CPB2 in this extracellular compartment. Levels of VEGF and CCL4 were increased in CSF of GBM and correlated with the levels of cleaved OPN. GBM cell lines were more adherent to OPN-R and OPN-L than OPN-FL. Adhesion to OPN altered gene expression, in particular genes involved with cellular processes, cell cycle regulation, death, and inflammation. OPN and its cleaved forms promoted motility of U-87 MG cells and conferred resistance to apoptosis. Although functional mutation of the RGD motif in OPN largely abolished these functions, OPNRAA-R regained significant cell binding and signaling function, suggesting that the SVVYGLR motif in OPN-R may substitute for the RGD motif if the latter becomes inaccessible. OPN cleavage contributes to GBM development by allowing more cells to bind in niches where they acquire anti-apoptotic properties.
Introduction
Primary glial neoplasms produce clinical symptoms that arise through complex interactions between neoplastic and normal cells. In this milieu, damage not only results from a mass effect but through the aberrant expression of many cytokines that cause increased vascular permeability, angiogenesis, tumor cell invasion, and inflammatory cell infiltration. Osteopontin (OPN),3 also known as early T cell activation gene 1 (Eta-1) and secreted phosphoprotein 1 (SPP1), is a major pleiotropic proinflammatory cytokine and extracellular matrix protein (1, 2). It is expressed by many different cell types, including activated T cells, macrophages, osteoblasts, and cardiac fibroblasts (3). OPN has been associated with cancer, including breast cancer, ovarian cancer, melanoma, and malignant glioblastoma (GBM) (4). In general, high OPN levels are associated with aggressive tumor behavior, correlate with the presence of metastasis, and predict poor prognosis in patients (5, 6). The extent of OPN immunohistological staining in primary breast cancer is inversely correlated with patient survival (7). Using subtractive hybridization, OPN is one of five key genes that differentiate highly invasive incurable GBM from the less aggressive lower grade astrocytoma (8). An increase in extracellular OPN was observed in human GBM (9).
OPN may play several potential roles in cancer, including promotion of cell proliferation, cell adhesion and migration, angiogenesis, and metastasis (4, 10, 11). Knockdown of OPN expression in a human GBM cell line by introduction of siRNA led to a reduction in cell proliferation, motility, and migration but no change in tumor-associated angiogenesis. Similar data were obtained by knockdown of OPN expression in human glioma cells (12). However, cleavage of OPN was not assessed in these studies (13).
Human OPN is a highly negatively charged, heavily glycosylated 44-kDa protein containing several conserved structural elements, including an aspartic acid-rich domain (Asp86–Asp89) that binds to hydroxyapatite in bone, a calcium-binding domain (Asp216–Ser228), and two heparin-binding domains (Tyr165–Phe174 and Asp298–Ser305) (10). In addition, OPN possesses an RGD site that mediates its attachment to cells via multiple integrins, including αvβ3, αvβ5, αvβ1, and α5β1, and two CD44-binding domains (Leu136–Thr155 and Asp298–Ser305) that mediate extracellular matrix interactions, thus conferring on OPN many cytokine-like functions, including chemoattraction and signal transduction (14–16). Of note, OPN has a conserved thrombin cleavage site at Arg168/Ser169 contiguous to the 159RGD161 site. We have previously shown that OPN binding to thrombin requires its engagement with both anion exosites I and II in thrombin, in a way typical for other physiological substrates of thrombin, and it is cleaved effectively at that site by thrombin, demonstrating that OPN is a bona fide substrate for thrombin (17). There are no other known thrombin cleavage sites within OPN that are cleaved at a rate that is physiologically relevant.
Thrombin cleavage exposes a new C terminus, 162SVVYGLR168, in the cleaved OPN (OPN-Arg or OPN-R) that interacts with α4β1 and α9β1 integrins in a non-RGD-dependent manner (18–24). Based on their ligands, integrin α4β1 and integrin α9β1 represent a defined subset of integrins within the broad integrin family. We have shown that plasma procarboxypeptidase 2 (pCPB2, also termed thrombin-activatable fibrinolysis inhibitor or TAFI), upon activation to CPB2, cleaves the C-terminal arginine from SVVYGLR, thereby converting OPN-R to OPN-Leu (OPN-L). Jurkat cells and synoviocytes have enhanced cell adhesion to OPN-R compared with full-length OPN (OPN-FL) that is reduced when OPN-R is converted to OPN-L by either CPB2 or CPN, the constitutively active plasma carboxypeptidase (25, 26). We postulate that the sequential cleavages of OPN by thrombin and CPN/CPB2 may up-regulate and down-regulate OPN′s inflammatory properties, respectively.
Although proteolytic processing of OPN clearly has an impact on its biological properties in vitro, it has been difficult to assess its in vivo relevance due to a lack of specific antibodies that distinguish the different OPN forms. We recently developed specific ELISAs for OPN-R and OPN-L and demonstrated that these cleaved OPN forms are markedly elevated in the synovial fluid of patients with rheumatoid arthritis but not osteoarthritis or psoriatic arthritis, validating its significance in vivo (25). Thrombin activation is increased in patients with GBM as shown by the elevation of prothrombin fragment F1 + 2 in their plasma (27), and intravascular thrombosis has been suggested to be involved in progression of astrocytomas to GBMs (28).
As there are no studies on the role of thrombin cleavage of OPN in the literature and, given the reported importance of thrombin and OPN in GBM, in this study, we examined cleaved OPN products in tissues and cerebrospinal fluid (CSF) from patients with GBM, analyzed their correlation with markers of coagulation, angiogenesis, and inflammation, and investigated if thrombin cleavage of OPN affects the behavior of GBM cells.
EXPERIMENTAL PROCEDURES
Cells and Tissue Samples
Human cell lines, DBTRG-05MG (glioblastoma), Hs 683 (glioma), T98G (glioblastoma), U-87 MG (glioblastoma, astrocytoma), and Jurkat (leukemic T-cell lymphoblast) were purchased from the American Type Culture Collection (ATCC, Manassas VA); 1321N1 (astrocytoma) cell line was from the European Collection of Cell Cultures (ECACC, Wiltshire, UK). 1321N1, Hs 683, and U-87 MG cells were maintained in DMEM with 10% heat-inactivated FBS and 50 units/ml penicillin and 50 mg/ml streptomycin. T98G cells were cultured in Eagle's minimal essential medium supplemented with 10% FBS, 1% nonessential amino acid, 1 mm sodium pyruvate. DBTRG-05MG cells were grown in RPMI 1640 medium with 10% FBS, 0.1 mm sodium hypoxanthine, 16 mm thymidine, 1 mm sodium pyruvate, whereas Jurkat cells were cultured in RPMI 1640 medium with 10% FBS, 1% nonessential amino acid, and 55 mm 2-mercaptoethanol. Stem cell-like U-87 MG (U-87 SC) cells were isolated by culture in defined serum-free DMEM/F-12 supplemented with N-2 supplement (Invitrogen), 20 ng/ml EGF, and 20 ng/ml basic FGF (Peprotech, Rocky Hill, NJ). The cells were fed every 3 days by adding fresh medium with growth factors. Rat neural stem cells were provided by Dr. Hiroyuki Sakata (Dept. of Neurosurgery, Stanford University School of Medicine). Frozen tissue specimens and CSF were obtained from the Stanford Brain Tumor Tissue Depository. These samples were collected after informed consent as approved by the Stanford Institutional Review Board under protocol IRB-8049. CSF samples were collected during times of diagnostic or therapeutic CSF sampling and were then aliquoted and frozen at −80 °C until used.
Determination of OPN-FL, OPN-R, and OPN-L in CSF and Tissue Samples by ELISA
Tissue samples obtained from GBM or epileptic brain tissue were thawed on ice. Following a 10-min centrifugation at 400 × g at 4 °C, samples were appropriately diluted in PBS with 2% BSA, pH 7.4. ELISAs for OPN and its cleaved fragments were performed as described (25). Briefly, a commercial antibody (MAB14331, R&D Systems, Minneapolis, MN) was used for the capture of OPN-FL, OPN-R, and OPN-L. OPN-FL was measured using the R&D Systems Quantikine OPN ELISA kit. Captured OPN-R and OPN-L were detected by specific anti-OPN-R or anti-OPN-L antibodies followed by peroxidase-conjugated goat anti-rabbit antibody. Recombinant OPN-R and OPN-L (0.625 ng/ml-50 ng/ml) were used to construct the calibration curves. Levels of thrombin-antithrombin complexes (TAT) were determined using a commercial ELISA kit (Assaypro, St. Charles, MO). VEGF, TNF-α, chemokine (C-C motif) ligand 3 (CCL3, also known as macrophage inflammatory protein 1α or MIP-1α), and CCL4 (MIP-1β) in CSF and cell culture samples were measured using R&D Systems DuoSet ELISA kit following the manufacturer's instructions.
Cell Adhesion Assay
Recombinant human OPN-FL, OPN-R, and OPN-L and RGD-mutated OPN-FL, OPN-R, and OPN-L, denoted as OPNRAA-FL, OPNRAA-R, and OPNRAA-L, respectively, generated as described previously (26), were coated overnight onto Nunclon DELTA 96-well black fluorescent plates (NUNC, Rochester, NY) at 100 nm in 0.1 m NaHCO3, pH 8.5. Wells were washed with Hanks' balanced salt solution with 50 mm Hepes, pH 7.5 (HBSS/Hepes), and blocked with HBSS/Hepes buffer containing 1% BSA for 1 h at room temperature (RT). Cells labeled with carboxyfluorescein diacetate succinimidyl ester cell tracer dye (Invitrogen) in HBSS/Hepes buffer with 0.2 mm MnCl2 were added (1 × 105 cells/well) and incubated for 2 h at 37 °C. After washing extensively with HBSS/Hepes buffer, the plate was read at an excitation wavelength of 485 nm and emission of 538 nm using a Fluoroskan Ascent plate reader (Thermo Scientific, Waltham, MA). The data were expressed as a percentage of the total input cells based on a standard curve generated from a known number of input labeled cells. The whole assay was repeated on at least three independent occasions for each cell line.
Protein Preparation and Western Blot Analysis
After 2 days culture, the cells were collected in radioimmunoprecipitation assay buffer with protease inhibitors (Roche Applied Science) by scraping and then homogenized by sonication. Lysates (10 μg) were separated by SDS-PAGE under reducing conditions and transferred to a PVDF membrane (Invitrogen). Blots were blocked with 5% skim milk in Tris-buffered saline (TBS) with 0.05% Tween (TBS-T) and then incubated with either anti-CD44 (8E2), anti-integrin antibodies (Cell Signaling Technology, Inc., Danvers, MA), or anti-integrin α9 (ab87995; Abcam, Cambridge, MA) in 5% BSA/TBS-T before development with peroxidase-conjugated secondary antibody. The immunoreactive bands were visualized using the ECL detection system (GE Healthcare). To confirm equal loading of samples, after the visualization, the blots were stained with Ponceau S.
Gene Expression Analysis
U-87 MG cells were bound to BSA and different forms of recombinant OPNs (OPN-FL, OPN-R, OPN-L, and OPNRAA-R) (26) for 2 h and washed extensively before being cultured in DMEM for 2 days at 37 °C. In parallel, a separate set of wells was tested as above for adhesion to the OPN forms to ensure that the cells used for the mRNA analysis were behaving as determined earlier. Because the number of cells adherent to BSA and OPNRAA-R was much less than that on the other OPNs, a larger number of BSA and OPNRAA-R wells were used so that the total number of cells used for RNA extraction was equivalent. Total RNA was isolated by direct lysis of adherent cells (Qiagen RNeasy Mini kit, Valencia, CA). The generation of labeled cRNA and its hybridization to Human Gene 1.0 ST Array (Affymetrix, Santa Clara, CA) were performed by the Stanford PAN facility. In one experiment, the RNA was analyzed on three-dimensional gene expression arrays (Toray Industries Inc., Kanagawa, Japan). Data were analyzed using the Partek Genomics Suite (Partek, St Louis, MO). Selected genes were assayed by real time PCR using predesigned and validated primer sets (Qiagen). The values were normalized to the expression of GAPDH. The data were expressed as mean ± S.D. of relative gene expression to BSA using the ΔΔCt method.
Migration Assay
The migration assay was carried out as described previously (29). Briefly 2 × 105 U-87 MG or T98G cells in serum-free DMEM were seeded in the transwell insert of a polycarbonate 24-well transwell plate with 8-μm pore size (Corning, Lowell, MA). Different concentrations of OPN-FL, OPN-R, and OPN-L were added to 600 μl of the same medium in the bottom well. After 4 h of incubation at 37 °C, the cells that passed through the membrane were labeled with CCK-8 (Dojindo, Kumamoto, Japan). The data were expressed as fold changes in cell number compared with the addition of PBS to the bottom well. The whole assay was repeated on at least three occasions for each protein.
Determination of Apoptosis
U-87 MG or T98G cells were bound to the different OPN forms, washed extensively, and incubated overnight in DMEM with 10% FBS before exposure for 2 days to 10 μm (U-87 MG) or 15 μm (T98G) thapsigargin (Sigma) or 30 μm (U-87 MG) or 10 μm (T98G) WP 1066 (Calbiochem). In some experiments U-87 MG cells were treated with 10 μm LY294002 (EMD Millipore, Billerica, MA) (14, 15) or 1 μm IKK VII (EMD Millipore) for 24 h before exposure to 30 μm WP 1066 for 2 days. Viable cells were determined by measuring absorbance of CCK-8 at 450 nm, and their overall viability was calculated by comparison with data from untreated cells.
Immunofluorescent Staining
4 × 104 cells of U-87 SC were seeded onto poly-l-ornithine/laminin-coated wells of Lab-TekTM II chambered coverglass (NUNC). On the following day, differentiation of U-87 SC cells was induced in DMEM/F-12 containing 1% FBS and 1 μm retinoic acid for 5 days (30). After fixation with 4% paraformaldehyde at RT for 20 min, the cells were permeabilized and blocked with PBS containing 0.1% Triton X-100 and 3% FBS for 20 min. The cells were then incubated with anti-Nestin (Millipore, Billerica, MA), anti-Tuj1 (Covance, Princeton, NJ), and anti-GFAP (DAKO, Carpinteria, CA), followed by visualization with Alexa Fluor-conjugated secondary antibody. For O4 staining (R&D Systems), live cells were stained. Hoechst 33342 (Sigma) was used to identify individual cells.
Statistical Analyses
Statistical analyses were performed using Prism version 4 (GraphPad Software, Inc., San Diego). Mann-Whitney Rank Sum test was used to compare the two groups. Spearman rank order correlations and linear regression analysis were used to assess correlations between groups. Experiments with more than two groups were analyzed by one-way ANOVA with post hoc analysis using Dunnett's using the BSA values as the control. Differences and correlations were considered significant if p < 0.05.
RESULTS
Conservation of the Thrombin Cleavage Site in OPN
OPN protein sequences were extracted from the ENSEMBL database for reptiles, birds, and mammals whose genomes were available. OPN sequences from 41 species were identified. All contained an RGD sequence homologous to the human RGD. These sequences were then aligned around the human 159RGD161 sequence, and a selection from various phyla is shown in Table 1. Arg168 that is the cleavage site for thrombin in humans is present in 31 of the sequences, and in another seven it has been substituted by lysine, a less good but still compatible with being a thrombin substrate. In only two species, elephant and Chinese softshell turtle, has the basic amino acid been substituted with a small neutral one, although the armadillo sequence is not complete (supplemental Table 1). Thus, the thrombin cleavage site has been conserved at least because birds and mammals split in evolution, suggesting selection pressure to maintain the cleavage and its physiological importance. Because of the involvement of the coagulation cascade and thrombin in particular in GBM (31, 32), we decided to investigate if OPN was cleaved by thrombin in GBM.
TABLE 1.
Sequence alignment of OPNs from selected species around the RGD-binding site
The amino acids in boldface are the RGD site, and the vertical arrow denotes the thrombin cleavage site.

Elevation of OPN-R and OPN-L in GBM Tissues and CSF Samples
Prompted by the observation that OPN is a key gene involved in GBM biology (5) and our recent study showing that the levels of OPN-R and OPN-L were markedly elevated in the synovial fluid of patients with rheumatoid arthritis (25), we hypothesized that cleavage of OPN plays a role in GBM. To test this, we first measured OPN-FL, OPN-R, and OPN-L in CSF samples from patients with GBM (n = 29) and non-GBM gliomas (n = 20, anaplastic oligodendroglioma 4, oligodendroglioma 4, gliomatosis cerebri 2, ependymoma 3, anaplastic astrocytoma 3, brain stem glioma 2, juvenile pilocytic astrocytoma and astrocytoma, 1 each). In addition, we determined OPN levels in 14 CSF samples from patients with systemic cancer (non-Hodgkin lymphoma 4, breast cancer 3, melanoma, non-small cell lung cancer, adenocarcinoma, rectal cancer, squamous cell carcinoma, lung cancer, and other 1 each) and 15 samples from patients with no evidence of cancer (multiple sclerosis 3, normal pressure hydrocephalus 2, sarcoid, Bell palsy, stroke, pseudotumor cerebri, ataxia and sensory loss, 1 each and other 4). OPN-FL levels were elevated in GBM, non-GBM gliomas, and systemic cancer CSF samples to a similar extent, all markedly elevated compared with noncancer CSF samples (Fig. 1A and Table 2). However, although their OPN-FL levels were similarly elevated, OPN-R and OPN-L levels were significantly elevated in GBM samples compared with systemic cancer samples (OPN-R, 129.6 ± 38.4 versus 30.7 ± 7.3 ng/ml, p = 0.008; OPN-L, 25.3 ± 6.1 versus 3.3 ± 0.9 ng/ml, p = 0.002, for GBM versus systemic cancer CSF samples, respectively), and the same was true for OPN-L, but not OPN-R, in non-GBM glioma CSF samples (57.2 ± 19.1 versus 3.3 ± 0.9 ng/ml, p = 0.0003, for non-GBM gliomas versus systemic cancer samples). The data suggest that increases in levels of OPN-R and OPN-L are a specific phenomenon in both GBM and non-GBM gliomas.
FIGURE 1.

Thrombin-cleaved fragments of OPN significantly increased in both GBM CSF and tissue. A, CSF samples from GBM patients (n = 29) were compared with CSF samples from non-GBM (n = 20), systemic cancer (n = 14), and noncancer patients (n = 15) for levels of OPN-FL (○), OPN-R (□), and OPN-L (▵). Bars represent the mean value of each group. Each point represents CSF from an individual patient. B, fraction of cleaved OPN was significantly increased in GBM and epilepsy samples compared with noncancer samples. Data were calculated from the results shown in A and shown as mean ± S.E. *, p < 0.05. C, levels of OPN-FL (●), OPN-R (■), and OPN-L (▴) in epileptic and GBM tissues determined by ELISA (n = 5 each).
TABLE 2.
Levels of different OPNs, VEGF, CCL3, and CCL4 in CSF from patients with GBM, non-GBM, systemic cancer, and noncancer
p values were compared with noncancer. NS means not significant.
| OPN-FL | OPN-R | OPN-L | Total OPN | VEGF | CCL3 | CCL4 | |
|---|---|---|---|---|---|---|---|
| ng/ml | ng/ml | ng/ml | ng/ml | pg/ml | pg/ml | pg/ml | |
| GBM (n = 29) | 833.6 ± 110.7 | 129.6 ± 38.4 | 25.3 ± 6.1 | 988.6 ± 141.0 | 1229.0 ± 320.8 | 3.5 ± 1.3 | 20.5 ± 5.8 |
| p < 0.0001 | p < 0.0001 | p = 0.0003 | p < 0.0001 | p = 0.0003 | NS | p = 0.0007 | |
| Non-GBM (n = 20) | 856.7 ± 160.7 | 87.6 ± 22.2 | 57.2 ± 19.1 | 1001.6 ± 179.7 | 450.1 ± 214.3 | 1.9 ± 0.7 | 17.4 ± 7.7 |
| NS | p = 0.0013 | p < 0.0001 | p = 0.0267 | NS | NS | p = 0.0161 | |
| Systemic cancer (n = 14) | 702.4 ± 176.1 | 30.7 ± 7.3 | 3.3 ± 0.9 | 736.4 ± 176.2 | 9.5 ± 7.8 | 1.1 ± 0.8 | 7.5 ± 3.4 |
| NS | NS | NS | NS | NS | NS | NS | |
| Noncancer (n = 15) | 283.6 ± 46.0 | 11.4 ± 3.9 | 2.3 ± 0.7 | 297.4 ± 47.0 | 1.2 ± 0.9 | 0 | 0 |
We calculated the percentage of total OPN that had been cleaved in these CSF samples and found that the mean was 22.6 ± 3.0% for GBM, 31.4 ± 6.0% for non-GBM, 18.5 ± 7.1% for systemic cancer, and 11.1 ± 2.7 for the cancer samples (cancer samples versus either GBM or non-GBM p < 0.05) (Fig. 1B). In several of the GBM and non-GBM samples, more than 50% of the total OPN had been cleaved. The levels of cleaved OPN in GBM and non-GBM were significantly different from the levels determined in the noncancer samples suggesting extensive thrombin activity in the CSF of glioma patients.
To identify the origin of the increases in thrombin-cleaved OPN present in the CSF, we assayed the levels of the different species of OPN in tissue homogenates from patients with epilepsy and GBM (Fig. 1B). OPN-FL and OPN-R levels are significantly elevated in human GBM tissues compared with brain tissues taken from patients with epilepsy as follows: OPN-FL, 507.9 versus 49.6 ng/ml, p = 0.008; OPN-R, 519.1 versus 87.3 pg/ml, p = 0.016, for GBM and epilepsy samples, respectively. The differences in OPN-L levels between these two groups did not reach statistical significance, although there was a trend for an increased level in GBM samples (1858.1 versus 617.6 pg/ml, p = 0.096). The source of the OPN could be from either the tumor itself and/or the surrounding stromal cells in response to the presence of the tumor.
Correlation of OPN-R and OPN-L with TAT Levels in GBM CSF Samples
To test our hypothesis that the high levels of OPN-R and OPN-L in GBM CSF are due to thrombin cleavage, we assessed another independent series of 30 CSF samples from patients with GBM for OPN-FL, OPN-R, OPN-L, and a marker of thrombin activity, TAT (Fig. 2A and Table 3). Using regression analysis and a Spearman correlation analysis, we noted a very high correlation between OPN-R and TAT levels (0.725, p < 0.0001), although there was no correlation between OPN-FL and TAT (0.022, p = not significant). Consistent with the fact that OPN-L requires an additional step for its production from OPN-R (i.e. cleavage by CPB2 or CPN), there was a lower but still significant correlation for OPN-L with TAT (0.464, p = 0.01).
FIGURE 2.

Thrombin-cleaved OPN in CSF from GBM patients correlates with TAT levels as well as VEGF, CCL3, and CCL4. A, CSF from an independent cohort of 30 GBM patients had the levels of OPN isoforms and TAT determined. Left panel, OPN-FL (●); center panel, OPN-R (■); right panel, OPN-L (▴). B, OPN-R (center panel) and OPN-L (right panel) levels in CSF from GBM patients vary more than OPN-FL levels (left panel). CSF was obtained from 10 GBM patients on three separate occasions at least 1 month apart, and the levels of OPN-FL, OPN-R, and OPN-L were determined. The x axis is marked with the occasion when the samples were acquired. C, VEGF and CCL4 levels were increased in GBM. Levels of VEGF (left panel), CCL3 (center panel), and CCL4 (right panel) in CSF from GBM (n = 24), non-GBM (n = 18), systemic cancer (n = 13), and noncancer patients (n = 9) were determined by ELISA. Bars represent the mean value of each group.
TABLE 3.
Spearman rank order correlations of levels of OPNs with TAT, VEGF, CCL3, and CCL4
NS means not significant.
| OPN-FL | OPN-R | OPN-L | Total OPN | |
|---|---|---|---|---|
| TAT correlation coefficient | 0.0223 | 0.7254 | 0.4637 | 0.2948 |
| p value | NS | p < 0.0001 | p = 0.0099 | NS |
| VEGF correlation coefficient | 0.1184 | 0.6864 | 0.5837 | 0.0738 |
| p value | NS | p < 0.0001 | p < 0.0001 | NS |
| CCL3 correlation coefficient | 0.1734 | 0.4446 | 0.4331 | 0.2603 |
| p value | NS | p = 0.0002 | p = 0.0003 | p = 0.0362 |
| CCL4 correlation coefficient | 0.1986 | 0.5229 | 0.3704 | 0.2933 |
| p value | NS | p < 0.0001 | p = 0.0024 | p = 0.0178 |
OPN-R and OPN-L Levels in GBM CSF Samples as a Potential Biomarker
The presence of such high levels of OPN-R and OPN-L in CSF from GBM patients raises the possibility that their levels could be used as a biomarker of disease activity. To address if the levels of OPN-R and OPN-L were stable over time, we assessed three CSF samples drawn at least 1 month apart from 10 individual patients with GBM. There was a fairly constant level of OPN-FL noted throughout all samples with a mean fold difference between highest and lowest inter-individual samples of 2.6 (Fig. 2B). In contrast, there was a much wider inter-individual distribution of OPN-R and OPN-L levels. The average variation of OPN-R levels was almost 7-fold higher than OPN-FL levels (p < 0.04). To assess if the levels of the different OPN isoforms correlated with markers of angiogenesis and inflammation, we then examined levels of VEGF as a marker of angiogenesis and TNF-α, CCL3 (MIP-1α), and CCL4 (MIP-1β) as markers of inflammation in GBM. The levels of VEGF and CCL4 were significantly elevated in GBM and non-GBM glioma patients compared with noncancer patients (Fig. 2C and Table 1). Notably, VEGF is significantly higher in GBM than non-GBM gliomas. In contrast, TNF-α was not detectable. Moreover, the levels of OPN-R and OPN-L were well correlated with VEGF, CCL3, and CCL4 (Table 2), suggesting that the levels of these markers were all related to disease activity.
Different Expression Profile of OPN Receptors in Human Glioma Cell Lines to Jurkat Cells
To assess if the increased levels of OPN and its cleavage products could affect the adhesion of GBM and non-GBM gliomas, we first determined the expression of integrins as well as CD44, an alternative adhesion molecule, in human GBM and non-GBM glioma cell lines by Western blotting (Fig. 3). Jurkat cells were used as a positive control because we had previously shown that they bound better to OPN-R, due to the exposure of the cryptic SVVYGLR integrin-binding motif (26). CD44 was strongly expressed in all the GBM and non-GBM cell lines, although it was barely present on Jurkat cells. The α5, αV, and β1 integrins were also strongly expressed in all the GBM and non-GBM glioma cell lines and to a lesser extent in Jurkat cells, although there was substantial heterogeneity in the expression of the α5 integrins. In contrast, the α4 and α9 integrins were highly expressed in Jurkat cells, supporting our previous report (26). High expression of β3 integrin was only observed in U-87 MG cells, whereas β5 integrin was not detectable in any of the cell lines tested.
FIGURE 3.

Expression pattern of integrins and CD44 in human glioma cells and Jurkat cells. Different glioma cells showed different patterns of integrin expression but all could be readily distinguished from Jurkat cells. Cell lysates were separated by SDS-PAGE before detection of adhesion molecules by Western blotting. Equal loading was demonstrated by visualizing the proteins with Ponceau S staining (bottom panel).
Enhanced Adhesion of Human GBM Cell Lines to OPN upon Proteolytic Cleavage
We then examined cell adhesion of three GBM cell lines (DBTRG-05MG, T98G, and U-87 MG) and two non-GBM glioma cell lines (1321N1 and Hs 683) to OPN isoforms (Fig. 4). DBTRG-05MG, T98G, and U-87 MG and 1321N1 cells showed an ∼1.5-fold increase in binding to immobilized OPN-R and OPN-L compared with OPN-FL, whereas Hs 683 showed no adhesion to any of the OPN isoforms. In contrast to Jurkat cells and synoviocytes (25), there was no significant change in binding when OPN-R was cleaved to OPN-L. Removing the RGD site by mutation in OPN-FL (OPNRAA-FL) abolished cell binding completely in all three GBM cell lines, indicating that cell binding was primarily driven by the RGD motif. In these cases, thrombin cleavage of the C terminus may enhance the accessibility of the RGD motif, accounting for the increased cell binding to OPN-R and OPN-L. Because there was not reduced cell binding to OPN-L, however, it suggests that the SVVYGLR motif in OPN-R does not play an important role in cell adhesion under these circumstances. However, when the RGD motif was mutated, it completely abolished the binding of DBTRG-05MG and T98G cells to OPNRAA-FL, but it markedly increased adhesion of DBTRG-05MG and T98G cells to OPNRAA-R, increasing it to a level equivalent to OPN-FL, suggesting that the SVVYGLR sequence could become functional and substitute for the RGD motif in its absence. When the C-terminal arginine was removed, cell binding to OPNRAA-L was abolished, consistent with the notion that the SVVYGLR sequence becomes the functional domain of OPN supporting cell adhesion in the absence of an intact RGD motif. In contrast to the three GBM cell lines, the non-GBM cell line 1321N1 retained significant cell binding to OPNRAA-L, in the absence of both RGD and SVVYGLR motifs, suggesting that an alternative cell adhesion molecule, perhaps CD44, is primarily responsible for binding to OPN in this particular cell line.
FIGURE 4.
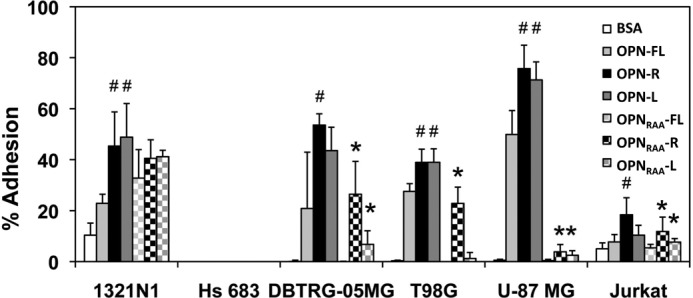
Adherence of human glioma cells to OPN-R and OPN-L compared with OPN-FL. Carboxyfluorescein diacetate succinimidyl ester-labeled cells were tested for their ability to bind to different forms of OPN. The data represent the mean ± S.D. of at least three independent experiments. #, p < 0.05 compared with OPN-FL; *, p < 0.05 compared with OPNRAA-FL by paired Student's t test.
OPN Binding Modulates Gene Expression in U-87 MG Cells
To investigate if binding of GBM cells to OPN alters their cellular phenotype, we then tested the gene expression profiles following adhesion to OPN by microarray analysis. Because adhesion of three adherent GBM cell lines to OPNs was exclusively through its RGD motif, the U-87 MG cell line was utilized. Samples from cells that bound to OPN forms were then compared with the BSA control, and those genes whose expression was changed by >1.5-fold with p < 0.001 were identified. The data are displayed as a heat map created by unsupervised sorting of the samples and genes. This resulted in the control samples being adjacent to each other and segregated from the cell samples bound to OPN, whereas gene expression in the U-87 MG cells bound to the different forms of OPN are sorted together (Fig. 5A and supplemental Table 2). This shows that attachment of U-87 MG cells to OPN, compared with a control matrix (BSA), results in modulation of gene expression. Somewhat surprisingly, there are only minor differences in gene expression induced by contact with OPN-FL, OPN-R, and OPN-L, implying that the intracellular signaling pathway in U-87 MG cells triggered by both OPN-FL and cleaved OPNs was primarily mediated through the RGD motif. It is notable that binding to OPNRAA-R gave a similar pattern to that observed in the three OPNs with an intact RGD motif, suggesting that the C-terminal SVVYGLR motif, in the absence of RGD, can signal via α9β1 and substitute for RGD signaling via the other integrins. To validate the data from the microarray analysis, the levels of a panel of genes that changed were determined by quantitative PCR. In all cases, the data on changes in mRNA levels were congruent between the microarray and quantitative PCR data (Fig. 5C).
FIGURE 5.
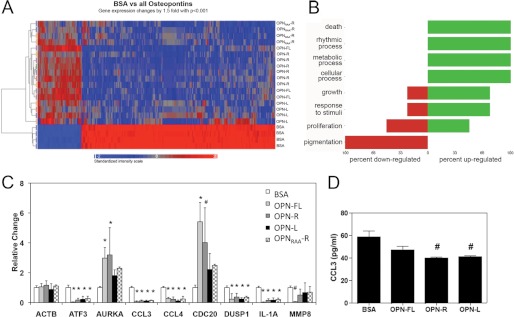
OPN binding altered gene expression in U-87 MG cells. A, heat map showing clustering of genes changed by >1.5-fold with p < 0.005 when U-87 MG cells bound to BSA were compared with U-87 MG cells bound to all OPNs determined by ANOVA. Both samples (y axis) and genes (x axis) were allowed to cluster unsupervised. The color scale of the log2 ratios is shown at the bottom. B, Gene Ontology classification. C, mRNA for nine genes shown were checked by quantitative PCR to validate the data from the microarray. The data were normalized to the levels of GAPDH and expressed as mean ± S.D. of relative gene expression to BSA. #, p < 0.05; *, p < 0.01 compared with BSA by paired Student's t test. D, determination of CCL3 protein level in 2 day-conditioned medium from U-87-MG cells bound to BSA and OPN by ELISA. #, p < 0.05 compared with BSA by paired Student's t test.
Annotation of the genes using Gene Ontology classification shows that the list of genes whose expression is altered with the highest probability is enriched in genes involved in cell death, cellular process, and cell growth (Fig. 5B). For example, mRNAs encoding aurora kinase A and CDC20, two kinases involved in cell cycle control and cell division, were increased by more than 3-fold when the cells were bound to OPN-FL and OPN-R (Fig. 5C). Interestingly, two key chemokines, CCL3 and CCL4, and one inflammatory cytokine, IL-1α, were identified as genes whose expression was reduced following binding of U-87 MG cells to OPN, compared with BSA, by ∼5-fold. Furthermore, decreased levels of CCL3 protein could be detected in the conditioned medium from U-87 cells adhered to OPN-R and OPN-L (Fig. 5D).
OPN Promoted U-87 MG Cell Migration and Conferred Resistance to Cell Apoptosis
Based on the data we obtained from the Gene Ontology classification of the genes whose expression was altered by binding to OPNs, we examined the effects of OPN on cell growth, VEGF production, cell migration, and apoptosis. When U-87 MG cells were treated with OPNs for 2 days, no changes were observed in terms of cell growth (Fig. 6A) and VEGF production (Fig. 6B). Chemotaxis assays showed dose-dependent enhancement of U-87 MG cell motility toward OPN-FL, OPN-R, and OPN-L without any significant difference among the OPNs, suggesting the effect was mediated through the RGD motif (Fig. 6C). Because OPN is known to confer resistance to apoptosis (13, 33), its effect on apoptosis was tested. Treatment with a STAT3 inhibitor, WP1066, induced significant apoptosis in U-87 MG cells (Fig. 6D). Binding to OPN conferred significant protection from apoptosis, with an ∼2.5-fold increase in viable cells compared with control BSA following WP1066 treatment. When another inducer of apoptosis, thapsigargin, a specific inhibitor of endoplasmic Ca2+-ATPase, was used, binding to OPN also resulted in protection from apoptosis with an ∼1.3-fold increase in viability (Fig. 6D).
FIGURE 6.

OPN promoted U-87 MG cell migration and conferred resistance to cell apoptosis. U-87 MG cells were treated with different concentrations of OPN-FL, OPN-R, and OPN-L (0 nm, white; 1 nm, light gray; 10 nm, dark gray; and 100 nm, black) for 2 days. A, cell proliferation was determined by CKK-8 assay. B, VEGF production was determined by assaying VEGF concentration in the conditioned media by ELISA, and the overall value normalized by the cell number was determined in A. C, dose-dependent enhancement of U-87 MG cell motility by OPN using the transwell chemotaxis assay. D, U-87 MG cells adhered to BSA (white), OPN-FL (light gray), OPN-R (black), and OPN-L (dark gray) are protected against apoptosis induced by 10 μm thapsigargin or 30 μm WP-1066. E, U-87 MG cells adhered to BSA (white), OPN-FL (light gray), OPN-R (black), and OPN-L (dark gray) are protected against apoptosis induced by 30 μm WP1066, even in the presence of 10 μm LY294002 or 1 μm IKK VII, inhibitors of PI3K and IKK, respectively. Their overall viability was calculated by comparison with data from untreated cells. Data are shown as mean ± S.D. and calculated from at least three independent experiments. *, p < 0.05; #, p < 0.01 compared with BSA by one-way ANOVA with post hoc Dunnett's.
Two signaling mechanisms have been reported upon binding of cells to OPN. These are the phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) pathway when the binding is mostly mediated via CD44 and IKK when the binding is RGD-mediated (14, 34). To investigate the mechanism by which OPN binding leads to protection from apoptosis, U-87 MG cells were treated with a PI3K inhibitor (LY294002) or an IKK inhibitor (IKK VII) in the presence or absence of WP1066 to induce apoptosis (Fig. 6E). LY294002 by itself induced some cell death that was reversed when the cells were bound to OPN. In the presence of WP1066, neither LY294002 nor IKK VII reversed the anti-apoptotic effects of binding to OPN. This suggests that neither of these pathways is involved in the mechanism leading to protection from apoptosis.
Similar to the cell migration results, there was no significant difference in viability between cells bound to OPN-FL, OPN-R, and OPN-L. The data suggest that OPN might play an important role as a chemoattractant and anti-apoptotic molecule in GBM pathogenesis, and this is largely mediated via the RGD motif.
Effects of OPN and Its Cleaved Forms on T98G and DBRTG-05MG Cells
To determine whether the observed effects of OPN and its cleaved forms on U-87 cells apply to other GBM cell lines, we investigated their effects on T98G and DBTRG-05MG cells. First, we determined whether different forms of OPN influenced growth rates in these cells. Neither cell line grew differently in the presence of OPN-FL, OPN-R, or OPN-L (Fig. 7, A and B). Similarly, there was no effect of OPN or its cleaved forms on their production of VEGF (Fig. 7, C and D), and thus the results were similar to those seen in U-87 cells. We observed dose-dependent enhanced cell migration of T98G cells to OPN-FL, but OPN-R and OPN-L did not have any significant chemotactic activity (Fig. 7E), suggesting that the enhanced chemotaxis observed in OPN-FL is not RGD-mediated. It is possible that in this case the chemotactic activity is primarily driven by the C-terminal CD44-binding domain that is removed upon thrombin cleavage. When apoptosis was induced by thapsigargin in T98G cells, resistance to apoptosis was conferred by the presence of OPN, with the greatest effect observed with OPN-FL and less but still significant protective effect with OPN-R and OPN-L (Fig. 7F); this pattern is similar to that previously observed in cultured synoviocytes (25). However, when apoptosis was induced by WP1066, both OPN-FL and the cleaved forms conferred an equal degree of protection (Fig. 7F), similar to that seen in U-87 cells. Thus the effects of OPN and its cleaved forms on T98G cells are broadly consistent with that observed in U-87 cells, with the differences likely reflecting the intrinsic cellular heterogeneity in different GBM cell lines.
FIGURE 7.

OPN promoted T98G cell migration and conferred resistance to cell apoptosis in T98G cells. T98G and DBTRG-05MG cells were treated with different concentrations of OPN-FL, OPN-R, and OPN-L (0 nm, white; 1 nm, light gray; 10 nm, dark gray; and 100 nm, black) for 2 days. A, T98G. B, DBTRG-05MG. Cell proliferation was determined by CKK-8 assay. C, T98G. D, DBTRG-05MG. VEGF production was determined by assaying VEGF concentration in the conditioned media by ELISA, and the overall value was normalized by the cell number determined in A or B. E, dose-dependent enhancement of T98G cell motility by OPN-FL using the transwell chemotaxis assay. F, T98G cells adhered to BSA (white), OPN-FL (light gray), OPN-R (black), and OPN-L (dark gray) are protected against apoptosis induced by 15 μm thapsigargin or 10 μm WP-1066. Their overall viability was calculated by comparison with data from untreated cells. Data are shown as mean ± S.D. *, p < 0.05, compared with BSA by one-way ANOVA with post hoc Dunnett's.
Cancer Stem Cell-like U-87 MG Cells Showed a Similar Pattern of Cell Adhesion to OPNs
Recent studies suggest that brain tumors have a subpopulation of cancer stem cells with the capacity for sustained self-renewal and tumor propagation (35, 36). Glioma stem cells contribute to therapeutic resistance and tumor angiogenesis. CD44 has been characterized as a marker for putative cancer stem cells in various types of solid tumors, such as breast (37), pancreas (38), prostate (39), and GBM. Fetal neural stem cells also express CD44 (40). Glioma stem cells share significant similarities with normal neural SCs, including the expression of stem cell markers (CD133, Nestin, Musashi, and Sox2) and the capacity to differentiate into the three CNS lineages, neurons, oligodendrocytes, and astrocytes. More recently, the CD44v6-OPN axis has been reported to regulate growth of brain tumor stem cells through Akt activation (41). These reports led us to investigate the effect on OPN and its cleaved forms on the behavior of cancer stem cell-like U-87 MG (U-87 SC). Neurosphere induction in U-87 MG was performed according to published reports (42, 43). U-87 SC cells in neurosphere culture condition were positive for Nestin and negative for the neuronal, oligodendrocyte, and astrocyte markers (Fig. 8A, top panel). When these cells were treated with 1 μm retinoic acid and 1% FBS for 5 days to induce cell differentiation, positive cells stained with a neuronal marker, Tuj-1, or an oligodendrocyte marker, O4, were observed, whereas nestin expression was reduced (Fig. 8A, middle panel). However, GFAP-positive cells, a marker for astrocytes, could not be detected. Using the same culture conditions, normal rat neural stem cells were differentiated into all three CNS lineages (Fig. 8A, bottom panel). Under these experimental conditions, the expression of nestin was much lower in U-87 SC cells than in the normal rat neural stem cells. We then tested if the stem cell-like GBM cells would now respond differently in adhesion to OPN by determining the ability of U-87 SC cells to bind to OPN and its cleaved forms. U-87 SC cells possessed a similar adhesion profile to U-87 MG cells, with ∼2-fold more U-87 SC cells bound to OPN-R and OPN-L than to OPN-FL, and cell adhesion was largely ablated by disruption of the RGD motif (Fig. 8B).
FIGURE 8.

U-87 SC cells were differentiated into Tuj-1-positive neuron and O4-positive oligodendrocyte and bound to OPN-R and OPN-L more than OPN-FL. A, characterization of U-87 SC cells in expansion (20 ng/ml EGF and basic FGF, upper panels) and differentiation conditions (1 μm retinoic acid and 1% FBS, middle panels). The cells were stained for neural stem cell (Nestin), neuron (Tuj-1), oligodendrocyte (O4), and astrocyte (GFAP) markers. The individual cells were visualized with Hoechst 33342. Control of rat neural stem cells under the same differentiation conditions is shown (bottom panels). B, adherence of U-87 MG and U-87 SC cells to OPN-R and OPN-L compared with OPN-FL. Data are shown as mean ± S.D. and calculated from at least three independent experiments.
DISCUSSION
In this study, we showed the following: 1) the levels of OPN-R and OPN-L are markedly elevated in both tissue and CSF of malignant glioma patients relative to systemic cancer and noncancer patients; 2) levels of VEGF and CCL4 (MIP-1β) are increased in CSF of GBM and significantly correlated with the levels of OPN-R and OPN-L, and 3) OPN and its cleaved forms induce cell migration and confer resistance to apoptosis in GBM cell lines. Cell binding to OPN, mediated by the RGD motif, is enhanced after OPN cleavage by thrombin, and the SVVYGLR motif in OPN-R may substitute for the RGD motif in cell binding and signaling when the latter becomes inaccessible. Taken together, our data suggest that unprocessed OPN-FL is up-regulated in the GBM tissue and CSF, the source of which may be either the GBM cells and/or the stromal cells surrounding the cancer. In the setting of tumor-induced tissue inflammation, thrombin is generated which then cleaves OPN to OPN-R. Either by the action of CPB-2, which is activated by thrombin bound to thrombomodulin, or by CPN as a result of vascular leakage from the plasma compartment, OPN-R is converted to OPN-L. OPN and its cleaved forms lead to migration of GBM cells to form a tumor cluster. Furthermore, OPN and its cleaved forms bound on extracellular matrix may provide a molecular niche for the cancer cells and confer a survival advantage by enhancing their resistance to apoptosis (Fig. 9). All three cellular processes, cell migration, adhesion, and resistance to apoptosis, are primarily driven by the RGD motif, with OPN-R and OPN-L more effective than OPN-FL in supporting cell adhesion, likely mediated by enhanced accessibility to the RGD motif, resulting in the generation of more apoptosis-resistant cells. Thus, thrombin cleavage of OPN leads to qualitatively similar (anti-apoptotic) but quantitatively different (enhanced) glial cell behavior. The C-terminal SVVYGLR in OPN-R may serve as an alternate cell binding and signaling motif when the RGD site becomes inaccessible.
FIGURE 9.

Model depicting the role of OPN isoforms in GBM. Both GBM and stromal cells secrete OPN, which is cleaved by thrombin or double cleaved by thrombin and CPB2 or CPN to generate OPN-R and OPN-L. All three OPN isoforms cause cell migration, increased cell adhesion, and inhibit apoptosis, thereby creating a niche that confers survival advantages on the GBM cells.
The wide fluctuations in levels of OPN-R in CSF that occur as a function of time in patients with GBM suggest that it may be used as a biomarker to assess disease burden and disease progression in gliomas. Considering the difficulties with interpreting imaging after standard radiochemotherapy treatment (i.e. pseudoprogression) (44, 45) and administration of bevacizumab (46), there is a pressing need for biomarkers that will accurately reflect tumor burden. The CSF may be the optimal body fluid to assess this issue because at least 20% is formed from intraparenchymal leakage and that modern neurosurgical techniques make chronic access relatively easy and safe. Both OPN-FL in serum and a peptide fragment of OPN in CSF have been suggested as potential markers (47, 48). However, measurements of OPN-R and OPN-L, along with OPN-FL, in CSF may represent a more optimal method of monitoring tumor burden over time. Combining this with determination of levels of other circulating markers such as VEGF and CCL4 (MIP-1β) could provide even more robust prognosis of GBM.
It is interesting that the GBM and non-GBM cell lines showed a spectrum of different adhesion patterns to OPN and its cleaved isoforms. The non-GBM cell line Hs 683 did not bind to OPN and its cleaved forms at all, although it expresses both CD44 and the relevant integrins. Because most integrins need to be activated, Hs 683 cell line might have a defect in signaling for integrin activation involving, for example, the protein-disulfide isomerase (49). In contrast, the other non-GBM cell line 1321N1 appeared to bind equally well to OPN and all its cleaved forms in an non-RGD-dependent manner, because its binding was not diminished in any of the OPN-RAA isoforms. In contrast to the glioma cell lines, Jurkat cells, derived from T cell leukemic lymphoblasts, bind more strongly to OPN-R than to OPN-FL, and its binding to OPN-L is reduced back to the level observed with OPN-FL, consistent with the notion that the C-terminal SVVYGLR in OPN-R provides a binding motif in addition to RGD. Interestingly, the ratio of CD44 to integrin expression is very much in favor of the integrins in Jurkat cells in comparison with the glioma cells, which may account for the ability to detect their enhanced cell binding to OPN-R.
All three GBM cell lines (DBTRG-05MG, T98G, and U-87 MG) showed a strong dependence on binding to the RGD motif within OPN because cell binding to OPNRAA-FL was completely abolished. This notion is further supported by gene expression analysis in the U-87 MG cells, where we can see clear differences in expression between BSA and OPNs but not among the different OPN forms. Thus, the RGD motif in OPN appears to play a dominant role in OPN-GBM cell interaction as well as activation of intracellular signaling, which contributes to GBM pathogenesis. In this environment, the C-terminal SVVYGLR sequence in OPN-R appears to play a secondary supplementary role. Mutation of the RGD motif leads to a complete loss in the ability of OPNRAA-FL to support cell adhesion. However, it is notable that OPNRAA-R, but not OPNRAA-L, is able to regain significant cell binding function, to a level equivalent to OPN-FL in two of the three GBM cell lines. It suggests that the newly exposed C-terminal SVYGLR motif in OPN-R can assume functional significance when the RGD motif is deficient or inaccessible. This is also consistent with the microarray data in which cell binding to OPNRAA-R gives a gene expression pattern similar to that observed in OPNs with an RGD motif, suggesting that signaling via α9β1, in this case in U-87 MG cells, can largely substitute for RGD signaling via the other integrins.
Disintegrin sequences were originally identified in some snake venoms and modulate cell migration by disrupting RGD-integrin-dependent cell adhesion (50). Disintegrin sequences are also present in endogenous ligands such as the a disintegrin and metalloprotease (ADAM) family of metalloproteases (51). It is possible that a disintegrin may render the RGD motif inaccessible in OPN-R, and under such a circumstance, the C-terminal SVVYGLR may serve as an alternative cell-binding and cell-signaling motif (52, 53).
OPN-R has recently been shown to be the dominant form of OPN responsible for the binding of hematopoietic stem cells and progenitors in the bone marrow niche (54). OPN, secreted from a primary tumor site, is capable of circulating to the host bone marrow where it mobilizes stromal and hematopoietic precursor cells, which then migrate to tumor cells at a second site to instigate their outgrowth (55). Thus, it is possible that one of the major functions of OPN-R is to augment the adhesion of tumor cells as well as hematopoietic and stromal precursor cells in these tumor-associated niches (56). To test the role of OPN and its cleaved forms in supporting the binding of glioma stem cells, U-87 SC cells were generated from U-87 MG cells in a defined serum-free neurosphere medium. This process promoted transition of adherent U-87 MG cells to neurosphere-like cells. The U-87 SC cells expressed Nestin, the neural stem cell marker, and possessed the potency to differentiate into the neuron and oligodendrocyte lineages but failed to differentiate into GFAP-positive astrocytes. Other differentiation conditions tested such as withdrawal of EGF and basic FGF, and leukemia inhibitory factor did not induce astrocyte differentiation (data not shown), suggesting that U-87 SC cells might express an alternative splice variant of GFAP, termed GFAPδ, that is not detected by anti-GFAP antibodies but that is found in human astrocytes from the sub-ventricular zone (40). In adhesion assays U-87 SC binding to OPN showed a similar profile to that of U-87 MG cells, with OPN-R and OPN-L ∼2-fold more effective than OPN-FL, consistent with the enhanced binding of hematopoietic SC to OPN-R in the bone marrow niche (40).
Along with our recent observation in rheumatoid arthritis synovial fluid, this is the second example of elevated OPN-R and OPN-L in human clinical samples. It is notable that they both occur in extravascular compartments, suggesting that they may result from local thrombin generation. The close correlation of the elevated levels of OPN-R with TAT in the CSF samples is consistent with that notion. There is increased expression of OPN in GBM and its associated microvasculature when compared with astrocytomas. Tissue factor and VEGF are also overexpressed and co-localized in these cell types (57). Thrombin immunoreactivity has been reported in GBM, and argatroban, a direct thrombin inhibitor, reduces glioma tumor mass and neurological deficits in a rat glioma model (32, 58), leading to the suggestion that the coagulation system can be a target for the treatment of malignant glioma (59). Thus, the production and cleavage of OPN by thrombin and CPB-2 may all occur locally. The resultant OPN-R and OPN-L would affect glioma growth and biology by generating more favorable niches for glioma cells.
Acknowledgments
We thank Dr. Heelo Sudo (Toray Industries Inc.) for performing three-dimensional GeneTM microarray, Dr. Peter N. Kao (Stanford University School of Medicine) for help with fluorescent microscopy, and Dr. Hiroyuki Sakata (Stanford University School of Medicine) for providing rat neural stem cells to us.
This work was supported, in whole or in part, by National Institutes of Health Grant HL057530.

This article contains supplemental Tables 1 and 2.
- OPN
- osteopontin
- OPN-FL
- full-length OPN
- OPN-L
- OPN-Leu
- OPN-R
- OPN-Arg
- CPB2
- carboxypeptidase
- CSF
- cerebrospinal fluid
- CCL
- chemokine (C-C motif) ligand
- GBM
- glioblastoma
- HBSS
- Hanks' balanced salt solution
- IKK
- IκB kinase
- MIP-1
- macrophage inflammatory protein 1
- Akt
- protein kinase B
- ANOVA
- analysis of variance
- GFAP
- glial fibrillary acidic protein
- TAT
- thrombin-antithrombin.
REFERENCES
- 1. Denhardt D. T., Noda M., O'Regan A. W., Pavlin D., Berman J. S. (2001) Osteopontin as a means to cope with environmental insults. Regulation of inflammation, tissue remodeling, and cell survival. J. Clin. Invest. 107, 1055–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Han M. H., Hwang S. I., Roy D. B., Lundgren D. H., Price J. V., Ousman S. S., Fernald G. H., Gerlitz B., Robinson W. H., Baranzini S. E., Grinnell B. W., Raine C. S., Sobel R. A., Han D. K., Steinman L. (2008) Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature 451, 1076–1081 [DOI] [PubMed] [Google Scholar]
- 3. Wang K. X., Denhardt D. T. (2008) Osteopontin. Role in immune regulation and stress responses. Cytokine Growth Factor Rev. 19, 333–345 [DOI] [PubMed] [Google Scholar]
- 4. El-Tanani M. K., Campbell F. C., Kurisetty V., Jin D., McCann M., Rudland P. S. (2006) The regulation and role of osteopontin in malignant transformation and cancer. Cytokine Growth Factor Rev. 17, 463–474 [DOI] [PubMed] [Google Scholar]
- 5. Minn A. J., Gupta G. P., Siegel P. M., Bos P. D., Shu W., Giri D. D., Viale A., Olshen A. B., Gerald W. L., Massagué J. (2005) Genes that mediate breast cancer metastasis to lung. Nature 436, 518–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van de Vijver M. J., He Y. D., van't Veer L. J., Dai H., Hart A. A., Voskuil D. W., Schreiber G. J., Peterse J. L., Roberts C., Marton M. J., Parrish M., Atsma D., Witteveen A., Glas A., Delahaye L., van der Velde T., Bartelink H., Rodenhuis S., Rutgers E. T., Friend S. H., Bernards R. (2002) A gene-expression signature as a predictor of survival in breast cancer. N. Engl. J. Med. 347, 1999–2009 [DOI] [PubMed] [Google Scholar]
- 7. Rudland P. S., Platt-Higgins A., El-Tanani M., De Silva Rudland S., Barraclough R., Winstanley J. H., Howitt R., West C. R. (2002) Prognostic significance of the metastasis-associated protein osteopontin in human breast cancer. Cancer Res. 62, 3417–3427 [PubMed] [Google Scholar]
- 8. Colin C., Baeza N., Bartoli C., Fina F., Eudes N., Nanni I., Martin P. M., Ouafik L., Figarella-Branger D. (2006) Identification of genes differentially expressed in glioblastoma versus pilocytic astrocytoma using suppression subtractive hybridization. Oncogene 25, 2818–2826 [DOI] [PubMed] [Google Scholar]
- 9. Atai N. A., Bansal M., Lo C., Bosman J., Tigchelaar W., Bosch K. S., Jonker A., De Witt Hamer P. C., Troost D., McCulloch C. A., Everts V., Van Noorden C. J., Sodek J. (2011) Osteopontin is up-regulated and associated with neutrophil and macrophage infiltration in glioblastoma. Immunology 132, 39–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Tuck A. B., Chambers A. F., Allan A. L. (2007) Osteopontin overexpression in breast cancer. Knowledge gained and possible implications for clinical management. J. Cell. Biochem. 102, 859–868 [DOI] [PubMed] [Google Scholar]
- 11. Formolo C. A., Williams R., Gordish-Dressman H., MacDonald T. J., Lee N. H., Hathout Y. (2011) Secretome signature of invasive glioblastoma multiforme. J. Proteome Res. 10, 3149–3159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jan H. J., Lee C. C., Shih Y. L., Hueng D. Y., Ma H. I., Lai J. H., Wei H. W., Lee H. M. (2010) Osteopontin regulates human glioma cell invasiveness and tumor growth in mice. Neuro-oncol. 12, 58–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lamour V., Le Mercier M., Lefranc F., Hagedorn M., Javerzat S., Bikfalvi A., Kiss R., Castronovo V., Bellahcène A. (2010) Selective osteopontin knockdown exerts anti-tumoral activity in a human glioblastoma model. Int. J. Cancer 126, 1797–1805 [DOI] [PubMed] [Google Scholar]
- 14. Lin Y. H., Yang-Yen H. F. (2001) The osteopontin-CD44 survival signal involves activation of the phosphatidylinositol 3-kinase/Akt signaling pathway. J. Biol. Chem. 276, 46024–46030 [DOI] [PubMed] [Google Scholar]
- 15. Denhardt D. T., Giachelli C. M., Rittling S. R. (2001) Role of osteopontin in cellular signaling and toxicant injury. Annu. Rev. Pharmacol. Toxicol. 41, 723–749 [DOI] [PubMed] [Google Scholar]
- 16. Wai P. Y., Kuo P. C. (2004) The role of osteopontin in tumor metastasis. J. Surg. Res. 121, 228–241 [DOI] [PubMed] [Google Scholar]
- 17. Myles T., Leung L. L. (2008) Thrombin hydrolysis of human osteopontin is dependent on thrombin anion-binding exosites. J. Biol. Chem. 283, 17789–17796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smith L. L., Giachelli C. M. (1998) Structural requirements for α9β1-mediated adhesion and migration to thrombin-cleaved osteopontin. Exp. Cell Res. 242, 351–360 [DOI] [PubMed] [Google Scholar]
- 19. Taooka Y., Chen J., Yednock T., Sheppard D. (1999) The integrin α9β1 mediates adhesion to activated endothelial cells and transendothelial neutrophil migration through interaction with vascular cell adhesion molecule-1. J. Cell Biol. 145, 413–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yokosaki Y., Matsuura N., Sasaki T., Murakami I., Schneider H., Higashiyama S., Saitoh Y., Yamakido M., Taooka Y., Sheppard D. (1999) The integrin α9β1 binds to a novel recognition sequence (SVVYGLR) in the thrombin-cleaved amino-terminal fragment of osteopontin. J. Biol. Chem. 274, 36328–36334 [DOI] [PubMed] [Google Scholar]
- 21. Barry S. T., Ludbrook S. B., Murrison E., Horgan C. M. (2000) A regulated interaction between α5β1 integrin and osteopontin. Biochem. Biophys. Res. Commun. 267, 764–769 [DOI] [PubMed] [Google Scholar]
- 22. Barry S. T., Ludbrook S. B., Murrison E., Horgan C. M. (2000) Analysis of the α4β1 integrin-osteopontin interaction. Exp. Cell Res. 258, 342–351 [DOI] [PubMed] [Google Scholar]
- 23. Bayless K. J., Davis G. E. (2001) Identification of dual α4β1 integrin binding sites within a 38-amino acid domain in the N-terminal thrombin fragment of human osteopontin. J. Biol. Chem. 276, 13483–13489 [DOI] [PubMed] [Google Scholar]
- 24. Green P. M., Ludbrook S. B., Miller D. D., Horgan C. M., Barry S. T. (2001) Structural elements of the osteopontin SVVYGLR motif important for the interaction with α4 integrins. FEBS Lett. 503, 75–79 [DOI] [PubMed] [Google Scholar]
- 25. Sharif S. A., Du X., Myles T., Song J. J., Price E., Lee D. M., Goodman S. B., Nagashima M., Morser J., Robinson W. H., Leung L. L. (2009) Thrombin-activatable carboxypeptidase B cleavage of osteopontin regulates neutrophil survival and synoviocyte binding in rheumatoid arthritis. Arthritis Rheum. 60, 2902–2912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Myles T., Nishimura T., Yun T. H., Nagashima M., Morser J., Patterson A. J., Pearl R. G., Leung L. L. (2003) Thrombin activatable fibrinolysis inhibitor, a potential regulator of vascular inflammation. J. Biol. Chem. 278, 51059–51067 [DOI] [PubMed] [Google Scholar]
- 27. Reynés G., Vila V., Martín M., Parada A., Fleitas T., Reganon E., Martínez-Sales V. (2011) Circulating markers of angiogenesis, inflammation, and coagulation in patients with glioblastoma. J. Neurooncol. 102, 35–41 [DOI] [PubMed] [Google Scholar]
- 28. Tehrani M., Friedman T. M., Olson J. J., Brat D. J. (2008) Intravascular thrombosis in central nervous system malignancies. A potential role in astrocytoma progression to glioblastoma. Brain Pathol. 18, 164–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yamaguchi Y., Du X. Y., Zhao L., Morser J., Leung L. L. (2011) Proteolytic cleavage of chemerin protein is necessary for activation to the active form, Chem157S, which functions as a signaling molecule in glioblastoma. J. Biol. Chem. 286, 39510–39519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hsieh J., Aimone J. B., Kaspar B. K., Kuwabara T., Nakashima K., Gage F. H. (2004) IGF-I instructs multipotent adult neural progenitor cells to become oligodendrocytes. J. Cell Biol. 164, 111–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Anand M., Brat D. J. (2012) Oncogenic regulation of tissue factor and thrombosis in cancer. Thromb. Res. 129, S46–S49 [DOI] [PubMed] [Google Scholar]
- 32. Hua Y., Tang L., Keep R. F., Schallert T., Fewel M. E., Muraszko K. M., Hoff J. T., Xi G. (2005) The role of thrombin in gliomas. J. Thromb. Haemost. 3, 1917–1923 [DOI] [PubMed] [Google Scholar]
- 33. Yan W., Qian C., Zhao P., Zhang J., Shi L., Qian J., Liu N., Fu Z., Kang C., Pu P., You Y. (2010) Expression pattern of osteopontin splice variants and its functions on cell apoptosis and invasion in glioma cells. Neuro-oncol. 12, 765–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rice J., Courter D. L., Giachelli C. M., Scatena M. (2006) Molecular mediators of αvβ3-induced endothelial cell survival. J. Vasc. Res. 43, 422–436 [DOI] [PubMed] [Google Scholar]
- 35. Singh S. K., Hawkins C., Clarke I. D., Squire J. A., Bayani J., Hide T., Henkelman R. M., Cusimano M. D., Dirks P. B. (2004) Identification of human brain tumour initiating cells. Nature 432, 396–401 [DOI] [PubMed] [Google Scholar]
- 36. Singh S. K., Clarke I. D., Terasaki M., Bonn V. E., Hawkins C., Squire J., Dirks P. B. (2003) Identification of a cancer stem cell in human brain tumors. Cancer Res. 63, 5821–5828 [PubMed] [Google Scholar]
- 37. Al-Hajj M., Wicha M. S., Benito-Hernandez A., Morrison S. J., Clarke M. F. (2003) Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. U.S.A. 100, 3983–3988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li C., Heidt D. G., Dalerba P., Burant C. F., Zhang L., Adsay V., Wicha M., Clarke M. F., Simeone D. M. (2007) Identification of pancreatic cancer stem cells. Cancer Res. 67, 1030–1037 [DOI] [PubMed] [Google Scholar]
- 39. Patrawala L., Calhoun T., Schneider-Broussard R., Li H., Bhatia B., Tang S., Reilly J. G., Chandra D., Zhou J., Claypool K., Coghlan L., Tang D. G. (2006) Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene 25, 1696–1708 [DOI] [PubMed] [Google Scholar]
- 40. Pollard S. M., Yoshikawa K., Clarke I. D., Danovi D., Stricker S., Russell R., Bayani J., Head R., Lee M., Bernstein M., Squire J. A., Smith A., Dirks P. (2009) Glioma stem cell lines expanded in adherent culture have tumor-specific phenotypes and are suitable for chemical and genetic screens. Cell Stem Cell 4, 568–580 [DOI] [PubMed] [Google Scholar]
- 41. Jijiwa M., Demir H., Gupta S., Leung C., Joshi K., Orozco N., Huang T., Yildiz V. O., Shibahara I., de Jesus J. A., Yong W. H., Mischel P. S., Fernandez S., Kornblum H. I., Nakano I. (2011) CD44v6 regulates growth of brain tumor stem cells partially through the AKT-mediated pathway. PLoS ONE 6, e24217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yao X. H., Ping Y. F., Chen J. H., Xu C. P., Chen D. L., Zhang R., Wang J. M., Bian X. W. (2008) Glioblastoma stem cells produce vascular endothelial growth factor by activation of a G-protein coupled formylpeptide receptor FPR. J. Pathol. 215, 369–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Annabi B., Laflamme C., Sina A., Lachambre M. P., Béliveau R. (2009) A MT1-MMP/NF-κB signaling axis as a checkpoint controller of COX-2 expression in CD133+ U87 glioblastoma cells. J. Neuroinflammation 6, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Scheinkestel C. D., Jones K., Myles P. S., Cooper D. J., Millar I. L., Tuxen D. V. (2004) Where to now with carbon monoxide poisoning? Emerg. Med. Australas. 16, 151–154 [DOI] [PubMed] [Google Scholar]
- 45. Brandsma D., Stalpers L., Taal W., Sminia P., van den Bent M. J. (2008) Clinical features, mechanisms, and management of pseudoprogression in malignant gliomas. Lancet Oncol. 9, 453–461 [DOI] [PubMed] [Google Scholar]
- 46. Norden A. D., Young G. S., Setayesh K., Muzikansky A., Klufas R., Ross G. L., Ciampa A. S., Ebbeling L. G., Levy B., Drappatz J., Kesari S., Wen P. Y. (2008) Bevacizumab for recurrent malignant gliomas. Efficacy, toxicity, and patterns of recurrence. Neurology 70, 779–787 [DOI] [PubMed] [Google Scholar]
- 47. Sreekanthreddy P., Srinivasan H., Kumar D. M., Nijaguna M. B., Sridevi S., Vrinda M., Arivazhagan A., Balasubramaniam A., Hegde A. S., Chandramouli B. A., Santosh V., Rao M. R., Kondaiah P., Somasundaram K. (2010) Identification of potential serum biomarkers of glioblastoma. Serum osteopontin levels correlate with poor prognosis. Cancer Epidemiol. Biomarkers Prev. 19, 1409–1422 [DOI] [PubMed] [Google Scholar]
- 48. Schuhmann M. U., Zucht H. D., Nassimi R., Heine G., Schneekloth C. G., Stuerenburg H. J., Selle H. (2010) Peptide screening of cerebrospinal fluid in patients with glioblastoma multiforme. Eur. J. Surg. Oncol. 36, 201–207 [DOI] [PubMed] [Google Scholar]
- 49. Lahav J., Gofer-Dadosh N., Luboshitz J., Hess O., Shaklai M. (2000) Protein-disulfide isomerase mediates integrin-dependent adhesion. FEBS Lett. 475, 89–92 [DOI] [PubMed] [Google Scholar]
- 50. Selistre-de-Araujo H. S., Pontes C. L., Montenegro C. F., Martin A. C. (2010) Snake venom disintegrins and cell migration. Toxins 2, 2606–2621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lu X., Lu D., Scully M. F., Kakkar V. V. (2007) Structure-activity relationship studies on ADAM protein-integrin interactions. Cardiovasc. Hematol. Agents Med. Chem. 5, 29–42 [DOI] [PubMed] [Google Scholar]
- 52. Qi B., Newcomer R. G., Sang Q. X. (2009) ADAM19/adamalysin 19 structure, function, and role as a putative target in tumors and inflammatory diseases. Curr. Pharm. Des. 15, 2336–2348 [DOI] [PubMed] [Google Scholar]
- 53. Stanton H., Melrose J., Little C. B., Fosang A. J. (2011) Proteoglycan degradation by the ADAMTS family of proteinases. Biochim. Biophys. Acta 1812, 1616–1629 [DOI] [PubMed] [Google Scholar]
- 54. Grassinger J., Haylock D. N., Storan M. J., Haines G. O., Williams B., Whitty G. A., Vinson A. R., Be C. L., Li S., Sørensen E. S., Tam P. P., Denhardt D. T., Sheppard D., Choong P. F., Nilsson S. K. (2009) Thrombin-cleaved osteopontin regulates hemopoietic stem and progenitor cell functions through interactions with α9β1 and α4β1 integrins. Blood 114, 49–59 [DOI] [PubMed] [Google Scholar]
- 55. McAllister S. S., Gifford A. M., Greiner A. L., Kelleher S. P., Saelzler M. P., Ince T. A., Reinhardt F., Harris L. N., Hylander B. L., Repasky E. A., Weinberg R. A. (2008) Systemic endocrine instigation of indolent tumor growth requires osteopontin. Cell 133, 994–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wels J., Kaplan R. N., Rafii S., Lyden D. (2008) Migratory neighbors and distant invaders. Tumor-associated niche cells. Genes Dev. 22, 559–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Takano S., Tsuboi K., Tomono Y., Mitsui Y., Nose T. (2000) Tissue factor, osteopontin, αvβ3 integrin expression in microvasculature of gliomas associated with vascular endothelial growth factor expression. Br. J. Cancer 82, 1967–1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hua Y., Tang L. L., Fewel M. E., Keep R. F., Schallert T., Muraszko K. M., Hoff J. T., Xi G. H. (2005) Systemic use of argatroban reduces tumor mass, attenuates neurological deficits, and prolongs survival time in rat glioma models. Acta Neurochir. Suppl. 95, 403–406 [DOI] [PubMed] [Google Scholar]
- 59. Ornstein D. L., Meehan K. R., Zacharski L. R. (2002) The coagulation system as a target for the treatment of human gliomas. Semin. Thromb. Hemost. 28, 19–28 [DOI] [PubMed] [Google Scholar]