

Background: Therapeutic values of Valeriana officinalis have been associated with sesquiterpenes whose biosynthetic origins have remained enigmatic.
Results: A cyclobutenyl intermediate in the catalytic cascade of valerena-1,10-diene synthase is reported.
Conclusion: A new class of sesquiterpene synthases for the biosynthesis of sesquiterpenes harboring isobutenyl functional groups is proposed.
Significance: Similar catalytic mechanisms from evolutionarily diverse organisms are proposed and portend sources for sesquiterpene diversity.
Keywords: Chemical Biology, Enzymes, Natural Products, Plant Biochemistry, Terpenoids
Abstract
Valerian is an herbal preparation from the roots of Valeriana officinalis used as an anxiolytic and sedative and in the treatment of insomnia. The biological activities of valerian are attributed to valerenic acid and its putative biosynthetic precursor valerenadiene, sesquiterpenes, found in V. officinalis roots. These sesquiterpenes retain an isobutenyl side chain whose origin has been long recognized as enigmatic because a chemical rationalization for their biosynthesis has not been obvious. Using recently developed metabolomic and transcriptomic resources, we identified seven V. officinalis terpene synthase genes (VoTPSs), two that were functionally characterized as monoterpene synthases and three that preferred farnesyl diphosphate, the substrate for sesquiterpene synthases. The reaction products for two of the sesquiterpene synthases exhibiting root-specific expression were characterized by a combination of GC-MS and NMR in comparison to the terpenes accumulating in planta. VoTPS7 encodes for a synthase that biosynthesizes predominately germacrene C, whereas VoTPS1 catalyzes the conversion of farnesyl diphosphate to valerena-1,10-diene. Using a yeast expression system, specific labeled [13C]acetate, and NMR, we investigated the catalytic mechanism for VoTPS1 and provide evidence for the involvement of a caryophyllenyl carbocation, a cyclobutyl intermediate, in the biosynthesis of valerena-1,10-diene. We suggest a similar mechanism for the biosynthesis of several other biologically related isobutenyl-containing sesquiterpenes.
Introduction
Valerian is a nutraceutical preparation from the roots of Valeriana officinalis and related species that is recommended for relief of tension, anxiety, and insomnia (1–3). V. officinalis grows as a wild herb in very diverse habitats around the globe, and valerian has been used for hundreds of years as a herbal remedy (4, 5). For example, it was used as a treatment to relieve the stress of air raids in England during WWII (6). The greatest biological efficacy of valerian has been correlated with freshly harvested and carefully dried root preparations and with the iridoid alkaloid and sesquiterpene content of these preparations (7). The valepotriates are epoxyiridoid esters with the dominant species being valtrate (8). Because of putative instability and water insolubility of the valepotriates, some investigations have suggested that the sesquiterpene compounds are more important for the sedative activity of valerian (8). The sesquiterpene derivatives valerenic acid and valeranone have both been shown to possess potent biological activities (9).
Valerenic acid is thought to be derived from valerena-1,10-diene, whereas valeranone might be derived from a germacrene precursor (Fig. 1). However, valerenadiene has been of particular interest to others studying related biologically active sesquiterpene compounds like tamariscol (10), tamariscene (11), and pacifigorgiol (12) (Fig. 1) because the biogenic origin of the isobutenyl substituent group common to these molecules has remained enigmatic. To investigate possible mechanisms for the biosynthesis of the unique valerenadiene scaffold (11), we have leveraged our recent assembly of transcriptomic (see Medicinal Plant Genomics Resource Internet website) and metabolomic (see Medicinal Plant Metabolomic Resource Internet website) resources for 14 different medicinal plant species including V. officinalis. Using these resources, we identified seven possible terpene synthase gene candidates, examined their tissue-specific expression profiles, and correlated these with the accumulation of sesquiterpenes, then functionally characterized the terpene synthase enzymes encoded by these genes in vitro and in vivo. During the course of this work, Pyle et al. (13) also reported a similar effort to identify sesquiterpene synthases from V. officinalis and described the characterization of a valerena-1,10-diene synthase. However, in the current work we have exploited the valerenadiene synthase to probe and validate an unusual catalytic cascade for the generation of isobutenyl constellations on a unique class of biologically active sesquiterpenes.
FIGURE 1.

Proposed pathways for the biosynthesis of the sesquiterpene valeranone (3) and valerenic acid (4) in V. officinalis and structural comparison to related hydrindane-type sesquiterpenes found in association with liverworts (Frullania tamarisci) and gorgonian sponges (Pacifigoria cf. adamsii). A, multistep pathways are depicted, the first step catalyzed by a sesquiterpene synthase (TPS) cyclizing FPP to either germacrene C (1) or valerena-1,10-diene (2) followed by internal ring formation and hydroxylation/reduction (valeranone) or successive hydroxylation/oxidation at one of the terminal methyls of the isobutenyl side chain (valerenic acid). B, tamariscene (5), tamarisol (6), and pacifigorgiol (7) appear to share a valerenadiene-like scaffold in common with valerenic acid and hence may share a similar biosynthetic origin as suggested by Paul et al. (11).
EXPERIMENTAL PROCEDURES
Plant Material
Authenticated V. officinalis L. (Valerianaceae) seeds were purchased from Richters Herbs (Ontario, Canada). The seeds were germinated in germination trays with Promix® potting mixture in an environmentally controlled greenhouse at the University of Mississippi Medicinal Plant Garden. The average temperature and humidity in the greenhouse were maintained at 70 °F and 60%, respectively, and natural light and fluorescent grow lights were used to maintain a minimum of 16 h of light per 24-h period. Two-week-old plantlets were transferred into larger pots with Promix® potting mixture. They were hand-watered and fertilized twice a month with Miracle Grow® all-purpose plant food. Three separate populations were maintained from three different seed accessions. The tissue harvesting was done from 1-year-old plants selected randomly. Young leaves (<25% expanded), mature leaves (fully expanded), petioles, and lateral roots were harvested for both reference and experiment samples with three replicates, frozen immediately in liquid nitrogen, and subsequently stored at −80 °C. The weight and photographic record of all samples were obtained.
Young leaves (5.0–15.0 mm) of Valeriana officinalis were used as explants for callus culture induction. Explants were surface-sterilized with 0.5% NaOCl (15% v/v bleach) and 0.1% Tween 20 for 20 min then washed extensively with sterile distilled water before inoculation onto Murashige and Skoog medium containing 3% (w/v) sucrose, 0.8% (w/v) Type E agar (Sigma) supplemented with 4.0 mg/liter napthalene acetic acid (NAA) in combination with 0.2 mg/liter thidiazuron for callus induction. The medium was adjusted to pH 5.7. The cultures were incubated at 25 ± 2 °C with a 16-h photoperiod under fluorescent light with 52 ± 5 μmol m−2 s−1 photon flux density. Thirty 5-day-old calli were collected for metabolomics and RNAseq analysis.
Metabolomics
One gram of freeze-dried tissue sample pulverized in liquid N2 was extracted for 3 h by shaking in 10 ml of chloroform:methanol (1:1 v/v) containing external standard propyl 4-hydroxybenzoate (10 μm) and antioxidant butylated hydroxytoluene (1 mg/ml). The mixture was filtered through pre-weighed Whatman filter paper, and the filtrate was used for time-of-flight mass spectrometry (LC-TOF MS) analysis. Filter paper containing plant residue was air-dried for 24 h, oven-dried at 40 °C for an additional 24 h, then weighed to determine dry weight of the plant tissue extracted.
Aliquots of the extracts were then analyzed using a nontargeted metabolomic approach based on ultrahigh performance liquid chromatography/TOF MS), electrospray ionization, and nonselective multiplexed collision-induced dissociation (14) that generates global accurate molecular and fragment mass information. Automated peak detection, alignment, and integration were performed using Waters MarkerLynx software, and peak areas were exported to a Microsoft Excel spreadsheet (for qualitative and quantitative assessments). Metabolite identities were confirmed by comparison of retention times and molecular and fragment masses to available authentic standards.
The spreadsheet data were incorporated into an extendable database linked to a search and display software interface, publically available at Medicinal Plant Metabolomic Resource. This interface enables metabolite searches by species, molecular mass ranges, and/or key terms. Analysis includes scatterplots for assessing replicability, means and standard errors of selected compound across multiple samples, and comparison of differences in metabolite levels across two samples with p values calculated according to the number of replicates via volcano plots and ratio plots.
Chemical Profiling of the Plant Tissues
The metabolite extracts were prepared from 300 mg of each tissue sample powdered in liquid nitrogen, then extracted with 5 ml of n-hexanes with shaking at room temperature for 2 h. The filtrate was then passed through a 0.25 × 2-cm silica column, concentrated to 100 μl under nitrogen gas, and then analyzed by GC-MS.
Transcriptome
RNA was isolated from callus, young leaves, mature leaves, lateral roots, and stem/petioles from three V. officinalis populations as described previously (15). cDNA libraries were constructed using the mRNA-Seq kit (Illumina, San Diego, CA) and sequenced to 35 bp in the single end mode or 76 bp in the paired end mode on the Illumina Genome Analyzer IIx platform at the Michigan State University Research Technology Support Facility. Reads were cleaned and assembled as described previously (15). Transcript assemblies were annotated for function using a combination of BLAST (16) sequence similarity results to UniRef100 (17) and Pfam (18) domain composition in ESTScan-predicted (19) peptide sequences. Custom perl scripts were used to remove low complexity sequences and potential contaminants in the transcriptome assembly based on sequence similarity to UniRef100 entries in the bacterial, fungal, virus, and viroid kingdoms as well as Arthropoda, Stramenopiles and human sequences. Expression abundances were estimated by mapping single end reads from each library to the reference transcriptome using TopHat (20) and Cufflinks (21) as described previously (15). Raw sequences are available in the NCBI Sequence Read Archive under accession number SRP005998. Assembled transcripts are available at Medicinal Plant Genomics Resource.
A total of 223,381,692 reads were generated from all libraries, and 138,222,300 (61.8%) were incorporated into the final de novo assembly. After removal of contaminants and low complexity sequences (1594), the assembly contained a total of 75,326 transcripts with an N50 (the contig size such that all contigs larger than that have 50% the bases of the assembly) of 1060 bp. Single and multiple isoform transcripts were present within the assembly, and a set of 35,556 unique transcripts (unigenes) was used for expression abundance estimations using TopHat (20) and Cufflinks (21).
DNA sequences described here are deposited in GenBankTM with the following accession numbers: VoTPS1,2 JX494699; VoTPS2, JX494700; VoTPS3, JX494701; VoTPS4, JX494702; VoTPS5, JX494703; VoTPS6, JX494704; VoTPS7, JX494705.
RT-PCR
Total RNA was isolated from Valeriana tissues using the RNeasy Plant Mini kit (Qiagen). First-strand cDNA was synthesized from 1 μg of total RNA using superscript II reverse transcriptase (Invitrogen) under conditions recommended by the manufacturer and used as a template for RT-PCR. To perform the PCR, the 50-μl reaction mixture contained 1.0 μl of the first-strand cDNA, 200 μm dNTPs, 1 unit of Ex Taq polymerase (TaKaRa), and 10 pmol of each specific primer sets. Primer pairs are given in supplemental Table S1. Forward and reverse primers included restriction sites for BamHII and NotI, respectively. These restriction sites were subsequently used to introduce the amplicons into the pET 28a expression vector (Novagen). The PCR protocol used was an initial denaturation at 98 °C for 3 min, then a cycling regime of 20–30 cycles at 98 °C for 10 s, 55 °C for 30 s, and 72 °C for 1 min. After completion of the PCR, aliquots of the reactions were examined by agarose gel electrophoresis.
Bacterial Expression/Enzyme Assays
The various RT-PCR amplification products were ligated into the pET 28a vector and transformed to Escherichia coli BL21 (DE). Cultures initiated from single colonies were cultured at 37 °C until an optical density of 0.7, then 0.5 mm isopropyl 1-thio-β-d-galactopyranoside was added, and the cultures were incubated for an additional 10 h at room temperature with shaking. Cells were then collected by centrifugation at 4,000 × g for 10 min, resuspended in lysis buffer of 50 mm NaH2PO4, pH 7.8, 300 mm NaCl, 10 mm imidazole, 1 mm MgCl2, 1 mm PMSF, 1% glycerol (v/v), and sonicated 5 times for 20 s, and the cleared supernatants (16,000 × g for 10 min at 4 °C) were stored at −80 °C before use.
Enzyme assays were initiated by mixing aliquots of the cleared supernatants with 250 mm Tris-HCl, pH 7.0, 50 mm MgCl2, and 30 μm geranyl diphosphate (0.5 μCi of [1-3H]GPP (∼20 Ci mmol−1, American Radiolabeled Chemicals) or 30 μm farnesyl diphosphate (0.5 μCi of [1-3H]FPP ( 20 Ci mmol−1, American Radiolabeled Chemicals) in a total reaction volume of 50 μl. Reactions were incubated at room temperature for 15 min and extracted with 150 μl of hexanes, unreacted prenyl diphosphates and prenyl alcohols were removed with silica gel, and incorporation of radioactivity was measured by scintillation counting.
For GC-MS profiling of the enzyme reaction products, 50 μl of cleared supernatant (0.8–1.0 μg protein/μl) was incubated in 250 mm Tris-HCl, pH 7.0, 50 mm MgCl2 with 10 μg of FPP for 2 h at room temperature, and the reaction mixture was then extracted twice with an equal volume of hexanes, concentrated 10-fold under nitrogen gas, and analyzed by GC-MS. GC conditions consisted of a Varian CP-3800 GC couple to a Varian Saturn 2200 MS/MS using a Supelco SLB-5ms fused silica capillary column (30 m × 0.25 mm × 0.25 μm film thickness). Initial oven temperature was set at 70 °C for 1 min followed by an 8 °C/min gradient to 200 °C, then 20 °C/min to 300 °C.
For enzyme kinetic studies, bacterial expressed enzymes were purified by their carboxyl-terminal hexahistidine tags using His-Select Cobalt affinity gel (Sigma) columns following the manufacturer's recommendations. The purified protein fractions were concentrated using Amicon Ultra centrifugation filter units and stored in 300 mm NaCl, 20 mm Tris-HCl, pH 7.5, 5 mm dithiothreitol, 2 mm MgCl2, 50% glycerol (v/v) at −80 °C. Protein concentrations were estimated by the Bradford assay (Bio-Rad) using bovine serum albumin as the standard.
NMR Studies
For yeast expression vector construction, the VoTPS1 and VoTPS7 genes were inserted into a pESC plasmid (Agilent Technologies) behind the constitutive GPD promoter. Yeast strain ZX178–08 was used for all the expression studies reported here. This strain was selected for its ability to meet its sterol needs by taking up exogenous sterols under aerobic conditions and further engineered to have a dispensable mevalonate biosynthetic pathway that can be redirected to sesquiterpene biosynthesis.3 40-ml seed train cultures of yeast harboring pESC-VoTPS1 or pESC-VoTPS7 were grown in SCE liquid medium (22) lacking leucine for 4 days then inoculated at 3% v/v of the seed train cultures into multiple 1-liter cultures and grown at 28 °C. When sesquiterpene production became saturated, approximately after 6 days, the fermentation broth was first incubated with an equal volume of acetone with shaking for 12 h at 28 °C then extracted with an equal volume of hexanes, and the hexanes extract were dried under nitrogen to a volume of ∼200 ml. The extract was then passed over a silica gel G60 column (20 × 160 mm), and the flow-through was collected. The flow-through of VoTPS1 afforded 6 mg of purified 2, whereas VoTPS7 required additional purification via an HPLC separation system (Develosil Packed Column, 20 × 250 mm, Nomura Chemical Co., Ltd.). Preparative scale separation of sesquiterpenes was conducted on a Waters Alliance 2695 system with an attached photodiode array in an isocratic mixture of 1:4000 methyl tert-butyl ether:hexanes.
For the isolation of sesquiterpenes from plant material, V. officinalis roots (78.85 g) were frozen and powdered in liquid nitrogen. The powdered material was distributed among eight 50-ml centrifuge tubes, and each sample was mixed thoroughly with 30 ml of n-hexane. The samples were then sonicated at room temperature for 30 min followed by centrifugation at 4000 rpm for 10 min, and the supernatants were collected and combined. The sonication extraction was repeated, and the combined supernatant was concentrated to 1 ml by a combination of rotoevaporation at 35 °C and drying under nitrogen gas. The sesquiterpenes were separated by silica chromatography (200–300 mesh, 21- × 1.5-cm inner diameter) using n-hexane as the solvent. Fractions of 3–4 ml were collected and evaluated by TLC and GC-MS. Compound 2, valerena-1,10-diene, was characterized from fractions 9–11, whereas compound 1, germacrene C, from fractions 21–23 was used for identification. NMR spectra were measured either on a Bruker 500MHz or Varian Vnmr 500 (1H, 500 MHz; 13C, 125MHz) spectrometer using hexane-d14 and CDCl3 as solvent.
Labeling Studies
To prepare 13C-labeled valerena-1,10-diene, six cultures of 0.5 liters of SCE-Leu liquid media were inoculated with seed train cultures as before and grown for 4 days. On the fourth day, 3.0 g of [1-13C]sodium acetate (Sigma) was added, and incubation was continued for an additional 48 h. The cultures were then harvested and extracted with acetone and hexane, and the hexane phase was fractionated over silica gel to isolate 6.5 mg of labeled 2. The labeled sample and 6.5 mg of unlabeled 2 were analyzed by 13C NMR under identical experimental conditions (standard s2pul pulse sequence with 340 scans and the delay and pulse width set to the manufacturer's default values). Enrichment values were determined by comparing the ratios of the peak heights between the two samples, then multiplying by 1.1% to give the percent enrichment relative to that occurring from the natural abundance of 13C (23, 24). For C-10 and C-11, which demonstrated Jc-c coupling in the labeled sample, the peak intensity at δc 126.391 ppm and δc 129.986 (for C-10 and C-11, respectively) were compared with the identical peak intensities corresponding to C-10 and C-11 in the unlabeled sample (see supplemental Fig. S8).
RESULTS
Computational and Molecular Screens for Terpene Synthases
To identify metabolic intermediates and the genes/enzymes that are required for their synthesis, it is useful to evaluate the site(s) of synthesis, which may be distinct from the site of accumulation. When considering the location of sesquiterpene biosynthesis versus accumulation of V. officinalis, we first sought to determine if the valerenadiene scaffold might accumulate in root tissue, thus suggesting that its site of synthesis might also be in this tissue. To do this, we searched the Medicinal Plant Metabolomics Resource, which contains metabolomics data and metadata, analysis, and visualization for 14 medicinal species including V. officinalis. The site provides a variety of linkages and methods for statistical analysis, visualization, and comparison among metabolite data, chemical information, and metadata. When the LC-TOF MS profile for eight different tissue types of V. officinalis, including leaves, flowers, developing and mature leaves, roots and callus cultures were screened for metabolites that yielded ions in the narrow range of m/z 203.1 to 203.2 (corresponding to a fragment ion of elemental formula C15H23+, derived from a downstream oxidized metabolite of valerenadiene), several metabolites meeting this criterion were found exclusively in the root samples, including one corresponding to an acetylated sesquiterpenoid diol that yielded multiple fragment ions supportive of this annotation (supplemental Fig. S1).
This was suggestive that the terpene synthase(s) responsible for the biosynthesis of the valerenadiene scaffold could in fact be localized to the root tissue. To evaluate this possibility, we next queried the V. officinalis de novo assembly transcriptome for type I sesquiterpene synthases using the well characterized premnaspirodiene synthase from Hyoscyamus muticus (HPS) (25) as the search query. Seven distinct contigs with sequence identity and similarity to HPS in excess of 29 and 46%, respectively, and appearing to represent full-length mRNAs encoding for proteins with greater than 490 amino acids were recovered (supplemental Fig. S2). These sequences were designated as VoTPS1 through -7. When the conceptual translations of each were aligned together with the HPS protein, several regions diagnostic for class I or clade “a” terpene synthases, TPS-a, like the DDXXD domain associated with the binding of a divalent cation co-factor (26, 27), were readily apparent (supplemental Fig. S2). In addition, VoTPS3 contained an extended amino-terminal sequence suggestive that this synthase might be targeted to the chloroplast compartment, consistent with a prediction score of 0.581 for the amino-terminal 88 amino acids serving as a chloroplast signal sequence peptide by the ChloroP 1.1 algorithm. Also important to note, due to the algorithm used in assembling the DNA sequence reads into contigs from RNA samples pooled from several reference plant tissue types, a second TPS, VoTPS4, nearly identical to VoTPS3 was identified. VoTPS4 is predicted to initiate translation at a second methionine 46 codons 3′ to the initiating methionine predicted for VoTPS3. Given the absolute sequence identity of these genes, we suspect these TPSs to be one and the same and henceforth designate the combined contig as VoTPS3/4.
If valerenadiene biosynthesis occurs in the roots of V. officinalis, then one would expect the mRNA encoding for the putative valerenadiene synthase to be represented, perhaps exclusively, in root tissue. The VoTPSs mRNA levels were thus examined in two ways. First, because of the quantitative nature of the RNA-seq methodology used to generate the tissue-specific transcriptomes, the relative abundance of each VoTPS transcripts can be quantified in each of the tissue types as the number of sequenced fragments mapping to a contig per kilobase of sequence per million reads (FRPM) (21) (Fig. 2). VoTPS3, its likely truncated form VoTPS4, and VoTPS6 all appear to be constitutively expressed across the various tissue types with VoTPS 3 expression being 10–20-fold greater than VoTPS6. In contrast, VoTPS1, -2, -5, and -7 exhibited expression patterns more specific to root tissue with the expression levels of VoTPS2 and -5 being ∼3–10-fold lower, respectively, than for VoTPS1 and -7.
FIGURE 2.

Expression profiles of VoTPS1 through 7 in the major tissue types of V. officinalis. In the upper table, the measure of terpene synthase gene expression is based on the number of sequenced fragments found for a particular contig per kilobase of sequence per million reads (FRPM) (21) (VoTPS1, contig 16506; VoTPS2, contig 75367; VoTPS3, contig 5803; VoTPS5, contig 13291; VoTPS6, contig 24,652; VoTPS7, contig 31,253). Orange highlighting indicates VoTPSs exhibiting constitutive expression, and green indicates VoTPSs showing preferential expression in root tissues. Color intensity is proportional to expression level. Independent biological replicates for tissue types are indicated as sample 1 or 2. In the lower panel, RNA was extracted from young leaves (YL), mature leaves (L), stems (S), and roots (R), converted to first strand cDNA, then utilized as the template for PCR reactions specific for each of the VoTPS genes (V1-V7) within the linear amplification range for each amplicon. PCR reactions with primers specific for actin mRNA were performed to judge the qualitative and quantitative quality of the RNA templates used.
The quantitative RNA-seq transcript compilation was also validated by a more traditional quantitative RT-PCR assay as well (Fig. 2). In these analyses, RNA was isolated for the various tissue types, converted to first strand cDNA, and then used as a template with specific PCR primer pairs for each VoTPS transcript under PCR conditions determined to be within a linear amplification range for all the amplicons (i.e. 20–26 cycles). Although the levels of VPS1–4 transcripts were consistent with the quantitative RNA-seq comparisons, transcript amounts for VoTPS5 and -7 were much more evident in stem and mature leaf tissue and directly comparable with those observed in roots.
Functional Characterization of VoTPS1 and VoTPS7
Although sequence alignments are generally sufficient to classify the VoTPS genes as terpene synthases, direct biochemical characterization is necessary to determine if these genes code for monoterpene or sesquiterpene synthases and certainly to ascribe a particular biochemical reaction to these enzymes. Thus, we first prepared cell-free extracts from E. coli expressing each of the VoTPS cDNAs and used these extracts for monoterpene and sesquiterpene synthase enzyme assays. In these assays the E. coli extracts were incubated with radiolabeled allylic diphosphate substrates, [1-3H]GPP or [1-3H]FPP, and the conversion of these substrates to hexane extractable products was determined (Fig. 3). The activities of these putative terpene synthases were examined relative to a negative (an extract prepared from E. coli harboring the expression plasmid without an inserted TPS gene) and positive (an extract prepared from E. coli expressing the previously characterized sesquiterpenes synthase gene encoding for HPS (25)) control. As evident in Fig. 3, very little monoterpene synthase activity was recorded for the negative control, but the HPS-positive control did yield some monoterpene product. This has been observed previously and attributed to the sesquiterpene synthases having larger activity site pockets that can accommodate smaller substrates. VoTPS1, -5, and -7 appear to have similar low level activities with GPP, whereas VoTPS3 and -4 were clearly capable of turning over significant amounts of GPP to hydrophobic extractable products. This information suggested that VoTPS3 and -4, although different by only 32 amino acids at the amino terminus of VoTPS3, might in fact be monoterpene synthases. In contrast, relative to the HPS positive control, VoTPS 1, 5, and 7 all exhibited very substantial sesquiterpene synthase activities with typical kinetic properties (supplemental Fig. S3).
FIGURE 3.

Substrate specificity of six VoTPSs. Cell-free extracts prepared from E. coli cells overexpressing the indicated terpene synthase genes were incubated with 30 μm [3H]GPP (A) or [3H]FPP (B) for 15 min before determining the amount of radiolabel incorporated into the hexanes extractable reaction products. pET corresponds to bacteria harboring a vector only negative control; HPS, bacteria expressing the Hyoscyamus premnaspirodiene synthase gene (25) as a positive sesquiterpene synthase control.
When the reaction products of these in vitro assays were profiled by GC-MS, three distinct compounds were observed for the VoTPS1 reaction, and one major compound with several possible minor, closely migrating compounds was observed for the VoTPS7-mediated reaction (Fig. 4 and supplemental Fig. S4). The reaction products of both these enzymes were also found in common with constituents found in extracts prepared from roots (Fig. 4D) but not from leaves or stems (Fig. 4, A–C). Although the mass spectra for each of the TPS reaction compounds exhibited signature ion patterns consistent with sesquiterpenes (supplemental Fig. S4), these MS patterns were not sufficient to determine the absolute compound identities. Hence, we overexpressed the VoTPS1 and -7 genes in a yeast line developed for high level accumulation of FPP and sesquiterpene biosynthesis to obtain sufficient amounts of the respective compounds for NMR analysis (supplemental Fig. S5).
FIGURE 4.

Comparison of hexane-extractable hydrocarbons that accumulate in various plant tissues to the reaction products generated by VoTPS1 and -7. GC traces of the terpenes extracted from V. officinalis young leaves (A), mature leaves (B), stems (C), and roots (D) in comparison to the extractable reaction products generated by VoTPS1 (E) and VoTPS7 (F) are shown. The VoTPS 1 and 7 genes were expressed in bacteria, and the bacterial lysates were then incubated with FPP before profiling the reaction products by GC-MS. The chromatograms are annotated for the elution behavior of germacrene C (1), valerena-1,10-diene (2), and an unknown sesquiterpene compound (3).
Structural Elucidation of Sesquiterpene Products from VoTPS1 and VoTPS7
Six-liter fermentations of recombinant yeast overexpressing the respective TPS genes were lysed in acetone, and then the hydrocarbon fraction was extracted three times with hexanes to isolate terpene components. The extracts were fractionated via silica gel chromatography to afford 20 mg of the sesquiterpene products produced by VoTPS7. Four sesquiterpenes were identified in the mixture based on GC-MS analyses, whereas the dominant peak constituted 90% of the total mixture, the second peak was 7%, and the third and fourth compounds were less than 3%. The two major compounds were further purified via HPLC to afford 12 and 1 mg, respectively.
The identity of the VoTPS7 products as germacrenes was supported by their degradation to elemenes via a characteristic Cope rearrangement in the GC-MS at high temperatures (28, 29). One of the minor compounds was identified as germacrene A based on identical retention time and mass spectral characteristics to a germacrene A standard provided by Robert M. Coates (University of Illinois) (30). Compound 1 isolated from root material was identified as germacrene C based on a combination of 1H,13C, 1H,1H gCOSY, 13C,1H HSQC, and 13C,1H HMBC spectral analyses (Fig. 5, Table 1). 1H NMR analyses revealed three olefinic protons, (δH 4.96, 5.26, 6.28), two singlet olefinic methyl groups (δH 1.20 and 1.61), and two geminal coupling methyl groups (δH 1.12 and 1.14). 1H,1H gCOSY analysis revealed four spin systems stretching from H-1 to H-2, H-4 to H-6, H-8 to H-9, H-11 to CH3-12 and CH3-13. 13C and 13C,1H HMBC correlations established the E-configuration of double bonds between C-2 and C-3 and between C-1 and C-10 (Fig. 5). Altogether, compound 1 isolate from plant tissue was identified as germacrene C, which was identical to compound 1 produced by yeast overexpressing the VoTPS7 gene based on GC retention time and the MS fragmentation pattern (supplemental Fig. S4). Minor amounts of germacrene A and lesser amounts of two other putative germacrenes, B and D, are also based on GC-MS assessments (30).
FIGURE 5.

Selected 1H,1H gCOSY and 13C,1H HMBC correlations for germacrene C (1) and valerena-1,10-diene (2).
TABLE 1.
1H and 13C NMR assignments of germacrene C (1) and valerena-1,10-diene (2)
Compound 1 was purified from root tissue, whereas compound 2 was purified from yeast cultures overexpressing the VoTPS1 gene.
| Position | Germacrene C (1)a |
Valerena 1,10 diene (2)b |
||
|---|---|---|---|---|
| δC (125 MHz) | δH (500 MHz) | δC (125 MHz) | δH (500 MHz) | |
| 1 | 123.6 | 6.28 (d, J = 9.2; 1H) | 136.2 | |
| 2 | 131.5 | 5.26 (d, J = 9.3; 1H) | 33.8 | 3.39 (dd, J = 9.7; 5.4; 1H) |
| 3a | 127.9 | 24.8 | 1.50 1.60 (m, overlap, 1H) | |
| 3b | 1.70 1.86 (m, overlap, 1H) | |||
| 4a | 41.4 | 2.22 (dt, J = 12.1, 3.3; 1H) | 28.9 | 1.30 1.40 (m, overlap, 1H) |
| 4b | 1.78 (td, J = 12.3, 5.2; 1H) | 1.70 1.80 (m, overlap, 1H) | ||
| 5a | 29.1 | 2.14 2.18 (m, overlap, 1H) | 33.8 | 1.93 (m, 1H) |
| 5b | 2.05 (ddd, J = 24.3, 12.4, 3.9; 1H) | |||
| 6 | 126.6 | 4.96 (br dd, J = 11.4, 4.6; 1H) | 47.6 | 2.91 (br s, 1H) |
| 7a | 142.3 | 26.8 | 1.30 1.40 (m, overlap, 1H) | |
| 7b | 1.30 1.40 (m, overlap, 1H) | |||
| 8a | 41.6 | 2.55 2.62 (m, 1H) | 37.7 | 2.16 2.25 (m, overlap, 1H) |
| 8b | 1.95 (td, J = 12.8, 4.8; 1H) | 2.16 2.25 (m, overlap, 1H) | ||
| 9a | 33.2 | 2.38 2.50 (m, overlap, 1H) | 128.6 | |
| 9b | 2.12 2.13 (m, overlap, 1H) | |||
| 10 | 145.3 | 126.4 | 5.46 (dd, J = 9.2; 1H) | |
| 11 | 38.4 | 2.38 2.50 (m, overlap, 1H) | 129.9 | |
| 12 | 23.6 | 1.14 (d, J = 3.5; 3H) | 26.3 | 1.68 (s, 3H) |
| 13 | 23.3 | 1.12 (d, J = 3.6; 3H) | 17.9 | 1.66 (s, 3H) |
| 14 | 17.8 | 1.61 (s, 3H) | 13.6 | 1.62 (s, 3H) |
| 15 | 21.8 | 1.20 (s, 3H) | 12.3 | 0.75 (d, J = 7; 3H) |
a Recorded on Bruker 500 MHz spectrometer in hexane d14, J in Hz.
b Recorded on a Varian J NMR 500 MHz spectrometer in CDCl3, J in Hz.
Compound 2 was produced by overexpression of VoTPS1 in a 6-liter yeast fermentation then extracted with acetone and hexane. Silica gel fractionation of the hexane extract afforded 6 mg of pure compound 2. Compound 2 was subjected to one- and two-dimensional NMR spectroscopic analyses, including 1H,1H gCOSY, 13C,1H HSQC, and 13C,1H HMBC analyses (Fig. 5, Table 1). A single olefinic proton (δH 5.46, d, J = 9 Hz) and three olefinic methyl singlets were evident from the 1H-NMR spectrum (δH 1.62, 1.66, 1.68). An additional aliphatic methyl group was found at δH 0.75 (δH 0.75, d, J = 7). The 1H,1H gCOSY revealed two spin systems, one stretching from H-15 to H-10 and another from H-6-to H-8. This indicated the possibility of a bicyclic terpenoid skeleton. The 13C NMR revealed three quarternary olefinic (δC 128.6, 129.9, 136.2) and one methine olefinic carbon (δC 126.4).
The 13C,1H HMBC spectrum illuminated the unusual carbon skeleton of compound 2. Correlations between the methyl groups at C-12 and C-13 and the H-10 olefinic proton revealed an unusual isobutenyl constellation attached at C-2. The position of the other olefinic methyl at 9-C could be established by a weak HMBC correlations between H-2 and C-9 and between the H-14 methyl protons and C-1 and C-9. Correlations between the H-15 methyl protons and C-4, C-5, and C-6 established the connectivity of the methyl group at C-5 (Fig. 5). Altogether, the one- and two-dimensional NMR data elucidated the structure of 2 as being valerena-1,10-diene. This assignment was further verified by comparison to published spectral data of valerena-1,10-diene from Paul et al. (11). Additionally, the one- and two-dimensional NMR data obtained for compound 2 (valerena-1,10-diene) isolated from V. officinalis root tissue, the VoTPS1 yeast-produced compound, and that prepared synthetically by Kitayama et al. (31) were identical, as were the GC retention time and MS fragmentation pattern (supplemental Figs. S4 and S6).
Elucidating a Catalytic Mechanism for Valerenadiene Synthase
The biogenic origin of valerenadiene and other related sesquiterpenes has remained enigmatic due to the conceptual difficulties in chemically rationalizing the generation of an isobutenyl side chain at C-2 of 2. The isobutenyl side chain could be hypothesized to simply stem from the unreacted “tail” end of FPP, but this view is complicated by the formation of the atypical 5,6-bicyclic ring system. Connolly et al. (10) addressed this mechanistic uncertainty in part by suggesting the possible involvement of a caryophyllene intermediate, and Paul et al. (11) proposed the possible involvement of a cyclopropyl intermediate. We have expanded on these possibilities by envisioning the formation of the isobutenyl side chain as arising from a ring opening of a caryophyllenyl carbocation intermediate followed by internal ring formation and migration of the isobutenyl substituent via either 1) a 1,2 isobutenyl migration or 2) through a cyclopropyl intermediate (Fig. 6).
FIGURE 6.
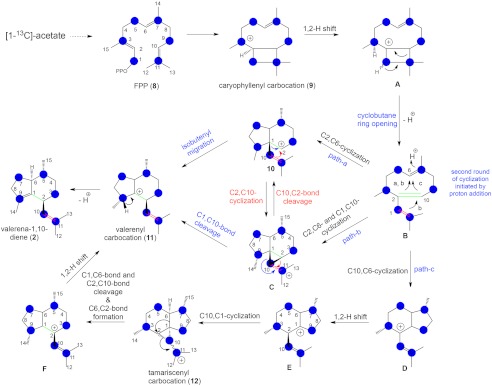
Proposed reaction mechanisms for the biosynthesis of valerena-1,10-diene catalyzed by VoTPS1 and the predicted labeling pattern that would arise from [1-13C]acetate being utilized by yeast overexpressing the VoTPS1 gene (blue spheres).
To test this model, we have taken advantage of the yeast heterologous expression system, noting that [1-13C]acetate would be preferentially incorporated into six positions of FPP as a result of the mevalonate biosynthetic pathway operating in yeast. It is well characterized that [1-13C]acetate can feed into the mevalonate pathway and become incorporated at positions 1 and 3 of the intermediate isopentenyl diphosphate (32). By following this biosynthetic logic, we envisage that feeding [1-13C]acetate to VoTPS1-expressing yeast would result in the predicted incorporation of 13C into the FPP substrate and the subsequent labeling of 2 at carbons 3, 5, 7, 9, 10, and 11 (Fig. 6). A 3-liter fermentation of yeast overexpressing VoTPS1 was grown in six 2-liter shake flasks to rapid growth phase before 500 mg of [1-13C]acetate was added to each flask. The yeast were grown for an additional 48 h, and the labeled valerena-1,10-diene hydrocarbon was purified from the culture. 6.5 mg of labeled 2 was isolated, and the measurement of its 13C NMR spectrum was compared with 6.5 mg of unlabeled 2 run under an identical set of spectroscopic conditions. Because control yeast (yeast that do not harbor any of the VoTPS genes) do not accumulate any sesquiterpenes (supplemental Fig. S5), parallel labeling and purification efforts with control yeast were not performed.
The 13C-labeled sample demonstrated enrichments of 3.2 to 8.0% for six carbons at positions C-3, C-5, C-7, C-9, C-10, and C-11 without any enrichment at other carbons as compared with the unlabeled control (Table 2). Most notably, the C-10- and C-11-labeled carbons evinced an additional J = 35 Hz coupling due to the 13C enrichment (supplemental Fig. S8). This suggests that the C-2 isobutenyl moiety of 2 does not arise “intact” from FPP but rather from some sort of caryophyllene rearrangement as proposed. This observation agrees with the predicted enrichment patterns envisaged in the hypothetical pathway.
TABLE 2.
13C NMR chemical shifts and relative percent enrichment of [1-13C]acetate incorporated into valerena 1,10 diene (2)
Enrichment calculations were determined by comparing peak heights of unlabeled authentic 2 with [1-13C]acetate incorporated into 2 run under identical spectrometric conditions. The ratio of labeled peak height to unlabeled peak height at each C was multiplied by 1.1 to express the relative percent enrichment above the natural abundance of 13C at that position according to Hirai et al. (23) and Takahashi and Tomita (24).
| Position | Valerena-1,10-diene (2) |
|
|---|---|---|
| δC(125 MHz) | Relative % enrichment | |
| 1 | 136.2 | 1.2 |
| 2 | 33.8 | 1.0 |
| 3 | 24.8 | 8.0 |
| 4 | 28.9 | 1.6 |
| 5 | 33.8 | 7.0 |
| 6 | 47.6 | 1.3 |
| 7 | 26.8 | 8.0 |
| 8 | 37.7 | 1.3 |
| 9 | 128.6 | 4.4 |
| 10 | 126.4 | 4.7 |
| 11 | 129.9 | 3.2 |
| 12 | 26.3 | 1.0 |
| 13 | 17.9 | 1.0 |
| 14 | 13.6 | 1.4 |
| 15 | 12.3 | 1.2 |
The apparent differential enrichment of ∼2.5-fold is not uncommon in such labeling experiments (33), and the same enrichment pattern (C-3, -5, and -7 showing greater enrichment than C-9, -10, and -11) was observed in an independent, replicated experiment. Although it is difficult to envision a biochemical mechanism to account for the differential incorporation of 13C at these positions from 1-13C acetate, a possible explanation relates to the physical environment of these labeled carbons themselves. C-3, -5, and -7 are hybridized to neighboring atoms via sp3 orbitals, whereas C-9, -10, and -11 are associated with sp2 orbitals. It is also not difficult to envision that the relaxation rates of these carbons in the different bond configurations could also be affected, and hence the relative intensity of their NMR signals could be dampened or enhanced accordingly.
DISCUSSION
Although the work presented here was initiated in large part to unravel the catalytic mechanism for valerenadiene, progress was greatly facilitated by the availability of transcriptomic and metabolomics datasets from the Medicinal Plant Consortium efforts (Medicinal Plant Metabolomic Resource, Medicinal Plant Genomics Resource). These resources allowed us to determine that at least one sesquiterpene hydrocarbon accumulated exclusively in root tissue, thus implicating roots as a primary site for sesquiterpene biosynthesis, a notion consistent with previous reports of diverse sesquiterpenoids found in association with roots of Valeriana and related plant species (11, 34, 35). Independent analysis of the hydrocarbon profile of leaves, stems, and roots (Fig. 4) confirmed and extended these observations. Three major sesquiterpenes hydrocarbons, including germacrene C and valerenadiene, and at least five minor sesquiterpene hydrocarbons accumulate specifically in root tissue. Screening of the V. officinalis transcriptome, assembled by combining the RNA-seq data generated from individual tissue types, revealed seven terpene synthase-like contigs that were all confirmed by independent RT-PCR experimentation. The contig pair VoTPS3/4 appears to represent a single gene highly expressed in all tissue types examined that encodes for a monoterpene synthase-like enzyme targeted to the chloroplast compartment. A second gene, VoTPS6, also exhibited a more constitutive expression profile based on the detection of sequence reads within the individual tissue-specific transcriptomes, albeit 10–20-fold lower than that for TPS3/4. Unfortunately, we were unable to functionally characterize the VoTPS6 enzyme because the heterologous expression conditions have yet to yield catalytically competent protein capable of utilizing GPP or FPP as a substrate. In contrast, VoTPS5 mRNA is present in all tissue types examined except young leaf and appears to encode for a sesquiterpene synthase activity based on a 5-fold greater activity with saturating amounts of FPP rather than GPP as substrate.
VoTPS1, -2, and -7 were of particular interest because the expression profiles of these mRNAs were 18–40-fold more specific to root tissue, the putative site of valerenadiene and valerenic acid biosynthesis, than to any other tissue. We have not been able to functional characterize the VoTPS2 enzyme activity because of insufficient expression of the corresponding cDNA in bacteria or yeast. In contrast, VoTPS7 was functionally characterized as a germacrene C synthase, which may be important for the biosynthesis and accumulation of valeranone (Fig. 1). Confirmation of such a suggestion, however, must await additional efforts to measure valeranone levels in roots of plants overexpressing or suppressing VoTPS7 gene expression.
Most importantly, we identified VoTPS1 as encoding for a functional valerana-1,10-diene synthase, as has Pyle et al. (13), which provided us with a means for probing the mechanistic features of this enigmatic reaction. The conversion of FPP to valerianadiene was recognized much earlier by Connolly et al. (10) and Paul et al. (11) as not likely occurring by mechanisms typically employed in the biosynthesis of many of the other classes of sesquiterpenes. More specifically, there is not an obvious mechanism for how the FPP molecule might be cyclized to yield the isobutenyl side chain. The precedence for bond formation between C10 or C11 (see FPP (4) numbering system, Fig. 6), which share a double bond, and the carbocation generated at C1 upon ionization of the diphosphate substituent group is well established (36). However, formation of a bond between the carbocation at C1 and C9 to yield an isobutenyl side chain has not been observed and is difficult, if not impossible, to envision chemically because C9 does not have a Pi orbital nor an electron available for formation of a new carbon-carbon bond. As an alternative, Pyle et al. (13) suggested that valerenadiene might arise from a macrocyclic germacene intermediate via a ring contraction mechanism. In this mechanism the initial germacrenyl ring arises when a bond between C10 and the carbocation at C1 forms, shifting the carbocation to the tertiary carbon of the isopropyl side chain. A 1,3 hydride shift to reposition the carbocation to C1 could then allow for a methylene migration of the bond between C9 and C10 to yield a new C9-C1 bond and formation of the isobutenyl side chain appended from C1. Besides considerable physical and energetic difficulties (i.e. hydride shift from the tertiary to secondary carbon), this mechanism is unlikely based on the [13C]acetate labeling experiments presented here. If the macrocyclic rearrangement occurred as proposed by Pyle et al. (13), C1 in the six-membered ring and the carbon bound to the methyl groups of the isobutenyl side chain should be enriched for 13C. Although the carbon bound to the methyl substituents was labeled, no label was incorporated into what would be C1 according the germacrene rearrangement model. Instead, the first and second carbons of the isobutenyl side chain were equally enriched with the 13C label. Based on the suggestion of Connolly et al. (10) for a possible involvement of a caryophyllene intermediate, a mechanism consistent with the observed labeling pattern has been developed (Fig. 6). In this mechanism C1 forms a bond with C11 of the originating FPP molecule in the formation of a caryophyllenyl carbocation followed by opening of the cyclobutyl ring to yield the desired isobutenyl side chain. Important to note, the two methylene carbons of the isobutenyl side are predicted to arise from C1 and C11 of the originating FPP and hence should become labeled when [1-13C]acetate is incorporated into FPP by the mevalonate biosynthetic pathway operating in yeast (32). This labeling pattern is indeed observed and consistent with the proposed caryophyllene reaction mechanism for valerena-1,10-diene synthase.
The proposed caryophyllenyl mechanism is also sufficient to account for the biosynthesis of pacifigorgiane sesquiterpenes (D) in sponges and tarmariscene sesquiterpenes (8) in liverworts (Fig. 6). The only difference is which carbons, C10 versus C2, participate in the formation of the fused, 5- and 6-membered ring system and the development of the cyclopropyl ring between carbons 1, 2, and 10 on the way to tamariscene formation. Confirmation of these predictions awaits the isolation and functional characterization of the corresponding liverwort and sponge terpene synthases, which should also provide new mechanistic insights into the evolution of these unique biochemical processes in very diverse and evolutionarily distant organisms.
This work was supported, in whole or in part, by National Institutes of Health Grant 1RC2GM092521 (to the Medicinal Plant Consortium).

This article contains supplemental Table S1 and Figs. S1—S8.
X. Zhuang and J. Chappell, unpublished data.
- VoTPS
- V. officinalis terpene synthase (TPS)
- GPP
- geranyl diphosphate
- HPS
- Hyoscyamus muticus premnaspirodiene synthase
- HSQC
- heteronuclear single quantum correlation
- HMBC
- heteronuclear multiple bond coherence
- FPP
- farnesyl diphosphate
- gCOSY
- gradient COSY.
REFERENCES
- 1. Bent S., Padula A., Moore D., Patterson M., Mehling W. (2006) Valerian for sleep. A systematic review and meta-analysis. Am. J. Med. 119, 1005–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Murphy K., Kubin Z. J., Shepherd J. N., Ettinger R. H. (2010) Valeriana officinalis root extracts have potent anxiolytic effects in laboratory rats. Phytomedicine 17, 674–678 [DOI] [PubMed] [Google Scholar]
- 3. Weeks B. S. (2009) Formulations of dietary supplements and herbal extracts for relaxation and anxiolytic action. Relarian (tm). Med. Sci. Monit. 15, RA256–RA262 [PubMed] [Google Scholar]
- 4. Houghton P. (1997) Valerian: The Genus Valeriana, Harwood Academic Publishers, Amsterdam, The Netherlands [Google Scholar]
- 5. Lefebvre T., Foster B. C., Drouin C. E., Krantis A., Livesey J. F., Jordan S. A. (2004) In vitro activity of commercial valerian root extracts against human cytochrome p450 3a4. J. Pharm. Pharm. Sci. 7, 265–273 [PubMed] [Google Scholar]
- 6. Grieve M. (1974) A Modern Herbal, pp. 824–830, Hafner Press, New York [Google Scholar]
- 7. Bos R., Woerdenbag H. J., Hendriks H., Zwaving J. H., DeSmet P. A. G. M., Tittel G., Wikstrom H. V., Scheffer J. J. C. (1996) Analytical aspects of phytotherapeutic valerian preparations. Phytochem. Anal. 7, 143–151 [Google Scholar]
- 8. Houghton P. J. (1999) The scientific basis for the reputed activity of valerian. J. Pharm. Pharmacol. 51, 505–512 [DOI] [PubMed] [Google Scholar]
- 9. Takemoto H., Yagura T., Ito M. (2009) Evaluation of volatile components from spikenard. Valerena-4,7(11)-diene is a highly active sedative compound. J. Nat. Med. 63, 380–385 [DOI] [PubMed] [Google Scholar]
- 10. Connolly J. D., Harrison L. J., Rycroft D. S. (1984) The structure of tamariscol, a new pacifigorgiane sesquiterpenoid alcohol from the liverwort Frullania tamarisci. Tetrahedron Lett. 25, 1401–1402 [Google Scholar]
- 11. Paul C., König W. A., Muhle H. (2001) Pacifigorgianes and tamariscene as constituents of Frullania tamarisci and Valeriana officinalis. Phytochemistry 57, 307–313 [DOI] [PubMed] [Google Scholar]
- 12. Izac R. R., Poet S. E., Fenical W., Vanengen D., Clardy J. (1982) The structure of pacifigorgiol, an ichthyotoxic sesquiterpenoid from the pacific gorgonian coral Pacifigorgia-cf-adamsii. Tetrahedron Lett. 23, 3743–3746 [Google Scholar]
- 13. Pyle B. W., Tran H. T., Pickel B., Haslam T. M., Gao Z., MacNevin G., Vederas J. C., Kim S. U., Ro D. K. (2012) Enzymatic synthesis of valerena-4,7(11)-diene by a unique sesquiterpene synthase from the valerian plant (Valeriana officinalis). FEBS J. 279, 3136–3146 [DOI] [PubMed] [Google Scholar]
- 14. Gu L., Jones A. D., Last R. L. (2010) Broad connections in the Arabidopsis seed metabolic network revealed by metabolite profiling of an amino acid catabolism mutant. Plant J. 61, 579–590 [DOI] [PubMed] [Google Scholar]
- 15. Góngora-Castillo E., Fedewa G., Yeo Y., Chappell J., DellaPenna D., Buell C. (2012) Genomic approaches for interrogating the biochemistry of medicinal plant species. Methods Enzymol. 517, 139–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997) Gapped blast and psi-blast. A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Suzek B. E., Huang H., McGarvey P., Mazumder R., Wu C. H. (2007) Uniref. Comprehensive and non-redundant uniprot reference clusters. Bioinformatics 23, 1282–1288 [DOI] [PubMed] [Google Scholar]
- 18. Punta M., Coggill P. C., Eberhardt R. Y., Mistry J., Tate J., Boursnell C., Pang N., Forslund K., Ceric G., Clements J., Heger A., Holm L., Sonnhammer E. L., Eddy S. R., Bateman A., Finn R. D. (2012) The pfam protein families database. Nucleic Acids Res. 40, D290–D301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Iseli C., Jongeneel C. V., Bucher P. (1999) Estscan. A program for detecting, evaluating, and reconstructing potential coding regions in est sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 138–148 [PubMed] [Google Scholar]
- 20. Trapnell C., Pachter L., Salzberg S. L. (2009) Tophat. Discovering splice junctions with rna-seq. Bioinformatics 25, 1105–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D. R., Pimentel H., Salzberg S. L., Rinn J. L., Pachter L. (2012) Differential gene and transcript expression analysis of rna-seq experiments with tophat and cufflinks. Nat. Protoc. 7, 562–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Takahashi S., Yeo Y., Greenhagen B. T., McMullin T., Song L., Maurina-Brunker J., Rosson R., Noel J. P., Chappell J. (2007) Metabolic engineering of sesquiterpene metabolism in yeast. Biotechnol. Bioeng. 97, 170–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hirai N., Yoshida R., Todoroki Y., Ohigashi H. (2000) Biosynthesis of abscisic acid by the non-mevalonate pathway in plants, and by the mevalonate pathway in fungi. Biosci. Biotechnol. Biochem. 64, 1448–1458 [DOI] [PubMed] [Google Scholar]
- 24. Takahashi K., Tomita F. (1983) Gilvocarcins, new antitumor antibiotics. 5. Biosynthesis of gilvocarcins. Incorporation of 13c-labeled compounds into gilvocarcin aglycones. J. Antibiot. 36, 1531–1535 [DOI] [PubMed] [Google Scholar]
- 25. Back K., Chappell J. (1995) Cloning and bacterial expression of a sesquiterpene cyclase from hyoscyamus-muticus and its molecular comparison to related terpene cyclases. J. Biol. Chem. 270, 7375–7381 [DOI] [PubMed] [Google Scholar]
- 26. Cao R., Zhang Y., Mann F. M., Huang C., Mukkamala D., Hudock M. P., Mead M. E., Prisic S., Wang K., Lin F.-Y., Chang T.-K., Peters R. J., Oldfield E. (2010) Diterpene cyclases and the nature of the isoprene fold. Proteins 78, 2417–2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chen F., Tholl D., Bohlmann J., Pichersky E. (2011) The family of terpene synthases in plants. A mid-size family of genes for specialized metabolism that is highly diversified throughout the kingdom. Plant J. 66, 212–229 [DOI] [PubMed] [Google Scholar]
- 28. Bowers W. S., Nishino C., Montgomery M. E., Nault L. R., Nielson M. W. (1977) Sesquiterpene progenitor, germacrene-a - alarm pheromone in aphids. Science 196, 680–681 [DOI] [PubMed] [Google Scholar]
- 29. Weinheimer A. J., Youngblood W. W., Washecheck P. H., Karns T. K. B., Ciereszko L. S. (1970) Isolation of the elusive (−)-germacrene A from the gorgonian eunicea-mammosa chemistry of coelenterates. Tetrahedron Lett. 7, 497–500 [Google Scholar]
- 30. Rising K. A., Starks C. M., Noel J. P., Chappell J. (2000) Demonstration of germacrene A as an intermediate in 5-epi-aristolochene synthase catalysis. J. Am. Chem. Soc. 122, 6526–6526 [Google Scholar]
- 31. Kitayama T., Kawabata G., Ito M. (2010) Concise synthesis of valerena-4,7(11)-diene, a highly active sedative, from valerenic acid. Biosci. Biotechnol. Biochem. 74, 1963–1964 [DOI] [PubMed] [Google Scholar]
- 32. Disch A., Rohmer M. (1998) On the absence of the glyceraldehyde 3-phosphate pyruvate pathway for isoprenoid biosynthesis in fungi and yeasts. FEMS Microbiol. Lett. 168, 201–208 [DOI] [PubMed] [Google Scholar]
- 33. Wu S., Schalk M., Clark A., Miles R. B., Coates R., Chappell J. (2006) Redirection of cytosolic or plastidic isoprenoid precursors elevates terpene production in plants. Nat. Biotechnol. 24, 1441–1447 [DOI] [PubMed] [Google Scholar]
- 34. Bos R., Hendriks H., Kloosterman J., Sipma G. (1986) Isolation of the sesquiterpene alcohol (−)-pacifigorgiol from Valeriana officinalis. Phytochemistry 25, 1234–1235 [Google Scholar]
- 35. Tanaka K., Komatsu K. (2008) Comparative study on volatile components of nardostachys rhizome. J. Nat. Med. 62, 112–116 [DOI] [PubMed] [Google Scholar]
- 36. Chappell J., Coates R. M. (2010). in Comprehensive Natural Products II-Chemistry and Biology (Ebizuka Y. ed.) pp. 609–641, Elsevier Ltd., Oxford, England [Google Scholar]