

Background: Many GPCRs that utilize β-arrestins differ with respect to downstream signaling and cellular consequences.
Results: Exchanging the C termini of two GPCRs switches the β-arrestin responses and relative affinities for the two receptors.
Conclusion: Sequences within the C termini of different GPCRs are important for determining the nature of β-arrestin recruitment and signaling.
Significance: These studies provide new insight regarding receptor-specific β-arrestin signals.
Keywords: Seven-helix Receptor, Arrestin, Cell Signaling, G-protein-coupled Receptors (GPCRs), MAP Kinases (MAPKs), Protease-activated Receptor
Abstract
β-Arrestins are multifunctional adaptor proteins that, upon recruitment to an activated G-protein-coupled receptor, can promote desensitization of G-protein signaling and receptor internalization while simultaneously eliciting an independent signal. The result of β-arrestin signaling depends upon the activating receptor. For example, activation of two Gαq-coupled receptors, protease-activated receptor-2 (PAR2) and neurokinin-1 receptor (NK1R), results in drastically different signaling events. PAR2 promotes β-arrestin-dependent membrane-sequestered extracellular signal-regulated kinase (ERK1/2) activation, cofilin activation, and cell migration, whereas NK1R promotes nuclear ERK1/2 activation and proliferation. Using bioluminescence resonance energy transfer to monitor receptor/β-arrestin interactions in real time, we observe that PAR2 has a higher apparent affinity for both β-arrestins than does NK1R, recruits them at a faster rate, and exhibits more rapid desensitization of the G-protein signal. Furthermore, recruitment of β-arrestins to PAR2 does not require prior Gαq signaling events, whereas inhibition of Gαq signaling intermediates inhibits recruitment of β-arrestins to NK1R. Using chimeric receptors in which the C terminus of PAR2 is fused to the N terminus of NK1R and vice versa and a critical Ser/Thr mutant of PAR2, we demonstrate that interactions between β-arrestins and specific phosphoresidues in the C termini of each receptor are crucial for determining the rate and magnitude of β-arrestin recruitment as well as the ultimate signaling outcome.
Introduction
β-Arrestins are pleiotropic adaptor proteins that can terminate heterotrimeric G-protein signaling, promote clathrin-mediated endocytosis, and bind numerous signaling proteins to enhance or inhibit their activities, downstream of many seven-transmembrane receptors. Interestingly, the downstream effects of β-arrestin recruitment vary significantly, depending on the receptor to which they were recruited. The molecular determinants of these differences remain poorly understood.
Protease-activated receptor-2 (PAR2)3 and neurokinin-1 receptor (NK1R) are both Gαq-coupled receptors that promote β-arrestin-dependent signal termination and activation of extracellular signal-regulated kinases 1 and 2 (ERK1/2) (1–5). The two receptors differ both in their mechanism of activation of ERK1/2 and the downstream consequences of ERK1/2 activity. PAR2 is activated by serine protease-mediated cleavage of its N terminus, which reveals a tethered ligand that then binds and activates the receptor. Peptides corresponding to the tethered ligand sequence (SLI(G/K)(V/L)NH2) and peptidomimetics, such as 2-furoyl-LIGRLNH2 (2fAP) can be used to specifically activate PAR2 in the absence of proteolytic cleavage (6–8). NK1R is activated by the neurotransmitter Substance P (SP), and the synthetically modified agonist [Sar-9,Met(O2)-11]SP (Sar-Met-SP) can specifically bind NK1R without activating NK2 and NK3 receptors. Both PAR2 and NK1R can activate Gαq, leading to Ca2+ mobilization and activation of protein kinase C (PKC) as well as recruitment of β-arrestin-1/2 (1–3, 9–11). However, although a large portion of PAR2 remains associated with β-arrestins and is degraded in lysosomes, NK1R dissociates from β-arrestins and recycles back to the cell surface (3, 9, 10). β-Arrestin-dependent ERK1/2 activation downstream of PAR2 results in sequestration of ERK1/2 at the membrane, leading to cell migration, whereas downstream of NK1R, ERK1/2 translocates to the nucleus to promote proliferation. Furthermore, despite the common involvement of Gαq and β-arrestins, the mechanisms by which the two receptors activate ERK1/2 are distinctly different. PAR2 can promote ERK1/2 activation by independent G-protein- and β-arrestin-dependent mechanisms, the former requiring Src and Ras and the latter requiring neither (1–3, 12). In contrast, NK1R requires input from both G-protein- and β-arrestin-dependent pathways for Src and Ras-dependent activation of ERK1/2, suggesting that PAR2 might recruit β-arrestins independent of G-protein coupling, whereas NK1R may rely on G-protein-mediated events for β-arrestin recruitment (1–3, 5, 13, 14).
The classic paradigm for β-arrestin recruitment to GPCRs suggests that β-arrestins are recruited after phosphorylation of the receptor C termini by second messenger kinases or G-protein-coupled receptor kinases (GRKs) (15–17). Studies have shown that broad-spectrum PKC inhibitors can block PAR2 desensitization, and both PKC and GRKs have been reported to play a role in NK1R down-regulation (2, 9, 18, 19). We hypothesized that the differences in ERK1/2 activation by the two receptors and the subsequent downstream events might be dependent upon interactions between β-arrestin-1/2 and the receptor C termini, which might in turn be dependent upon the phosphorylation state of the receptors. To address this possibility, we switched the C-tails of the two receptors and examined Ca2+ mobilization, β-arrestin recruitment kinetics, ERK1/2 activation and localization, and two downstream signaling events: proliferation and cell migration.
EXPERIMENTAL PROCEDURES
Materials
All chemicals were from Sigma unless otherwise stated. The following primary antibodies were used for Western blotting, immunostaining, or in-cell Western assays: mouse monoclonal antibodies to FLAG M2 (Sigma, 1:100 for in-cell Western, 1:250 for IF); rabbit anti-phosphocofilin (Cell Signaling, 1:1000 for WB); mouse anti-total cofilin (BD Biosciences, 1:1000 for WB); rabbit anti-phospho-ERK1/2 and mouse anti-total ERK1/2 (Cell Signaling, 1:1000 for WB and in-cell Western, 1:250 for IF); mouse monoclonal antibody to EEA-1 (BD Biosciences, 1:250 for IF); rabbit anti-LAMP1 (Santa Cruz Biotechnology, Inc. (Santa Cruz, CA), 1:250 for IF). Alexa546-tagged secondary antibodies to mouse and rabbit were obtained from Invitrogen (1:500 for IF). IRDye®680- and IRDye®800-tagged secondary antibodies (1:45,000 for WB) were from Rockland. 2-Furoyl-LIGRLO-NH2, Sar-Met-SP, U73122, and BAPTA-AM were purchased from Tocris. FLAG-tagged PAR2WT and PAR2S363A/T366A in the pBJ1 vector, Renilla luciferase-tagged β-arrestin-1 and -2 constructs, and FLAG-tagged β-arrestin-1 and -2 constructs were obtained as gifts from Dr. JoAnn Trejo (University of California San Diego, La Jolla, CA), Dr. Michel Bouvier (University of Montreal), and Dr. Robert Lefkowitz (Duke University Medical Center), respectively. FLAG-tagged PAR2WT and PAR2S363A/T366A were subcloned from the pBJ1 vector into the p-eYFP-N1 vector using HindIII and BamHI. Human PAR2 and NK1R, cloned into eYFP-N1 or eGFP-N1, at BamHI and HindIII sites have been described previously (3, 10, 20). The C termini of PAR2 (nucleotide 1081 to the end) and NK1R (nucleotide 1227 to the end) were amplified by PCR with primers containing N-terminal EcoRI and C-terminal BamHI sites, digested, and ligated into eGFP-N1 to generate NK1RC-GFP and PAR2C-GFP. The N termini of PAR2 (nucleotides 1–1080) and NK1R (nucleotides 1–1226) were amplified by PCR with N-terminal HindIII and C-terminal EcoRI sites, digested, and ligated into EcoRI/HindIII-digested NK1RC-GFP and PAR2C-GFP, respectively. Sequences and reading frames across the fusion junction were confirmed by ABI sequencing.
Cell Culture and Transfection
Human embryonic kidney 293 (HEK293), mouse embryonic fibroblasts (MEFs) from wild type and β-arrestin-1/2 double knock-out mice (from Robert Lefkowitz), and Chinese hamster ovary (CHO) cell lines were maintained as described previously (3). All plasmids were stably transfected in CHO cells using Lipofectamine and Plus reagent (Invitrogen) following the manufacturer's protocols. Stable CHO cell lines were selected by flow cytometry and were not single cell clonal cell lines. Transient transfections were carried out using FuGene (Roche Applied Science) following the manufacturer's protocols.
Single Cell Calcium Response Measurements
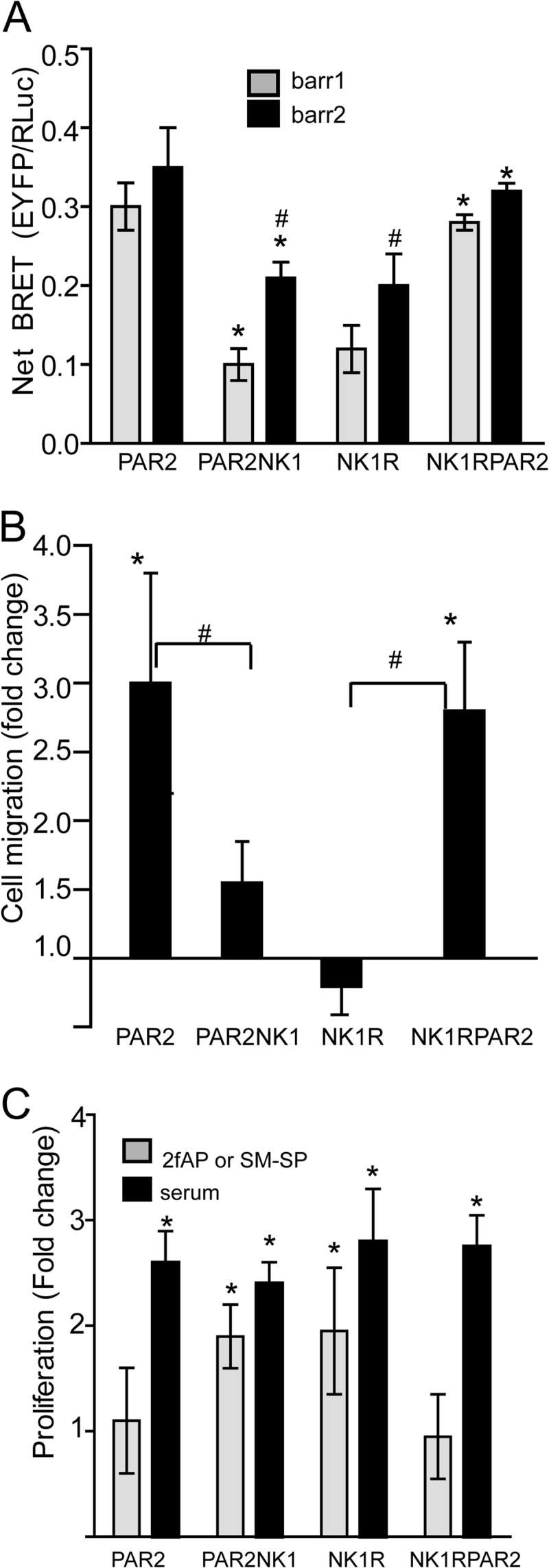
Cells were preloaded with Fura-2, washed, and mounted in a perfusion chamber on the stage of a Nikon TE300 microscope, and Ca2+ mobilization was determined as described previously (3). Briefly, either 2fAP or Sar-Met-SP was added to the bath, and fluorescence was detected in individual cells using a Nikon video camera and a video microscopy program (Metafluor). Fluorescence was quantified at 340- and 380-nm excitation and 510-nm emission. The ratio of the fluorescence at the two excitation wavelengths, which is proportional to the [Ca2+], was determined. To determine maximum Ca2+ levels, readings were followed by the addition of the calcium ionophore ionomycin. For determination of the amount of calcium response (or the duration of signaling), areas under the calcium response curves were determined using Kaleidagraph (version 4.0), and total [Ca2+] released was calculated using the Grynkiewitz equation,
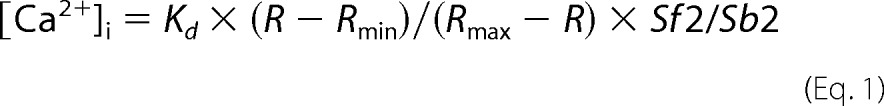 |
where Kd (for Ca2+ binding to Fura-2/AM) = 220 nm, R = A340/A380 at each time point, Rmin = A340/A380 under Ca2+-free conditions, Rmax = A340/A380 at saturating conditions (with the addition of ionomycin), Sf2 represents the base-line fluorescence at 340 nm without Fura, and Sb2 represents the base-line fluorescence at 340 nm with Fura.
Bioluminescence Resonance Energy Transfer (BRET)
YFP-tagged receptor constructs were transiently co-expressed with either β-arrestin-1-luciferase or β-arrestin-2-luciferase in HEK293 cells. 48 h post-transfection, the cells were treated with appropriate concentrations of 2fAP or Sar-Met-SP and 5 μm coelenterazine. For dose curves, coelenterazine was added, and readings were taken 15 min after agonist stimulation. For kinetic measurements, agonist and coelenterazine were added simultaneously, and readings were initiated at that time. Pharmacological inhibitors were added at appropriate concentrations, and the cells were incubated at 37 °C 10 min prior to the addition of 2fAP. Light emission was detected (460–500 nm for RLuc and 510–550 nm for YFP) using a TRISTAR LB941 multilabel plate reader from Berthold Technologies. BRET signal was calculated as the ratio of the light emitted by eYFP and the light emitted by luciferase. For a negative control, cells transfected with the β-arrestin-luciferase construct alone were used to determine the background. The ratio observed in β-arrestin luciferase-only-transfected cells was subtracted from that observed in the presence of YFP-tagged receptors to give the net BRET values. Half-lives (t½) of the kinetics reactions were determined from five separate experiments. For titration curves, β-arrestin was held constant and co-transfected with increasing amounts of YFP-tagged receptor; BRET readings were taken for each acceptor/donor ratio as described for dose curves but using a constant concentration of 1 μm 2fAP. Protein expression levels were confirmed by luciferase emission and YFP fluorescence, respectively.
In-cell Western Assays
For pERK assays, cells were seeded into 96-well plates, serum-starved overnight, and then stimulated with 2fAP or Sar-Met-SP for 0–60 min, after which they were fixed, blocked, and incubated with anti-rabbit pERK and anti-mouse total ERK1/2 overnight at 4 °C. For cell surface receptor detection, HEK293 cells were transiently transfected with empty vector, FLAG-tagged PAR2WT, or PAR2S363A/T366A, transferred to 24-well plates, fixed, blocked, and incubated with mouse anti-FLAG monoclonal antibody. Following incubation with IRDye680 and IRDye800-conjugated secondary antibodies, the plates were scanned using the LI-COR Odyssey imaging system, and the integrated intensities of the wells were quantified using the LI-COR software.
Immunofluorescence and Confocal Microscopy
For the receptor internalization assays, 3 × 104 CHO cells stably expressing GFP-tagged receptors were cultured overnight on collagen-coated coverslips. When required, the cells were transiently transfected with DsRed-ERK1/2 or FLAG-tagged β-arrestin-1 or -2. 36 h post-transfection, medium was changed to serum-free DMEM, and the cells were treated with appropriate agonists (i.e. 1 μm 2fAP or 100 nm Sar-Met-SP) for 0–120 min, fixed, and blocked, and immunostaining was carried out as described previously (3).
Cell Migration Assay
CHO cells stably expressing GFP-tagged PAR2, NK1R, PAR2-NK1R, NK1R-PAR2, and HEK293 cells transiently transfected with FLAG-tagged PAR2WT or PAR2S363A/T366A were used in these assays. 3 × 104 cells were seeded onto collagen-coated transwell supports (8-μm pore size) and allowed to attach for 2 h at 37 °C and then treated with 10 μm 2fAP or 100 nm Sar-Met-SP, added to the lower chambers, for 4 h. Non-migratory cells were removed with a cotton swab, filters were stained with crystal violet, and the total number of cells that migrated to the bottom was quantified counting in four fields of vision under the ×20 objective of a Nikon phase-contrast microscope. Alternatively, cells were grown to confluence in 35-mm dishes and serum-starved overnight, and a wound was generated by scratching across a monolayer with a 10-μl pipette tip. After treatment with or without 1 μm 2fAP for 6–24 h, cell migration into the wound area was monitored using the ×4 objective of a Nikon Eclipse TE2000U microscope. The wound area was quantified using ImageJ software (wound area at time 0 minus the wound area at each time point). The migration index was calculated as a -fold change in area covered by cells at the 0 min time point over that at the 24 h time point.
Proliferation Assay
104 CHO cells expressing GFP-tagged PAR2, NK1R, PAR2-NK1R, and NK1R-PAR2 were seeded onto 35-mm dishes, attached for 2 h, and serum-starved overnight and then treated with 2fAP, Sar-Met-SP, or serum (positive control) for 48 h. Cells were detached, stained with propidium iodide, and resuspended in 1 ml of flow cytometry buffer. Cells per ml were determined using a Beckman flow cytometer. Cell number was determined by multiplying the number of propidium iodide-excluding GFP-positive cells times the total volume. Untransfected cells were used to determine the gating strategy for identifying GFP-positive cells.
RESULTS
The C Termini of PAR2 and NK1R Determine β-Arrestin-dependent Desensitization and Signaling Patterns
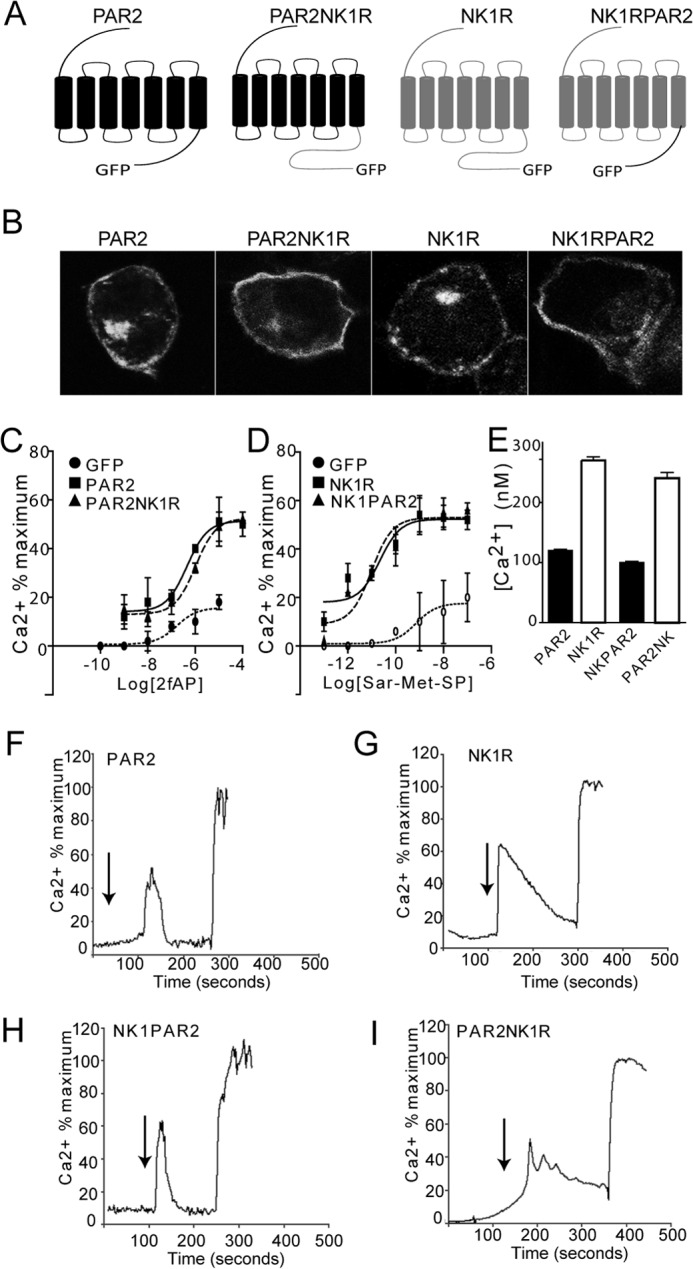
To determine the importance of the receptor C termini in directing specific β-arrestin-dependent signaling events, we constructed chimeric receptors, in which either the cytoplasmic C terminus of PAR2 was fused to the N terminus of NK1R at the seventh transmembrane domain (NK1R-PAR2) or the C terminus of NK1R was fused to the N terminus of PAR2 (PAR2-NK1R) (Fig. 1A). They were expressed in CHO cells, and agonist-induced Ca2+ mobilization was determined using Fura-2/AM (PAR2 and PAR2-NK1R received 2fAP, and NK1R and NK1R-PAR2 received Sar-Met-SP). Mobilization of Ca2+ from intracellular stores is an early signaling event downstream of Gαq-coupled receptors, such as PAR2 and NK1R, and is rapidly terminated upon β-arrestin recruitment. Thus, Ca2+ mobilization is often used as a read-out for Gαq-coupled receptor activation and for examining defects in desensitization (1, 3, 15, 21). The chimeric receptors, like their wild type C-terminal parent receptors, are expressed at the cell surface (Fig. 1B), colocalize with β-arrestins upon activation (supplemental Fig. S1), and are able to mobilize calcium in response to agonist in a dose-dependent fashion, demonstrating that all four receptors are functional (Fig. 1, C and D). No significant differences in the dose responses of chimeric receptors and the corresponding parent receptors were observed (Fig. 1, C and D). As observed previously, the duration of the NK1R-induced Ca2+ signal was longer than that following PAR2 activation, resulting in a 2.2-fold increase in the total concentration of intracellular Ca2+ (Fig. 1E). The chimeric receptors display Ca2+ mobilization patterns similar to their C-terminal parent (i.e. PAR2-NK1R (Fig. 1I) displays a prolonged 2fAP-induced Ca2+ response, and NK1-PAR2 (Fig. 1H) displays a transient Sar-Met-SP-induced response). All four receptors also demonstrated agonist-induced internalization (Fig. 2). As reported previously, PAR2 internalized rapidly, as did NK1R-PAR2, colocalizing with EEA1 after 5 min (supplemental Fig. S2, A and C) and with lysosomal marker LAMP1 after 1 h of 2fAP treatment (supplemental Fig. S3). NK1R and PAR2-NK1R colocalized with EEA1 at 15 min (supplemental Fig. S2, B and D). Unlike PAR2 and NK1R-PAR2, NK1R and PAR2-NK1R colocalized with EEA1 at 15 and 30 min of agonist treatment, showing very little colocalization with LAMP1 (supplemental Figs. S2 (B and D) and S3). Thus, the C terminus of the two receptors is also important for determining the pattern of internalization.
FIGURE 1.
Mobilization of intracellular Ca2+ by PAR2, NK1R, and C-terminal chimeras. A, schematic representation of PAR2, NK1R, PAR2-NK1R, and NK1R-PAR2 generated by cloning the C-terminal tail of PAR2 to the seventh transmembrane domain of NK1R and vice versa. B, confocal micrographs showing cell surface expression of GFP-tagged PAR2, PAR2-NK1R, NK1R, and NK1R-PAR2 in stably transfected CHO cells. C and D, Ca2+ mobilization (expressed as a percentage of ionomycin-induced signal) in response to activation of PAR2 and PAR2-NK1R with 1 μm 2fAP (C) or activation of NK1R or NK1R-PAR2 with 100 nm Sar-Met-SP (D), n = 4. Cells transfected with GFP alone are included in each as a negative control. E, average total cytosolic Ca2+ mobilized by activation of each receptor, calculated from the area under the curves shown in F–I. PAR2- and NK1R-PAR2-induced Ca2+ mobilization was significantly different from that induced by NK1R and PAR2-NK1R, as determined by Tukey t tests (p = 0.01, n = 4). F–I, representative traces of Ca2+ mobilization responses to activation of PAR2 (F), NK1R (G), NK1R-PAR2 (H), or PAR2-NK1R (I). Error bars, S.E.
FIGURE 2.
Agonist-induced internalization of PAR2, NK1R, and C-terminal chimeras. CHO cells expressing GFP-tagged PAR2, PAR2-NK1R, NK1R-PAR2, or PAR2-NK1R were treated with 1 μm 2fAP or 100 nm Sar-Met-SP on ice and then incubated at 37 °C for 0–120 min, fixed, and observed by confocal microscopy. Scale bar, 10 μm. Images are representative of three independent experiments.
Both PAR2 and NK1R activate ERK1/2 through G-protein- and β-arrestin-dependent pathways, but although activated ERK1/2 downstream of PAR2 is primarily found at the membrane and in the cytoplasm, downstream of NK1R, activated ERK1/2 is primarily nuclear (1–3, 5). Western blot analysis with anti-pERK shows that the chimeric receptors were also able activate ERK1/2 in response to the appropriate agonist (Fig. 3, A–E). To examine the subcellular localization of ERK1/2, cells expressing each of the four GFP-tagged receptors were treated with or without 2fAP or Sar-Met-SP, stained for phospho-ERK (Fig. 3F) or total ERK1/2 (supplemental Fig. S4A), and examined by confocal microscopy. Additionally, cells expressing each receptor were co-transfected with DsRed-ERK2, treated with 2fAP or Sar-Met-SP, and examined by confocal microscopy (supplemental Fig. S4B). As previously reported, in cells expressing PAR2, phospho-ERK1/2 was primarily observed in the cytoplasm and near the plasma membrane after activation, whereas in cells expressing NK1R, most of the phospho-ERK1/2 translocated into the nucleus (Fig. 3F). After treatment with Sar-Met-SP, activated ERK1/2 distribution in cells expressing NK1R-PAR2 was predominantly cytoplasmic and near the plasma membrane (Fig. 3F), whereas 2fAP treatment of cells expressing PAR2-NK1R resulted in nuclear translocation of ERK1/2 (Fig. 3F). Total ERK1/2 localization showed a similar pattern of localization (supplemental Fig. S4).
FIGURE 3.

Subcellular localization of activated ERK1/2 is determined by the receptor C terminus. A–D, cells expressing wild type or chimeric receptors were treated with the indicated amount of 2fAP or Sar-Met-SP, and cell lysates were analyzed by SDS-PAGE followed by Western blotting for phosphorylated and total ERK1/2. A, representative Western blots of PAR2 (A), NK1R (B), PAR2-NK1R (C), and NK1R-PAR2 (D) are shown. E, graph of mean ± S.E. (error bars) phospho-ERK (normalized to total ERK), plotted as -fold increase over base line (n = 3). F, cells expressing each receptor and treated as described for A–D were stained for pERK, and images were taken by confocal microscopy. TOPRO3 was used to identify nuclei. Images from 0 and 60 min of 2fAP are shown. The arrows indicate cytoplasmic/membrane staining of pERK. Scale bar, 10 μm.
The mechanism of β-arrestin-dependent ERK1/2 activation is also different between the two receptors (Fig. 4A). It was previously shown that downstream of PAR2, the G-protein- and β-arrestin-dependent ERK1/2 activation pathways work independently of each other. Furthermore, although G-protein activation and Ca2+ mobilization lead to Ras- and Src-dependent ERK1/2 activation, β-arrestin-dependent ERK1/2 activation is independent of Src and Ras (2, 5, 12). In contrast, both G-protein- and β-arrestin-dependent NK1R activation are Ras-dependent, suggesting that β-arrestin and Ca2+-signaling pathways may be cooperative rather than independent (1, 2). We first confirmed these mechanistic differences in ERK1/2 activation by examining sensitivity to various inhibitors of Gαq signaling as well as β-arrestin deletion. As previously reported, activation of ERK1/2 downstream of PAR2 and NK1R was decreased by 70 and 95%, respectively, in embryonic fibroblasts from β-arrestin-1/2−/− mice (MEFβarrDKO) compared with WT cells (MEFwt) (Fig. 4, B and C). In MEFwt, PAR2-induced ERK1/2 activation was decreased by only 30% after the addition of either the Ca2+-chelating agent BAPTA-AM or the Src inhibitor SU6566, suggesting that the G-protein/Ca2+-independent, β-arrestin-dependent pathway is the major mechanism for ERK1/2 activation in MEFs, and the G-protein/Ca2+ pathway plays a more minor role (Fig. 4D). In MEFβarrDKO, the remaining ERK1/2 activation downstream of PAR2 was abolished by BAPTA-AM and SU6566, suggesting that the β-arrestin-independent ERK1/2 activation pathway is dependent upon the classical G-protein signaling pathway (Fig. 4E). In contrast, activation of ERK1/2 by NK1R was inhibited by greater than 95% by both BAPTA-AM and SU6566 (Fig. 4F), confirming that ERK1/2 activation downstream of NK1R requires cooperation of β-arrestin-dependent and G-protein-dependent pathways. In MEFβarrDKO, because there was no statistically significant ERK1/2 activation, no further effect of BAPTA-AM or SU6566 was observed (Fig. 4G). The chimeric receptors behaved similarly to their C-terminal parent receptors. Activation of ERK1/2 was reduced by 70% in MEFβarrDKO expressing NK1R-PAR2 (similar to what was observed with WT PAR2), and pretreatment with BAPTA-AM or SU6566 abolished the remaining ERK1/2 activation in MEFβarrDKO (Fig. 4, H and I). Pretreatment with BAPTA-AM (Fig. 4H) or SU6566 (Fig. 4I) reduced ERK1/2 activation by 30% in MEFwt expressing NK1R-PAR2. As with NK1R, BAPTA-AM abolished ERK1/2 activation in both MEFwt and MEFβarrDKO expressing PAR2-NK1R. Pretreatment with BAPTA-AM or SU6566 abolished ERK1/2 activation by PAR2-NK1R in MEFwt, and no ERK1/2 activation was observed in MEFβarrDKO expressing PAR2-NK1R (Fig. 4, H and I).
FIGURE 4.

PAR2 and NK1R activate ERK1/2 by distinct β-arrestin-dependent mechanisms. A, schematic depicting signaling to ERK1/2 by PAR2 and NK1R. B and C, mouse embryonic fibroblasts from wild type (MEFwt) or β-arrestin-1/2 knock-out mice (MEFβarrDKO) were transfected with either PAR2 (B) or NK1R (C). Cells were treated with 2fAP or Sar-Met-SP, and ERK1/2 activation was determined by in-cell Western, using pERK and total ERK antibodies. Normalized ERK1/2 activation is graphed as -fold increase over base line (pERK/total ERK in untreated cells). *, statistically significant increase in ERK1/2 phosphorylation compared with base line (p < 0.01, n = 5). ERK1/2 phosphorylation was significantly lower in MEFβarrDKO at all time points (p = 0.001, n = 5). D–G, in-cell Western analysis of ERK1/2 activation in MEFwt (D and F) or MEFβarrDKO (E and G) transfected with either PAR2 (D and E) or NK1R (F and G) after pretreatment with either vehicle (DMSO), BAPTA-AM (to block Ca2+), or SU6566 (Src inhibitor). ERK1/2 phosphorylation was significantly reduced in SU6566- and BAPTA-AM-treated cells compared with vehicle-treated cells at all time points in NK1R-transfected cells and at 5–30 min in PAR2-transfected cells (p < 0.05, n = 5). H, graph of mean maximal agonist-stimulated ERK1/2 activation after pretreatment with vehicle or BAPTA-AM in MEFwt or MEFβarrDKO expressing PAR2, NK1R, PAR2-NK1R, or NK1R-PAR2. I, graph of mean maximal agonist-stimulated ERK1/2 activation in MEFwt, transfected with respective receptor, after pretreatment with SU6566. #, statistically significant difference between vehicle and SU6566- or BAPTA-AM-treated cells. *, statistically significant difference between METwt and MEFβarrDKO (p < 0.05, n = 5). Error bars, S.E.
PAR2 and NK1R Recruit β-Arrestins with Different Apparent Affinity and Kinetics
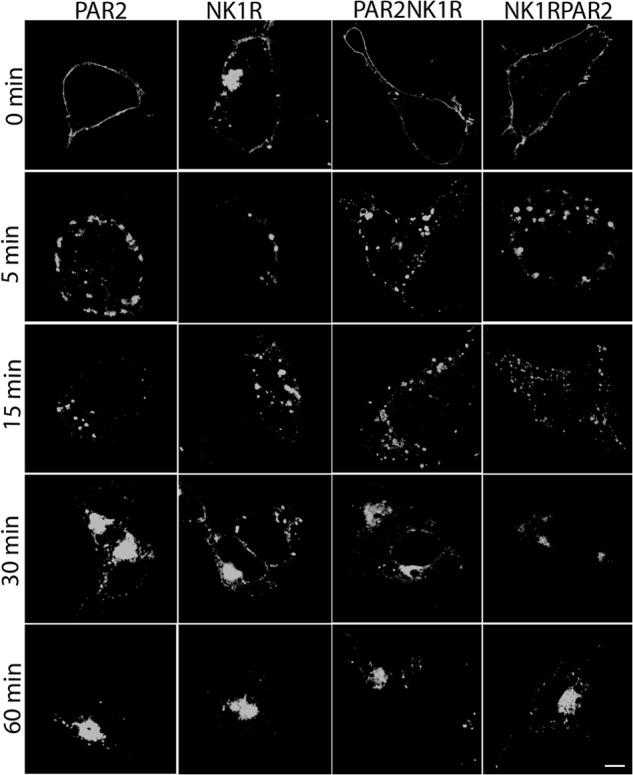
The data described thus far suggest that the C termini of the two receptors determine the mechanism of ERK1/2 activation and subsequent subcellular localization. Although both receptors have been shown to interact with β-arrestins by multiple groups (1, 2, 4, 10, 22, 23), the differences in downstream signaling events suggest that they might direct distinct patterns of β-arrestin recruitment. To examine the more subtle differences in recruitment of β-arrestin-1 and -2 to PAR2 and NK1R, BRET assays were employed. First, agonist dose responses of β-arrestin recruitment to each receptor at 15 min were determined, which revealed no significant differences in the agonist dose required to recruit β-arrestin to either PAR2 or NK1R (Fig. 5, A and B). We then examined the kinetics of recruitment by examining BRET in response to a single dose of agonist over 20 min. Agonist stimulation of PAR2 led to rapid recruitment of both β-arrestin-1 and -2 at equivalent rates (Fig. 5, C–E). In contrast, recruitment of both β-arrestins to NK1R was 2-fold slower than for PAR2 (Fig. 5, C–E), and recruitment of β-arrestin-1 to NK1R was significantly slower than recruitment of β-arrestin-2. Another observation apparent in both the dose response and kinetic assays was that net BRET values for β-arrestin-1 and -2 recruitment to PAR2 were higher than those for NK1R, which could reflect a higher affinity of both β-arrestins for PAR2 or a conformational difference in the receptor/β-arrestin conformation that results in a greater distance between the luciferase and YFP tags. To distinguish between these possibilities, we monitored net BRET as a function of the acceptor/donor ratio (receptor-YFP/β-arrestin-luciferase) and determined the acceptor-donor ratio at which half-maximal BRET (BRET50) is observed. These studies revealed several key differences between the two receptors. First, BRET50 values for NK1R/β-arrestin interactions were higher than those for PAR2/β-arrestin interactions (nearly 2-fold for β-arrestin-2 and 2.7-fold for β-arrestin-1), suggesting that PAR2 has a higher relative affinity for both β-arrestins than NK1R. Second, although no significant difference in BRET50 was observed for interactions between either β-arrestin-1 or -2 and PAR2, the BRET50 value was significantly higher for β-arrestin-1/NK1R than for β-arrestin-2/NK1R, suggesting that NK1R preferentially binds β-arrestin-2 over β-arrestin-1 (Fig. 5F). Consistent with our data demonstrating a requirement for the C terminus in a number of β-arrestin-dependent events, the chimeric receptors displayed β-arrestin recruitment rates similar to their C-terminal parent (i.e. PAR2-NK1R preferentially recruited β-arrestin-2, whereas NK1R-PAR2 recruited both equally) (Fig. 6A).
FIGURE 5.

β-arrestin is recruited to PAR2 and NK1R with different kinetics and binding affinities. HEK293 cells were transiently co-transfected with PAR2-YFP or NK1R-YFP and β-arrestin-1- or β-arrestin-2-Rluc. A and B, mean maximal net BRET in response to increasing concentrations of 2fAP or Sar-Met-SP (net BRET = eYFP/Rluc observed with receptor minus eYFP/Rluc observed with β-arrestin-Rluc alone), n = 6. C and D, net BRET values were monitored over a period of 20 min after the agonist addition. Traces represent mean values from five independent experiments. E, mean ± S.E. (error bars) half-lives of β-arrestin-1/2 recruitment to PAR2 or NK1R (n = 5). *, significant difference between NK1R and PAR2, p < 0.05, n = 5. F, cells were transfected with a constant amount of β-arrestin-1 or β-arrestin-2-luc (donors) and increasing amounts of PAR2-YFP or NK1R-YFP (acceptors) and treated with the respective agonists for 15 min. Net BRET is graphed as a function of acceptor/donor. G, average BRET50 and BRETmax values calculated from three independent experiments. #, significant difference between β-arrestin-1 and -2. *, significant difference between PAR2 and NK1R, p < 0.01, n = 5.
FIGURE 6.
β-Arrestin-dependent cellular effects of the chimeric receptors are similar to their respective C-terminal parent. A, maximal net BRET signal was determined for PAR2, NK1R, PAR2-NK1R, or NK1R-PAR2 with either β-arrestin-1 or -2 as described in the legend to Fig. 5. *, significant difference from N-terminal parent; #, significant difference from β-arrestin-1 (p < 0.05, n = 3). B, cells transfected with each of the four receptors were seeded onto transwell filters. Cell migration is expressed as a mean ± S.E. (error bars) increase in number of cells that migrated after 2fAP or Sar-Met-SP treatment compared with untreated cells. *, significant increase in cell migration; #, significant difference between bracketed groups (p < 0.01, n = 4). C, cells transfected with each receptor serum-starved and then treated with or without 2fAP, Sar-Met-SP, or serum (positive control) for 24 h. Mean ± S.E. increase in total cell number is shown. *, significant increase in proliferation (p < 0.01, n = 4).
Cellular Consequence of β-Arrestin-dependent Signaling Is Determined by the C Terminus of Each Receptor
Both cofilin and membrane-sequestered ERK1/2 activities are important for β-arrestin-dependent chemotaxis downstream of PAR2, whereas nuclear ERK1/2 activation is essential for NK1R-stimulated proliferation (5, 13, 14, 24). NK1R does not promote cofilin activation (supplemental Fig. S5), further demonstrating that the two receptors utilize β-arrestins for different signaling pathways. To determine whether swapping the C termini of the two receptors also affected the cellular events resultant from β-arrestin engagement, we looked at proliferation and cell migration downstream of all four receptors. As previously reported, activation of PAR2 promoted a 4-fold increase in cell migration; however, NK1R did not significantly affect cell migration (Fig. 6B). Conversely, activation of NK1R-PAR2 increased cell migration by 3-fold, whereas activation of PAR2-NK1R did not (Fig. 6B). Similarly, NK1R and PAR2-NK1R, but not PAR2 and NK1R-PAR2, promoted a 2-fold increase in proliferation (Fig. 6C). Thus, the ultimate cellular consequence of β-arrestin recruitment to PAR2 and NK1R is determined by the C terminus of each receptor.
Recruitment of β-Arrestins to PAR2 but Not to NK1R Is Independent of Gαq Signaling
Although PAR2 can promote β-arrestin-dependent signaling independent of Gαq, NK1R appears to require integration of both Gαq and β-arrestin signaling to activate ERK1/2. To determine whether the two receptors differ in their requirement for initial Gαq coupling, we examined β-arrestin-1/2 recruitment in the presence of pharmacological inhibitors of phospholipase Cβ (U73122) and PKC (GFX) and a chelator of intracellular Ca2+ (BAPTA-AM) using BRET. Both β-arrestin-1 and -2 were recruited to PAR2, following agonist stimulation in the presence of all three inhibitors (Fig. 7A). On the contrary, all three inhibitors led to a significant reduction in recruitment of β-arrestins to NK1R (Fig. 7B).
FIGURE 7.

Inhibition of Gαq signaling does not inhibit β arrestin-1 or -2 recruitment to agonist-stimulated PAR2. HEK 293 cells transiently transfected with PAR2-eYFP (A) or NK1R-eYFP (B) and β-arrestin-1/2-Rluc were pretreated with BAPTA-AM (30 μm, 10 min), GFX (10 μm, 10 min), and U73122 (10 μm, 10 min) and then stimulated with 2fAP (A) (1 μm, 20 min) or Sar-Met-SP (B) (100 nm, 20 min). Net BRET values with and without agonist treatment were determined and graphed as the change in net BRET in the presence and absence of agonist (Induced Net BRET). *, p < 0.05. **, p < 0.002. Shown is a graph of mean ± S.E. (error bars) -fold increase in cell migration in response to 2fAP in cells expressing PAR2WT or PAR2S363A/T366A. Migration was significantly reduced in response to mutant compared with WT PAR2 (p < 0.01, n = 3).
Previous studies have suggested that mutation of two putative PKC phosphorylation sites in PAR2 (Ser-363 and Thr-366; PAR2S363A/T366A) inhibits stable colocalization of β-arrestin with PAR2, receptor desensitization, and internalization and membrane activation of ERK1/2. This mutant receptor robustly promoted nuclear translocation of activated ERK1/2 and proliferation, whereas the wild type receptor did so only weakly (2). Because inhibition of PKC did not abolish recruitment of β-arrestin to PAR2, we examined whether more subtle features of β-arrestin/receptor interactions were affected by mutation of these residues. Both receptors are expressed on the cell surface, and both are capable of recruiting β-arrestin-1 and -2 at the same agonist dose (Fig. 8, A and B). Kinetic BRET assays revealed that the rate of recruitment to PAR2S363A/T366A was reduced 1.5- and 2-fold for β-arrestin-1 and -2, respectively, compared with the wild type PAR2 (Fig. 8, C and D). Furthermore, BRET50 values were increased for the phosphomutant, suggesting that it had a lower relative affinity for both β-arrestins than the wild type receptor. The BRETmax value was also decreased, indicating that the nature of the receptor/β-arrestin interactions was different such that the acceptor and donor tags were in closer proximity with the wild type receptor. To examine whether PAR2S363A/T366A was deficient in other aspects of β-arrestin-dependent PAR2 signaling, we examined cofilin dephosphorylation and cell migration. Consistent with its decreased ability to stably recruit β-arrestins, PAR2S363A/T366A promoted a transient increase in cofilin phosphorylation, which returned to base line within 10 min, and no cofilin dephosphorylation (activation) was observed (Fig. 9, A and B). In contrast, activation of PAR2WT resulted in a 50% dephosphorylation of cofilin (Fig. 9, A and B). Similarly, whereas wild type PAR2 promoted a 2-fold increase in cell migration, PAR2S363A/T366A did not promote cell migration (Fig. 9C).
FIGURE 8.

β-Arrestin recruitment to PAR2S363A/T366A versus PAR2. A, cell surface expression of N-terminally FLAG-tagged PAR2WT-YFP and PAR2S363A/T366A-YFP was determined by in-cell Western assays using anti-FLAG. *, p < 0.05, n = 3. B–F, BRET assays were performed with FLAG-tagged PAR2-YFP or PAR2S363A/T366A-YFP and β-arrestin-1/2-Rluc. Net BRET ratio was estimated in response to incremental doses of 2fAP. B, inset, percentage of maximal BRET response to the increasing 2fAP doses. C, net BRET response was quantified over time in response to 1 μm 2fAP. D, mean ± S.E. half-lives (t½) of BRET kinetics are shown as mean ± S.E. (error bars) (significant differences between bracketed groups: *, p < 0.03; **, p < 0.008; n = 3). E, titration curves monitoring net BRET in response to varying acceptor/donor ratios were performed as in Fig. 5F. F, BRET50 and BRETmax for PAR2WT or PAR2S363A/T366A were computed from E. Both values were significantly different, reduced in PAR2S363A/T366A compared with PAR2WT (p < 01, n = 3).
FIGURE 9.
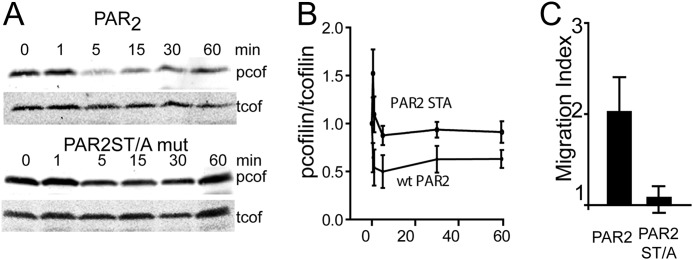
PAR2S363A/T366A fails to promote cofilin dephosphorylation and cell migration. A, representative Western blot of 2fAP-stimulated phosphocofilin and total cofilin levels in lysates from cells transfected with PAR2WT or PAR2S363A/T366A. B, mean ± S.E. (error bars) phosphocofilin levels (normalized to total cofilin levels) are graphed as a function of time (phosphocofilin levels in response to PAR2S363A/T366A were significantly elevated at all time points, compared with PAR2WT, p < 0.01, n = 3). C, graph of mean ± S.E. -fold increase in cell migration in response to 2fAP in cells expressing PAR2WT or PAR2S363A/T366A. Migration was significantly reduced in response to mutant compared with WT PAR2 (p < 0.01, n = 3).
DISCUSSION
Since the discovery of β-arrestin-dependent signaling, it has become apparent that not all GPCRs elicit the same signals upon recruitment of β-arrestins. The studies presented here demonstrate that differences in β-arrestin-dependent signaling are determined in part by specific interactions between β-arrestins and the C terminus of each receptor. Here we used two Gαq-coupled receptors, PAR2 and NK1R, both of which utilize β-arrestins for activation of ERK1/2 but do so by distinct mechanisms with different outcomes, to examine the receptor features underlying these signaling differences. We provide evidence that by swapping the C termini of the two receptors, we can force PAR2 agonists to induce a β-arrestin-dependent signaling pattern similar to that of NK1R and NK1R agonists to induce a signaling pattern similar to that of PAR2. This result suggests that each receptor may have a unique “β-arrestin fingerprint” that ultimately determines the specific scaffolds formed on the receptor-bound β-arrestin as well as the downstream signaling events. This model would predict that interactions with different GPCR C-terminal residues may induce subtle differences in β-arrestin conformation that then expose distinct sets of binding partner sites.
In these studies, we compared and contrasted different β-arrestin functions: 1) recruitment to each receptor, 2) duration of Gαq response, 3) signaling events (e.g. ERK1/2 activation mechanism and cofilin activation), and 4) functional responses (e.g. cell migration and proliferation). The results of these comparisons suggest that PAR2, which promotes β-arrestin-dependent membrane-associated ERK1/2 activity, cofilin activation, and chemotaxis, rapidly recruits β-arrestins and demonstrates equal apparent affinities for both β-arrestin-1 and -2. In contrast, NK1R, which promotes β-arrestin-dependent nuclear ERK1/2 activity and proliferation, does not activate cofilin and preferentially recruits β-arrestin-2 over β-arrestin-1. Furthermore, recruitment of both β-arrestins to NK1R occurs significantly more slowly, and with a lower apparent affinity, than to PAR2. These results allow us to make certain predictions regarding the relationship between β-arrestin recruitment patterns and downstream effects.
Ca2+ assays performed in this study specifically measure intracellular Ca2+, because EGTA is included in the medium bathing the cells, and are thus considered a read-out of Gαq-dependent signaling. We have shown that activated NK1R promotes a prolonged release of intracellular Ca2+ compared with PAR2, which ultimately results in a higher concentration of total Ca2+ released into the cytosol. We have previously demonstrated that, although both β-arrestins are required for full receptor down-regulation, genetic deletion of β-arrestin-1 but not β-arrestin-2, results in a prolonged Ca2+ signal downstream of PAR2 (3). Thus, the lower apparent affinity of NK1R for β-arrestin-1 and the slower recruitment kinetics of both β-arrestins to NK1R are consistent with the longer duration of the Ca2+ signal. Further supporting this model, in which the pattern of β-arrestin recruitment determines the signal duration, the chimeric receptor containing the N terminus of PAR2 and the C terminus of NK1R showed the same pattern of β-arrestin recruitment as NK1R, along with a prolonged Ca2+ signal. Conversely, the chimeric receptor containing the N terminus of NK1R and the C terminus of PAR2 had a short Ca2+ signal and a β-arrestin recruitment pattern similar to PAR2. Because β-arrestins directly uncouple G-protein/GPCR signals as well as facilitate clathrin-mediated endocytosis, these results suggest that residues within the C terminus of each receptor determine the relative affinities for each β-arrestin, which in turn affects how efficiently the G-protein signal is terminated.
A more puzzling distinction between PAR2 and NK1R signaling that is common to many GPCRs is the observation that although both receptors require β-arrestins for full activation of ERK1/2, they do so by different mechanisms with drastically different cellular responses. PAR2 utilizes separate G-protein-/Ca2+-dependent and β-arrestin-dependent mechanisms, whereas NK1R requires both Ca2+ and β-arrestins, and the β-arrestin signal induced by NK1R is not independent from the G-protein signal (1, 2). Here we demonstrate that recruitment of both β-arrestins to PAR2 occurs even in the presence of inhibitors of the G-protein signaling arm, whereas recruitment to NK1R is impaired by inhibition of the Gαq effector phospholipase Cβ or by chelation of intracellular Ca2+. Furthermore, PAR2 promotes formation of a stable complex containing β-arrestins and the entire ERK1/2 module (Raf, MEK1, and ERK1/2), leading to Src-independent, membrane-associated ERK1/2 activity and cell migration (2, 5, 13). In contrast, NK1R promotes formation of a transient complex containing β-arrestins and Src but not Raf, leading to nuclear translocation of ERK1/2 and proliferation (1). Here we show that the subcellular localization of activated ERK1/2 downstream of the chimeric receptors and the ultimate consequence of ERK1/2 activation (e.g. proliferation versus chemotaxis) follow that of the C-terminal parent.
The traditional model for GPCR signaling predicts that receptor phosphorylation, by GRKs or second messenger kinases, creates binding sites for β-arrestins. Indeed, studies have suggested that C-terminal phosphorylation of both PAR2 and NK1R is essential for receptor desensitization, internalization, and ERK1/2 activation (2, 19, 25). Although regulation of NK1R/β-arrestin recruitment and internalization by GRKs has been demonstrated, similar GRK-mediated regulation of PAR2 has only been hypothesized (9, 10). Regulation of both receptors by PKC phosphorylation has been suggested by the fact that desensitization and internalization of both are sensitive to broad-spectrum PKC inhibitors, and mutation of putative PKC phosphorylation sites in both receptors hinders receptor desensitization and internalization (2, 18, 19). However, when BRET was used to more closely monitor real-time β-arrestin interactions with a mutant PAR2 previously shown to be defective in β-arrestin colocalization and PKC-induced desensitization, we observed that β-arrestin-1 and -2 are both recruited to this mutant receptor but with a significantly slower rate and lower apparent affinity. These data suggest that interactions between phosphorylated Ser-363 and Thr-366 in the C-tail of PAR2 strengthen its interaction with β-arrestins but are not necessary for initial recruitment. Consistent with the decreased apparent affinity for β-arrestins, PAR2S363A/T366A also fails to promote cofilin dephosphorylation or cell migration, which are hallmarks of β-arrestin-dependent signaling downstream of PAR2. Importantly, differences in the C-terminal phosphorylation pattern of GPCRs may contribute to the stability of their respective interactions with β-arrestins and, thus, to the differences in their downstream signaling patterns. There are a number of GPCRs that appear to elicit similar G-protein-independent, β-arrestin-dependent signaling as PAR2. Likewise, there are receptors that, like NK1R, promote β-arrestin-dependent recruitment of Src and nuclear ERK1/2 activation and proliferation (26). Other studies using chimeric angiotensin and vasopressin receptors have suggested that the stability of the ERK1/2 scaffolding complexes is dependent upon the C termini of the receptors (27). Our previous studies had demonstrated that the native β-arrestin·ERK1/2 complex formed in response to PAR2 was sufficiently stable to remain intact on a size exclusion column, whereas the complex formed in response to NK1R required cross-linking in order to survive the purification process (1, 2). These studies suggested that the PAR2-associated complex is significantly more stable than that formed downstream of NK1R. Thus, the differences in the β-arrestin signaling pathways elicited by PAR2 and NK1R may be dependent upon the stability of the complex formed, which previous studies demonstrated is dependent upon interactions between β-arrestins and receptor C termini (27). This hypothesis is further supported by the observation in the BRET studies presented here that β-arrestins display a higher affinity for PAR2 compared with NK1R, and the pattern of β-arrestin recruitment is dependent upon the receptor C terminus. Interestingly, the PAR2 phosphomutant, which was previously shown to be deficient in desensitization and β-arrestin ERK1/2 localization (2), displays a lower β-arrestin affinity and slower recruitment kinetics than the wild type receptor. Like the NK1R, it does not promote cofilin activation or cell migration, but it promotes proliferation to a greater extent than the wild type receptor. Although there are studies indicating phosphoreceptor-binding sites on β-arrestins (28, 29), it is likely that there are receptor-specific differences that are yet to be appreciated, which may contribute to more subtle differences in signaling depending upon the phosphorylation state of the receptor. These studies also suggest that differences in the phosphorylation patterns of different GPCRs influence the stability of β-arrestin interactions. Ultimately, differences in receptor phosphorylation may in turn affect which binding domains on β-arrestin are exposed, resulting in association of certain putative binding partners and exclusion of others. Recent studies using a β-arrestin biosensor to detect gross changes in conformation suggested that biased agonists of several GPCRs, capable of eliciting β-arrestin-dependent signaling in the absence of G-protein coupling, promote a conformation that is distinct from that elicited by standard agonists (30, 31). Thus, differences in β-arrestin conformation in response to recruitment to PAR2 versus NK1R might underlie the differences in signaling we observe. Ultimately, these subtle differences in β-arrestin/receptor interactions lead to dramatic differences in cellular responses (e.g. proliferation versus cell migration).
Acknowledgment
We thank Dr. Michel Bouvier (University of Montreal) for assistance in setting up BRET assays.
This work was supported, in whole or in part, by National Institutes of Health Grant R01GM066151 (to K. A. D.).

This article contains supplemental Figs. S1–S5.
- PAR2
- protease-activated receptor-2
- NK1R
- neurokinin-1 receptor; Substance P
- GRK
- G-protein-coupled receptor kinase
- BAPTA-AM
- 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid acetoxymethyl ester
- eYFP and eGFP
- enhanced YFP and GFP, respectively
- 2fAP
- 2-furoyl-LIGRLNH2
- BRET
- bioluminescence resonance energy transfer
- Sar-Met-SP
- [Sar-9,Met(O2)-11]SP
- MEFwt
- wild type MEF
- PAP2WT
- wild type PAP2
- MEF
- mouse embryo fibroblast
- MEFβarrDKO
- MEF from β-arrestin-1/2−/− mice
- IF
- immunofluorescence
- WB
- Western blot
- pERK
- phospho-ERK.
REFERENCES
- 1. DeFea K. A., Vaughn Z. D., O'Bryan E. M., Nishijima D., Déry O., Bunnett N. W. (2000) The proliferative and antiapoptotic effects of substance P are facilitated by formation of a β-arrestin-dependent scaffolding complex. Proc. Natl. Acad. Sci. U.S.A. 97, 11086–11091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. DeFea K. A., Zalevsky J., Thoma M. S., Déry O., Mullins R. D., Bunnett N. W. (2000) β-Arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell Biol. 148, 1267–1281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kumar P., Lau C. S., Mathur M., Wang P., DeFea K. A. (2007) Differential effects of β-arrestins on the internalization, desensitization and ERK1/2 activation downstream of protease activated receptor-2. Am. J. Physiol. Cell Physiol. 293, C346–C357 [DOI] [PubMed] [Google Scholar]
- 4. Stalheim L., Ding Y., Gullapalli A., Paing M. M., Wolfe B. L., Morris D. R., Trejo J. (2005) Multiple independent functions of arrestins in the regulation of protease-activated receptor-2 signaling and trafficking. Mol. Pharmacol. 67, 78–87 [DOI] [PubMed] [Google Scholar]
- 5. Ge L., Ly Y., Hollenberg M., DeFea K. (2003) A β-arrestin-dependent scaffold is associated with prolonged MAPK activation in pseudopodia during protease-activated receptor-2-induced chemotaxis. J. Biol. Chem. 278, 34418–34426 [DOI] [PubMed] [Google Scholar]
- 6. Al-Ani B., Hansen K. K., Hollenberg M. D. (2004) Proteinase-activated receptor-2. Key role of amino-terminal dipeptide residues of the tethered ligand for receptor activation. Mol. Pharmacol. 65, 149–156 [DOI] [PubMed] [Google Scholar]
- 7. Lovgren A. K., Kovacs J. J., Xie T., Potts E. N., Li Y., Foster W. M., Liang J., Meltzer E. B., Jiang D., Lefkowitz R. J., Noble P. W. (2011) β-Arrestin deficiency protects against pulmonary fibrosis in mice and prevents fibroblast invasion of extracellular matrix. Sci. Transl. Med. 3, 74ra23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nystedt S., Emilsson K., Wahlestedt C., Sundelin J. (1994) Molecular cloning of a potential proteinase activated receptor. Proc. Natl. Acad. Sci. U.S.A. 91, 9208–9212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McConalogue K., Corvera C. U., Gamp P. D., Grady E. F., Bunnett N. W. (1998) Desensitization of the neurokinin-1 receptor (NK1-R) in neurons. Effects of substance P on the distribution of NK1-R, Gαq/11, G-protein receptor kinase-2/3, and β-arrestin-1/2. Mol. Biol. Cell 9, 2305–2324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McConalogue K., Déry O., Lovett M., Wong H., Walsh J. H., Grady E. F., Bunnett N. W. (1999) Substance P-induced trafficking of β-arrestins. The role of β-arrestins in endocytosis of the neurokinin-1 receptor. J. Biol. Chem. 274, 16257–16268 [DOI] [PubMed] [Google Scholar]
- 11. Morris D. R., Ding Y., Ricks T. K., Gullapalli A., Wolfe B. L., Trejo J. (2006) Protease-activated receptor-2 is essential for factor VIIa and Xa-induced signaling, migration, and invasion of breast cancer cells. Cancer Res. 66, 307–314 [DOI] [PubMed] [Google Scholar]
- 12. Seatter M. J., Drummond R., Kanke T., Macfarlane S. R., Hollenberg M. D., Plevin R. (2004) The role of the C-terminal tail in protease-activated receptor-2-mediated Ca2+ signalling, proline-rich tyrosine kinase-2 activation, and mitogen-activated protein kinase activity. Cell. Signal. 16, 21–29 [DOI] [PubMed] [Google Scholar]
- 13. Ge L., Shenoy S. K., Lefkowitz R. J., DeFea K. (2004) Constitutive protease-activated receptor-2-mediated migration of MDA MB-231 breast cancer cells requires both β-arrestin-1 and -2. J. Biol. Chem. 279, 55419–55424 [DOI] [PubMed] [Google Scholar]
- 14. Zoudilova M., Kumar P., Ge L., Wang P., Bokoch G. M., DeFea K. A. (2007) β-arrestin-dependent regulation of the cofilin pathway downstream of protease-activated receptor-2. J. Biol. Chem. 282, 20634–20646 [DOI] [PubMed] [Google Scholar]
- 15. Böhm S. K., Grady E. F., Bunnett N. W. (1997) Regulatory mechanisms that modulate signalling by G-protein-coupled receptors. Biochem. J. 322, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Luttrell L. M., Lefkowitz R. J. (2002) The role of β-arrestins in the termination and transduction of G-protein-coupled receptor signals. J. Cell Sci. 115, 455–465 [DOI] [PubMed] [Google Scholar]
- 17. Ferguson S. S., Downey W. E., 3rd, Colapietro A. M., Barak L. S., Ménard L., Caron M. G. (1996) Role of β-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science 271, 363–366 [DOI] [PubMed] [Google Scholar]
- 18. Böhm S. K., Khitin L. M., Grady E. F., Aponte G., Payan D. G., Bunnett N. W. (1996) Mechanisms of desensitization and resensitization of proteinase-activated receptor-2. J. Biol. Chem. 271, 22003–22016 [DOI] [PubMed] [Google Scholar]
- 19. Déry O., Defea K. A., Bunnett N. W. (2001) Protein kinase C-mediated desensitization of the neurokinin 1 receptor. Am. J. Physiol. Cell Physiol. 280, C1097–C1106 [DOI] [PubMed] [Google Scholar]
- 20. Ramachandran R., Mihara K., Mathur M., Rochdi M. D., Bouvier M., Defea K., Hollenberg M. D. (2009) Agonist-biased signaling via proteinase activated receptor-2. Differential activation of calcium and mitogen-activated protein kinase pathways. Mol. Pharmacol. 76, 791–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Exton J. H. (1994) Phosphoinositide phospholipases and G proteins in hormone action. Annu. Rev. Physiol. 56, 349–369 [DOI] [PubMed] [Google Scholar]
- 22. Yu Y. J., Arttamangkul S., Evans C. J., Williams J. T., von Zastrow M. (2009) Neurokinin 1 receptors regulate morphine-induced endocytosis and desensitization of μ-opioid receptors in CNS neurons. J. Neurosci. 29, 222–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Déry O., Thoma M. S., Wong H., Grady E. F., Bunnett N. W. (1999) Trafficking of proteinase-activated receptor-2 and β-arrestin-1 tagged with green fluorescent protein. β-Arrestin-dependent endocytosis of a proteinase receptor. J. Biol. Chem. 274, 18524–18535 [DOI] [PubMed] [Google Scholar]
- 24. Zoudilova M., Min J., Richards H. L., Carter D., Huang T., DeFea K. A. (2010) β-Arrestins scaffold cofilin with chronophin to direct localized actin filament severing and membrane protrusions downstream of protease-activated receptor-2. J. Biol. Chem. 285, 14318–14329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ricks T. K., Trejo J. (2009) Phosphorylation of protease-activated receptor-2 differentially regulates desensitization and internalization. J. Biol. Chem. 284, 34444–34457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Defea K. (2008) β-Arrestins and heterotrimeric G-proteins. Collaborators and competitors in signal transduction. Br. J. Pharmacol. 153, S298–S309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tohgo A., Choy E. W., Gesty-Palmer D., Pierce K. L., Laporte S., Oakley R. H., Caron M. G., Lefkowitz R. J., Luttrell L. M. (2003) The stability of the G protein-coupled receptor-β-arrestin interaction determines the mechanism and functional consequence of ERK activation. J. Biol. Chem. 278, 6258–6267 [DOI] [PubMed] [Google Scholar]
- 28. Goodman O. B., Jr., Krupnick J. G., Santini F., Gurevich V. V., Penn R. B., Gagnon A. W., Keen J. H., Benovic J. L. (1996) β-Arrestin acts as a clathrin adaptor in endocytosis of the β2-adrenergic receptor. Nature 383, 447–450 [DOI] [PubMed] [Google Scholar]
- 29. Gurevich V. V., Dion S. B., Onorato J. J., Ptasienski J., Kim C. M., Sterne-Marr R., Hosey M. M., Benovic J. L. (1995) Arrestin interactions with G protein-coupled receptors. Direct binding studies of wild type and mutant arrestins with rhodopsin, β2-adrenergic, and m2 muscarinic cholinergic receptors. J. Biol. Chem. 270, 720–731 [DOI] [PubMed] [Google Scholar]
- 30. Shukla A. K., Violin J. D., Whalen E. J., Gesty-Palmer D., Shenoy S. K., Lefkowitz R. J. (2008) Distinct conformational changes in β-arrestin report biased agonism at seven-transmembrane receptors. Proc. Natl. Acad. Sci. U.S.A. 105, 9988–9993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nobles K. N., Xiao K., Ahn S., Shukla A. K., Lam C. M., Rajagopal S., Strachan R. T., Huang T. Y., Bressler E. A., Hara M. R., Shenoy S. K., Gygi S. P., Lefkowitz R. J. (2011) Distinct phosphorylation sites on the β2-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci. Signal. 4, ra57. [DOI] [PMC free article] [PubMed] [Google Scholar]