

Background: Mast cells modulate events in wound healing.
Results: Shorter forms of perlecan are produced by mast cells via proteolytic processing and alternative splicing, which contain domain V and functional endorepellin.
Conclusion: The production of these shorter forms modulates endothelial cell adhesion, proliferation, and migration.
Significance: Mast cells produce specific forms of perlecan that affect endothelial cell behavior.
Keywords: Alternative Splicing, Angiogenesis, Basement Membrane, Extracellular Matrix, Mast Cell, Proteoglycan Structure, Proteolytic Enzymes, Wound Healing, Perlecan
Abstract
Mast cells are derived from hematopoietic progenitors that are known to migrate to and reside within connective and mucosal tissues, where they differentiate and respond to various stimuli by releasing pro-inflammatory mediators, including histamine, growth factors, and proteases. This study demonstrated that primary human mast cells as well as the rat and human mast cell lines, RBL-2H3 and HMC-1, produce the heparan sulfate proteoglycan, perlecan, with a molecular mass of 640 kDa as well as smaller molecular mass species of 300 and 130 kDa. Utilizing domain-specific antibodies coupled with N-terminal sequencing, it was confirmed that both forms contained the C-terminal module of the protein core known as endorepellin, which were generated by mast cell-derived proteases. Domain-specific RT-PCR experiments demonstrated that transcripts corresponding to domains I and V, including endorepellin, were present; however, mRNA transcripts corresponding to regions of domain III were not present, suggesting that these cells were capable of producing spliced forms of the protein core. Fractions from mast cell cultures that were enriched for these fragments were shown to bind endothelial cells via the α2β1 integrin and stimulate the migration of cells in “scratch assays,” both activities of which were inhibited by incubation with either anti-endorepellin or anti-perlecan antibodies. This study shows for the first time that mast cells secrete and process the extracellular proteoglycan perlecan into fragments containing the endorepellin C-terminal region that regulate angiogenesis and matrix turnover, which are both key events in wound healing.
Introduction
Perlecan, a ubiquitous and modular heparan sulfate (HS)2 proteoglycan, has been hypothesized to have roles in a wide variety of biological and pathological processes. The HS decorating the N-terminal domain has been shown to bind to many heparin-binding growth factors (1) and to modulate the signaling activities of some of the members of the FGF family, most notably FGF1 and -2 (2), FGF10 (3), and FGF18 (4), with FGF7 (5) and FGF18 (6) also interacting with the protein core. The C-terminal region of the protein core contains the α2β1 integrin binding site in the LG3 region of domain V. Fragments arising from the action of BMP1/Tolloid-like proteases that contain this integrin site have been referred to as endorepellin due to its anti-angiogenic activities in vitro (7). This same region of perlecan has also been shown to interact with the major VEGF receptor, VEGFR2, supporting the idea that perlecan can control cell adhesion in concert with VEGF signaling while also being involved in HS-mediated growth factor signaling (8).
The process of wound healing involves a series of well orchestrated phases commencing with coagulation and hemostasis. This is followed by an inflammatory phase, where neutrophils and macrophages migrate into the transitional matrix, which then encourages the fibroblasts to proliferate and produce extracellular matrix. Finally, a remodeling or resolution phase occurs, where the matrix is turned over and the wound bed contracts (9). A minor population of granulocytes that are related to neutrophils in that they share a primordial cell resident in the bone marrow are basophils that contain distinct basophilic granules. It is thought that these cells give rise to the tissue-resident mast cells that are distributed throughout the skin, lung, and mucosa of the intestine, where they are key cells in IgE-mediated allergic inflammation, such as immediate type hypersensitivity reactions and the response to other pathogens. Increased numbers of mast cells have been associated with fibrotic conditions, such as scleroderma of the skin (10), the fibrotic response induced around tumors (11), and bleomycin-induced fibrosis of the lungs of rats (12). When activated, mast cells degranulate and release mediators that include histamine, cytokines, and growth factors stored in their granules bound to the proteoglycan serglycin (13).
It is thought that serglycin is decorated with the highly sulfated form of HS known as heparin and that the high charge density is required to package the proteases effectively and control their proteolytic activity when released into the tissues (14). Mast cells have been hypothesized to have important roles in wound healing, where they degrade the extracellular matrix and release angiogenic peptides and cause contraction of the wound bed via the action of specific proteases, such as chymases and tryptases (15). Mast cells have been shown previously to synthesize laminin, type IV collagen, and perlecan. However, the biological function of this phenomenon remained unknown and was hypothesized to contribute to the fibrotic response in tissues (16).
This study has demonstrated that human primary mast cells as well as the rat (17) and human (HMC-1) mast cell lines synthesize perlecan, which was cleaved into smaller fragments by a range of proteases also produced by the cells. This paper also shows evidence to suggest that these cells produce alternatively spliced forms of perlecan that originate via splicing events in domain I. These fragmented and shorter forms included the intact C-terminal region of the protein core, known as endorepellin, which had the ability to modulate angiogenesis, a major factor in successful wound healing.
EXPERIMENTAL PROCEDURES
Chemicals were purchased from Sigma-Aldrich unless stated otherwise.
Primary Human Mast Cell Culture
Primary human lung mast cells were obtained under ethics approval for the supply of lung tissue from the Sydney South West Area Health Service and for their isolation from the lung samples from the Human Ethics Committee of the University of Sydney. These were cultured, and mast cells were isolated, resuspended at 1 × 106/ml, and activated with IgE/anti-IgE and sonicated to lyse the cells after 24 h (18).
Culture of Mast Cell Lines, RBL-2H3 and HMC-1
The rat basophilic leukemia cell line, RBL-2H3, and the human mast cell line, HMC-1, were cultured in RPMI 1640 medium containing 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) penicillin/streptomycin at 37 °C in 5% CO2. Conditioned medium was collected every 3 days and stored at −20 °C until required. Both of these cell lines have been characterized substantially with respect to their cell markers, identifying them as suitable model cell lines for either human (19) or rat (17) mast cells, respectively. Mast cells were activated by adding a protein kinase C activator, 50 nm phorbol 12-myristate 13-acetate (PMA), and a calcium ionophore, 500 nm A23187, 0.1% (v/w) BSA, and 1 mm CaCl2 in Siriganian buffer (119 mm NaCl, 5 mm KCl, 25 mm PIPES, 5.6 mm dextrose, 0.4 mm MgCl2, pH 7.25) to the cells for 2–24 h at 37 °C.
Proteoglycan Enrichment
Anion exchange chromatography using a diethylaminoethyl column (1-ml DEAE-Sepharose Fast Flow, GE Healthcare) attached to an FPLC (AKTA purifier, GE Healthcare) was used to enrich medium conditioned by HMC-1 or RBL-2H3 cells for proteoglycans that were eluted with 1 m NaCl, 20 mm Tris base, 10 mm EDTA, pH 7.5. Some samples were reloaded onto the DEAE column and eluted with a linear gradient of 0.1–1.0 m NaCl, 20 mm Tris base, 10 mm EDTA, pH 7.5, over 36 column volumes. DEAE-enriched fractions were analyzed for protein concentration using a Coomassie Blue protein assay (Thermo Scientific, Rockford, IL).
Endoglycosidase Digestion
Samples were digested with 50 milliunits/ml proteinase-free chondroitinase ABC (Seikagaku Corp., Tokyo, Japan) in 0.1 m Tris acetate, pH 8, at 37 °C for 16 h to determine the presence of chondroitin sulfate (CS). Samples were digested with 50 milliunits/ml of both heparinase I and III (Seikagaku Corp.), diluted in 10 mm Tris-HCl, pH 7.4, at 37 °C for 16 h to determine the presence of HS.
ELISA
HMC-1 or RBL-2H3 cells were activated for 2 h, the cell supernatant was removed, and the proteoglycans were enriched as described above using DEAE chromatography. Medium from non-activated cells was also enriched for proteoglycans prior to ELISA by using DEAE chromatography as described above. Samples (100 μg/ml) (some were treated with endoglycosidases as indicated) were coated onto high binding 96-well ELISA plates for 2 h at room temperature. Wells were rinsed twice with Dulbecco's phosphate-buffered saline (DPBS), pH 7.4, followed by blocking with 0.1% (w/v) casein in DPBS for 1 h at room temperature. Wells were rinsed with DPBS containing 1% (v/v) Tween 20 (PBST), followed by incubation with primary antibodies diluted in 0.1% (w/v) casein in DPBS for 2 h at room temperature. Primary antibodies used included mouse monoclonal anti-keratan sulfate (KS) antibody (clone 5-D-4, gift from Prof. Bruce Caterson, Cardiff University, supernatant 1:500) (20), mouse monoclonal anti-chondroitin sulfate type A and C antibody (clone CS-56, ascites 1:2500), mouse monoclonal anti-chondroitin sulfate type A and D antibody (clone LY-111, Seikagaku Corp., 2 μg/ml), mouse monoclonal anti-chondroitin sulfate type D antibody (clone MO-225, Seikagaku Corp., 2 μg/ml), mouse monoclonal anti-heparan sulfate antibody (clone F58-10E4, Seikagaku Corp., 1 μg/ml), mouse monoclonal anti-heparin antibody (clone 2Q546, U.S. Biological (Marblehead, MA), 2 μg/ml), mouse monoclonal anti-perlecan domain I (clone A76, AbCam (Cambridge, MA), 2 μg/ml), mouse monoclonal anti-perlecan domain III (clone 7B5, supernatant 1:500), rat monoclonal anti-perlecan domain IV antibody (clone A7L6, AbCam, 2 μg/ml), mouse monoclonal anti-perlecan domain V (clone A74, AbCam, 2 μg/ml), rabbit polyclonal anti-endorepellin antibody (1:1000), rabbit polyclonal anti-perlecan antibody (ab906; gift from Prof. Marie Dziadek (Garvan Institute, Sydney, Australia), ascites 1:1000), and rabbit polyclonal anti-serglycin antibody (gift from Prof. Theocharis Achilleas (University of Patras, Greece), ascites 1:1000) (21). Wells were rinsed with PBST, followed by incubation with biotinylated secondary antibodies, anti-mouse IgG, goat anti-rat Ig (Dako, Campbellfield, Australia), or donkey anti-rabbit IgG (GE Healthcare, 1:1000) diluted in 0.1% (w/v) casein in DPBS for 1 h at room temperature, rinsed with PBST, and then incubated with streptavidin-horseradish peroxidase (HRP) (GE Healthcare, 1:500) for 30 min at room temperature. Binding of the antibodies to the samples was detected using the colorimetric substrate, 2 mm 2,2′-azino-di-3-ethylbenzthiazoline sulfonic acid in 0.5 m sodium citrate, pH 4.6, and absorbance was measured at 405 nm.
Immunocytochemistry
RBL-2H3 mast cells were seeded onto microscope slides (SuperFrost Ultraplust, Lomb Scientific, Taren Point, Australia) at a density of 1 × 105 cells/ml and cultured for 3 days. If cells were to be activated, they were incubated with the activation buffer for 2 h. HMC-1 cells, a suspension cell line, were seeded onto slides that had been precoated with 10 μg/ml fibronectin for 1 h at 37 °C to promote the attachment of the HMC-1 cells. Slides were rinsed twice with 50 mm Tris-HCl, pH 7.6, containing 0.15 m NaCl (TBS), 4% (w/v) paraformaldehyde for 15 min at room temperature and permeabilized with 300 mm sucrose, 50 mm NaCl, 3 mm MgCl2, 2 mm HEPES, 0.5% Triton X-100, pH 7.2, for 5 min on ice. Slides were then blocked with 1% (w/v) BSA in TBS containing 0.05% (w/v) Tween 20 (TBST) for 1 h at room temperature, followed by incubation with primary antibodies diluted in 1% (w/v) BSA in TBST for 16 h at 4 °C. Primary antibodies used included are described above as well as mouse monoclonal anti-HS antibody (clone HepSS-1, Seikagaku Corp., 1 μg/ml) and mouse monoclonal anti-CS antibody (clone 7D4, gift from Prof. Bruce Caterson, supernatant 1:1000). The slides were then rinsed three times in TBST and incubated with biotinylated anti-mouse Ig secondary antibody (1:500 dilution) for 1 h at room temperature. The slides were then rinsed again with TBST before incubation with streptavidin-FITC (GE Healthcare, 1:250 dilution) for 30 min at room temperature, followed by four rinses in TBST. The slides were then counterstained with 4′,6-diamidino-2-phenylindole, dilactate (DAPI) (Invitrogen, 1 μg/ml) in DPBS for 10 min in the dark and rinsed four times with the deionized water before imaging using fluorescence microscopy (Zeiss Axioskop Mot Mat 2, Sydney, Australia).
Flow Cytometry
HMC-1 cells were analyzed for the expression of HS, CS, heparin, serglycin, and perlecan by flow cytometry. Briefly, HMC-1 cells suspended in sterile DPBS at a cell concentration of 1 × 106 cells/ml were analyzed in their unactivated or activated states. Activated mast cells were prepared by incubating with a combination of 100 nm PMA and 500 nm A23187 in sterile DPBS for 2 h at 37 °C. The activation was stopped by placing the cells on ice, followed by centrifugation at 1200 rpm for 5 min. The pellet was resuspended with sterile DPBS. Both unactivated and activated cells were fixed with 4% (w/v) paraformaldehyde for 15 min at room temperature, followed by washing with DPBS. Cells were then resuspended with the permeabilizing solution for 5 min on ice, washed with DPBS, and centrifuged at 1200 rpm for 5 min. Cells were blocked with 1% (w/v) BSA in DPBS for 30 min at room temperature. 500 μl of cell suspension (5 × 105 cells) was incubated with primary antibodies for 30 min at room temperature, followed by washing with DPBS and centrifugation at 1200 rpm for 5 min. Primary antibodies used included are described above as well as mouse monoclonal anti-HS antibody (clone JM403, Sapphire Bioscience (Sydney, Australia), 1 μg/ml) and a rabbit polyclonal anti-perlecan antibody (CCN-1, 1:5000) that was raised against immunopurified endothelial cell-derived perlecan (22). Control samples were prepared by incubating cells with isotype controls, mouse Ig (G + M) or rabbit IgG (1:500 in 1% BSA in DPBS). Cells were then incubated with FITC-conjugated secondary antibodies (1:500 in 1% (w/v) BSA in DPBS) for 30 min at room temperature and analyzed using a flow cytometer (BD Biosciences). For each sample, data were acquired for 1 × 104 gated events, and fluorescence intensity was displayed along with the number of cells.
Western Blot Analysis
Human coronary arterial endothelial cell (HCAEC)-derived perlecan was immunopurified by anion exchange and monoclonal antibody affinity chromatography, as described previously (22). DEAE-enriched samples and immunopurified HCAEC perlecan (5 μg/lane) (selected samples were treated with endoglycosidases) were electrophoresed on 3–8% Tris acetate SDS-polyacrylamide gels (Invitrogen) using 50 mm Tris base, 50 mm Tricine, 0.1% (w/v) SDS, pH 8.3, at 160 V for 60 min. Samples were transferred to an Immobilon P membrane (Merck) using 5 mm Bicine, 5 mm BisTris, 0.2 mm EDTA, 50 μg/ml SDS, 10% (v/v) methanol, pH 7.2, in a semidry blotter (Invitrogen) at 300 mA and 20 V for 60 min. The membrane was blocked with 1% (w/v) BSA in TBST for 2 h at room temperature, followed by incubation with primary antibody diluted in 1% (w/v) BSA/TBST for 2 h at room temperature. Membranes were rinsed with TBST, incubated with either HRP-conjugated goat anti-rabbit IgG or sheep anti-mouse Ig antibodies (Merck, 1:50,000), for 45 min at room temperature, and rinsed with TBST and TBS before being imaged using the Super Signal West Femto reagent kit (Thermo Scientific, Rockford, IL) and x-ray film.
N-terminal Sequencing
Fractions eluted from the linear gradient elution DEAE column, which were identified to be rich in perlecan as determined by ELISA, were analyzed by N-terminal sequencing. The fractions were pooled and concentrated using a Microcon YM-2 centrifugal filter unit (Merck). The protein concentration was estimated using a BCA assay, and a minimum of 10 pm protein was electrophoresed on a 3–8% Tris acetate NuPAGE SDS-polyacrylamide gel and then transferred to a PVDF membrane using a semidry transfer system. The proteins on the membrane were stained with Coomassie Blue R-250, which detects between 10 and 30 ng of protein/band. After the membrane was air-dried, each band was cut from the membrane and analyzed at the Australian Proteome Analysis Facility (Macquarie University).
Expression and Purification of Recombinant Perlecan Domain V
Perlecan domain V (2346 bp, exons 79–97) was amplified by PCR using mRNA of HCAECs as a template and the following oligonucleotides: forward, 5′-ATAAGCTTCTGATGCTGCCCTCAGTCCGACCCCA-3′, containing an HindIII site; reverse, 5′-ATGCTAGCCAACGAGGGGCAGGGGCGTGTGTT-3′, containing an NheI site. The amplified fragment was then cloned into CEFLsec vectors, which encompassed the sequence of the BM40 signal peptide, polyhistidine tag, and neomycin phosphotransferase. Subsequently, the cloned vectors were transfected into human embryonic kidney 293 (HEK293) cells using Lipofectamine® 2000 transfection reagent (Invitrogen). A stably transfected HEK293 pool was selected with Geneticin in serum-containing medium for 2 weeks or until mock transfectants were no longer viable. Recombinant perlecan domain V was purified using DEAE anion exchange chromatography as described above, followed by nickel affinity chromatography, HisTrap HP (GE Healthcare).
Endothelial Cell Adhesion
96-well tissue culture plates were coated with HCAEC-derived perlecan (10 μg/ml), recombinant perlecan domain V (10 μg/ml), and DEAE-enriched HMC-1 conditioned medium (20 μg/ml) for 16 h at 4 °C. Wells were then blocked with 1% (w/v) BSA in PBS for 1 h at 37 °C. Selected wells were treated with 0.01 unit/ml heparinase III, 0.1 unit/ml chondroitinase ABC, or both glycosidases for 1 h at 37 °C. The wells were then rinsed twice with PBS. Selected wells were then incubated with antibodies against perlecan, either the rabbit polyclonal antibody against endorepellin or a mouse monoclonal perlecan protein core (clone 5D7-2E4, Merck, 2 μg/ml) for 1 h at 37 °C. Endothelial cells (HCAEC, passage 4) were seeded at a density of 1 × 104 cells/well for 3 h at 37 °C. To investigate the role of integrin α2β1 in endothelial cell adhesion to perlecan, cells were pretreated with a mouse monoclonal antibody against α2β1 (clone BHA2.1, Merck, 1 μg/ml) for 20 min at 37 °C prior to seeding into perlecan-coated wells for 3 h at 37 °C. Cells were rinsed with PBS and fixed with 4% (w/v) paraformaldehyde in PBS for 15 min at 37 °C. Wells were then blocked with 1% (w/v) BSA in PBS for 5 min at 37 °C and then incubated with rhodamine-phalloidin (1:200, Invitrogen) in 1% (w/v) BSA in PBS for 1 h at 37 °C. Cells were rinsed three times with 0.5% (w/v) Tween 20 in PBS for 5 min. Cells were then counterstained with DAPI (1:1000 in 1% (w/v) BSA in PBS) for 10 min at 37 °C. Cells were imaged using a fluorescence microscope (Zeiss Axioskop Mot Mat 2), and cells were counted using ImageJ version 1.43u.
Endothelial Cell Proliferation
C11-STH cells were cultured in basal medium (M199 culture medium supplemented with 20% (v/v) FBS, 1% (v/v) penicillin/streptomycin, 1 mm NaHCO3, 10 mm HEPES, 1 mm sodium pyruvate, 100 μg/ml endothelial cell growth supplement (BD Biosciences), and 3 μg/ml heparin as described previously (23). 24-well tissue culture plates were coated with 5 μg/ml fibronectin and incubated for 1 h at 37 °C. Unbound fibronectin was then washed away with DPBS, and C11-STH cells were seeded at a density of 1 × 104 cells/well. Each well was supplemented with basal medium or basal medium with one of the following additives: 20 ng/ml FGF2, 20 ng/ml VEGF165, and HMC-1 releasate from activated cells. Cell proliferation was monitored at 1, 3, 5, and 7 days after cell seeding using crystal violet. At each time point, cells were washed twice with sterile DPBS containing 1 mm MgCl2 and 1 mm CaCl2, followed by fixation with 10% (v/v) formaldehyde in PBS for 20 min at room temperature. After two washes with deionized water, the fixed cells were air-dried for 1 h. 0.1% (w/v) crystal violet solution in 200 mm MES was then added to the wells, and the cells were stained for 30 min at room temperature. The plate was rinsed with deionized water four times and air-dried for 1 h. 100 μl of 10% (v/v) acetic acid was added into each well, the plate was placed on a rocker for 5 min, and then the absorbance of solubilized crystal violet at 590 nm was measured using a plate reader.
Endothelial Cell Migration
Tissue culture dishes were coated with 10 μg/ml fibronectin for 1 h at 37 °C. Dishes were then washed with DPBS, and C11-STH endothelial cells were seeded at a density of 5 × 104 cells/dish in basal medium and grown to confluence. Cell monolayers were “scratched” with a sterile cell scraper or a sterile P500 pipette tip to create an area devoid of cells, which was rinsed twice with sterile DPBS. The location of the “scratch” was marked on the underside of the dish using a pen, and images were taken using a phase-contrast microscope. The DPBS was removed from the wells, and then cells were incubated in basal medium or basal medium supplemented with one of the following additives: 20 ng/ml FGF2, 20 ng/ml VEGF165, and HMC-1 releasate from cells incubated in the presence of activation buffer for 24 h, concentrated 5-fold, and diafiltered using 5000 molecular weight cut-off membranes (Microcon YM-2 centrifugal filter unit, Merck) and then rediluted 1:5 back into endothelial growth medium. Cell migration was visualized under a phase-contrast microscope at 0, 2.5, 5, 24, and 28 h after the scratch was generated. Cell migration was quantified by measuring the area of the scratch at each of these time points and presented as the percentage of cells migrated compared with the area of the scratch at time 0.
RT-PCR and Quantitative Real-time PCR (qPCR)
Total RNA was isolated from HMC-1 cells and HCAEC using TRI Reagent (1 ml/107 cells) and then treated with DNase using the RQ1 RNase-free DNase kit (Promega, Madison, WI) to remove contaminating DNA. Subsequently, 1 μg of RNA was transcribed into cDNA using oligo(dT) primer mixer (ProtoScript® M-MuLV First Strand cDNA synthesis kit, NewEngland Labs, GeneSearch Pty. Ltd., Arundel, Australia) and amplified during 35 cycles by PCR utilizing perlecan domain-specific primers (Table 1). The reactants were cycled at 95 °C for 1 min, 60 °C for 1 min, and 72 °C for 1.5 min to enable denaturation, annealing, and extension, respectively. PCR products were then separated on 1% (w/v) agarose gel at 60 V for 1 h in TBE buffer (89 mm Tris base, 89 mm boric acid, and 2 mm EDTA, pH 8). The gels were stained with GelRed (Jomar Diagnostics, Stepney, Australia) for 30 min and then visualized under UV light. Each PCR experiment was repeated at least three times.
TABLE 1.
Primers for PCR amplification of HSPG2 cDNA (accession number NM_005529)
| Domain | Exons | Primer sequences (5′–3′) | PCR product size | Mast cell qPCR product DNA sequencing |
|
|---|---|---|---|---|---|
| Alignment with HSPG2 (nucleotides) | Sequence match | ||||
| bp | % | ||||
| I | 2 and 3 | Forward: CCATGGGCTGAGGGCATACG | 175 | ||
| Reverse: TCCAGCGCATTTGGCTTGCT | |||||
| 2–5 | Forward: CCATGGGCTGAGGGCATACG | 333 | |||
| Reverse: ACACTGACAACCTGGTCTCCGGG | |||||
| 2–7 | Forward: CCATGGGCTGAGGGCATACG | 510 | |||
| Reverse: GGCACTGTGCCCAGGCGTCGGAACT | |||||
| 3–7 | Forward: ACCTGGGCAGTGGGGACCTG | 403 | 280–483 | 97 | |
| Reverse:GCCTCCGTGCAGGCTCTTGG | |||||
| III | 29–36 | Forward:ACCCCACCTGTGATGCGTGCTC | 796 | ||
| Reverse: GTGGCCCGGATCAGGAGCTCAT | |||||
| 35–37 | Forward: CGATGTGCAGATCAGGGC | 454 | |||
| Reverse: CTGGCATTGCGAGCAGG | |||||
| V | 87–97 | Forward: GTGTGACAGTGACCACCCCCT | 1406 | ||
| Reverse: CTACGAGGGGCAGGGGCGT | |||||
| 89–94 | Forward: GTGTCAGTGAATGGCAAACG | 578 | 12314–12855 | 99 | |
| Reverse: CTGAAGACAAGGTGCCCG | |||||
For qPCR, 1 μl of cDNA was mixed with 0.5 μl of forward and reverse primers (10 μm each) and 10 μl of Power SYBR Green PCR Master Mix (Applied Biosystems, Mulgrave, Australia), followed by the addition of RNase-free water to make up to 20 μl/sample. The GAPDH was used as an endogenous control and to normalize the perlecan domain PCR results. Samples were subject to 40 reactions using the ABI StepOneTM real-time PCR system.
Statistical Significance
Student's t test (for two samples, assuming equal variance) was used to compare statistical significance. Results of p < 0.05 were considered significant.
RESULTS
The types of glycosaminoglycans produced by the RBL-2H3 cell line were determined by analyzing medium conditioned by these cells in culture as well as the material released by the cells upon activation with PMA and A23187. Analysis by ELISA showed that the proteoglycan-enriched fraction from both non-activated and activated cells contained the major glycosaminoglycan types, KS, CS, heparin, and HS (Fig. 1A). Mast cell activation led to a slight decrease in cell-associated KS and CS, which was reflected by an increase of these same glycosaminoglycans in the relative proteoglycan-enriched fractions. Concomitantly, there was a relative increase in cell-associated heparin, suggesting that not all of the heparin contained in the α-granules is released upon activation.
FIGURE 1.

Rat and human mast cell lines, RBL-2H3 and HMC-1, synthesize glycosaminoglycans. A, the presence of KS, CS, and HS and heparin in the proteoglycan-enriched fraction, isolated from medium conditioned by non-activated and activated (treated with PMA and A23187) RBL-2H3 cells, was identified by ELISA using monoclonal antibodies against KS (5-D-4), CS types A and C (CS-56), CS types A and D (LY-111), CS type D (MO-225), HS (F58-10E4), and heparin (2Q546). Wells were coated with 50 μl of a solution with a protein concentration of 100 μg/ml (absorbance readings were per 5 μg of coated protein). All absorbance readings were corrected for background and presented as mean ± S.D. (error bars) (n = 3). B, immunolocalization of glycosaminoglycans in cultures of RBL-2H3 isolated under non-activated and activated conditions. The presence of HS (HepSS1; A and B), heparin (2Q546; C and D), and CS (CS-56 and 7-D-4) was determined using antibodies against each of these glycosaminoglycans and visualized using FITC-conjugated secondary antibodies shown in green. Nuclei were stained with DAPI as shown in blue. Scale bars, 10 μm. C, the presence of KS, CS, and HS and heparin in the DEAE-enriched medium conditioned by non-activated and activated (treated with PMA) HMC-1 cells was identified by ELISA using the same set of monoclonal antibodies as described above: KS (5-D-4), CS types A and C (CS-56), CS types A and D (LY-111), CS type D (MO-225), HS (F58-10E4), and heparin (2Q546). All absorbance readings were corrected for background and presented as mean ± S.D. (n = 3). D, flow cytometry analyses were performed to detect the presence of HS (JM-403), CS (CS-56), and heparin (2Q546) by non-activated and activated HMC-1 cells. These experiments were controlled by comparing the readings obtained from the specific antibodies with those obtained using an irrelevant isotype antibody.
The distribution of the glycosaminoglycans in non-activated and activated RBL-2H3 cells was assessed, and the immunolocalization patterns obtained for HS, heparin, and CS were similar in non-activated RBL-2H3 cells, with the cells showing punctuate intracellular staining reminiscent of cytoplasmic granules, consistent with their identification as mast cells (Fig. 1B, HS, Heparin, and CS). The immunolocalization patterns for HS and CS appeared less granular for activated RBL-2H3 cells, whereas the staining pattern for the heparin epitopes appeared more granular in the activated cells compared with the non-activated cells. This suggested that the heparin, HS, and CS epitopes were not randomly distributed throughout the α-granules and were decorating the protein core of serglycin to bind and modulate the function of the mediators.
The glycosaminoglycans produced by the human mast cell line, HMC-1, were also analyzed for comparison with the rat mast cell line. The proteoglycan-enriched fraction isolated from medium conditioned by non-activated or activated HMC-1 cells showed a different distribution of glycosaminoglycans than the RBL-2H3 cells, using the same set of antibodies. The HMC-1 cells did not have immunoreactive KS or HS in either fraction, but the fraction did contain both CS and heparin (Fig. 1C). In order to analyze the cell-associated glycosaminoglycans, flow cytometry was used due to the fact that the HMC-1 cells are loosely adherent, preventing analysis by immunocytochemistry. CS immunoreactivity was found to be increased in the activated HMC-1 cells, whereas there was no change in the presence of heparin between non-activated and activated cells (Fig. 1D). The HS epitope (JM-403) was detected at higher levels in activated cells, which suggested that not only were the structures of the glycosaminoglycans different between the non-activated and activated cells, but those responsible for binding the mediators in the granules were different.
Serglycin was detected in the proteoglycan-enriched fraction of non-activated HMC-1 culture medium, whereas it was not detected in a similarly treated fraction from the RBL-2H3 culture medium (Fig. 2A). Serglycin was, however, detected in RBL-2H3 cells with a granular pattern similar to that shown for the glycosaminoglycans in these cells (Fig. 2B, upper panels). The staining pattern for serglycin also became more granular in activated cells (Fig. 2B) in a similar fashion to heparin (Fig. 1B).
FIGURE 2.

RBL-2H3 and HMC-1 cells in culture produce the proteoglycans serglycin and perlecan. A, the presence of serglycin and perlecan in DEAE-enriched medium conditioned by non-activated cells was identified by ELISA using a polyclonal anti-serglycin antibody as well as monoclonal antibodies against perlecan domains I (A76), III (7B5), IV (A7L6), and V (A74) and a polyclonal anti-perlecan antibody (ab906). All absorbance readings were corrected for background and data presented as mean ± S.D. (error bars) (n = 3). B, immunolocalization of serglycin (polyclonal anti-serglycin) and perlecan (A7L6) in non-activated and activated RBL-2H3 cells was visualized using FITC-conjugated secondary antibodies shown in green. Nuclei were stained with DAPI, as shown in blue. Scale bars, 10 μm. C, flow cytometry of serglycin (polyclonal anti-serglycin) and perlecan (CCN-1) produced by non-activated and activated HMC-1 cells compared with an isotype control. D, immunolocalization of perlecan (A7L6) and serglycin (polyclonal anti-serglycin) produced by HMC-1 cells plated on fibronectin and visualized using FITC-conjugated secondary antibodies shown in green. Nuclei were stained with DAPI, as shown in blue. Scale bars, 10 μm.
Perlecan was detected in the proteoglycan-enriched fractions isolated from non-activated culture medium from both cell lines (Fig. 2A). The rat perlecan reacted with the polyclonal anti-perlecan antibodies as well as the antibodies against perlecan protein core domains III, IV, and V. Perlecan protein core domains I, IV, and V were detected in the proteoglycan-enriched fraction from the HMC-1 cells, whereas this material reacted weakly with the domain III-specific antibodies (Fig. 2A). Flow cytometry was used to examine the HMC-1 cells for the presence of perlecan, which was significantly above the amount of signal obtained from the isotype control in both activated and non-activated cells (Fig. 2C). Perlecan was detected in both non-activated and activated RBL-2H3 cells with a staining pattern that was relatively evenly spread throughout the cytoplasm (Fig. 2B). A similar pattern was observed in the HMC-1 cells when they were plated onto fibronectin in order to adhere them to the surface and to minimize the loss of cells throughout the staining procedure (Fig. 2D). This treatment had no deleterious effects on the cells, and serglycin was also detected under these same conditions (Fig. 2D).
Together, these data demonstrated that non-activated and activated mast cells synthesized and secreted perlecan, whereas the variable detection of perlecan protein core domains warranted further investigation because this may have been due to protease activity in the medium of the non-activated cells. The proteoglycan-enriched fraction from non-activated HMC-1 cells was probed for the presence of perlecan by Western blotting and found to contain immunoreactive bands of ∼460 kDa as well as bands at 130, 200, and 300 kDa (Fig. 3, lane A), which confirmed that HMC-1 cells produced the full-length protein core of perlecan at 460 kDa as well as smaller molecular mass species. Analysis of primary human lung mast cell lysates gave a similar pattern of immunoreactivity, suggesting that the primary human mast cells also produced full-length protein core at an approximate molecular mass of 460 kDa as well as fragments of similar molecular masses (Fig. 3, lane B). These molecular masses were not detected in samples of immunopurified endothelial cell-derived perlecan samples, which gave a single smear in excess of 460 kDa (Fig. 3, lane C) and suggested that the HMC-1-derived perlecan contained less glycosaminoglycan-decorated protein core than the endothelial cell-derived form. The proteoglycan-enriched fraction from each of the two mast cell lines as well as immunopurified perlecan derived from HCAECs was analyzed by Western blotting and probed with the domain-specific antibodies for the presence of perlecan protein core domains (Fig. 4). Both the non-activated RBL-2H3 and HMC-1 proteoglycan-enriched fractions were reactive with antibodies against domains I, IV, and V and gave similar banding patterns, whereas both fractions were negative against the domain III antibody (Fig. 4, A and B, III). The domain I-specific antibody detected a major band at 300 kDa in the HMC-1 proteoglycan-enriched fraction, whereas it also reacted, albeit weakly, with the band at 130 kDa in the RBL-2H3 proteoglycan-enriched fraction (Fig. 4, A and B, I). The A7L6 antibody that binds to domain IV of perlecan reacted with the band at 300 kDa as well as higher molecular mass species in the region of ∼640 kDa, which corresponded to the expected molecular weight of intact perlecan decorated with glycosaminoglycans (Fig. 4A, IV). This same reactivity was not as evident in the RBL-2H3 sample (Fig. 4B, IV), which is not surprising, given that A7L6 is a rat antibody. The domain V antibody did not react with the intact perlecan (∼640 kDa) in either the HMC-1 or RBL-2H3 proteoglycan-enriched fraction (Fig. 4, A and B, V), but it reacted with the bands of approximate molecular masses of 130 and 300 kDa. Each of the perlecan domain-specific antibodies reacted with immunopurified HCAEC perlecan of ∼640 kDa. Together, these data suggested that the mast cell lines produced intact perlecan and that the 130-kDa fragment reacted with the anti-domain V antibody (A74) in HMC-1 samples as well as reacting with the anti-domain I antibody in RBL-2H3, suggesting that this band contained both ends of the protein core. The 300 kDa band contained the N-terminal domain I, domain IV, and the C-terminal domain V, which supported the hypothesis that cleavage of the protein core within sites of domain IV could give rise to fragments of this size from either end of the protein core. The 300 kDa band, however, was not reactive with the anti-domain III antibody (7B5) and fragmentation of the perlecan protein core, suggesting that it did not contain the 7B5 epitope and hence that portion domain III, whereas it could have contained most of domain IV and all of domain V. A schematic describing these findings and showing the potential 130- and 300-kDa fragments is shown in Fig. 5. This schematic is also annotated with the relative positions of the antibody epitope used in the experiments described in the paper as well as the relative positions of the endorepellin and LG3 fragments and the recombinantly expressed domain V.
FIGURE 3.
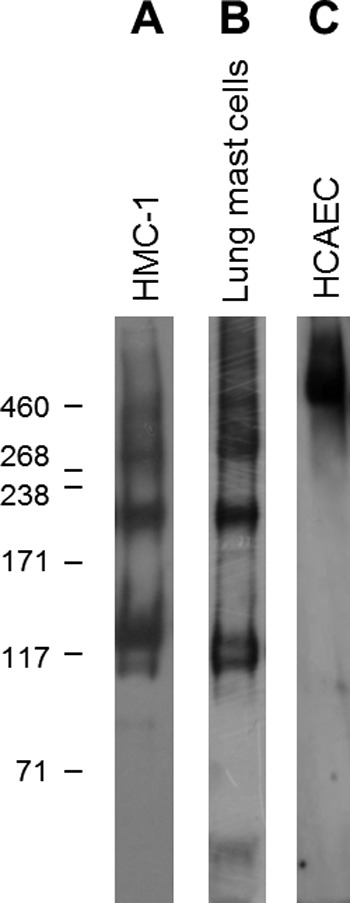
Detection of perlecan produced by HMC-1 cells, primary lung mast cells, and immunopurified HCAEC perlecan. HMC-1 conditioned medium was enriched for proteoglycans by passing it over a DEAE anion exchange column and eluting with NaCl (A). Cell lysate was prepared from primary human lung mast cells using sonication as described under “Experimental Procedures” (B). Perlecan immunopurified from HCAEC conditioned medium was used as a positive control (C). The membrane was probed for the presence of perlecan using polyclonal anti-perlecan antiserum (CCN-1).
FIGURE 4.
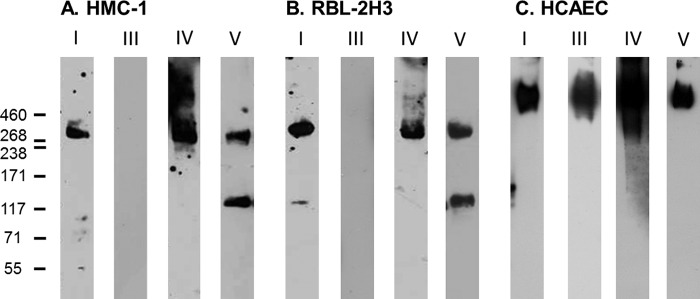
Detection of perlecan protein core domains produced by HMC-1 (A), RBL-2H3 (B), and HCAEC (C) cells. Conditioned medium from each of the cell lines was enriched for proteoglycans by DEAE anion exchange chromatography and eluting with NaCl. Samples were probed for the presence of perlecan domains I (A76), III (7B5), IV (A7L6), and V (A74).
FIGURE 5.

Schematic of perlecan. Exons and bases in each domain of perlecan are denoted at the top. Amino acids within each domain as well as the glycosaminoglycan attachment sites are denoted as well, and the potential binding sites for monoclonal antibodies used in Figs. 2, 4, and 6 are shown in yellow. A76 is a domain I antibody (37), 7B5 is a domain III antibody (33), A7L6 is a domain IV antibody (58) and A74 is a domain V antibody (37). The binding sites for A76, 7B5, and A74 were determined by alignment of the human and mouse perlecan amino acid sequences. Amino acid sequence alignment for human and rat perlecan yielded several possible binding sites for A7L6 throughout domain IV. Recombinantly expressed regions of domain V are shown in red, including endorepellin (59) and the domain V used in Fig. 7 as well as the LG3 peptide (38). Possible mast cell perlecan fragments produced by proteases are indicated at the bottom.
The HMC-1-derived perlecan was analyzed further by taking the proteoglycan-enriched conditioned medium from non-activated cultures, reloading it onto an anion exchange column, and eluting with a gradient of salt from 0.1 to 1 m NaCl over 36 column volumes (Fig. 6A). The fractions were interrogated for the presence of perlecan and serglycin by ELISA, which showed that the chromatography had separated these two proteoglycans present in the proteoglycan-enriched fraction (Fig. 6B). The polyclonal perlecan antibody, monoclonal anti-domain V, and a polyclonal endorepellin antibody all had significant reactivity with the same range of fractions (Fig. 6B). HMC-1-derived perlecan eluted from the column earlier in the gradient than HMC-1-derived serglycin, supporting the idea that the perlecan carried less charge than HMC-1-derived serglycin (Fig. 6B). The type of glycosaminoglycans that decorated the HMC-1 derived perlecan was analyzed by treating the proteoglycan-enriched fractions with a mixture of heparinase I and III and/or chondroitinase ABC. Both the 300 and 130 kDa bands were reactive with the domain V antibody that shifted in their molecular mass by ∼20 kDa after digestion with chondroitinase ABC (Fig. 6C, Domain V, denoted with Case ABC +), suggesting that the C-terminal fragments were decorated with CS. Incubation with either heparinase I and III or chondroitinase B had no effect on molecular weight, indicating that these fragments were not decorated with HS or dermatan sulfate (Fig. 6C, Domain V). These results were confirmed when the same samples were treated in the same way and probed with the polyclonal endorepellin antibody (Fig. 6C, Endorepellin).
FIGURE 6.

Isolation of endorepellin-containing fragments produced by HMC-1 cells. A, elution profile of medium conditioned by HMC-1 cells subject to a linear elution gradient DEAE chromatography from 0.10 to 1.0 m NaCl over 36 column volumes. B, elution profile of perlecan domain V (A74 and endorepellin), polyclonal anti-perlecan (ab906), and anti-serglycin, which eluted between 0.1 and 1.0 m NaCl. C, fractions that eluted between 0.4 and 0.6 m NaCl were probed for the presence of perlecan domain V (A74) and endorepellin (polyclonal anti-endorepellin) before and after either heparinase I and III (H'ase I/III), chondroitinase ABC (C'ase ABC), or chondroitinase B (C'ase B) digestion by Western blotting.
To determine whether the 300- and 130-kDa fragments were generated by proteases, N-terminal sequence analysis of the fragments was performed. No peptide sequences corresponding to perlecan were identified from the analysis of the 300 kDa band, whereas 15 sequences were identified from the 130 kDa band (Table 2). Of the 15 peptide sequences identified in the sample containing the 130-kDa protein, eight of them were located in domain IV, which is the largest domain in perlecan, two each from domains II and III, and one peptide sequence from domain V. One potential peptide identified was from the signal peptide, suggesting that it may have been present in the medium due to cell lysis. Also included in Table 2 is a list of mast cell-derived proteases that have cleavage site specificity aligning with the N-terminal sequences. Of interest was the peptide SVHGP because cleavage at the N-terminal serine residues will give rise to a C-terminal fragment with an approximate molecular mass of 130 kDa, suggesting that chymase, MMPs, and cathepsins were capable of generating this fragment. A peptide with the sequence SVPLS was also identified, which corresponded to amino acid residues 4060–4064 in domain V.
TABLE 2.
Amino acid sequences identified by N-terminal sequencing analysis of the 130 kDa band detected in DEAE-enriched condition medium shown in Fig. 4A
Amino acid sequences were aligned by cycle order and identified by a BLAST protein search. Amino acid sequences shown in boldface type aligned with the 130-kDa fragment calculated from the C terminus.
| Perlecan Protein core domain | Amino acid sequence | Amino acid positions | P1–P1′ | Mast cell proteases |
||
|---|---|---|---|---|---|---|
| Cathepsins | MMPs | Others | ||||
| GALLLA | 7–12 | A–G | S | 2, 3 | ||
| II | EAEFA | 201–205 | T–E | |||
| SEGGR | 457–461 | T–S | G, L | 2, 9, 11 | ||
| III | VLQSLE | 685–690 | Q–V | 2, 9, 13 | ||
| SPGQPS | 1048–1053 | P–S | 2, 3, 9 | |||
| IV | SVPLAA | 1499–1504 | S–S | S | 2, 3 | |
| AELLV | 1759–1763 | E–A | B, D, E, L | 2, 3 | ||
| APSKP | 1766–1770 | R–A | B, L | 1, 2 | ||
| PPQAR | 1997–2001 | L–P | D, E | 2 | ||
| SASLA | 2542–2546 | S–S | S | 2, 3 | ||
| SVHGP | 3107–3111 | L–S | D, E, G | 9 | Chymase | |
| ALGDP | 3512–3516 | L–A | D, G, S | |||
| VELAD | 3545–3549 | H–V | 2, 3 | |||
| VLLLV | 3568–3572 | H–V | 2, 3 | |||
| V | SVPLS | 4060–4064 | P–S | 2, 3, 9 | ||
Given that the two mast cell lines produce and process perlecan into fragments that contain domain V, which react with an anti-endorepellin antibody, the biological role of these fragments in cell adhesion was investigated. Endothelial cells adhered to wells coated with intact perlecan, and their number increased when the heparan sulfate chains were removed (Fig. 7A). The cells adhered to a similar extent to recombinant domain V, with their numbers also increasing in a statistically significant fashion when the HS and CS chains were removed (Fig. 7A). Of interest was the finding that the endothelial cells adhered to the proteoglycan-enriched fraction from HMC-1 medium to the same extent as they did to intact perlecan and recombinant domain V (Fig. 7A). The cell adhesive activity residing in the endothelal perlecan protein core was partially inhibited by antibodies against endorepellin or perlecan (Fig. 7B), whereas the cell adhesive activity in the recombinant domain V was only partially inhibited by the anti-endorepellin antibodies (Fig. 7B). The anti-perlecan antibody used in this assay (5D7-2E4) does not react with recombinant domain V, which suggested that there might be another site within the intact perlecan protein core outside of domain V that modulates cell adhesion events or is synergistic with the integrin binding site within domain V. The cell adhesive activity residing in the proteoglycan-enriched fraction was mostly inhibited by the anti-endorepellin antibodies, and the pattern of inhibition was similar to that demonstrated for the recombinant domain V (Fig. 7B). A major proportion of the cell adhesive activity demonstrated was via α2β1 integrins because function-blocking antibodies inhibited approximately half of the activity of both intact perlecan and recombinant domain V (Fig. 7C). Of interest was the observation that a major proportion of the cell adhesive activity residing in the HMC-1 proteoglycan-enriched fraction was also blocked by the same anti-α2β1 antibodies (Fig. 7C), which suggested that the HMC-1-enriched fractions contained biologically active fragments containing domain V and functional α2β1 adhesion sites.
FIGURE 7.

Perlecan supports endothelial cell adhesion via domain V and integrin α2β1. A, endothelial cell adhesion to HCAEC perlecan, HCAEC perlecan treated with heparinase III (H'ase III) to remove its HS chains, recombinant perlecan domain V, recombinant perlecan domain V treated with heparinase III and chondroitinase ABC (C'ase ABC), and DEAE-enriched medium conditioned by HMC-1 cells (0.4–0.6 m NaCl fraction). B, cell adhesion to perlecans pretreated with antibodies against endorepellin (anti-endorepellin) or perlecan (5D7-2E4) before the addition of endothelial cells. Data are presented as percentage of cell adhesion compared with cell adhesion to perlecans in the absence of antibodies as shown in A. C, cell adhesion to perlecans when cells were pretreated with antibodies against the integrin α2β1 (BHA2.1). Data are presented as the percentage of cell adhesion compared with cell adhesion to perlecans in the absence of antibodies as shown in A. *, significant differences (p < 0.05) compared with cell adhesion for each of the respective perlecans shown in A. Error bars, S.D.
The effect of the HMC-1-derived perlecan and its fragments on the proliferation and/or migration of endothelial cells was investigated. The activated HMC-1 supernatant stimulated the proliferation of endothelial cells to the same extent as VEGF165 but not to the same degree as adding exogenous FGF2 (Fig. 8A). Migration was measured as a component of the expansion of endothelial cells in “scratch assays.” The activated HMC-1 supernatant had a significant stimulatory effect on the migration of endothelial cells (Fig. 8, A–E). The effect observed was similar to that obtained by adding FGF2 to the cultures and significantly better than that achieved by adding VEGF165 (Fig. 8, B and E, images labeled FGF2). A significant amount of the migration activity present in the activated HMC-1 supernatant was inhibited by the addition of antibodies that blocked the function of either FGF2 or VEGF165, suggesting that these growth factors were present in the HMC-1 supernatant and were involved either directly or indirectly in the stimulation of endothelial cell migration (Fig. 8C). When the migration assay was performed in the presence of activated HMC-1 supernatant and antibodies against perlecan, the stimulatory activity in the HMC-1 supernatant was inhibited (Fig. 8D). The activity was blocked by the addition of any of the anti-perlecan antibodies tested, which was controlled for by the addition of an isotype control antibody that had no effect. The stimulatory activity present in activated HMC-1 supernatant was also inhibited by the addition of the anti-endorepellin antibodies (Fig. 8, D and E, Activated HMC-1 supernatant + anti-endorepellin), suggesting that it is not only the turnover of endogenous endothelal cell-derived perlecan that was involved in the migration process but also that the stimulatory activity residing in the HMC-1 supernatant was due to endorepellin-containing fragments of perlecan.
FIGURE 8.

Perlecan promotes endothelial cell migration. A, proliferation of endothelial cells over 7 days in the presence of medium supplemented with FGF2, VEGF165, or activated HMC-1 supernatant compared with cells exposed to medium only. Cell proliferation was measured by crystal violet and presented as mean ± S.D. (error bars) (n = 3). B, endothelial cell migration measured over 28 h in the presence of medium supplemented with FGF2, VEGF165, or activated HMC-1 supernatant compared with cells exposed to medium only. Cell migration was measured by a scratch assay and monitoring the migration of cells into the scratched area. Data are presented as percentage of cell migration, which indicates the scratched area at a given time as a proportion of the initial scratched area. C, endothelial cell migration measured over 28 h in the presence of medium supplemented with activated HMC-1 supernatant, activated HMC-1 supernatant, and anti-FGF2 antibodies (AB1435) or anti-VEGF165 antibodies (ab38473) compared with cells exposed to medium only. Cell migration was measured by a scratch assay and monitoring the migration of cells into the scratched area. Data are presented as percentage of cell migration, which indicates the scratched area at a given time as a proportion of the initial scratched area. D, endothelial cell migration measured over 28 h in the presence of medium supplemented with activated HMC-1 supernatant, activated HMC-1 supernatant, and anti-perlecan antibodies (clone 5D7-2E4 or 7B5 or anti-endorepellin), or anti-perlecan antibodies only (clone 5D7-2E4 or 7B5 or anti-endorepellin) compared with cells exposed to medium only. Cell migration was measured by a scratch assay and monitoring the migration of cells into the scratched area. Data are presented as percentage of cell migration, which indicates the scratched area at a given time as a proportion of the initial scratched area. E, phase-contrast images of the scratched area as times 0, 5, and 28 h after the scratch was generated for endothelial cells exposed to medium supplemented with FGF2, activated HMC-1 supernatant, activated HMC-1 supernatant, and anti-endorepellin compared with cells exposed to medium only. Scale bar, 150 μm.
Analysis of the mRNA for perlecan expressed by HMC-1 cells was analyzed initially by standard RT-PCR over 35 cycles and then with qPCR. The domain-specific primer sets used in these experiments were designed to span exons 2 and 7 from the N terminus (domain I), exons 29–37 from the laminin-like region of the protein core in domain III, and exons 87–97 from the C terminus (domain V) (Table 1). These domains were selected because they matched the domain-specific monoclonal antibodies utilized in the experiments described in Fig. 4. mRNA from HCAECs was used to confirm the presence of transcripts from all three domains and as a control demonstrating successful priming (Fig. 9). Using standard RT-PCR, similar amounts of transcripts were generated from endothelial cell-derived mRNA for all three domain I primer sets at the expected sizes (compare Table 1 and Fig. 9A). Transcripts generated from mRNA isolated from the HMC-1 cells demonstrated a shorter than expected product using the exon 2 and 3 primer set for domain I, no product at all for the exon 2–5 primers, and a shorter than expected product by almost 300 bp for the exon 2–7 primers of domain I (Fig. 9A, HMC-1). These data indicated that although exons 2, 3, 6, and 7 were present, because the exon 7 reverse primer spanned the exon/intron boundaries between exons 6 and 7, there were splicing events occurring affecting exons 4 and 5. DNA sequence analysis of the HMC-1 product using the exon 3–7 primer set indicated a 97% sequence alignment with the HSPG2 gene (Table 1). The sizes of the major PCR products generated from the endothelium-derived mRNA using the two domain III primer sets were as expected, although there was some evidence of smaller minor smears in both cases (compare Table 1 with Fig. 9B, HCAEC). However, no transcripts were detected for HMC-1-derived mRNA using the same two primer sets (Fig. 9B, HMC-1), indicating that regions of domain III were spliced out in the HMC-1 cells. The use of the exon 87–97 primer set produced a major band of the expected size as well as minor smaller sized bands, whereas the exon 89–94 primer set produced a single product at the expected size (compare Table 1 with Fig. 9C, HCAEC). These same primers produced single products at the expected sizes for HMC-1 mRNA (Fig. 9C, HMC-1). DNA sequence analysis of the HMC-1 product generated using the exon 89–94 primer set indicated a 99% sequence alignment with the HSPG2 gene (Table 1). The reduced intensity of the PCR product for HMC-1 cells compared with the endothelial cells suggested that there were different amounts of transcripts between the cell types, which were further investigated by qPCR using the exon 89–94 primer set. By normalizing the cycle times for the primer set to the GAPDH cycle time in each cell type, an estimate of the -fold change in domain expression between the two different cell types was obtained. There were ∼60 times fewer transcripts corresponding to domain V in the HMC-1 mRNA samples compared with the HCAEC mRNA and 1000 times fewer transcripts corresponding to domain I.
FIGURE 9.
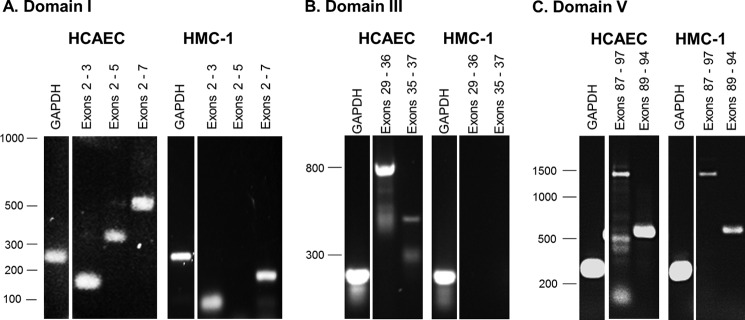
mRNA expression of perlecan using domain specific primers. mRNA derived from both HMC-1 cells and HCAECs was isolated and used to generate cDNA, which was amplified using domain-specific primers and electrophoresed on 1% (w/v) agarose gels. Products from the GAPDH primer set were electrophoresed on each gel to indicate that the same amount of cDNA was loaded on each gel for each cell type. A, PCR products for domain I primer sets, including exons 2 and 3 (77 bp), 2–5 (333 bp), and 2–7 (510 bp). B, PCR products for domain III primer sets, including exons 29–36 (796 bp) and 35–37 (454 bp). C, PCR products for domain V primer sets, including exons 87–97 (1406 bp) and 89–94 (578 bp).
DISCUSSION
Both of the mast cell lines, HMC-1 and RBL-2H3, used in this study synthesized HS, heparin, and CS, which all localized to the intracellular granules in a similar fashion to the proteoglycan, serglycin. The localization of these glycosaminoglycans (GAGs) did not change significantly after the activation of these cells in vitro, which is known to release the serglycin-GAG-bound mediators. It has been well established in the literature that the predominant proteoglycan present in the intracellular granules of mast cells is serglycin and that the major function of the GAG chains decorating its protein core is the packaging of mediators, such as proteases like tryptase, within the granules (24, 25). The expression of serglycin has been shown to be tightly linked to the differentiation of mast cells together with the expression of Golgi membrane-anchored enzymes responsible for the addition of the GAG side chains (26). Mast cells isolated from human lung synthesized significant amounts of heparin containing a high proportion of the highly sulfated disaccharides 2SO4IdoA-6SO4GlcNSO4 and 2SO4IdoA/GlcA-3SO46SO4GlcNSO4 characteristic of this type of GAG as well as CS. The major CS disaccharide is GlcA-4SO46SO4GalNAc, which is also known as CS type E (27, 28). Human gastric mucosal mast cells (29) and the RBL-2H3 cell line have been shown previously to synthesize a similar type of CS with similar GAG composition (30, 31) but not heparin. This supports the idea that these cells have the ability to switch GAG type depending upon their microenvironment, which was reproduced in vitro by co-culturing mast cells with fibroblasts (32).
Both HMC-1 and RBL-2H3 cells synthesized perlecan, which was localized intracellularly probably in the Golgi. Unlike serglycin, it was not stored in granules intracellularly and was localized most likely to the Golgi, where it was secreted into the medium. Mast cells have been shown previously to produce the major components of basement membranes, including perlecan, using a cDNA probe that recognizes a region in domain III of the protein core (16). In the studies described here, we were unable to generate a product corresponding to domain III using mRNA from mast cells and did not demonstrate reactivity of the mast cell-derived perlecan with an antibody against domain III (33). This domain of the perlecan protein core has been shown to be present in basement membranes of most of the major organs (34).
Perlecan produced by the primary mast cells and mast lines was detected as the full-length proteoglycan with a protein core of 460 kDa and ∼120 kDa of GAG chains, giving a total molecular mass of ∼580–600 kDa, and bands at 300 and 130 kDa that were both reactive with a domain V-specific monoclonal antibody as well as a polyclonal anti-endorepellin antibody. The 130 kDa band was also decorated with ∼20–30 kDa of CS. N-terminal sequencing of fragments within the 130 kDa band showed that it was a mix of peptides corresponding to sequences in domains II, III, IV, and V, suggesting the presence of the perlecan protein core. The production of perlecan with various molecular mass species similar to those described in this paper have been described previously for human (35) and bovine (35, 36) cartilage extracts, but the fragments were not characterized with respect to what proteases might be responsible for the cleavage patterns demonstrated. N-terminal sequencing of the bands described here revealed many putative cleavage sites of most of the major classes of proteases, including the serine and cysteine proteases as well as the metalloproteinases. Perlecan has been shown previously to be cleaved proteolytically into fragments by thrombin and MMP3 (37), which is supported by the data presented in this paper showing multiple potential MMP3 cleavage sites distributed throughout the protein core and most notably throughout domains III, IV, and V. The cleavage of a recombinant form of domain V referred to as endorepellin by BMP-1/Tolloid-like metalloprotease (38) or cathepsin L (39) has been shown to release the LG3 fragment, which was first identified in the urine of end stage renal failure patients (40) and has since been described in the urine of mine workers (41) and the plasma of both control and breast cancer patients (42). In this study, however, N-terminal peptide sequences corresponding to either endorepellin or the LG3 peptide were not identified.
The 300- and 130-kDa mast cell-derived perlecan fragments were reactive against the anti-endorepellin antibodies, suggesting that these fragments contained domain V that has been shown to interact with the α2β1 integrin (43) and more recently with the VEGF receptor type 2 (8). Fractions enriched for these fragments promoted the adhesion of endothelial cells via the α2β1 integrin and their proliferation to the same extent as either endothelial cell-derived perlecan or recombinant domain V. Initially, endorepellin was described as an anti-angiogenic fragment due to its inhibitory activity toward the α2β1 integrin site when added in a soluble form to the medium of endothelial cell cultures. However, recently, it has been shown to be adherent for platelets via the same α2β1 integrin (44) and pro-angiogenic in the brain after ischemic injury in rats (45), which was due to the lack of the α2β1 integrin in the brain microvascular endothelial cells. When the α2 integrin subunit was reintroduced into the brain microvascular endothelial cells, endorepellin was anti-angiogenic (45), suggesting that the effects of endorepellin are tissue- and cell context-specific. The α2β1 integrin binding site within domain V has been shown to be present in the LG3 region (43), which was released into the culture medium of apoptotic endothelial cells, which itself had anti-apoptotic effects on fibroblasts that were linked to the activation of Src and Fyn/phosphatidylinositol 3-kinases but independent of the activation of focal adhesion kinase (46). More recently, the LG3 fragment has been shown to have a similar anti-apoptotic activity toward mesenchymal stem cells (47) as well as stimulating the migration of vascular smooth muscle cells (48) via ERK1/2-dependent pathways. The study described here demonstrated that mast cells produced alternatively spliced forms of perlecan as well as processing full-length perlecan into fragments that were important for the migration of endothelial cells because the addition of anti-perlecan antibodies to the mast cell supernatants inhibited the stimulatory activity present in this fraction. This was most likely due to the binding of the protein core by the antibodies, which prevented the fragmentation.
Examination of the mast cell-derived mRNA in this study showed that there were relatively fewer transcripts for perlecan from HMC-1 mRNA compared with HCAEC. The perlecan gene, HSPG2, is arranged into 97 exons (49) that are translated in order into the protein domains of the core (50). The HSPG2 gene was shown to be under the control of a single promoter ∼450 bp in the 5′ direction from the translation start site, which was shown to be regulated by transcription factors downstream of TGFβ signaling (51). Alternative splicing of the HSPG2 gene has been reported in the NCBI database, Aceview, as well as during murine development (52). A shorter variant arising from splicing events in domain I was identified previously and referred to as “miniperl” in the NCBI Nucleotide database. However, none of the fragments described above have been characterized with respect to potential biological functions, and none have predicted molecular masses that aligned with the molecular masses of the fragments described herein. It is possible to speculate that the perlecan gene in HMC-1 cells has undergone mRNA splicing events resulting in transcripts missing regions of domains I and III, which may then result in perlecan protein cores that correspond to the sizes of the protein fragments detected in this study. The C. elegans homologue of the HSPG2 gene, Unc-52, has been shown to be alternatively spliced, producing shorter truncated forms lacking the C terminus (53). This splicing event was shown to be controlled by the Mec-8 (54) and Smu-1 (55) genes. A human homologue of Smu-1 has been described and termed fZAP57, which is ∼62% identical in nucleotide sequence. It has been localized to the nucleus and has RNA binding motifs suggesting that it might be part of the spliceosome (56). However, it awaits further investigation to determine if it performs this function in human cells and whether it is up-regulated in mast cells.
It has been hypothesized that the residence time of mast cells in wound sites affects the outcome by modulating events in the resolution phase that involve contraction of collagen in the wound bed. It was thought that this was mostly due to tryptase, which degrades the extracellular matrix and promotes angiogenesis. However, co-culturing mast cells and fibroblasts led to the contraction of collagen gels that was independent of tryptase activity (57), suggesting that other factors were responsible. The LG3 domain, an integral part of the C-terminal domain of perlecan is anti-apoptotic for fibroblasts (46), which might result in an increased output of the extracellular matrix. This study shows that mast cells produce and process perlecan into shorter forms that can modulate endothelial cell adhesion and migration and supports the hypothesis that mast cells promote fibrosis by producing pro-fibrotic forms of perlecan. Given that mast cells are important to wound healing and that their prolonged presence may cause keloid scars, an understanding of what maintains their numbers will be fundamental to controlling the scarring seen at sites of wounding.
Acknowledgments
We acknowledge the technical assistance of Dr. Bonny Tsoi and Michael Lee (Graduate School of Biomedical Engineering, University of New South Wales) as well as Dr. Christine Chuang (Heart Research Institute, Sydney, Australia).
This work was supported by partial funding from the Australian Research Council under the Linkage Project (Grant LP0776293).
- HS
- heparan sulfate
- PMA
- phorbol 12-myristate 13-acetate
- CS
- chondroitin sulfate
- DPBS
- Dulbecco's PBS
- KS
- keratan sulfate
- HCAEC
- human coronary arterial endothelial cell
- Bicine
- N,N-bis(2-hydroxyethyl)glycine
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- MMP
- matrix metalloproteinase
- GAG
- glycosaminoglycan
- qPCR
- quantitative real-time PCR.
REFERENCES
- 1. Whitelock J. M., Iozzo R. V. (2005) Heparan sulfate. A complex polymer charged with biological activity. Chem. Rev. 105, 2745–2764 [DOI] [PubMed] [Google Scholar]
- 2. Knox S., Melrose J., Whitelock J. (2001) Electrophoretic, biosensor, and bioactivity analyses of perlecans of different cellular origins. Proteomics 1, 1534–1541 [DOI] [PubMed] [Google Scholar]
- 3. Patel V. N., Knox S. M., Likar K. M., Lathrop C. A., Hossain R., Eftekhari S., Whitelock J. M., Elkin M., Vlodavsky I., Hoffman M. P. (2007) Heparanase cleavage of perlecan heparan sulfate modulates FGF10 activity during ex vivo submandibular gland branching morphogenesis. Development 134, 4177–4186 [DOI] [PubMed] [Google Scholar]
- 4. Chuang C. Y., Lord M. S., Melrose J., Rees M. D., Knox S. M., Freeman C., Iozzo R. V., Whitelock J. M. (2010) Heparan sulfate-dependent signaling of fibroblast growth factor 18 by chondrocyte-derived perlecan. Biochemistry 49, 5524–5532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mongiat M., Taylor K., Otto J., Aho S., Uitto J., Whitelock J. M., Iozzo R. V. (2000) The protein core of the proteoglycan perlecan binds specifically to fibroblast growth factor-7. J. Biol. Chem. 275, 7095–7100 [DOI] [PubMed] [Google Scholar]
- 6. Smith S. M., West L. A., Hassell J. R. (2007) The core protein of growth plate perlecan binds FGF-18 and alters its mitogenic effect on chondrocytes. Arch. Biochem. Biophys. 468, 244–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Woodall B. P., Nyström A., Iozzo R. A., Eble J. A., Niland S., Krieg T., Eckes B., Pozzi A., Iozzo R. V. (2008) Integrin α2β1 is the required receptor for endorepellin angiostatic activity. J. Biol. Chem. 283, 2335–2343 [DOI] [PubMed] [Google Scholar]
- 8. Goyal A., Pal N., Concannon M., Paul M., Doran M., Poluzzi C., Sekiguchi K., Whitelock J. M., Neill T., Iozzo R. V. (2011) Endorepellin, the angiostatic module of perlecan, interacts with both the α2β1 integrin and vascular endothelial growth factor receptor 2 (VEGFR2). A dual receptor antagonism. J. Biol. Chem. 286, 25947–25962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Diegelmann R. F., Evans M. C. (2004) Wound healing. An overview of acute, fibrotic and delayed healing. Front. Biosci. 9, 283–289 [DOI] [PubMed] [Google Scholar]
- 10. Hawkins R. A., Claman H. N., Clark R. A., Steigerwald J. C. (1985) Increased dermal mast cell populations in progressive systemic sclerosis. A link in chronic fibrosis? Ann. Intern. Med. 102, 182–186 [DOI] [PubMed] [Google Scholar]
- 11. Samoszuk M., Kanakubo E., Chan J. K. (2005) Degranulating mast cells in fibrotic regions of human tumors and evidence that mast cell heparin interferes with the growth of tumor cells through a mechanism involving fibroblasts. BMC Cancer 5, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Goto T., Befus D., Low R., Bienenstock J. (1984) Mast cell heterogeneity and hyperplasia in bleomycin-induced pulmonary fibrosis of rats. Am. Rev. Respir. Dis. 130, 797–802 [DOI] [PubMed] [Google Scholar]
- 13. Artuc M., Hermes B., Steckelings U. M., Grützkau A., Henz B. M. (1999) Mast cells and their mediators in cutaneous wound healing. Active participants or innocent bystanders? Exp. Dermatol. 8, 1–16 [DOI] [PubMed] [Google Scholar]
- 14. Braga T., Grujic M., Lukinius A., Hellman L., Abrink M., Pejler G. (2007) Serglycin proteoglycan is required for secretory granule integrity in mucosal mast cells. Biochem. J. 403, 49–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ng M. F. (2010) The Role of Mast Cells in Wound Healing. Int. Wound J. 7, 55–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thompson H. L., Burbelo P. D., Gabriel G., Yamada Y., Metcalfe D. D. (1991) Murine mast cells synthesize basement membrane components. A potential role in early fibrosis. J. Clin. Invest. 87, 619–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Passante E., Ehrhardt C., Sheridan H., Frankish N. (2009) RBL-2H3 cells are an imprecise model for mast cell mediator release. Inflamm. Res. 58, 611–618 [DOI] [PubMed] [Google Scholar]
- 18. Alkhouri H., Hollins F., Moir L. M., Brightling C. E., Armour C. L., Hughes J. M. (2011) Human lung mast cells modulate the functions of airway smooth muscle cells in asthma. Allergy 66, 1231–1241 [DOI] [PubMed] [Google Scholar]
- 19. Nilsson G., Blom T., Kusche-Gullberg M., Kjellén L., Butterfield J. H., Sundström C., Nilsson K., Hellman L. (1994) Phenotypic characterization of the human mast-cell line HMC-1. Scand. J. Immunol. 39, 489–498 [DOI] [PubMed] [Google Scholar]
- 20. Caterson B., Christner J. E., Baker J. R. (1983) Identification of a monoclonal antibody that specifically recognized corneal and skeletal keratan sulfate. Monoclonal antibodies to cartilage proteoglycan. J. Biol. Chem. 258, 8848–8854 [PubMed] [Google Scholar]
- 21. Theocharis A. D., Seidel C., Borset M., Dobra K., Baykov V., Labropoulou V., Kanakis I., Dalas E., Karamanos N. K., Sundan A., Hjerpe A. (2006) Serglycin constitutively secreted by myeloma plasma cells is a potent inhibitor of bone mineralisation in vitro. J. Biol. Chem. 281, 35116–35128 [DOI] [PubMed] [Google Scholar]
- 22. Whitelock J. M., Graham L. D., Melrose J., Murdoch A. D., Iozzo R. V., Underwood P. A. (1999) Human perlecan immuno-purified from different endothelial cell sources has different adhesive properties for vascular cells. Matrix Biol. 18, 163–178 [DOI] [PubMed] [Google Scholar]
- 23. Knox S., Merry C., Stringer S., Melrose J., Whitelock J. (2002) Not all perlecans are created equal. Interactions with fibroblast growth factor (FGF) 2 and FGF receptors. J. Biol. Chem. 277, 14657–14665 [DOI] [PubMed] [Google Scholar]
- 24. Matsumoto R., Sali A., Ghildyal N., Karplus M., Stevens R. L. (1995) Packaging of proteases and proteoglycans in the granules of mast cells and other hematopoietic cells. A cluster of histidines on mouse mast cell protease 7 regulates its binding to heparin serglycin proteoglycans. J. Biol. Chem. 270, 19524–19531 [DOI] [PubMed] [Google Scholar]
- 25. Henningsson F., Hergeth S., Cortelius R., Abrink M., Pejler G. (2006) A role for serglycin proteoglycan in granular retention and processing of mast cell secretory granule components. FEBS J. 273, 4901–4912 [DOI] [PubMed] [Google Scholar]
- 26. Duelli A., Rönnberg E., Waern I., Ringvall M., Kolset S. O., Pejler G. (2009) Mast cell differentiation and activation is closely linked to expression of genes coding for the serglycin proteoglycan core protein and a distinct set of chondroitin sulfate and heparin sulfotransferases. J. Immunol. 183, 7073–7083 [DOI] [PubMed] [Google Scholar]
- 27. Stevens R. L., Avraham S., Gartner M. C., Bruns G. A., Austen K. F., Weis J. H. (1988) Isolation and characterization of a cDNA that encodes the peptide core of the secretory granule proteoglycan of human promyelocytic leukemia HL-60 cells. J. Biol. Chem. 263, 7287–7291 [PubMed] [Google Scholar]
- 28. Thompson H. L., Schulman E. S., Metcalfe D. D. (1988) Identification of chondroitin sulfate E in human lung mast cells. J. Immunol. 140, 2708–2713 [PubMed] [Google Scholar]
- 29. Gilead L., Livni N., Eliakim R., Ligumsky M., Fich A., Okon E., Rachmilewitz D., Razin E. (1987) Human gastric mucosal mast cells are chondroitin sulphate E-containing mast cells. Immunology 62, 23–28 [PMC free article] [PubMed] [Google Scholar]
- 30. Katz H. R., Austen K. F., Caterson B., Stevens R. L. (1986) Secretory granules of heparin-containing rat serosal mast cells also possess highly sulfated chondroitin sulfate proteoglycans. J. Biol. Chem. 261, 13393–13396 [PubMed] [Google Scholar]
- 31. Seldin D. C., Austen K. F., Stevens R. L. (1985) Purification and characterization of protease-resistant secretory granule proteoglycans containing chondroitin sulfate di-B and heparin-like glycosaminoglycans from rat basophilic leukemia cells. J. Biol. Chem. 260, 11131–11139 [PubMed] [Google Scholar]
- 32. Levi-Schaffer F., Austen K. F., Gravallese P. M., Stevens R. L. (1986) Coculture of interleukin 3-dependent mouse mast cells with fibroblasts results in a phenotypic change of the mast cells. Proc. Natl. Acad. Sci. U.S.A. 83, 6485–6488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Murdoch A. D., Liu B., Schwarting R., Tuan R. S., Iozzo R. V. (1994) Widespread expression of perlecan proteoglycan in basement membranes and extracellular matrices of human tissues as detected by a novel monoclonal antibody against domain III and by in situ hybridization. J. Histochem. Cytochem. 42, 239–249 [DOI] [PubMed] [Google Scholar]
- 34. Couchman J. R., Ljubimov A. V., Sthanam M., Horchar T., Hassell J. R. (1995) Antibody mapping and tissue localization of globular and cysteine-rich regions of perlecan domain III. J. Histochem. Cytochem. 43, 955–963 [DOI] [PubMed] [Google Scholar]
- 35. Melrose J., Roughley P., Knox S., Smith S., Lord M., Whitelock J. (2006) The structure, location, and function of perlecan, a prominent pericellular proteoglycan of fetal, postnatal, and mature hyaline cartilages. J. Biol. Chem. 281, 36905–36914 [DOI] [PubMed] [Google Scholar]
- 36. West L., Govindraj P., Koob T. J., Hassell J. R. (2006) Changes in perlecan during chondrocyte differentiation in the fetal bovine rib growth plate. J. Orthop. Res. 24, 1317–1326 [DOI] [PubMed] [Google Scholar]
- 37. Whitelock J. M., Murdoch A. D., Iozzo R. V., Underwood P. A. (1996) The degradation of human endothelial cell-derived perlecan, and release of bound bFGF, by stromelysin, plasmin and heparanases. J. Biol. Chem. 271, 10079–10086 [DOI] [PubMed] [Google Scholar]
- 38. Gonzalez E. M., Reed C. C., Bix G., Fu J., Zhang Y., Gopalakrishnan B., Greenspan D. S., Iozzo R. V. (2005) BMP-1/Tolloid-like metalloproteases process endorepellin, the angiostatic C-terminal fragment of perlecan. J. Biol. Chem. 280, 7080–7087 [DOI] [PubMed] [Google Scholar]
- 39. Cailhier J. F., Sirois I., Laplante P., Lepage S., Raymond M. A., Brassard N., Prat A., Iozzo R. V., Pshezhetsky A. V., Hébert M. J. (2008) Caspase-3 activation triggers extracellular cathepsin L release and endorepellin proteolysis. J. Biol. Chem. 283, 27220–27229 [DOI] [PubMed] [Google Scholar]
- 40. Oda O., Shinzato T., Ohbayashi K., Takai I., Kunimatsu M., Maeda K., Yamanaka N. (1996) Purification and characterization of perlecan fragment in urine of end-stage renal failure patients. Clin. Chim. Acta 255, 119–132 [DOI] [PubMed] [Google Scholar]
- 41. Parker T. J., Sampson D. L., Broszczak D., Chng Y. L., Carter S. L., Leavesley D. I., Parker A. W., Upton Z. (2012) A fragment of the LG3 peptide of endorepellin is present in the urine of physically active mining workers. A potential marker of physical activity. PLoS One 7, e33714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chang J. W., Kang U. B., Kim D. H., Yi J. K., Lee J. W., Noh D. Y., Lee C., Yu M. H. (2008) Identification of circulating endorepellin LG3 fragment. Potential use as a serological biomarker for breast cancer proteomics. Clin. Appl. 2, 23–32 [DOI] [PubMed] [Google Scholar]
- 43. Bix G., Fu J., Gonzalez E. M., Macro L., Barker A., Campbell S., Zutter M. M., Santoro S. A., Kim J. K., Höök M., Reed C. C., Iozzo R. V. (2004) Endorepellin causes endothelial cell disassembly of actin cytoskeleton and focal adhesions through α2β1 integrin. J. Cell. Biol. 166, 97–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bix G., Iozzo R. A., Woodall B., Burrows M., McQuillan A., Campbell S., Fields G. B., Iozzo R. V. (2007) Endorepellin, the C-terminal angiostatic module of perlecan, enhances collagen-platelet responses via the alpha2beta1-integrin receptor. Blood 109, 3745–3748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee B., Clarke D., Al Ahmad A., Kahle M., Parham C., Auckland L., Shaw C., Fidanboylu M., Orr A. W., Ogunshola O., Fertala A., Thomas S. A., Bix G. J. (2011) Perlecan domain V is neuroprotective and proangiogenic following ischemic stroke in rodents. J. Clin. Invest. 121, 3005–3023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Laplante P., Raymond M. A., Labelle A., Abe J., Iozzo R. V., Hébert M. J. (2006) Perlecan proteolysis induces an α2β1 integrin- and Src family kinase-dependent anti-apoptotic pathway in fibroblasts in the absence of focal adhesion kinase activation. J. Biol. Chem. 281, 30383–30392 [DOI] [PubMed] [Google Scholar]
- 47. Soulez M., Sirois I., Brassard N., Raymond M. A., Nicodème F., Noiseux N., Durocher Y., Pshezhetsky A. V., Hébert M. J. (2010) Epidermal growth factor and perlecan fragments produced by apoptotic endothelial cells co-ordinately activate ERK1/2-dependent antiapoptotic pathways in mesenchymal stem cells. Stem Cells 28, 810–820 [DOI] [PubMed] [Google Scholar]
- 48. Soulez M., Pilon E. A., Dieudé M., Cardinal H., Brassard N., Qi S., Wu S. J., Durocher Y., Madore F., Perreault C., Hébert M. J. (2012) The perlecan fragment LG3 is a novel regulator of obliterative remodeling associated with allograft vascular rejection. Circ. Res. 110, 94–104 [DOI] [PubMed] [Google Scholar]
- 49. Cohen I. R., Grässel S., Murdoch A. D., Iozzo R. V. (1993) Structural characterization of the complete human perlecan gene and its promoter. Proc. Natl. Acad. Sci. U.S.A. 90, 10404–10408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Murdoch A. D., Dodge G. R., Cohen I., Tuan R. S., Iozzo R. V. (1992) Primary structure of the human heparan sulfate proteoglycan from basement membrane (HSPG2/perlecan). A chimeric molecule with multiple domains homologous to the low density lipoprotein receptor, laminin, neural cell adhesion molecules, and epidermal growth factor. J. Biol. Chem. 267, 8544–8557 [PubMed] [Google Scholar]
- 51. Iozzo R. V., Pillarisetti J., Sharma B., Murdoch A. D., Danielson K. G., Uitto J., Mauviel A. (1997) Structural and functional characterization of the human perlecan gene promoter. Transcriptional activation by transforming growth factor-β via a nuclear factor 1-binding element. J. Biol. Chem. 272, 5219–5228 [DOI] [PubMed] [Google Scholar]
- 52. Joseph S. J., Ford M. D., Barth C., Portbury S., Bartlett P. F., Nurcombe V., Greferath U. (1996) A proteoglycan that activates fibroblast growth factors during early neuronal development is a perlecan variant. Development 122, 3443–3452 [DOI] [PubMed] [Google Scholar]
- 53. Mullen G. P., Rogalski T. M., Bush J. A., Gorji P. R., Moerman D. G. (1999) Complex patterns of alternative splicing mediate the spatial and temporal distribution of perlecan/UNC-52 in Caenorhabditis elegans. Mol. Biol. Cell. 10, 3205–3221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lundquist E. A., Herman R. K., Rogalski T. M., Mullen G. P., Moerman D. G., Shaw J. E. (1996) The mec-8 gene of C. elegans encodes a protein with two RNA recognition motifs and regulates alternative splicing of unc-52 transcripts. Development 122, 1601–1610 [DOI] [PubMed] [Google Scholar]
- 55. Spike C. A., Shaw J. E., Herman R. K. (2001) Analysis of smu-1, a gene that regulates the alternative splicing of unc-52 pre-mRNA in Caenorhabditis elegans. Mol. Cell. Biol. 21, 4985–4995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Spartz A. K., Herman R. K., Shaw J. E. (2004) SMU-2 and SMU-1, Caenorhabditis elegans homologs of mammalian spliceosome-associated proteins RED and fSAP57, work together to affect splice site choice. Mol. Cell. Biol. 24, 6811–6823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Sköld C. M., Ohkuni Y., Liu X. D., Numerof R., Rennard S. I. (2001) Co-cultured human mast cells stimulate fibroblast-mediated contraction of collagen gels. Inflammation 25, 47–51 [DOI] [PubMed] [Google Scholar]
- 58. Tapanadechopone P., Hassell J. R., Rigatti B., Couchman J. R. (1999) Localization of glycosaminoglycan substitution sites on domain V of mouse perlecan. Biochem. Biophys. Res. Commun. 265, 680–690 [DOI] [PubMed] [Google Scholar]
- 59. Mongiat M., Sweeney S. M., San Antonio J. D., Fu J., Iozzo R. V. (2003) Endorepellin, a novel inhibitor of angiogenesis derived from the C terminus of perlecan. J. Biol. Chem. 278, 4238–4249 [DOI] [PubMed] [Google Scholar]