

Background: Arrestin-1 with enhanced binding to unphosphorylated active rhodopsin (Rh*) has therapeutic potential.
Results: Manipulation of the rhodopsin binding surface of arrestin-1 greatly increases its binding to Rh*.
Conclusion: Stable arrestin-1 with high binding to Rh* can be engineered with and without the ability to self-associate.
Significance: The affinity of arrestin-1 for Rh* and its propensity to oligomerize can be independently changed by targeted mutagenesis.
Keywords: 7-Helix Receptor, Arrestin, Protein Design, Protein Engineering, Retina, Signal Transduction
Abstract
Arrestin-1 preferentially binds active phosphorylated rhodopsin. Previously, a mutant with enhanced binding to unphosphorylated active rhodopsin (Rh*) was shown to partially compensate for lack of rhodopsin phosphorylation in vivo. Here we showed that reengineering of the receptor binding surface of arrestin-1 further improves the binding to Rh* while preserving protein stability. In mammals, arrestin-1 readily self-associates at physiological concentrations. The biological role of this phenomenon can only be elucidated by replacing wild type arrestin-1 in living animals with a non-oligomerizing mutant retaining all other functions. We demonstrate that constitutively monomeric forms of arrestin-1 are sufficiently stable for in vivo expression. We also tested the idea that individual functions of arrestin-1 can be independently manipulated to generate mutants with the desired combinations of functional characteristics. Here we showed that this approach is feasible; stable forms of arrestin-1 with high Rh* binding can be generated with or without the ability to self-associate. These novel molecular tools open the possibility of testing of the biological role of arrestin-1 self-association and pave the way to elucidation of full potential of compensational approach to gene therapy of gain-of-function receptor mutations.
Introduction
Rod photoreceptors mediate night vision in vertebrates. Rods demonstrate remarkable single photon sensitivity (1) and unusual reproducibility of a single photon response (2–4). These features require biochemically perfect signaling cascade with virtually no noise, enormous amplification, and extremely rapid shutoff. Light activates rhodopsin, which then sequentially catalyzes GDP/GTP exchange on numerous molecules of its cognate G protein transducin (5). Within 30–60 ms, active rhodopsin (Rh*)2 is phosphorylated by GRK1, whereupon visual arrestin-13 specifically binds light-activated phosphorylated rhodopsin (P-Rh*), ensuring timely signaling shutoff (6, 7).
Genetic defects in this rapid and efficient two-step shutoff mechanism invariably result in visual disorders. Loss-of-function mutations in arrestin-1 (8) or GRK1 (9) cause Oguchi disease, a form of stationary night blindness. Mutations in rhodopsin that lead to the loss of GRK1 phosphorylation sites result in retinal degeneration (10). Although in principle loss-of-function mutations can be cured by gene replacement therapy, in the case of gain-of-function defects, such as the loss of phosphorylation sites resulting in unquenchable signaling, viable therapeutic strategies remain to be developed.
We have recently proposed a compensational approach using enhanced arrestin-1 mutant that binds Rh* with much higher affinity than wild type (WT) arrestin-1 to reduce excessive signaling by unphosphorylated rhodopsin (11, 12). Although, the first proof-of-principle study showed that compensational gene therapy works in vivo, it also revealed the limitations of existing phosphorylation-independent arrestin-1 mutants (11). Enhanced arrestin-1 improved functional performance of phosphorylation-deficient rods, facilitated their recovery, and prolonged their survival, but “compensated” shutoff was still significantly slower than in WT animals with normal Rh* phosphorylation (11). Thus, improved enhanced arrestin-1 mutants with higher affinity binding to Rh* and sufficient stability for in vivo expression have to be designed.
Mammalian arrestin-1 at physiological concentrations found in rods (13–15) robustly self-associates, forming dimers and tetramers (16–18). Only monomeric arrestin-1 binds rhodopsin (18), because receptor binding surfaces in the tetramer and both possible dimers are shielded by sister subunits (19). Arrestin-1 is the only signaling protein in the visual system that forms inactive “storage” oligomers, and the biological significance of this phenomenon is unclear (20). To elucidate the role of arrestin-1 self-association in photoreceptors, the consequences of the replacement of WT arrestin-1 with a constitutively monomeric mutant in vivo must be determined. In addition, it is unknown whether oligomerization needs to be preserved in a therapeutically effective enhanced mutant with high Rh* affinity. These in vivo experiments require a stable constitutively monomeric version of arrestin-1 as well as enhanced mutants both with normal and absent ability to oligomerize.
Here we report extensive redesign of the rhodopsin binding surface of arrestin-1 that yielded mutants with significantly higher affinity for unphosphorylated Rh*. We identified further enhanced arrestins, constitutively monomeric forms, and a mutant that combines inability to self-associate with high Rh* binding, all of which are stable. These proteins are promising candidates for in vivo evaluation of the potential of compensational gene therapy and for elucidation of the role of arrestin-1 oligomerization.
EXPERIMENTAL PROCEDURES
Materials
[γ-32P]ATP, [14C]leucine, and [3H]leucine were from PerkinElmer Life Sciences. All restriction and DNA modifying enzymes were from New England Biolabs (Ipswich, MA). Rabbit reticulocyte lysate was from Ambion (Austin, TX), and SP6 RNA polymerase was prepared as described (21). Cell culture reagents and media were from Mediatech (Manassas, VA) or Invitrogen. All other reagents were from Sigma.
Mutagenesis and Plasmid Construction
For in vitro transcription mouse arrestin-1 (generous gift of Dr. Cheryl Craft, University of California) was subcloned into pGEM2 (Promega, Madison, WI) with “idealized” 5-UTR (21) between NcoI and HindIII sites as described (70). All mutations were introduced in transcription construct by PCR using the strategy previously described (23). All constructs were confirmed by dideoxy sequencing. For the expression in HEK293 cells, coding sequences with 5′-UTR were excised from pGEM2 constructs using EcoRI and HindIII restriction sites and subcloned into pcDNA3 vector (Invitrogen) with a modified multiple cloning site as described previously (24, 25). In vitro transcription, translation, and preparation of phosphorylated and unphosphorylated rhodopsin were performed as described recently (26, 27).
A direct binding assay was performed as described (26). Briefly, 1 nm arrestin-1 (50 fmol) was incubated with 0.3 μg of P-Rh* in 50 μl of 50 mm Tris-HCL, pH 7.4, 100 mm potassium acetate, 1 mm EDTA, 1 mm DTT for 5 min at 37 °C under room light. Samples were cooled on ice, and bound and free arrestin-1 was separated at 4 °C by gel filtration on a 2-ml column of Sepharose 2B-CL. Arrestin-1 eluting with rhodopsin-containing membranes was quantified by liquid scintillation counting. Nonspecific “binding” was determined in samples where rhodopsin was omitted and subtracted.
In Vitro Arrestin Stability Assay
Translated radiolabeled arrestin-1 was incubated for 0.5–4 h at the indicated temperatures and cooled on ice. The binding of arrestin-1 in these samples to P-Rh* was compared with that of control sample kept on ice as described above except that 2 nm arrestin-1 (100 fmol/sample) was used.
Cell-based Stability Assay
HEK293 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% serum as described (28). Cells were transfected with 3 μg of DNA per well of a 6-well plate using Lipofectamine according to manufacturer's instructions. After 36 h cells were serum-starved overnight and then kept at 37 °C (control) or at 40 °C for 3 h. Cells were washed with PBS and non-enzymatically detached from plates with 2 × 500 μl of Versene. Cells were pelleted by centrifugation at 4 °C for 10 min at 300 rpm (Eppendorf centrifuge 5417R) and resuspended in 400 μl of 50 mm Tris-HCl, pH 7.4, 100 mm NaCl, 1 mm EDTA, 1 mm DTT supplemented with Complete protease inhibitors mixture (Roche Applied Science) and 1 mm PMSF. Cells were lysed by sonication (Sonic Dismembrator Model 500, Fisher Scientific) 3 × 5 s at 30% power. Unbroken cells and debris were pelleted by centrifugation at 4 °C for 10 min at 14,000 rpm (Eppendorf centrifuge 5417R), and the supernatant was frozen in aliquots and stored at −80 °C until used. Total protein was measured using Bradford reagent (Bio-Rad), and the expression of different forms of arrestin-1 was determined by Western blot with anti-arrestin rabbit polyclonal antibody (14). To determine functionality of soluble arrestins, equal volumes of lysates (10 μl) were incubated with or without (nonspecific) 1 μg of P-Rh* in 50 μl of 50 mm Tris-HCL, pH 7.4, 100 mm potassium acetate, 1 mm EDTA, 1 mm DTT for 5 min at 37 °C in room light. The samples were cooled on ice, loaded onto 0.1 ml of 0.2 m sucrose in binding buffer, and centrifuged 20 min at 100,000 rpm in Beckman TL-120 tabletop ultracentrifuge (rotor TLA120.1). Supernatants were removed, and pellets were dissolved in 50 μl of SDS sample buffer. One-tenth of each sample along with one-tenth of the input were subjected to SDS-PAGE and analyzed by Western blot with anti-arrestin antibody (14) followed by HRP-conjugated anti-rabbit secondary antibody, with detection by SuperSignal Pico (Pierce). Nonspecific binding (the amount of arrestin-1 pelleted in the absence of rhodopsin, likely due to aggregation, was subtracted from binding to P-Rh*. Protein survival was calculated as the ratio of the fraction of arrestin-1 in lysate of cells incubated at 40 °C that specifically bound to P-Rh* to the bound fraction in lysates of control cells kept at 37 °C.
Arrestin-1 Purification and Analysis of Its Self-association
Mouse arrestin-1 mutants were expressed in Escherichia coli and purified essentially as described (29). All light-scattering measurements were made with a DAWN EOS detector coupled to an Optilab T-rEX refractometer (Wyatt Technologies) after gel filtration on a QC-PAK GFC 300 column (7.8 mm inner diameter × 15 cm) (Tosoh Bioscience). The arrestin samples (100 μl) at different concentrations were incubated in fresh 5 mm DTT for 30 min at room temperature to disrupt covalent inter-arrestin disulfide bonds and injected onto the column at 25 °C at a flow rate of 0.6 ml/min in 50 mm MOPS, 100 mm NaCl, pH 7.2. The column did not resolve oligomeric species but simply acted as a filter to remove highly scattering particulates. Light scattering at 7 angles (72–126°), absorbance at 280 nm, and refractive index (at 658 nm) for each sample were taken for a narrow slice centered at the peak of the elution profile (18). The experimental weight-averaged molecular weight values were obtained from the protein concentration and light scattering data using ASTRA 5.3.4.20 software (Wyatt Technologies). The weight-averaged molecular weight data were analyzed using the two-step monomer-dimer-tetramer (MDT) model,
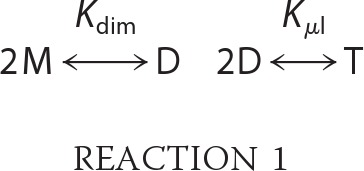 |
where M, D, and T are monomer, dimer, and tetramer, respectively. The data for arrestin-1 carrying F86A/F198A mutations did not fit the MDT model and was fitted using the monomer-dimer model instead. Details of the analysis have been previously described (30, 70). Except where noted, the equilibrium constants are given in terms of the corresponding dissociation constants, KD,dimer (KD,dim) and KD,tetramer (KD,tet). The errors in equilibrium constants were determined from least-squares fitting of the data to MDT or monomer-dimer model taking into account an estimated error of ± 1 kDa in the computed values of the average molecular weight (70).
RESULTS
The first enhanced arrestin-1 mutant used in vivo to compensate for the defect in rhodopsin phosphorylation was preactivated simply by the elimination of one of the conformational constraints that holds arrestin-1 in its basal state (11). Triple alanine substitution of the bulky hydrophobic residues anchoring the C-tail (yielding arrestin-1-3A mutant) promotes its detachment, thereby facilitating arrestin-1 transition into a high affinity rhodopsin binding state and arrestin-1 binding to Rh* (31). The replacement of WT arrestin-1 in GRK1-deficient mice with the arrestin-1-3A mutant resulted in an ∼3-fold acceleration of Rh* shutoff but did not bring the recovery rate to the WT level (11). Single cell recording from rods expressing the arrestin-1-3A mutant suggested that it allowed Rh* reactivation (11). Thus, arrestin-1-3A complexes with Rh* were not stable enough to survive longer than it takes Rh* to decay to opsin.
Elimination of Phosphate Binding Positive Charges Does Not Enhance Arrestin-1 Binding to Rh*
Multiple positive charges in bovine arrestin-1, including Lys-14, Lys-15 (32), Arg-18 (33), Lys-163, Lys-166, Lys-167, Arg-171, and Arg-175 (34) were shown to bind rhodopsin-attached phosphates. Mouse arrestin-1 also carries all of these positive charges: Lys-15, Lys-16, Arg-19, Lys-164, Lys-167, Lys-168, Arg-172, and Arg-176. Therefore, we hypothesized that a mismatch between the presence of these positive charges on the receptor binding surface of arrestin-1 and the absence of negatively charged phosphates on the C terminus of Rh* destabilized the complex. To test this idea, we replaced phosphate binding lysines and arginines (Fig. 1A) with neutral hydrophilic glutamine with hydrogen-bonding capability, introducing K15Q, K16Q, R19Q, R172Q, K164Q, K167Q, and K168Q mutations. Although R176E mutation per se in mouse arrestin-1 significantly increases the binding to Rh*, it was not introduced because previously we found that it greatly reduces protein stability (11). Neutralizing mutations were introduced, singly or in groups, in the context of the two arrestin-1 mutants that were successfully expressed in transgenic mice, arrestin-1-3A and arrestin-1-(1–377) (11, 13, 14, 35). As expected, on both backgrounds, mutations eliminating phosphate binding charges significantly reduced arrestin-1 ability to interact with dark P-Rh (Fig. 1). Combinations K15Q/K16Q and K15Q/K16Q/R19Q also reduced the binding to P-Rh*, but only R172Q mutation marginally enhanced Rh* binding (Fig. 1, B, C, and D). These data suggest that the presence of phosphate binding-positive charges is not the main factor destabilizing the complex of enhanced arrestin-1 with Rh*.
FIGURE 1.

Elimination of phosphate-binding residues does not improve the binding of phosphorylation-independent arrestin-1 mutants to Rh*. A, the structural model of mouse arrestin-1 (based on bovine arrestin-1 crystal structure PDB 1CF1 (37)) shows surface residues mutated in this study (blue, positive charges; red, negative charge). B–D, shown is the binding of WT mouse arrestin-1 and indicated N-domain (residues 1–177) mutants, where positively charged phosphate binding residues were replaced with glutamine, constructed on the background of either arrestin-1-(L374A/V375A/F376A) (3A) or truncated arrestin-1-(1–377) (Tr) to four functional forms of rhodopsin. The colors of the bars are: white, dark P-Rh; black, P-Rh*; horizontally striped, dark Rh; green, Rh*. The means ± S.D. of two experiments, each performed in duplicate, are shown. Note that R172Q is the only mutation that marginally increases Rh* binding of parental L374A/V375A/F376A and truncated arrestin-1-(1–377) mutants.
Selected Mutations on Arrestin-1 Surface Engaged by Other Parts of the Receptor Increase Rh* Binding but Reduce Thermal Stability
Several mutations on the receptor binding surface of the C-domain, such as K257E and E346K, were previously shown to increase bovine arrestin-1 binding to Rh* in the context of WT protein (36). Therefore, we introduced homologous mutations K258E and E347K in mouse arrestin-1 (Fig. 2A). These substitutions proved to act additively with the 3A mutation and the deletion of the C-tail, enhancing arrestin-1 binding to P-Rh* (Fig. 2A), and especially to Rh* well beyond the level achieved by the 3A mutation or C-terminal deletion alone. Because the effects of all mutations tested were essentially the same on the background of arrestin-1-3A and arrestin-1-(1–377) (Figs. 1, B, C, and D, and 2A), we focused on arrestin-1-3A, which showed higher expression levels in transgenic mice (14). On arrestin-1-3A background the addition of R172Q mutation to K258E, E347K, or K258E/E347K combination further increased Rh* binding without appreciably affecting the interaction with P-Rh* (Fig. 2B). The best combination (3A+K258E/E347K/R172Q) yielded the level of Rh* binding virtually indistinguishable from that of WT arrestin-1 to P-Rh* (Fig. 2B).
FIGURE 2.

Reengineering of the rhodopsin binding surface of the C-domain improves Rh* binding of phosphorylation-independent arrestin-1 base mutants but reduces protein stability. A and B, shown is binding of WT mouse arrestin-1 and the indicated C-domain (residues 190–360) mutants constructed on the background of either arrestin-1-(L374A,V375A,F376A) (3A) or truncated arrestin-1-(1–377) (Tr) to four functional forms of rhodopsin. Note that K258E and E347K mutations and their combination significantly increase Rh* binding of parental enhanced mutants. The colors of the bars are: white, dark P-Rh; black, P-Rh*; horizontally striped, dark Rh; green, Rh*.Bars are color-coded, as in Fig. 1. Statistical significance of the differences in Rh* binding (p < 0.05), as compared with parental L374A/V375A/F376A or truncated arrestin-1-(1–377) mutant, is indicated by green star. C, to determine protein stability, indicated translated arrestins were incubated for 30 or 60 min at 39 °C, and their specific binding to P-Rh* was compared with that of controls kept on ice (0 min). Note that substitutions, which further increase Rh* binding, significantly reduce the stability of parental L374A/V375A/F376A and truncated arrestin-1-(1–377) mutants. The means ± S.D. of two-three experiments, each performed in duplicate, are shown in all panels.
Because many activating mutations reduce arrestin-1 stability (11, 31) and only sufficiently stable proteins can be successfully expressed in vivo at physiologically relevant levels (11), next we tested the stability of these mutants (Fig. 2C). To this end, radiolabeled WT mouse arrestin-1 and mutants produced in cell-free translation were incubated at elevated temperature for varying periods of time, and then their ability to specifically bind P-Rh* was compared with that of the control samples kept on ice. WT arrestin-1 as well as the 3A and truncated (1–377) mutants did not lose any activity after 60 min of incubation at 39 °C (Fig. 2C). Mutations E347K, K258E, and their combination somewhat reduced arrestin-1 stability on both backgrounds, and the addition of R172Q to any mutation or combination further reduced arrestin-1 stability (Fig. 2C). Thus, the most potent ligands of Rh* in this series are less stable than the 3A mutant successfully used in vivo (11).
Neutralization of Surface Charges Is Less Detrimental for Stability than Charge Reversal
Surface residues in soluble proteins often stabilize the structure via intramolecular interactions with neighboring side chains. Therefore, we analyzed the available crystal structure of highly homologous bovine arrestin-1 (37) and then ascertained that the residues of interest are conserved in the mouse protein (Fig. 3A shows mouse residue numbers). R172Q reduced the stability of all arrestin-1 mutants tested (Fig. 2). Arg-172 appears to form a salt bridge with Glu-149, which in turn contacts Lys-151. The R172Q mutation is likely to weaken or destroy the interaction of this residue with Glu-149, which would in this case become fully engaged by Lys-151. We reasoned that replacing Lys-151 with aspartic acid would likely prevent that, making the interaction via H-bond between Glu-149 and Gln-172 more likely. Importantly, the homologous K150E mutation in bovine arrestin-1 did not significantly affect the binding to P-Rh* or other forms of rhodopsin (36). Another reason the R172Q mutation could reduce the stability is inappropriate H-bonding of Gln with some of the neighboring residues. We reasoned that replacing Arg-172 with an alanine should solve this problem. The K258E and E347K mutations also appear to be detrimental for arrestin-1 stability (Fig. 2). Judging by bovine arrestin-1 structure, Lys-258 forms a salt bridge with Asp-139 (Fig. 3A), which would be destroyed by K258E mutation introducing a negative charge that repulses Asp-139. The replacement of Lys-258 with a neutral glutamine would prevent the repulsion and likely create an opportunity for H-bonding to preserve the stabilizing effect of the salt bridge in the WT protein. Glu-347 forms a salt bridge with Lys-333 and H-bond with Thr-335 (Fig. 3A). Lys-347, introduced by E347K mutation, would repulse Lys-333; in addition, its longer side chain allows the end amino group to reach all the way to Ser-345. The modeling shows that replacement of Glu-347 with glutamine, which has exactly the same geometry and no charge, would prevent both repulsion of Lys-333 and unnatural H-bonding to Ser-345. The replacement of Glu-347 with histidine, which has only a partial positive charge at neutral pH and shorter side chain, would moderate the repulsion of Lys-333 and also prevent H-bonding with Ser-345.
FIGURE 3.

Design of stable arrestin-1 mutants with high Rh* binding. A, a structural model of mouse arrestin-1 (based on bovine arrestin-1 crystal structure 1CF1 (37)) show the residues mutated in this series. B and C, shown is binding of WT mouse arrestin-1 and the indicated mutants constructed on the background of arrestin-1-(L374A,V375A,F376A) (3A) to P-Rh* (black bars) and Rh* (green bars). D, indicated translated arrestins were incubated for 30, 60, or 120 min at 39 °C, and their specific binding to P-Rh* was compared with that of controls kept on ice (0 min). Note that charge neutralization mutations preserve the stability much better than charge reversals. The means ± S.D. of two experiments each performed in duplicate are shown in all panels.
To test these predictions, on the arrestin-1-3A background we added K151D mutation to the R172Q/E347K and R172Q/K258E combinations, made the R172A/E347K combination to compare with R172Q/E347K, and generated three “milder” versions of the K258E/E347K combination: K258E/E347Q, K258E/E347H, and K258Q (Fig. 3B). Because two additional mutations, R291E and Q328K, increased the binding of bovine arrestin-1 to Rh* (36), we constructed homologous mutants of mouse arrestin-1: R292E, its less drastic version R292Q, and H329K (note that in WT mouse protein bovine Gln-328 is replaced with His-329). The data show that the H329K and R292E mutations increase Rh* binding, whereas the R292Q does not; the R172A mutation is less effective than the R172Q; the K151D mutation decreases Rh* binding of any combination it is added to; the combinations K258Q/E347K and K258E/E347H work as well as the original K258E/E347K (Fig. 3B). Based on these data, we generated several combinations of the most effective mutations and constructed the K258Q/E347H, K258Q/E347H/R292E, K258Q/E347H /H329K, and K258Q/E347H/R292E/H329K combination mutants on the arrestin-1-3A background (Fig. 3C). We found that R292E does not act additively with the K258Q/E347H combination, whereas in the context of the K258Q/E347H/H329K mutant it actually reduces Rh* binding (Fig. 3C). Interestingly, the H329K mutation significantly increased the binding of the K258Q/E347H mutant to both P-Rh* and Rh*. Moreover, the absolute level of the K258Q/E347H/H329K mutant binding to Rh* is even higher than achieved with P-Rh* by WT arrestin-1, making it by far the most potent Rh* ligand constructed (Fig. 3C).
Next, we tested the stability of the combination mutants yielding high Rh* binding, extending the incubation time to 2 h to make the test more rigorous (Fig. 3D). The results showed that the R172Q mutation reduces the stability of the K258E/E347H combination to the same extent as that of K258E/E347K (compare Figs. 2C and 3D). We also confirmed that the K258E/E347K combination is significantly less stable than parental arrestin-1-3A and found that both the K258Q/E347H and K258Q/E347H/H329K mutants essentially retain the stability of arrestin-1-3A. Considering that the arrestin-1-3A was successfully expressed in transgenic mice at fairly high levels (11), these data suggest that both the K258Q/E347H and K258Q/E347H/H329K mutants might be stable enough for in vivo expression.
Mutant Stability in Cells Follows the Same Pattern as in the in Vitro Assay
To closer mimic the intracellular environment that these proteins will be facing in rod photoreceptors, we expressed several arrestins in HEK293 cells. We chose the K258Q/E347H+3A mutant, the K258Q/E347H/H329K+3A mutant, with stable WT arrestin-1 and the arrestin-1-3A mutant as positive controls as well as the much less stable K258E/E347H/R172Q+3A mutant for comparison. Three hours before lysis one plate with cells expressing each mutant was kept at 37 °C (control), whereas a second plate was incubated at 40 °C. The cells were lysed and centrifuged to remove debris and aggregated proteins. To measure possible loss due to aggregation during the 3-h incubation at 40 °C, 2 μg of total protein from each cell lysate was analyzed by Western blot with rabbit polyclonal anti-arrestin antibodies (14). We found a slight reduction of soluble arrestin-1 level in the lysates of cells incubated at 40 °C, which in no case exceeded that observed for WT protein (Fig. 4A). Next, we tested the ability of these proteins to bind P-Rh* in our standard extensively characterized direct binding assay (26, 27, 31–36, 38–43) using lysates of cells expressing various mutants (Fig. 4B). Arrestin-containing lysates were incubated for 5 min at 37 °C with or without 1 μg of P-Rh in room light, and rhodopsin-containing membranes were pelleted by centrifugation. We compared the total amount of arrestin-1 added (input (I)) with that bound to P-Rh* (Rh) or non-specifically aggregated (ns) in samples containing no rhodopsin. In all cases at least half of the arrestin added bound P-Rh*, and very little was pelleted in the absence of rhodopsin (Fig. 4B). All mutants performed similarly to WT, with the exception of the K258E/E347H/R172Q+3A mutant, which demonstrated increased nonspecific aggregation and lower specific binding, in agreement with its reduced stability in the in vitro survival test (Fig. 3D). The ratio of specifically bound fraction of each arrestin from cells incubated at 40 and 37 °C was used to estimate protein survival (Fig. 4B). This parameter was found to be ∼100% for WT arrestin-1 and 3A mutant, >80% for the K258Q/E347H+3A mutant, and lower for the other two mutants (Fig. 4B). These results match in vitro survival test (Fig. 3D) remarkably well. Both tests suggest that the K258Q/E347H+3A mutant, with significantly higher Rh* binding than 3A (Fig. 3C), demonstrates stability comparable to the 3A mutant (Figs. 3D and 4B).
FIGURE 4.

Several mutants with the highest Rh* binding are stable in cellular environment. A, indicated forms of mouse arrestin-1 were expressed in HEK293 cells. For the last 3 h before lysis the cells were kept either at regular (37 °C) or elevated (40 °C) temperature. The levels of soluble arrestin-1 in lysates were determined by Western blot (upper panel) and expressed as percent of the level of WT arrestin-1 in cells kept at 37 °C (lower bar graph). B, functional activity of soluble arrestin-1 in lysates is shown. Equal volumes of lysates (10 μl) were incubated with (Rh) or without (ns) 1 μg of P-Rh*, and bound arrestin-1 was separated from free by centrifugation as described under “Experimental Procedures.” One-tenth of each pellet and input (I) was analyzed by Western blot (upper panel). Protein survival was calculated as the ratio of the fraction of arrestin-1 in lysate of cells incubated at 40 °C that specifically bound to P-Rh* to the bound fraction in lysates of control cells kept at 37 °C. Nonspecific binding (the amount of arrestin-1 pelleted in the absence of rhodopsin, likely due to aggregation) was subtracted from specific binding to P-Rh*. The means ± S.D. of two independent experiments, each quantified in two blots, are shown in both panels.
Elimination of Arrestin-1 Self-association Is Compatible with Enhanced Rh* Binding
Arrestin-1 that retains normal ability to bind P-Rh* but does not self-associate is necessary to test the biological role of its oligomerization in photoreceptors. Therefore, we performed both cell-based and in vitro stability tests with the two previously designed constitutively monomeric mouse arrestin-1 mutants, F86A/F198A and F86A/F198A/A349V (19, 70). We found that these mutants express at least as well as WT arrestin-1 in HEK293 cells, and both survive 3 h of incubation of cells at 40 °C at least as well as WT (Fig. 5A). The fraction of the two mutants in cell lysates that specifically bound P-Rh* was similar to that of WT arrestin-1 (Fig. 5B). Both constitutively monomeric mutants showed essentially the same ∼100% survival, estimated as the ratio of specifically bound fraction of WT and mutant arrestin-1 from cells incubated at 40 and 37 °C (Fig. 5B). In full agreement with these data, the in vitro testing at 39 °C of both constitutive monomers demonstrated even higher stability than 3A mutant (Fig. 5C). Thus, in contrast to activating mutations, substitutions disabling self-association of arrestin-1 do not appear to adversely affect protein stability (Figs. 3–5).
FIGURE 5.

High stability of constitutively monomeric forms of arrestin-1. A, indicated forms of mouse arrestin-1 were expressed in HEK293 cells. For the last 3 h before lysis the cells were kept either at regular (37 °C) or elevated (40 °C) temperatures. The levels of soluble arrestin-1 in lysates were determined by Western blot (upper panel) and expressed as percent of the level of WT arrestin-1 in cells kept at 37 °C (lower bar graph). B, functional activity of soluble arrestin-1 in lysates was determined as described under “Experimental Procedures” and in legend to Fig. 4. Protein survival was calculated as the ratio of the fraction of arrestin-1 in the lysate of cells incubated at 40 °C that specifically bound to P-Rh* to the bound fraction in lysates of control cells kept at 37 °C. Quantification in panels A and B represents the means of two independent experiments, each quantified in two blots. ns, without P-Rh*. C, the indicated translated arrestins were incubated for 60, 120, or 240 min at 39 °C, and their specific binding to P-Rh* was compared with that of controls kept on ice (0 min). The means ± S.D. of two independent experiments are shown.
Mutations preventing oligomerization of arrestin-1 introduced on WT background were shown not to affect the binding to rhodopsin and microtubules (70). On the basis of the multifunctionality of arrestin proteins (44–46) and demonstrated feasibility of changing individual functions without affecting others (28, 47–49), it was suggested that combinatorial mutagenesis can generate arrestins with essentially any desired set of functional characteristics (12, 46). To test this idea, we combined the double mutation F86A/F198A that renders arrestin-1 constitutively monomeric with several mutations enhancing binding to Rh*, including 3A, K258Q/E347H+3A and K258Q/E347H/H329K+3A, and compared these proteins with parental mutants where only a single function was changed (Fig. 6). We found that although the addition of F86A/F198A to enhancing mutations does not appreciably affect P-Rh* interaction, the binding to Rh* of constitutively monomeric-enhanced arrestins was somewhat reduced compared with the parental forms where the same activating mutations were introduced on WT background (Fig. 6A). However, arrestin-1 carrying combinations of K258Q/E347H+3A and K258Q/E347H/H329K+3A with F86A/F198A largely retained its high binding to unphosphorylated Rh* (Fig. 6A). Finally, to determine the suitability of these mutants for in vivo testing, we performed harsher in vitro stability test, measuring the survival of these proteins at 42 °C (Fig. 6B). At this temperature even WT arrestin-1 shows some inactivation, whereas the arrestin-1-3A mutant, which was successfully expressed in rods of transgenic mice, lost ∼50% of activity by 4 h (Fig. 6B). These conditions also revealed the difference between the two constitutive monomers: F86A/F198A is almost as stable as WT, whereas F86A/F198A/A349V demonstrated ∼50% survival, similar to 3A (Fig. 6B). The F86A/F198A double mutation somewhat reduced the stability of the less robust K258Q/E347H/H329K+3A, and it did not significantly affect the survival of K258Q/E347H+3A (Fig. 6B). This test revealed that the stability of K258Q/E347H+3A on both WT and constitutively monomeric background is comparable to that of 3A mutant. Thus, both forms of enhanced K258Q/E347H+3A as well as constitutively monomeric mutants are suitable candidates for in vivo expression.
FIGURE 6.

Enhanced binding to Rh* can be achieved with and without ability to self-associate. A, the binding of WT mouse arrestin-1 and the indicated mutants to P-Rh* (black bars) and Rh* (white bars) are shown. B, to rigorously compare protein stability, indicated translated arrestins were incubated for 60, 120, or 240 min at 42 °C, and their specific binding to P-Rh* was compared with that of controls kept on ice (0 min). The means ± S.D. of two independent experiments performed in duplicate are shown in both panels.
Because the effect of the F86A/F198A double mutation on arrestin-1 self-association was only tested on the WT background (70), we determined the ability to oligomerize of the enhanced K258Q/E347H+3A mutant and the combination of K258Q/E347H+3A with F86A/F198A. To this end both proteins were expressed in E. coli and purified, and their self-association was quantitatively tested by multiangle light scattering (18, 19, 70). We found that the K258Q/E347H+3A mutant retained the ability to form dimers and tetramers (Fig. 7). However, its self-association was reduced relative to WT mouse arrestin-1 (70). Although the data were readily fitted to the MDT model, log Kdim and log Ktet were 3.35 ± 0.05 and 3.85 ± 0.28, which translates into KD,dim and KD,tet of 447 ± 7 and 141 ± 10 μm, respectively. Similar to what we previously found for mouse arrestin-1-F86A/F198A, the data for the K258Q/E347H/H329K+3A + F86A/F198A combination mutant (Fig. 7) could not be fitted by the MDT model but were fitted by a simpler monomer-dimer model, with log Kdim = 2.43 + 0.16, which corresponds to KD,dim = 3,715 ± 245 μm. Thus, on both WT (70) and the K258Q/E347H+3A background (Fig. 7), the F86A/F198A double mutation reduced dimerization about 10-fold and totally eliminated tetramerization.
FIGURE 7.

Self-association of engineered mouse arrestin-1 mutants. A, the structural model of mouse arrestin-1 (based on bovine arrestin-1 crystal structure 1CF1 (37)) shows two residues mutated to enhance Rh* binding (Lys-258 and Glu-347) in red and two phenylalanines (Phe-86 and Phe-198) mutated to alanines to block self-association in blue. B, the average molecular weight of the K258Q/E347H+3A (red circles) and K258Q/E347H+3A+F86A/F198A (blue circles) mouse arrestin-1 mutants as a function of total arrestin-1 concentration was determined from the light scattering data (symbols). The fit of the data to the MDT model (dotted lines) was obtained as described (18). Note that K258Q/E347H+3A+F86A/F198A mutant showed no detectable tetramerization, so that the resulting fit describes monomer-dimer equilibrium.
The data (Figs. 6 and 7) show that self-association and binding to unphosphorylated Rh* are not completely independent of each other, which could be expected as the mutations necessary to change both functions localize to the concave sides of the two arrestin-1 domains (Fig. 7A). Nonetheless, targeted reengineering of mouse arrestin-1 yielded new forms where two of its functional characteristics, the level of Rh* binding, and the ability to self-associate, were differentially affected, generating expected combinations.
DISCUSSION
Rhodopsin is a prototypical class A G protein-coupled receptor (GPCR). GPCRs are the largest and arguably the most structurally and functionally diverse family of signaling proteins in mammals (50, 51), with ∼800 subtypes in humans and other primates and much larger numbers in most mammals (SEVENS database). Mutations in different GPCRs underlie a variety of genetic disorders (52). In particular, about a dozen gain-of-function mutations resulting in excessive receptor signaling underlie disorders ranging from retinal degeneration (10) to dwarfism, hypo- and hyper-thyroidism, and several forms of cancer (52). In some cases, constitutive activity of the receptor results in its excessive phosphorylation and association with arrestin. Constitutive desensitization of human vasopressin receptor mutant underlies nephrogenic diabetes insipidus (53), whereas in the case of rhodopsin it results in night blindness or retinal degeneration (54).
Despite technical challenges, the treatment strategy for loss-of-function mutations is conceptually straightforward; gene replacement therapy introducing a functional version of the affected protein is the most logical approach, as was recently demonstrated in the case of Leber congenital amaurosis associated with deficit of RPE65 (55–58). However, gain-of-function mutations pose a conceptual problem; these mutations are dominant, which means that the second perfectly normal allele does not help. Theoretically, mutant mRNA can be targeted by appropriately designed ribozyme that spares normal mRNA. The ribozyme must be extremely efficient, as even low expression of non-phosphorylatable rhodopsin induces retinal degeneration (59). In addition, it has to be very selective, because mutant and normal mRNA sometimes differ by as little as a single nucleotide, as in case of frameshift mutations (10). The combination of these characteristics may not be practical.
The only alternative proposed so far is compensation: balancing a receptor that signals too much with hyperactive arrestin or a receptor that cannot be phosphorylated with a phosphorylation-independent arrestin mutant (for review, see Refs. 12 and 46). However logical this sounds, a recent proof-of-principle study testing this approach in the visual system in vivo demonstrated that existing enhanced arrestins are not effective enough to achieve full compensation (11). Here we describe extensive targeted redesign of the rhodopsin-binding surface of arrestin-1 to generate mutant forms with much higher affinity for unphosphorylated Rh* that have a potential to achieve more effective phosphorylation-independent shutoff. We found that the combination of a triple alanine substitution that forcibly detached the arrestin C-tail from the body of the molecule and facilitated arrestin transition into the active high affinity receptor binding state (31, 60–62) with a re-engineered rhodopsin binding surface yields mutants that bind Rh* better than WT arrestin-1 binds its natural target, P-Rh* (Fig. 2). However, many activating mutations reduce thermal stability of the protein (Figs. 2, 3, 4, and 6). We found that less drastic changes are not as detrimental for protein stability (Figs. 2 and 3). In particular, replacement of charge reversal with charge neutralization, such as changing K258E to K258Q or E347K to E347H, preserves most of the gain in Rh* binding while improving protein survival at elevated temperatures (Figs. 3 and 4). Similarly, the H329K mutation, which moderately increases the positive charge in this position, does not adversely affect arrestin-1 stability (Fig. 3). Moreover, the K258Q/E347H+3A mutant shows equally impressive stability in vitro and in cells subjected to elevated temperatures (Figs. 3, 4, and 6), which makes it a good candidate for in vivo testing and a promising tool for gene therapy of genetic disorders associated with defects in rhodopsin phosphorylation. It should be noted that we only tested the binding of these mutants to unphosphorylated light-activated WT rhodopsin. Additional tests with actual disease-causing rhodopsin mutants, particularly the truncated form lacking the C terminus (10), are necessary to fully evaluate therapeutic potential of enhanced phosphorylation-independent arrestins.
Bovine, mouse, and human arrestin-1 robustly self-associate at physiological concentrations, forming dimers and tetramers (70). Yet only monomeric arrestin-1 can bind P-Rh* (18). Arrestin-1 is present in rods and cones at very high concentrations (13–15), exceeding those achieved by non-visual arrestins in any cell (63, 64) by 4 orders of magnitude. Its propensity to oligomerize in different mammalian species suggests that this phenomenon must be biologically important. The most straightforward approach to elucidation of the role of arrestin-1 self-association in photoreceptor physiology is the replacement of WT protein with a constitutively monomeric mutant that retains all other normal arrestin-1 functions. We showed that mouse arrestin-1 with mutations disabling its self-association is stable enough for this purpose. Moreover, we showed that deficient oligomerization can be successfully combined with increased ability to bind unphosphorylated Rh*, identifying stable enhanced mutants that can be generated with or without the ability to self-associate. These are the first proof-of-principle results demonstrating the feasibility of engineering proteins with desired combinations of functional characteristics. Depending on the role of arrestin-1 self-association in photoreceptors, the most therapeutically efficient enhanced mutants could be those that either self-associate or lack this ability. Molecular tools described here will help to answer these important biological questions.
Remarkable sequence similarity in the arrestin family (45) along with conservation of the three-dimensional structure among all four vertebrate subtypes (33, 37, 65, 66) suggests that homologous substitutions in non-visual arrestins will likely similarly enhance the binding of arrestin-2 and -3 to unphosphorylated forms of their cognate GPCRs. Enhanced forms of arrestin-2 and -3 proved to be useful tools to study GPCR signaling (60, 61, 67) and trafficking (47). The versions of inherently promiscuous non-visual arrestins with increased specificity for individual receptor subtypes (23, 27, 68) have therapeutic potential in disorders associated with excessive GPCR signaling (12, 20, 46).
Targeted redesign of signaling proteins and regulatory pathways (11, 12, 22, 28, 46, 48, 69) has been proposed as a novel therapeutic approach for diseases where signaling imbalance underlies the pathology. However, activating mutations often destabilize the basal conformation, thereby reducing protein stability. This is a critical difficulty in developing more active mutants engineered to rebalance cell signaling. Here we demonstrate that protein reengineering guided by high resolution structure allows one to create mutants with desired functional characteristics that retain a significant proportion of the stability of parental WT protein perfected over millions of years by evolution.
Acknowledgments
We are grateful to Dr. Cheryl M. Craft for mouse arrestin-1 cDNA and Dr. Rosalie K. Crouch and NEI, National Institutes of Health, for 11-cis-retinal.
This work was supported, in whole or in part, by National Institutes of Health Grants EY011500, GM077561, and GM081756 (to V. V. G.), and EY05216 (to W. L. H.), and GM079419 and GM095633 (to T. M. I.). This work was also supported by and the Jules Stein Professorship Endowment (to W. L. H.).
We use systematic names of arrestin proteins: arrestin-1 (historic names S-antigen, 48-kDa protein, visual, or rod arrestin), arrestin-2 (β-arrestin or β-arrestin1), arrestin-3 (β-arrestin2 or hTHY-ARRX), and arrestin-4 (cone or X-arrestin; for unclear reasons its gene is called arrestin 3 in the HUGO database).
- Rh*
- active rhodopsin
- P-Rh*
- light-activated phosphorylated rhodopsin
- MDT
- monomer-dimer-tetramer
- GPCR
- G protein-coupled receptor.
REFERENCES
- 1. Baylor D. A., Lamb T. D., Yau K. W. (1979) Responses of retinal rods to single photons. J. Physiol. 288, 613–634 [PMC free article] [PubMed] [Google Scholar]
- 2. Rieke F., Baylor D. A. (1998) Origin of reproducibility in the responses of retinal rods to single photons. Biophys. J. 75, 1836–1857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Caruso G., Bisegna P., Lenoci L., Andreucci D., Gurevich V. V., Hamm H. E., DiBenedetto E. (2010) Kinetics of rhodopsin deactivation and its role in regulating recovery and reproducibility of rod photoresponse. PLoS Comput. Biol. 6, e1001031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Caruso G., Bisegna P., Andreucci D., Lenoci L., Gurevich V. V., Hamm H. E., DiBenedetto E. (2011) Identification of key factors that reduce the variability of the single photon response. Proc. Natl. Acad. Sci. U.S.A. 108, 7804–7807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Burns M. E., Baylor D. A. (2001) Activation, deactivation, and adaptation in vertebrate photoreceptor cells. Annu. Rev. Neurosci. 24, 779–805 [DOI] [PubMed] [Google Scholar]
- 6. Krispel C. M., Chen D., Melling N., Chen Y. J., Martemyanov K. A., Quillinan N., Arshavsky V. Y., Wensel T. G., Chen C. K., Burns M. E. (2006) RGS expression rate-limits recovery of rod photoresponses. Neuron 51, 409–416 [DOI] [PubMed] [Google Scholar]
- 7. Gross O. P., Burns M. E. (2010) Control of rhodopsin active lifetime by arrestin-1 expression in mammalian rods. J. Neurosci. 30, 3450–3457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fuchs S., Nakazawa M., Maw M., Tamai M., Oguchi Y., Gal A. (1995) A homozygous 1-base pair deletion in the arrestin gene is a frequent cause of Oguchi disease in Japanese. Nat. Genet. 10, 360–362 [DOI] [PubMed] [Google Scholar]
- 9. Yamamoto S., Sippel K. C., Berson E. L., Dryja T. P. (1997) Defects in the rhodopsin kinase gene in the Oguchi form of stationary night blindness. Nat. Genet. 15, 175–178 [DOI] [PubMed] [Google Scholar]
- 10. Apfelstedt-Sylla E., Kunisch M., Horn M., Ruther K., Gerding H., Gal A., Zrenner E. (1993) Ocular findings in a family with autosomal dominant retinitis pigmentosa and a frameshift mutation altering the carboxyl terminal sequence of rhodopsin. Br. J. Ophthalmol. 77, 495–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Song X., Vishnivetskiy S. A., Gross O. P., Emelianoff K., Mendez A., Chen J., Gurevich E. V., Burns M. E., Gurevich V. V. (2009) Enhanced arrestin facilitates recovery and protects rods lacking rhodopsin phosphorylation. Curr. Biol. 19, 700–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gurevich V. V., Gurevich E. V. (2010) Custom-designed proteins as novel therapeutic tools? The case of arrestins. Expert. Rev. Mol. Med. 12, e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hanson S. M., Gurevich E. V., Vishnivetskiy S. A., Ahmed M. R., Song X., Gurevich V. V. (2007) Each rhodopsin molecule binds its own arrestin. Proc. Natl. Acad. Sci. U.S.A. 104, 3125–3128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Song X., Vishnivetskiy S. A., Seo J., Chen J., Gurevich E. V., Gurevich V. V. (2011) Arrestin-1 expression level in rods. Balancing functional performance and photoreceptor health. Neuroscience 174, 37–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Strissel K. J., Sokolov M., Trieu L. H., Arshavsky V. Y. (2006) Arrestin translocation is induced at a critical threshold of visual signaling and is superstoichiometric to bleached rhodopsin. J. Neurosci. 26, 1146–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schubert C., Hirsch J. A., Gurevich V. V., Engelman D. M., Sigler P. B., Fleming K. G. (1999) Visual arrestin activity may be regulated by self-association. J. Biol. Chem. 274, 21186–21190 [DOI] [PubMed] [Google Scholar]
- 17. Imamoto Y., Tamura C., Kamikubo H., Kataoka M. (2003) Concentration-dependent tetramerization of bovine visual arrestin. Biophys. J. 85, 1186–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hanson S. M., Van Eps N., Francis D. J., Altenbach C., Vishnivetskiy S. A., Arshavsky V. Y., Klug C. S., Hubbell W. L., Gurevich V. V. (2007) Structure and function of the visual arrestin oligomer. EMBO J. 26, 1726–1736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hanson S. M., Dawson E. S., Francis D. J., Van Eps N., Klug C. S., Hubbell W. L., Meiler J., Gurevich V. V. (2008) A model for the solution structure of the rod arrestin tetramer. Structure 16, 924–934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gurevich V. V., Hanson S. M., Song X., Vishnivetskiy S. A., Gurevich E. V. (2011) The functional cycle of visual arrestins in photoreceptor cells. Prog. Retin. Eye Res. 30, 405–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gurevich V. V. (1996) Use of bacteriophage RNA polymerase in RNA synthesis. Methods Enzymol. 275, 382–397 [DOI] [PubMed] [Google Scholar]
- 22. Elowitz M., Lim W. A. (2010) Build life to understand it. Nature 468, 889–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gimenez L. E., Kook S., Vishnivetskiy S. A., Ahmed M. R., Gurevich E. V., Gurevich V. V. (2012) Role of receptor-attached phosphates in binding of visual and non-visual arrestins to G protein-coupled receptors. J. Biol. Chem. 287, 9028–9040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Song X., Raman D., Gurevich E. V., Vishnivetskiy S. A., Gurevich V. V. (2006) Visual and both non-visual arrestins in their “inactive” conformation bind JNK3 and Mdm2 and relocalize them from the nucleus to the cytoplasm. J. Biol. Chem. 281, 21491–21499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Song X., Gurevich E. V., Gurevich V. V. (2007) Cone arrestin binding to JNK3 and Mdm2. Conformational preference and localization of interaction sites. J. Neurochem. 103, 1053–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vishnivetskiy S. A., Raman D., Wei J., Kennedy M. J., Hurley J. B., Gurevich V. V. (2007) Regulation of arrestin binding by rhodopsin phosphorylation level. J. Biol. Chem. 282, 32075–32083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vishnivetskiy S. A., Gimenez L. E., Francis D. J., Hanson S. M., Hubbell W. L., Klug C. S., Gurevich V. V. (2011) Few residues within an extensive binding interface drive receptor interaction and determine the specificity of arrestin proteins. J. Biol. Chem. 286, 24288–24299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Breitman M., Kook S., Gimenez L. E., Lizama B. N., Palazzo M. C., Gurevich E. V., Gurevich V. V. (2012) Silent scaffolds. Inhibition of c-Jun N-terminal kinase 3 activity in cell by dominant-negative arrestin-3 mutant. J. Biol. Chem. 287, 19653–19664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Gurevich V. V., Benovic J. L. (2000) Arrestin. Mutagenesis, expression, purification, and functional characterization. Methods Enzymol. 315, 422–437 [DOI] [PubMed] [Google Scholar]
- 30. Hanson S. M., Vishnivetskiy S. A., Hubbell W. L., Gurevich V. V. (2008) Opposing effects of inositol hexakisphosphate on rod arrestin and arrestin2 self-association. Biochemistry 47, 1070–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gurevich V. V. (1998) The selectivity of visual arrestin for light-activated phosphorhodopsin is controlled by multiple nonredundant mechanisms. J. Biol. Chem. 273, 15501–15506 [DOI] [PubMed] [Google Scholar]
- 32. Vishnivetskiy S. A., Schubert C., Climaco G. C., Gurevich Y. V., Velez M.-G., Gurevich V. V. (2000) An additional phosphate-binding element in arrestin molecule. Implications for the mechanism of arrestin activation. J. Biol. Chem. 275, 41049–41057 [DOI] [PubMed] [Google Scholar]
- 33. Sutton R. B., Vishnivetskiy S. A., Robert J., Hanson S. M., Raman D., Knox B. E., Kono M., Navarro J., Gurevich V. V. (2005) Crystal structure of cone arrestin at 2.3 Å. Evolution of receptor specificity. J. Mol. Biol. 354, 1069–1080 [DOI] [PubMed] [Google Scholar]
- 34. Gurevich V. V., Benovic J. L. (1995) Visual arrestin binding to rhodopsin. Diverse functional roles of positively charged residues within the phosphorylation-recognition region of arrestin. J. Biol. Chem. 270, 6010–6016 [DOI] [PubMed] [Google Scholar]
- 35. Nair K. S., Hanson S. M., Mendez A., Gurevich E. V., Kennedy M. J., Shestopalov V. I., Vishnivetskiy S. A., Chen J., Hurley J. B., Gurevich V. V., Slepak V. Z. (2005) Light-dependent redistribution of arrestin in vertebrate rods is an energy-independent process governed by protein-protein interactions. Neuron 46, 555–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hanson S. M., Gurevich V. V. (2006) The differential engagement of arrestin surface charges by the various functional forms of the receptor. J. Biol. Chem. 281, 3458–3462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hirsch J. A., Schubert C., Gurevich V. V., Sigler P. B. (1999) The 2.8 Å crystal structure of visual arrestin. A model for arrestin's regulation. Cell 97, 257–269 [DOI] [PubMed] [Google Scholar]
- 38. Vishnivetskiy S. A., Paz C. L., Schubert C., Hirsch J. A., Sigler P. B., Gurevich V. V. (1999) How does arrestin respond to the phosphorylated state of rhodopsin? J. Biol. Chem. 274, 11451–11454 [DOI] [PubMed] [Google Scholar]
- 39. Vishnivetskiy S. A., Hirsch J. A., Velez M.-G., Gurevich Y. V., Gurevich V. V. (2002) Transition of arrestin into the active receptor-binding state requires an extended interdomain hinge. J. Biol. Chem. 277, 43961–43968 [DOI] [PubMed] [Google Scholar]
- 40. Gurevich V. V., Benovic J. L. (1992) Cell-free expression of visual arrestin. Truncation mutagenesis identifies multiple domains involved in rhodopsin interaction. J. Biol. Chem. 267, 21919–21923 [PubMed] [Google Scholar]
- 41. Gurevich V. V., Benovic J. L. (1993) Visual arrestin interaction with rhodopsin. Sequential multisite binding ensures strict selectivity toward light-activated phosphorylated rhodopsin. J. Biol. Chem. 268, 11628–11638 [PubMed] [Google Scholar]
- 42. Gurevich V. V., Benovic J. L. (1997) Mechanism of phosphorylation-recognition by visual arrestin and the transition of arrestin into a high affinity binding state. Mol. Pharmacol. 51, 161–169 [DOI] [PubMed] [Google Scholar]
- 43. Gurevich V. V., Dion S. B., Onorato J. J., Ptasienski J., Kim C. M., Sterne-Marr R., Hosey M. M., Benovic J. L. (1995) Arrestin interactions with G protein-coupled receptors. Direct binding studies of wild type and mutant arrestins with rhodopsin, β2-adrenergic, and m2 muscarinic cholinergic receptors. J. Biol. Chem. 270, s731. [DOI] [PubMed] [Google Scholar]
- 44. Gurevich V. V., Gurevich E. V. (2006) The structural basis of arrestin-mediated regulation of G-protein-coupled receptors. Pharmacol. Ther. 110, 465–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gurevich E. V., Gurevich V. V. (2006) Arrestins. Ubiquitous regulators of cellular signaling pathways. Genome Biology 7, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gurevich V. V., Gurevich E. V. (2012) Synthetic biology with surgical precision: targeted reengineering of signaling proteins. Cell. Signal. 24, 1899–1908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pan L., Gurevich E. V., Gurevich V. V. (2003) The nature of the arrestin x receptor complex determines the ultimate fate of the internalized receptor. J. Biol. Chem. 278, 11623–11632 [DOI] [PubMed] [Google Scholar]
- 48. Seo J., Tsakem E. L., Breitman M., Gurevich V. V. (2011) Identification of arrestin-3-specific residues necessary for JNK3 kinase activation. J. Biol. Chem. 286, 27894–27901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Coffa S., Breitman M., Spiller B. W., Gurevich V. V. (2011) A single mutation in arrestin-2 prevents ERK1/2 activation by reducing c-Raf1 binding. Biochemistry 50, 6951–6958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kristiansen K. (2004) Molecular mechanisms of ligand binding, signaling, and regulation within the superfamily of G-protein-coupled receptors. Molecular modeling and mutagenesis approaches to receptor structure and function. Pharmacol. Ther. 103, 21–80 [DOI] [PubMed] [Google Scholar]
- 51. Gurevich V. V., Gurevich E. V. (2008) Rich tapestry of G protein-coupled receptor signaling and regulatory mechanisms. Mol. Pharmacol. 74, 312–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schöneberg T., Schulz A., Biebermann H., Hermsdorf T., Römpler H., Sangkuhl K. (2004) Mutant G-protein-coupled receptors as a cause of human diseases. Pharmacol. Ther. 104, 173–206 [DOI] [PubMed] [Google Scholar]
- 53. Barak L. S., Oakley R. H., Laporte S. A., Caron M. G. (2001) Constitutive arrestin-mediated desensitization of a human vasopressin receptor mutant associated with nephrogenic diabetes insipidus. Proc. Natl. Acad. Sci. U.S.A. 98, 93–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rim J., Oprian D. D. (1995) Constitutive activation of opsin: interaction of mutants with rhodopsin kinase and arrestin. Biochemistry 34, 11938–11945 [DOI] [PubMed] [Google Scholar]
- 55. Bainbridge J. W., Smith A. J., Barker S. S., Robbie S., Henderson R., Balaggan K., Viswanathan A., Holder G. E., Stockman A., Tyler N., Petersen-Jones S., Bhattacharya S. S., Thrasher A. J., Fitzke F. W., Carter B. J., Rubin G. S., Moore A. T., Ali R. R. (2008) Effect of gene therapy on visual function in Leber's congenital amaurosis. N. Engl. J. Med. 358, 2231–2239 [DOI] [PubMed] [Google Scholar]
- 56. Cideciyan A. V., Aleman T. S., Boye S. L., Schwartz S. B., Kaushal S., Roman A. J., Pang J. J., Sumaroka A., Windsor E. A., Wilson J. M., Flotte T. R., Fishman G. A., Heon E., Stone E. M., Byrne B. J., Jacobson S. G., Hauswirth W. W. (2008) Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc. Natl. Acad. Sci. U.S.A. 105, 15112–15117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hauswirth W. W., Aleman T. S., Kaushal S., Cideciyan A. V., Schwartz S. B., Wang L., Conlon T. J., Boye S. L., Flotte T. R., Byrne B. J., Jacobson S. G. (2008) Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector. Short-term results of a phase I trial. Hum. Gene Ther. 19, 979–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Maguire A. M., Simonelli F., Pierce E. A., Pugh E. N., Jr., Mingozzi F., Bennicelli J., Banfi S., Marshall K. A., Testa F., Surace E. M., Rossi S., Lyubarsky A., Arruda V. R., Konkle B., Stone E., Sun J., Jacobs J., Dell'Osso L., Hertle R., Ma J. X., Redmond T. M., Zhu X., Hauck B., Zelenaia O., Shindler K. S., Maguire M. G., Wright J. F., Volpe N. J., McDonnell J. W., Auricchio A., High K. A., Bennett J. (2008) Safety and efficacy of gene transfer for Leber's congenital amaurosis. N. Engl. J. Med. 358, 2240–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Chen J., Makino C. L., Peachey N. S., Baylor D. A., Simon M. I. (1995) Mechanisms of rhodopsin inactivation in vivo as revealed by a COOH-terminal truncation mutant. Science 267, 374–377 [DOI] [PubMed] [Google Scholar]
- 60. Kovoor A., Celver J., Abdryashitov R. I., Chavkin C., Gurevich V. V. (1999) Targeted construction of phosphorylation-independent β-arrestin mutants with constitutive activity in cells. J. Biol. Chem. 274, 6831–6834 [DOI] [PubMed] [Google Scholar]
- 61. Celver J., Vishnivetskiy S. A., Chavkin C., Gurevich V. V. (2002) Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J. Biol. Chem. 277, 9043–9048 [DOI] [PubMed] [Google Scholar]
- 62. Kim M., Vishnivetskiy S. A., Van Eps N., Alexander N. S., Cleghorn W. M., Zhan X., Hanson S. M., Morizumi T., Ernst O. P., Meiler J., Gurevich V. V., Hubbell W. L. (2012) Conformation of receptor-bound visual arrestin. Proc. Natl. Acad. Sci. U.S.A. 109, 18407–18412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Gurevich E. V., Benovic J. L., Gurevich V. V. (2002) Arrestin2 and arrestin3 are differentially expressed in the rat brain during postnatal development. Neuroscience 109, 421–436 [DOI] [PubMed] [Google Scholar]
- 64. Gurevich E. V., Benovic J. L., Gurevich V. V. (2004) Arrestin2 expression selectively increases during neural differentiation. J. Neurochem. 91, 1404–1416 [DOI] [PubMed] [Google Scholar]
- 65. Han M., Gurevich V. V., Vishnivetskiy S. A., Sigler P. B., Schubert C. (2001) Crystal structure of β-arrestin at 1.9 Å, Possible mechanism of receptor binding and membrane Translocation. Structure 9, 869–880 [DOI] [PubMed] [Google Scholar]
- 66. Zhan X., Gimenez L. E., Gurevich V. V., Spiller B. W. (2011) Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual subtypes. J. Mol. Biol. 406, 467–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Celver J. P., Lowe J., Kovoor A., Gurevich V. V., Chavkin C. (2001) Threonine 180 is required for G-protein-coupled receptor kinase 3- and β-arrestin 2-mediated desensitization of the μ-opioid receptor in Xenopus oocytes. J. Biol. Chem. 276, 4894–4900 [DOI] [PubMed] [Google Scholar]
- 68. Gimenez L. E., Vishnivetskiy S. A., Baameur F., Gurevich V. V. (2012) Manipulation of very few receptor discriminator residues greatly enhances receptor specificity of non-visual arrestins. J. Biol. Chem. 287, 29495–29505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lim W. A., Pawson T. (2010) Phosphotyrosine signaling. Evolving a new cellular communication system. Cell 142, 661–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kim M., Hanson S. M., Vishnivetskiy S. A., Song X., Cleghorn W. M., Hubbell W. L., Gurevich V. V. (2011) Robust self-association is a common feature of mammalian visual arrestin-1. Biochemistry 50, 2235–2242 [DOI] [PMC free article] [PubMed] [Google Scholar]