

Background: MreB, a bacterial actin homolog, is required for the maintenance of cell shape in E. coli.
Results: E. coli MreB has been purified, and its polymerization has been characterized.
Conclusion: E. coli MreB is capable of forming filament bundles in vitro.
Significance: The ability to investigate E. coli MreB function in vitro will help answer significant questions about its function in vivo.
Keywords: Actin, ATP, Dynamic Light Scattering, Electron Microscopy (EM), Escherichia coli, Membrane, Actin Homolog, Bacterial Cell Shape, Polymerization
Abstract
The actin homolog MreB is required in rod-shaped bacteria for maintenance of cell shape and is intimately connected to the holoenzyme that synthesizes the peptidoglycan layer. The protein has been reported variously to exist in helical loops under the cell surface, to rotate, and to move in patches in both directions around the cell surface. Studies of the Escherichia coli protein in vitro have been hampered by its tendency to aggregate. Here we report the purification and characterization of native E. coli MreB. The protein requires ATP hydrolysis for polymerization, forms bundles with a left-hand twist that can be as long as 4 μm, forms sheets in the presence of calcium, and has a critical concentration for polymerization of 1.5 μm.
Introduction
MreB was discovered as a gene product required for the maintenance of the rod-like shape of Escherichia coli (1). Subsequently, bioinformatic analyses indicated that the protein possessed five conserved amino acid motifs called the actin superfamily fold, which is common to a group of ATPases (2). However, it was not until the demonstration that a GFP-tagged Bacillus subtilis protein formed actin-like helical filaments in vivo (3) and that the Thermotoga maritima protein polymerized into filaments in vitro and its crystal structure was similar to that of F-actin (4) that it became generally accepted that bacteria might have an actin-like cytoskeleton.
These discoveries sparked considerable interest in the possibility that bacteria might use force-generating mechanisms similar to the microtubule spindle to segregate their chromosomes. Indeed, it has been demonstrated definitively that ParM, a member of the MreB family encoded by the low-copy number E. coli plasmid R1, does just that (5, 6). Unengaged ParM filaments in the cytoplasm are in a state of dynamic instability; however, when bound to plasmid DNA via interaction with ParR bound to the parS DNA sequence on the plasmid, the filaments elongate, pushing the daughter DNA molecules to opposite ends of a rod-shaped cell (7).
The case for such a mechanism with respect to bacterial chromosomal DNA segregation is problematic, at best. Initial reports were supportive of MreB-driven chromosomal movement. Overproduction of an ATPase-defective MreB variant in E. coli elicited chromosome segregation and cell division defects as well as mislocalized origin and terminus regions (8). In addition, bipolar migration of origins could be blocked by affecting MreB function in both E. coli (9) and Caulobacter crescentus (10). We reported that MreB affected the ability of topoisomerase IV to separate topologically linked DNA molecules in vitro (11). Other studies have shown that inhibition of MreB polymerization by the drug A22 did not affect either bulk DNA or origin separation (12) and that independent segregation of the two arms of the ori region did not require active MreB polymerization (13). Also, as explained below, our current preparations of MreB do not affect topoisomerase IV.
It is, however, quite clear that the role of MreB in maintaining cell shape is mediated via its interaction with proteins involved in synthesis of the cell envelope. Although details differ somewhat between Gram-negative and Gram-positive organisms, considerable evidence indicates that MreB is part of a large complex of proteins involved in peptidoglycan synthesis in both types of bacteria and in wall teichoic proteins in Gram-positive species. In Gram-negative organisms, MreB interacts with its operon partner MreC, Mur proteins, which synthesize cytoplasmic precursors, and cell wall assembly proteins such as Pbp2, RodA, and RodZ (14). In addition, a recent study demonstrates that in Gram-positive B. subtilis, MreB also interacts with Tag proteins, which are involved in wall teichoic acid synthesis (15). When tagged with fluorescent proteins, many of these proteins display helical type patterns dependent upon functional MreB (14).
The actual form of MreB in the cell is currently an open question. Initial immunofluorescence microscopy (3) and epifluorescent observation of MreB fusions to fluorescent proteins (16) indicated that MreB formed a spiral-like pattern under the cell surface. A GFP fusion to E. coli MreB was shown to form linear bundles in Schizosaccharomyces pombe (17). However, electron cryotomography, which can detect ParM filament bundles in E. coli (18), does not detect long helical filaments encircling the cells (19). Analysis of GFP-MreB fusions in B. subtilis using total internal reflection fluorescence microscopy showed that when only a small cross-section of the cell surface was illuminated, MreB appeared to move in patches in either direction around the cell circumference (20, 21), suggesting that the helical patterns observed previously using epifluorescence microscopy might be an artifact of the depth of field captured. Finally, an MreB-RFP sandwich fusion has been shown to rotate around the long axis of the cell dependent on assembly of the peptidoglycan layer (22).
Analysis of MreB filamentation in vitro has proven difficult. An N-terminal amphipathic helix that is necessary for binding of E. coli MreB to the membrane, which is not present in MreB proteins from Gram-positive organisms, causes the protein to aggregate (23). We have established a purification protocol that yields E. coli MreB in a nonaggregated, nondenatured, native form. We report here a characterization of the polymerization properties of this protein, which can form filament bundles longer than 4 μm.
EXPERIMENTAL PROCEDURES
Purification of MreB
pET3c-mreB was constructed by amplifying the mreB ORF from E. coli DNA using the primers 5′-CGACATATGTTGAAAAAATTTCGTGGCATG-3′ and 5′-GACAGCTTATCATCGATAAGCTTTAATGC-3′, which contained an NdeI and HindIII restriction enzyme site, respectively, digesting the PCR fragment with these enzymes, and ligating the fragment to similarly digested pET3c (Novagen) DNA. The primary structure of the expressed protein was: MLKKFRGMFSNDLSIDLGTANTLIYVKGQGIVLNEPSVVAIRQDRAGSPKSVAAVGHDAKQMLGRTPGNIAAIRPMKDGVIADFFVTEKMLQHFIKQVHSNSFMRPSPRVLVCVPVGATQVERRAIRESAQGAGAREVFLIEEPMAAAIGAGLPVSEATGSMVVDIGGGTTEVAVISLNGVVYSSSVRIGGDRFDEAIINYVRRNYGSLIGEATAERIKHEIGSAYPGDEVREIEVRGRNLAEGVPRGFTLNSNEILEALQEPLTGIVSAVMVALEQCPPELASDISERGMVLTGGGALLRNLDRLLMEETGIPVVVAEDPLTCVARGGGKALEMIDMHGGDLFSEE.
Plasmid pET3c-mreB was transformed into C41(λDE3), and five colonies from a fresh plate were inoculated into 500 ml of LB medium in a 2-liter flask containing 100 μg/ml ampicillin and grown for 5 h at 37 °C. This culture was then used to inoculate 20 liters of medium in a New Brunswick Scientific Bioflo 4500 fermentor and grown for an additional 2 h at 37 °C, at which time the temperature was reduced to 20 °C and growth was continued for an additional 12 h. The cells were harvested using a Sharples continuous flow centrifuge, resuspended to 50% (w/v) in 50 mm Tris-HCl (pH 8.0 at 4 °C), 10% sucrose, frozen in liquid N2, and stored at −80 °C. The culture was not induced with isopropyl-1-thio-β-d-galactopyranoside.
Cells (230 g) from three fermentor runs were thawed rapidly in a 37 °C water bath with continuous, gentle stirring, distributed into 30-ml ultracentrifuge tubes, and put on ice. All subsequent procedures were conducted at 4 °C. The cell suspension was made 50 mm KCl, 20 mm EDTA, 5 mm DTT, and 0.1% Triton X-100. Tubes were mixed gently by inverting, incubated at 37 °C for 3 min, and then put on ice for 20 min. The cell suspension was then centrifuged at 100,000 × g (34,000 rpm in a Sorvall T1250 rotor) for 70 min. The supernatant (fraction 1, 330 ml, 4.5 g of protein) was made 0.07% in Polymin-P by slow addition with stirring from a 1% solution. The resulting suspension was stirred for 10 min, and the pellet was cleared by centrifugation in a Sorvall SS34 rotor at 19,000 rpm for 10 min. Saturated and neutralized (NH4)2SO4 was added to the supernatant to a final concentration of 35% saturation, the suspension was stirred for 1 h, and the pellet was cleared by centrifugation in a Sorvall SS34 rotor at 19,000 rpm for 30 min. The supernatant (fraction 2, 345 ml, 3.8 g of protein) was dialyzed against 10 volumes of buffer A-S (50 mm Tris-HCl (pH 8.0 at 4 °C), 5 mm DTT, 0.1 mm EDTA, and 10% sucrose) plus 50 mm KCl for 3 h and then diluted with buffer A-S to a conductivity equivalent to 50 mm KCl. Fraction 2 was applied to a 200-ml column of Q-Sepharose (GE Healthcare) that had been equilibrated with buffer A-S plus 50 mm KCl. The column was washed with 2 column volumes of buffer A-S plus 150 mm KCl and then eluted with a 10-column volume gradient of 150–500 mm KCl in buffer A-S. Fractions containing MreB (eluting at 280 mm KCl) were identified by SDS-PAGE analysis, pooled, and dialyzed overnight against 10 volumes of buffer A-S plus 100 mm KCl (fraction 3, 270 ml, 400 mg of protein). Fraction 3 was applied to a 75-ml column of hydroxyapatite (60:17, Bio-Rad DNA grade Bio-Gel hydroxyapatite:Whatman CF-11 cellulose) that had been equilibrated with buffer A-S plus 100 mm KCl. The column was washed with 2 column volumes of buffer A-S plus 50 mm (NH4)2S04 and eluted with a 10-column volume gradient of 50–300 mm (NH4)2SO4 in buffer A-S. Fractions containing MreB (eluting at 160 mm (NH4)2SO4) were identified by SDS-PAGE analysis, pooled, and dialyzed against 30 volumes of buffer A (50 mm Tris-HCl (pH 8.0 at 4 °C), 5 mm DTT, 0.1 mm EDTA, 10% glycerol) plus 50 mm KCl (fraction 4, 95 ml, 30 mg of protein). Fraction 4 was applied to a 5-ml HiTrap Heparin HP FPLC column (GE Healthcare) that had been equilibrated with buffer A plus 50 mm KCl. The column was washed with 2.5 column volumes of equilibration buffer and eluted with a 10-column volume gradient of 50–500 mm KCl in buffer A. Fractions containing MreB (eluting at 190 mm KCl) were identified by SDS-PAGE analysis, pooled, and dialyzed overnight against 2 liters of MreB storage buffer (50 mm Tris-HCl (pH 8.0 at 4 °C), 5 mm DTT, 0.1 mm EDTA, 100 mm KCl, and 20% glycerol), divided into aliquots, frozen in liquid N2, and stored at −80 °C (fraction 5, 3.3 ml, 1.9 mg of protein).
MreB Polymerization and ATPase Assay
Standard reaction mixtures (60 μl) contained 50 mm Tris-HCl (pH 7.0 at 37 °C), 5 mm DTT, 2 mm MgCl2, 50 mm KCl, 2 mm ATP, 10% glycerol, and the indicated concentrations of MreB. Reaction mixtures were assembled at room temperature from components stored on ice. ATP was added last, and the reaction mixture was either incubated at 37 °C as indicated or immediately transferred to a cuvette and inserted into a Brookhaven Instruments 90 Plus Particle Size Analyzer. Dynamic light scattering (DLS)2 was measured at a 90° angle using a 658-nm laser and DLSW software at a sampling rate of 30 HZ. In the plots displayed, each data point represents the average of 10 consecutive data readouts.
ATPase assays were performed using the same reaction buffer with 4 μm MreB except that 83 nm [γ-32P]ATP was included and the reaction volume was 20 μl. Reactions were incubated at 37 °C for 60 min and terminated by the addition of EDTA to 10 mm. An aliquot (2 μl) was spotted on a PEI-cellulose thin layer plate (EMD Chemicals), and the plate was developed with 1 m HCOOH, 0.5 m LiCl. The plate was dried, exposed to a phosphorimaging screen and imaged using a Fujifilm FLA 7000 phosphorimaging device. The fraction of ATP converted to Pi was calculated.
Electron Microscopy
Polymerized sample (10 μl) was placed on 400-mesh copper grids coated with Formvar carbon film (Electron Microscopy Sciences) for 1 min at room temperature. Liquid was then removed by blotting (a piece of filter paper was touched to the side of the grid). The grids were washed with three drops of polymerization buffer (without glycerol) for 1 min, liquid was removed, the grids were covered with three drops of 2% uranyl acetate for 1 min, liquid was removed, and the grids were air-dried for 5 min. Samples were visualized using either a JEOL JEM-1200EX or a JEM-1400 electron microscope at 80 kV.
RESULTS AND DISCUSSION
Purification of MreB
E. coli MreB used in a previous study from this laboratory (11) was purified in the continual presence of ATP. These preparations of MreB gave short filaments in thick bundles. In addition, the ORF used for those studies contained a 20-amino acid N-terminal extension that was thought to possibly represent a signal sequence (24). In our search for more reliable preparations of MreB, we eliminated this N-terminal extension from the ORF and tried various tags to allow overexpression and quick purification of the protein. None of these preparations formed visible filaments in the electron microscope (EM), instead yielding amorphous masses that, nevertheless, would generate convincing DLS signals in a nucleotide-dependent manner. About this time, Salje et al. (23) reported their finding that the N terminus of MreB formed an amphipathic helix that could bind the protein to membranes in a vesicle-pelleting assay. These authors also reported difficulty purifying the full-length protein because of aggregation and reported that they could purify a stable version if they deleted this helix. Based on this study and our own difficulties in purifying the protein, we concluded that our previous preparations were likely to be, for the most part, denatured (11). We posited that one might be able to avoid the aggregation/denaturation problem by not overexpressing the protein to the huge degree inherent in the use of the bacteriophage T7 systems (25) and by using conventional column chromatography steps. This proved to be the case. However, the current preparations, which, as shown below, filament reliably, do not interact with the ParC subunit of topoisomerase IV nor affect the activity of topoisomerase IV. Our previous observations (11) may have resulted from interaction between ParC and a surface of MreB not exposed in the fully native protein.
We overexpressed MreB slightly by using the pET3c expression vector and the C41(λDE3) strain (25) without induction of the T7 RNA polymerase. The C41(λDE3) strain produces some T7 RNA polymerase in the absence of isopropyl-1-thio-β-d-galactopyranoside induction, so the net effect is a substantially decreased extent of overproduction when compared with when isopropyl-1-thio-β-d-galactopyranoside is used. We also lowered the growth temperature of the cells once they had reached early log phase to slow the rate of protein production and mitigate intermolecular aggregation of the unfolded protein chains. As a result, MreB was overproduced by only 9-fold when compared with similarly treated cells that did not contain the pET3C-mreB plasmid (Fig. 1A).
FIGURE 1.

Purification of MreB. A, overproduction of MreB. The upper panel shows a Coomassie Blue-stained SDS gel of extracts (5 μg) prepared from cells grown as described under “Experimental Procedures” and carrying either the overexpression plasmid or pET3C-mreB. Purified MreB (1 μg) is shown as a reference. The bottom panel is a Western blot using anti-MreB antisera of a similar gel. In this case, 50 μg of protein from the culture carrying the expression vector was electrophoresed through the gel, whereas only 5 μg of protein from the culture carrying the pET3c-mreB plasmid was used. MreB marker was at 50 ng. Marker proteins were Precision Plus Protein WesternC standards (Bio-Rad). B, purification of MreB. Top panel, Coomassie Blue-stained gel with 5 μg of fraction 1, 5 μg of fraction 2, 2.5 μg of fraction 3, 2 μg of fraction 4, and 1 μg of fraction 5 as indicated. Bottom panel, Western blot with 1 μg of fraction 1, 1 μg of fraction 2, 0.5 μg of fraction 3, 0.1 μg of fraction 4, and 0.05 μg of fraction 5, as indicated. Marker proteins were as in panel A. Images were captured with a Kodak Image Station 4000R Pro and analyzed using Fujifilm Image Gauge software.
Because there was no simple assay for MreB activity, we followed purification of the protein using SDS-PAGE and Western blotting (Fig. 1B). MreB was purified from the cleared lysate by sequential column chromatography on Q-Sepharose, hydroxyapatite, and heparin-agarose. The final preparation was judged to be 95% homogeneous for MreB (Fig. 1B). The two bands migrating slightly faster then the major band of MreB are positive in Western blots with anti-MreB antisera, and we assume that they are proteolytic degradation products. Furthermore, mass spectrometric analysis of tryptic peptides from both these minor bands, as well as the major band, indicated MreB as the primary component.
Polymerization Characteristics of MreB
Increasing DLS signal and pelleting from solution are routinely used to score MreB polymerization. Without confirmation of the formation of filaments by EM, conclusions reached from these assays must be considered problematic. We have many preparations of MreB that score well in these assays but do not produce filaments, just aggregates. All of our assays were confirmed by EM.
The rate of MreB polymerization under standard conditions (pH 7.0, 37 °C) increased in a protein concentration-dependent manner (see Fig. 3A). A typical EM field of filaments formed at 4 μm MreB (the concentration used routinely) is shown in Fig. 2A. A distinct lag was evident in the DLS signals for polymerization, suggesting that polymerization proceeded with a nucleation step. MreB formed bundles of filaments in the presence of Mg2+ and ATP (Fig. 2, A, E, and F), and occasionally, a clear left-handed twist to the bundles could be observed (Fig. 2E). MreB filament bundles did not display much curvature and did not form the ring-like structures observed previously for the T. maritima protein (4, 26). The minimum number of filaments in a bundle could not be ascertained, although occasional high-resolution images appeared to show 2–4 filaments minimally per bundle (Fig. 2G). We have been unable to establish polymerization conditions that yielded single filaments of MreB. MreB bundles could be quite long, over 4 μm in length (Fig. 2, C and D).
FIGURE 3.

Concentration dependence of the rate of MreB polymerization. A, DLS traces of MreB polymerization under standard conditions at 1.0 μm (red), 2.0 μm (light green), and 4.0 μm (green) as described under “Experimental Procedures.” The black trace indicates no MreB. Error bars indicate S.D. kcps, kilo-counts per second. B, critical concentration determination. MreB at 1.0, 2.0, 4.0, and 8.0 μm was polymerized under standard conditions overnight. Light scattering was then measured for 5 min. Error bars indicate S.D.
FIGURE 2.

Filament and sheet formation by MreB. A, a typical EM field of MreB filaments formed in the presence of Mg2+ and ATP. B, a typical EM field of MreB filaments formed in the presence of Mg2+ and GTP. C, distribution of lengths of MreB filament bundles formed in the presence of Mg2+ and ATP. Molecules from 20 different experiments were measured. D, a long filament bundle (3.8 μm) formed in the presence of Mg2+ and ATP. E, a filament bundle formed in the presence of Mg2+ and ATP that displays a clear twist. F, MreB filament bundles formed in the presence of Mg2+ and ATP. G, a close-up view of an MreB filament formed in the presence of Mg2+ and ATP. H, spherical aggregates of MreB formed in the presence of Mg2+ but in the absence of nucleotide. I, spherical aggregates of MreB formed in the presence of Mg2+ and AMP-PNP. J, MreB sheets formed in the presence of Ca2+ and ATP. K, MreB sheets formed in the presence of Ca2+ and GTP.
The critical concentration for MreB polymerization was determined to be 1.5 μm by allowing MreB at various concentrations to polymerize overnight at 37 °C, determining the extent of light scattering, and extrapolating the trace to zero light scattering (Fig. 3B). This value is considerably different from the 3 nm value determined by Esue et al. (26) and closer to the 0.5 μm value determined by Bean and Amann (27) (where no EM was reported) and the 1 μm value determined by Popp et al. (28) for the T. maritima protein. We have not observed any filament formation in the EM at concentrations in the range of 1 μm, supporting the higher value for the critical concentration. The intracellular concentration of the T. maritima protein has been estimated at 5.6 μm (26).
The rate of MreB polymerization was quite sensitive to the concentration of monovalent salt (Fig. 4). Because of the presence of KCl in the storage buffer and the concentration of our final fraction, we could not examine MreB polymerization at lower than 25 mm KCl. The rate of polymerization was robust between 25 and 75 mm KCl, but decreased dramatically at 100 mm KCl and was very low at 200 mm KCl, although filaments could be observed at the latter salt concentrations if polymerization was conducted for 30 min (see below). There is some inconsistency in the reported effect of monovalent salt on polymerization of the T. maritima protein. van den Ent et al. (4) and Popp et al. (28) did not observe significant inhibition, whereas Esue et al. (26) and Bean and Amann (27) did. Polymerization of the B. subtilis protein was also reported to be sensitive to salt (29).
FIGURE 4.

The rate of MreB polymerization is sensitive to the concentration of monovalent salt. MreB (4 μm) polymerization was measured by DLS under standard conditions except that the reaction mixtures contained 25 mm KCl (light green), 50 mm KCl (green), 75 mm KCl (orange), 100 mm KCl (pink), and 200 mm KCl (blue). Error bars indicate S.D. kcps, kilo-counts per second.
MreB required nucleoside triphosphate for polymerization (Fig. 5A). GTP supported polymerization at about one-third the initial rate of ATP. CTP and UTP were not very efficient in supporting polymerization. GTP-MreB filament bundles were essentially indistinguishable from ATP-MreB filament bundles, although some observers in the laboratory thought that the GTP-MreB filament bundles displayed a slightly higher extent of curvature (Fig. 2B). The rate of ATP hydrolysis by MreB was 0.07 ± 0.01 ATP hydrolyzed/min/MreB at 50 mm KCl, conditions where filaments will form, and 0.17 ± 0.01 ATP hydrolyzed/min/MreB at 200 mm KCl, conditions where filaments will form but may be unstable (see below). (ATPase rates are the average of three determinations.) This observation suggests either that turnover of ADP and Pi is, in general, slow, and was enhanced by the added salt, or that release of ADP and Pi from the filaments is slow. Because the ATPase activity was low and we were limited by the amount of MreB we could introduce to our assays, we could not measure polymerization and ATP hydrolysis simultaneously. The ATPase rate reported here is similar to that reported for the T. maritima protein by Esue et al. (26).
FIGURE 5.

Nucleotide selectivity of MreB polymerization. A, MreB polymerization was measured by DLS under standard conditions except that the reaction mixtures contained ATP (green), GTP (light green), no nucleotide (black), CTP (orange), or UTP (pink). Error bars indicate S.D. B, a comparison of the rate of MreB polymerization as measured by DLS in the presence of either ATP (green) or ADP (black). kcps, kilo-counts per second. Error bars indicate S.D.
No MreB filament formation was observed with ADP (Fig. 5B). Interestingly, an increase in DLS signal to nearly the same extent observed in the presence of ATP occurred after a long lag when MreB was incubated in the absence of nucleotide (Fig. 5A). Similar results were observed with the nonhydrolyzable ATP analog AMP-PNP (data not shown). However, filament formation was not supported under either of these conditions. Instead, we observed large, spherical aggregates in the EM under these conditions (Fig. 2, H and I), which could clearly account for the increase in light scattering. Similar aggregates were reported to form in the presence of either AMP-PNP or GMP-PNP with the T. maritima protein (28). This lack of correspondence between the DLS signal and physical observation of filaments in the EM presumably accounts for the variability in the reports of nucleotide requirements for MreB polymerization. Reports that T. maritima (27), B. subtilis (29), and Chlamydophila pneumoniae (30) MreB polymerization did not require nucleotide or nucleotide hydrolysis were based solely on DLS results.
Although MreB formed filament bundles in the presence of Mg2+, the protein formed extensive sheets in the presence of Ca2+ with either ATP (Fig. 2J) or GTP (Fig. 2K). These sheets were similar to those observed for the T. maritima protein (28), although those were observed in the presence of either Mg2+ or Ca2+. Sheets formed with the E. coli protein tended to role up at one end (Fig. 2, J and K). Power spectra analysis (31) of a small MreB bundle (Fig. 6A and a sheet (Fig. 6B) showed strong reflections at 49 and 50.5 Å, respectively. By comparison, ATP filaments of the T. maritima protein displayed a strong reflection at 51 Å (4), whereas GTP and GDP sheets showed reflections at 51 and 50 Å, respectively (28).
FIGURE 6.
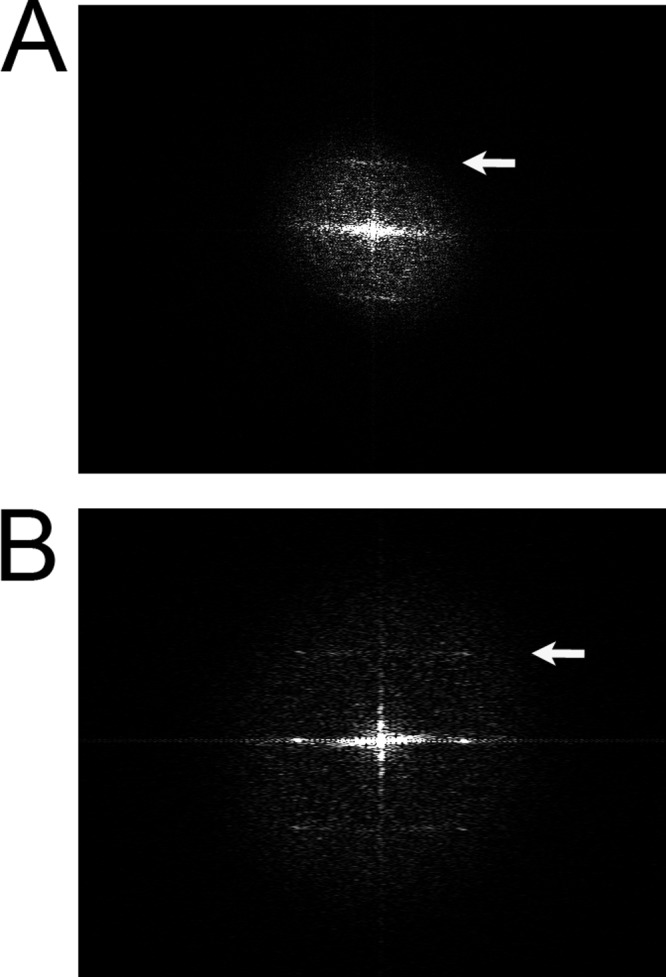
Power spectra analysis of MreB bundles and sheets. A, power spectrum of a small bundle. B, power spectrum of a sheet. The arrows point to strong reflections at 49 Å in panel A and 50.5 Å in panel B.
Conclusions
Our results demonstrate that E. coli MreB can indeed polymerize into filament bundles that are long enough to span the longitudinal axis of an E. coli cell growing rapidly in rich medium. This finding lends some support to the concept that there is a network of actin-like filaments in bacterial cells. We found the filaments formed to be stable for several days at 4 °C. However, the rate of polymerization was quite sensitive to monovalent salt in the concentration range expected in vivo. Filaments formed at 100 mm KCl were similar in appearance to those that formed under standard conditions (Fig. 7A). However, filaments that formed at 150 mm KCl (Fig. 7B) and 200 mm KCl (Fig. 7C) appeared unstable. Thus, the active network in the cell may be more unstable than what we observe in vitro. Of course, it is also possible that MreB filaments are stabilized by one or some of the many other proteins that interact with it. With the ability to purify E. coli MreB in a form that filaments readily, these questions can now be investigated in vitro.
FIGURE 7.

Effect of increasing salt on MreB bundles. A, an MreB bundle formed under standard conditions in the presence of 100 mm KCl. B, an MreB bundle formed under standard conditions in the presence of 150 mm KCl. C, an MreB bundle formed under standard conditions in the presence of 200 mm KCl.
Acknowledgments
We thank Edward Egelman (University of Virginia) for the power spectra analysis shown in Fig. 6, Nina Lampen for assistance with the electron microscopy, and Piet de Boer for a critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant GM34558 (to K. J. M.).
- DLS
- dynamic light scattering
- AMP-PNP
- adenosine 5′-(β,γ-imino)triphosphate
- GMP-PNP
- 5′-guanylyl imidodiphosphate.
REFERENCES
- 1. Wachi M., Doi M., Tamaki S., Park W., Nakajima-Iijima S., Matsuhashi M. (1987) Mutant isolation and molecular cloning of mre genes, which determine cell shape, sensitivity to mecillinam, and amount of penicillin-binding proteins in Escherichia coli. J. Bacteriol. 169, 4935–4940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bork P., Sander C., Valencia A. (1992) An ATPase domain common to prokaryotic cell cycle proteins, sugar kinases, actin, and hsp70 heat shock proteins. Proc. Natl. Acad. Sci. U.S.A. 89, 7290–7294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jones L. J., Carballido-López R., Errington J. (2001) Control of cell shape in bacteria: helical, actin-like filaments in Bacillus subtilis. Cell 104, 913–922 [DOI] [PubMed] [Google Scholar]
- 4. van den Ent F., Amos L. A., Löwe J. (2001) Prokaryotic origin of the actin cytoskeleton. Nature 413, 39–44 [DOI] [PubMed] [Google Scholar]
- 5. Møller-Jensen J., Borch J., Dam M., Jensen R. B., Roepstorff P., Gerdes K. (2003) Bacterial mitosis: ParM of plasmid R1 moves plasmid DNA by an actin-like insertional polymerization mechanism. Mol. Cell 12, 1477–1487 [DOI] [PubMed] [Google Scholar]
- 6. Møller-Jensen J., Jensen R. B., Löwe J., Gerdes K. (2002) Prokaryotic DNA segregation by an actin-like filament. EMBO J. 21, 3119–3127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Garner E. C., Campbell C. S., Mullins R. D. (2004) Dynamic instability in a DNA-segregating prokaryotic actin homolog. Science 306, 1021–1025 [DOI] [PubMed] [Google Scholar]
- 8. Kruse T., Møller-Jensen J., Løbner-Olesen A., Gerdes K. (2003) Dysfunctional MreB inhibits chromosome segregation in Escherichia coli. EMBO J. 22, 5283–5292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kruse T., Blagoev B., Løbner-Olesen A., Wachi M., Sasaki K., Iwai N., Mann M., Gerdes K. (2006) Actin homolog MreB and RNA polymerase interact and are both required for chromosome segregation in Escherichia coli. Genes Dev. 20, 113–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gitai Z., Dye N. A., Reisenauer A., Wachi M., Shapiro L. (2005) MreB actin-mediated segregation of a specific region of a bacterial chromosome. Cell 120, 329–341 [DOI] [PubMed] [Google Scholar]
- 11. Madabhushi R., Marians K. J. (2009) Actin homolog MreB affects chromosome segregation by regulating topoisomerase IV in Escherichia coli. Mol. Cell 33, 171–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karczmarek A., Martínez-Arteaga R., Baselga R. M., Alexeeva S., Hansen F. G., Vicente M., Nanninga N., den Blaauwen T. (2007) DNA and origin region segregation are not affected by the transition from rod to sphere after inhibition of Escherichia coli MreB by A22. Mol. Microbiol. 65, 51–63 [DOI] [PubMed] [Google Scholar]
- 13. Wang X., Sherratt D. J. (2010) Independent segregation of the two arms of the Escherichia coli ori region requires neither RNA synthesis nor MreB dynamics. J. Bacteriol. 192, 6143–6153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. White C. L., Gober J. W. (2012) MreB: pilot or passenger of cell wall synthesis? Trends Microbiol. 20, 74–79 [DOI] [PubMed] [Google Scholar]
- 15. Kawai Y., Marles-Wright J., Cleverley R. M., Emmins R., Ishikawa S., Kuwano M., Heinz N., Bui N. K., Hoyland C. N., Ogasawara N., Lewis R. J., Vollmer W., Daniel R. A., Errington J. (2011) A widespread family of bacterial cell wall assembly proteins. EMBO J. 30, 4931–4941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vats P., Rothfield L. (2007) Duplication and segregation of the actin (MreB) cytoskeleton during the prokaryotic cell cycle. Proc. Natl. Acad. Sci. U.S.A. 104, 17795–17800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Srinivasan R., Mishra M., Murata-Hori M., Balasubramanian M. K. (2007) Filament formation of the Escherichia coli actin-related protein, MreB, in fission yeast. Curr. Biol. 17, 266–272 [DOI] [PubMed] [Google Scholar]
- 18. Salje J., Zuber B., Löwe J. (2009) Electron cryomicroscopy of E. coli reveals filament bundles involved in plasmid DNA segregation. Science 323, 509–512 [DOI] [PubMed] [Google Scholar]
- 19. Swulius M. T., Chen S., Jane Ding H., Li Z., Briegel A., Pilhofer M., Tocheva E. I., Lybarger S. R., Johnson T. L., Sandkvist M., Jensen G. J. (2011) Long helical filaments are not seen encircling cells in electron cryotomograms of rod-shaped bacteria. Biochem. Biophys. REs. Comm. 407, 650–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Garner E. C., Bernard R., Wang W., Zhuang X., Rudner D. Z., Mitchison T. (2011) Coupled, circumferential motions of the cell wall synthesis machinery and MreB filaments in B. subtilis. Science 333, 222–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Domínguez-Escobar J., Chastanet A., Crevenna A. H., Fromion V., Wedlich-Söldner R., Carballido-López R. (2011) Processive movement of MreB-associated cell wall biosynthetic complexes in bacteria. Science 333, 225–228 [DOI] [PubMed] [Google Scholar]
- 22. van Teeffelen S., Wang S., Furchtgott L., Huang K. C., Wingreen N. S., Shaevitz J. W., Gitai Z. (2011) The bacterial actin MreB rotates, and rotation depends on cell-wall assembly. Proc. Natl. Acad. Sci. U.S.A. 108, 15822–15827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Salje J., van den Ent F., de Boer P., Löwe J. (2011) Direct membrane binding by bacterial actin MreB. Mol. Cell 43, 478–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Doi M., Wachi M., Ishino F., Tomioka S., Ito M., Sakagami Y., Suzuki A., Matsuhashi M. (1988) Determinations of the DNA sequence of the mreB gene and of the gene products of the mre region that function in formation of the rod shape of Escherichia coli cells. J. Bacteriol. 170, 4619–4624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Studier F. W., Rosenberg A. H., Dunn J. J., Dubendorff J. W. (1990) Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 185, 60–89 [DOI] [PubMed] [Google Scholar]
- 26. Esue O., Cordero M., Wirtz D., Tseng Y. (2005) The assembly of MreB, a prokaryotic homolog of actin. J. Biol. Chem. 280, 2628–2635 [DOI] [PubMed] [Google Scholar]
- 27. Bean G. J., Amann K. J. (2008) Polymerization properties of the Thermotoga maritima actin MreB: roles of temperature, nucleotides, and ions. Biochemistry 47, 826–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Popp D., Narita A., Maeda K., Fujisawa T., Ghoshdastider U., Iwasa M., Maéda Y., Robinson R. C. (2010) Filament structure, organization, and dynamics in MreB sheets. J. Biol. Chem. 285, 15858–15865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mayer J. A., Amann K. J. (2009) Assembly properties of the Bacillus subtilis actin, MreB. Cell Motil. Cytoskeleton 66, 109–118 [DOI] [PubMed] [Google Scholar]
- 30. Gaballah A., Kloeckner A., Otten C., Sahl H. G., Henrichfreise B. (2011) Functional analysis of the cytoskeleton protein MreB from Chlamydophila pneumoniae. PLoS One 6, e25129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Frank J., Radermacher M., Penczek P., Zhu J., Li Y., Ladjadj M., Leith A. (1996) SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 116, 190–199 [DOI] [PubMed] [Google Scholar]