

Background: Transplant patients treated with cyclosporine A, an inhibitor of calcineurin, frequently develop metabolic complications.
Results: Mice lacking calcineurin Aβ develop metabolic complications. A cell autonomous mechanism with altered cAMP-PKA signaling is found.
Conclusion: Calcineurin plays a role in metabolism via regulating phosphodiesterases.
Significance: Potentiated cAMP signaling provides an alternative mechanism for the metabolic complications in transplant patients.
Keywords: Calcineurin, Cyclic AMP (cAMP), Immunosuppression, Metabolism, Phosphodiesterases, Protein degradation
Abstract
Insulin resistance, hyperlipidemia, and cardiovascular complications are common dysregulations of metabolic syndrome. Transplant patients treated with immunosuppressant drugs such as cyclosporine A (CsA), an inhibitor of calcineurin phosphatase, frequently develop similar metabolic complications. Although calcineurin is known to mediate insulin sensitivity by regulating β-cell growth and adipokine gene transcription, its role in lipid homeostasis is poorly understood. Here, we examined lipid homeostasis in mice lacking calcineurin Aβ (CnAβ−/−). We show that mice lacking calcineurin Aβ are hyperlipidemic and develop age-dependent insulin resistance. Hyperlipidemia found in CnAβ−/− mice is, in part, due to increased lipolysis in adipose tissues, a process mediated by β-adrenergic G-protein-coupled receptor signaling pathways. CnAβ−/− mice also exhibit additional pathophysiological phenotypes caused by the potentiated GPCR signaling pathways. A cell autonomous mechanism with sustained cAMP/PKA activation is found in CnAβ−/− mice or upon CsA treatment to inhibit calcineurin. Increased PKA activation and cAMP accumulation in CnAβ−/− mice, however, are sensitive to phosphodiesterase inhibitor. Indeed, we show that calcineurin regulates degradation of phosphodiesterase 3B, in addition to phosphodiesterase 4D. These results establish a role for calcineurin in lipid homeostasis. These data also indicate that potentiated cAMP signaling pathway may provide an alternative molecular pathogenesis for the metabolic complications elicited by CsA in transplant patients.
Introduction
Metabolic syndrome includes a broad range of pathologies ranging from insulin resistance and hyperlipidemia to hypertension, all of which contribute, in part, to the epidemic rise in obesity, diabetes, and cardiovascular diseases. Metabolic syndrome is also frequently found in transplant patients treated with immunosuppressant drugs cyclosporine A (CsA)4 or tacrolimus (1, 2). Suppression of the immune function by CsA during and after transplant surgery minimizes the incidence of host versus graft in transplant patients and increases the success of organ transplantation. Many of these transplant patients, however, develop post-transplantation metabolic complications, most evidently diabetes, even without a previous history of such pathologies. Indeed, elevated triglyceride levels were observed in healthy human and rodents upon receiving CsA (3, 4), demonstrating the adverse effects of CsA in lipid homeostasis. In addition to metabolic complications, nephropathy and neurotoxicity are also commonly found in transplant patients treated with CsA.
Current models suggest that CsA disrupts the mitochondrial permeability transition pore function and causes cellular dysfunction in β-cells (5). CsA also inhibits calcineurin (6), a Ca2+-calmodulin dependent protein phosphatase. Calcineurin holoenzyme composes of calcineurin A catalytic subunit (CnA), calcineurin B regulatory subunit (CnB), and calmodulin for calcium binding (7–10). Two isoforms of CnA (CnAα and CnAβ) and one isoform of CnB (CnB1) are present in somatic cells. CnAγ and CnB2 are expressed mainly in germ cells. CnAα seems to localize in the nucleus, whereas CnAβ was found in the cytoplasm (11, 12). Calcineurin is also present in microsomal membrane fractions, although its function remains elusive. Differential expression of CnAα and CnAβ in different tissues further contributes to their distinct role in various biological processes. For example, CnAα−/− mice die within 3 weeks after birth, whereas CnAβ null mice are fertile and viable, and defects in thymus and heart are revealed only upon challenge (13–17). Targeted disruption of CnB1 (hence no calcineurin phosphatase activity in somatic cells) is embryonic lethal (18), demonstrating that calcineurin activity is required for normal growth and differentiation. Pancreas specific deletion of CnB1 has been reported recently (19), and the loss of calcineurin in pancreas leads to hypoinsulinaemia, which is in part due to decreased β-cell mass and proliferation.
A well recognized function of calcineurin is dephosphorylation of transcription factor Nuclear Factor of Activated T Cells (NFAT) (20, 21). The calcineurin-NFAT signaling is critical for cytokine gene expression in immune cells (22, 23). The calcineurin-NFAT signaling also regulates pancreatic β-cell growth and adipokine gene transcription (19, 24, 25). Upon calcineurin inhibition by CsA, dysregulated pancreatic function and loss of insulin sensitivity can account for the metabolic complications found in transplant patients.
Improper lipid homeostasis, however, can also lead to metabolic dysregulations—a phenomenon known as lipotoxicity (26, 27). Excess circulating levels of triglyceride and free fatty acids cause accumulation of lipids in nonadipose tissues, which then initiates abnormal signaling cascade and cellular dysfunctions. These include impaired insulin receptor signaling and defects in translocation of GLUT4 glucose transporter in skeletal muscle (28). In pancreas, lipid deposition in β-cells reduces insulin secretion, resulting in hyperglycemia and insulin resistance (29). Lipotoxicity can also lead to cardiovascular diseases, liver malfunctions, and kidney failure (30, 31). Thus, dysregulated lipid homeostasis contributes significantly to metabolic complications.
Here, we uncover a role of calcineurin in lipid homeostasis. We find that CnAβ−/− mice are hyperlipidemic, in part, because of increased lipolysis, a process mediated by β-adrenergic G-protein-coupled receptor signaling pathway. A cell autonomous mechanism with sustained cAMP/PKA activation is found in CnAβ−/− mice or upon CsA treatment to inhibit calcineurin. We show that calcineurin regulates degradation of phosphodiesterase 3B (PDE3B), in addition to PDE4D, which we found previously (32). Together, these results provide an alternative molecular pathogenesis for the metabolic complications elicited by CsA in transplant patients. We propose that metabolic complications found in transplant patients treated with CsA may be a consequence not only of pancreatic dysregulations via the insulin pathway, but also of potentiation of Gs-coupled signaling pathway caused by sustained elevation of cAMP.
EXPERIMENTAL PROCEDURES
Mice
Animal experiments were performed in accordance with the guidelines of Albert Einstein College of Medicine Institute of Animal Studies. CnAβ−/− mice were generated as described previously by insertion of a neomycin-resistant gene to disrupt exon 2 (14). CnAβ−/− mice were backcrossed with C57BL/6 mice at least ten times, and 8-week-old mice were used for experiments except as indicated. The mice were fed ad libitum with regular chow (Research Diets, Inc.), except when fasted for experiments. Back-crossed CnAβ−/− mice were also used for isolation of mouse embryonic fibroblasts (MEFs).
Reagents
Expression vectors for FLAG-tagged PDE3B have been described previously (33). Point mutations on PDE3B were generated by QuikChange protocol and sequenced. The antibodies used were: FLAG M2 (Sigma; F3185), calcineurin A (Santa Cruz Biotechnology; sc6124 and sc17808), phospho-PKA substrates antibodies (Cell Signaling; 9621), phospho-CREB (Cell Signaling; 9198), HSL (Cell Signaling; 4107), and phospho-HSL (Cell Signaling; 4126). Tubulin antibody (clone E7) was obtained from the monoclonal antibody facility at the University of Iowa. Polyclonal antibodies against PDE3B were described previously (34). Phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX), calcineurin inhibitor CsA, and Triton WR1339 were obtained from Calbiochem, Sigma, and/or Fisher Scientific. Circulating triglyceride was measured from serum using a serum triglyceride determination kit from Sigma (TR0100). Circulating free fatty acid and cholesterol was measured from serum using a nonesterified fatty acid kit from Roche (11 383 175 001) and a cholesterol E kit (439-17501) from Wako, respectively. Glucose was measured from mouse tail vein using a glucometer (Truetest System). Insulin was measured by using a sensitive rat insulin radioimmunoassay kit from LINCO Research.
Cell Culture
MEFs were prepared from day 13.5/14.5 embryos after trypsin digestion as described previously (24). MEFs were cultured in Dulbecco modified Eagle's medium supplemented with 10% fetal calf serum, 2 mm l-glutamine, penicillin (100 units/ml), and streptomycin (100 μg/ml). 3T3/L1 cells were differentiated as described as previously (24). The cells were harvested in Triton lysis buffer (20 mm Tris, pH 7.4, 134 mm NaCl, 2 mm EDTA, 25 mm β-glycerophosphate, 2 mm NaPPi, 10% glycerol, 1% Triton X-100, 1 mm phenylmethylsulfonyl fluoride, 1 mm benzamidine, and 10 μg/ml leupeptin).
Metabolic Tests
CnAβ+/+ and CnAβ−/− male mice (12 and 8 weeks old) were fasted for 4–6 h before intraperitoneal injection with a bolus of insulin (0.75 unit/kg) for insulin tolerance tests (24). Serum glucose was measured from tail veil using a glucometer (True Test System) at 0, 15, 30, 45, 60, 90, and 120 min after the insulin injection. For glucose tolerance tests, the mice were orally gavaged with a bolus of glucose solution (2.5 g/kg). Serum glucose from the tail vein was measured with a glucometer at 0, 15, 30, 45, 60, 90, and 120 min after the gavage. At time 0, 15, 30, and 60 min, blood was also collected to determine insulin release by radioimmunoassays. Lipoprotein profile was determined by FPLC protocol as described previously (35). Approximately 150 μl of pooled serum from five CnAβ+/+ or CnAβ−/− male mice was loaded onto Superose 6 columns (Amersham Biosciences; analytical grade). Serum was passed at a flow rate of 0.3 ml/min, and 500 μl of eluate was collected in each fraction. Serum cholesterol and triglyceride content in each fraction was determined by colorimetric assays using Cholesterol E (Wako) and serum triglyceride determination kit (Sigma), respectively.
β-Adrenergic Receptor Activation
CnAβ+/+ and CnAβ−/− mice were injected intraperitoneally with β3-agonist CL316243 (CL; 0.1 mg/kg; Tocris Bioscience). White and brown adipose tissues were harvested at 0, 15, 30, and 45 min after CL316243 injection to determine the levels of PKA substrates phosphorylation. To determine the levels of free fatty acids and triglyceride levels, blood was collected at 0, 1, 2, 3, and 4 h after CL316243 injection. To determine the mean arterial blood pressure (MABP) of conscious mice (36), CnAβ+/+ and CnAβ−/− mice were anesthetized, and the catheter from the PA-C10 radiotelemeter pressure transducer (DSI, St. Paul, MN) was surgically inserted into the left carotid artery, whereas the transmitter was subcutaneously sutured on the mice. After 1 week of recovery, the mice were individually housed, and general adrenergic receptor agonist isoproterenol (Iso; 2 mg/kg) was injected. The mouse cages were placed on a receiver, and the MABP of conscious mice was monitored before and after the isoproterenol injection. MABP was recorded every 20 s for 10 s for 100 min at the times indicated. To determine β-adrenergic receptor/PKA activation ex vivo, epididymal adipose depots were harvested, minced, and challenged with CL316,243 (1 μm) or isoproterenol (1 μm) for the times indicated. Extracts prepared were subjected to immunoblotting analysis using phospho-PKA motif antibodies.
Cyclic AMP and PDE Assays
Serum-starved CnAβ+/+ and CnAβ−/− MEFs were stimulated with Iso (10 μm) for 5 or 15 min before harvest to determine the level of cAMP and PDE activity. Cyclic AMP was measured from cells after isoproterenol stimulation using a colorimetric enzyme immunoassay kit (Cayman Chemicals; number 581001). The PDE assay was performed as described previously (37). PDE inhibitor IBMX was added at final concentrations of 0.5 mm before the reaction.
Semiquantitative RT-PCR
Total RNA was isolated from epididymal fat depots using TRIzol reagents as described previously (24). The primers for PCR amplifications are the following: PDE3B, 5′-ATCCACCTGCAGTTTGTTCC-3′ and 5′-GCAAAAGATCGGGACCTACA-3′; PDE4D, 5′-CAAGTTTCAGGCTGGCTTTC-3′ and 5′-GTCCCATGTGTGACAAGCAC-3′; PDE4A, 5′-CCTGCCTGACAAGTTCCAAT-3′ and 5′-AGCAGAGATGACGGCAGAAT-3′; PDE4B, 5′-ATCACCTTGCTGTGGGATTC-3′ and 5′-AACCAACCTGGGATTTTTCC-3′; PDE7A, 5′-TGCTGCAGACGTTACTCAGG-3′ and 5′-AGCACATTTCAAGGCCATCT-3′; and actin, 5′-CAGTAATCTCCTTCTGCATCC-3′ and 5′-TTTGAGACCTTCAACACCCC-3′.
Statistical Analysis
The data are shown as the means ± S.E. Student's t test was performed, and values of p < 0.05 were considered to be significant.
RESULTS
Age-dependent Insulin Resistance in CnAβ−/− Mice
To investigate the role of calcineurin in metabolic regulation, we examined insulin sensitivity and glucose handling of mice lacking calcineurin Aβ (CnAβ−/−). Unlike the early postnatal lethality of CnAα−/− mice (15), CnAβ−/− mice were viable, fertile, and overtly normal (14). Thus, CnAβ−/− mice provide a defined model platform for investigating the systemic role of calcineurin in metabolic regulation. Insulin tolerance tests indicated that insulin-stimulated glucose uptake was reduced in 12-week-old CnAβ−/− mice (Fig. 1A). Glucose tolerance tests also indicated impaired responses to glucose challenge (Fig. 1B), supporting reduced insulin sensitivity in 12-week-old CnAβ−/− mice. Insulin-stimulated glucose uptake and glucose handling, however, were not significantly different in 8-week-old CnAβ−/− and CnAβ+/+ mice (Fig. 1, D and E). The area under the curve was calculated, further indicating the significant difference of the age-dependent reduction in insulin sensitivity and impairment in glucose handling between the CnAβ−/− and CnAβ+/+ mice (Fig. 1, A, B, D, and E). Together, these data indicate age-dependent insulin resistance in CnAβ−/− mice.
FIGURE 1.

CnAβ−/− mice exhibit age-dependent insulin resistance. 12-week-old (A–C) and 8-week-old (D–F) CnAβ−/− and CnAβ+/+ mice (n = 9) were subjected to insulin tolerance test (ITT) (A and D) and glucose tolerance test (GTT) (B, C, E, and F). The glucose (A, B, D, and E) and insulin (C and F) levels were measured at the indicated times and are presented. The area under the curve was also determined. *, p < 0.05; #, p < 0.005.
The delay in glucose clearance in CnAβ−/− mice could be due to improper insulin secretion by the pancreatic β-cells. For example, the calcineurin/NFAT signaling has been implicated in β-cell growth and function (38). We determined the insulin levels of CnAβ+/+ and CnAβ−/− mice during the glucose tolerance tests (Fig. 1, C and F). At both 8 and 12 weeks old, insulin release was, paradoxically, elevated in CnAβ−/− mice (Fig. 1, C and F). Increased secretion of insulin in CnAβ−/− mice, however, agrees with reduced glucose-induced insulin release in transgenic model of expression of calcineurin in pancreas (39). Together, these data indicate that insulin/glucose homeostasis may not completely account for the age-dependent metabolic dysregulation in CnAβ−/− mice.
Hyperlipidemic in CnAβ−/− Mice
We further examined other metabolic parameters of the 12- and 8-week-old CnAβ−/− mice (Fig. 2). After an overnight fast, circulating levels of triglyceride, cholesterol, and free fatty acids were significantly elevated in CnAβ−/− mice (Fig. 2, A–C and F–H). Increased triglyceride, free fatty acid, and cholesterol levels were found in 12- (Fig. 2, A–C) and 8-week-old (Fig. 2, F–H) CnAβ−/− mice. Indeed, elevated levels of triglyceride and cholesterol in 8-week-old CnAβ−/− mice corresponded to the increase in VLDL/LDL and HDL in lipoprotein profile, respectively (Fig. 2, K and L). Basal levels of glucose and insulin, however, were not significantly altered in 12- (Fig. 2, D and E) and 8-week-old (Fig. 2, I and J) CnAβ−/− mice. Together, these data indicate that hyperlipidemia found in CnAβ−/− mice, which was detected as early as 8 weeks after birth, also contributed to the age-dependent insulin/glucose dysregulation.
FIGURE 2.

CnAβ−/− mice are hyperlipidemic. A–J, levels of triglyceride (A and F), cholesterol (B and G), free fatty acids (FFA) (C and H), glucose (D and I), and insulin (E and J) of 12-week-old (A–E) and 8-week-old (F–J) CnAβ−/− and CnAβ+/+ mice (n = 10) were determined after a 16-h overnight fast. K and L, pooled serum isolated from five fasted CnAβ−/− or CnAβ+/+ mice were fractionated by FPLC and analyzed for triglyceride (K) and cholesterol (L) content in their lipoproteins. *, p < 0.05; #, p < 0.005.
Lipoprotein Clearance, Triglyceride Synthesis, and Lipid Mobilization in CnAβ−/− and CnAβ+/+ Mice
Imbalance of lipoprotein clearance, triglyceride synthesis, and/or mobilization (lipolysis) could account for the hyperlipidemia found in CnAβ−/− mice. We assessed whether triglyceride accumulation was due to lipid production/mobilization using Triton WR1339 (Fig. 3A), a nonionic detergent that coats lipoproteins and prevents access of lipoprotein lipases for the hydrolysis of triglyceride. Hence, in the presence of Triton WR1339, catabolism of circulating triglyceride is blocked, and lipoprotein clearance is inhibited. As expected, injection of Triton WR1339 increased triglyceride accumulation (Fig. 3A). CnAβ−/− mice, however, exhibited further elevated TG levels as compared with the CnAβ+/+ mice (Fig. 3A). Because the mice were fasted throughout the assay, circulating triglyceride were not derived from dietary lipids but from endogenous sources. These data suggest that changes in endogenous lipid production/mobilization likely account for the triglyceride accumulation in CnAβ−/− mice.
FIGURE 3.

Lipoprotein clearance, triglyceride synthesis, and lipid mobilization in CnAβ−/− and CnAβ+/+ mice. A, triglyceride accumulation in CnAβ−/− and CnAβ+/+ mice (n = 6) was determined upon injection of Triton WR1339 (500 mg/kg), which inhibits lipid clearance. The levels of triglyceride were determined at the times indicated and presented. B, mRNA in liver of 8-week-old CnAβ−/− and CnAβ+/+ mice (n = 6) were amplified by quantitative RT-PCR to determine the expression of key SREBP target genes involved in de novo triglyceride/cholesterol synthesis. The number of cycles required to reach threshold was represented as relative mRNA expression. Fold difference of 1 is equivalent to 2-fold change. *, p < 0.05; #, p < 0.005. C and D, the levels of triglyceride (C) and free fatty acids (FFA; D) of CnAβ−/− and CnAβ+/+ mice (n = 6) upon fasting were determined at the times indicated.
We also assessed whether de novo synthesis of triglyceride and cholesterol, under the master regulation of the sterol regulatory element-binding protein (SREBP) family of transcription factors (40), was affected in CnAβ−/− mice (Fig. 3B). Induced expression of SREBP target genes was frequently correlated with increased accumulation of triglyceride and cholesterol. There was, however, no significant difference in the expression of SREBP target genes in CnAβ+/+ and CnAβ−/− mice (Fig. 3B). These data indicate that de novo synthesis of triglyceride and cholesterol may not account for the hyperlipidemia found in CnAβ−/− mice.
Lipid mobilization via lipolysis and re-esterification (41) is controlled, in part, by the β-adrenergic G-protein-coupled receptor (GPCR) signaling pathways. For example, upon fasting, catecholamines released from the sympathetic/parasympathetic nervous systems stimulate the GPCR signaling pathways in adipocytes to induce lipolysis (42, 43). Notably, a dysfunctional β-adrenergic system can cause hyperlipidemia in many models of obesity (44, 45).
Next, we determined the accumulation of triglyceride and free fatty acids upon fasting (Fig. 3, C and D). Starting at 8 h after fasting, CnAβ−/− mice displayed significantly elevated levels of triglyceride as compared with the CnAβ+/+ mice (Fig. 3C). Accumulation of free fatty acids in CnAβ−/− mice was also higher than CnAβ+/+ mice and reached a peak at ∼12 h after fasting (Fig. 3D). The levels of free fatty acids were subsequently declined, which could be due to utilization and consumption as energy or re-esterification of free fatty acids into triglyceride (46). Nonetheless, these data indicate that β-adrenergic GPCR signaling pathway, which mediates lipolysis, may be potentiated in CnAβ−/− mice.
Potentiated Response in β-Adrenergic Receptor Signaling Pathway in CnAβ−/− Mice
The plausible potentiation of the GPCR signaling pathways could also account for the seemingly paradoxical elevated glucose-induced insulin release found in CnAβ−/− mice (Fig. 1, C and F), a process mediated by second messenger cAMP (47). Furthermore, CnAβ−/− mice exhibited additional pathophysiological phenotypes at old age, including increased incidence of seizure, aberrant bladder control, and abnormal weight loss (Fig. 4, A–C). These age-related changes in CnAβ−/− mice may also accrue from the potentiated GPCR signaling pathways.
FIGURE 4.

CnAβ−/− mice exhibited potentiated response in β-adrenergic receptor signaling pathway. A, number of incidences of seizure found in 6–12-month-old CnAβ−/− and CnAβ+/+ mice during their body weight measurement was recorded. B, amount of urine retained in bladder of CnAβ−/− and CnAβ+/+ mice (n = 5) after sedation was determined. C, body weight of CnAβ−/− and CnAβ+/+ mice (n = 10–15) was determined at the times indicated. D and E, the levels of triglyceride (D) and free fatty acids (FFA; E) of CnAβ−/− and CnAβ+/+ mice (n = 6) upon injection of β3-adrenergic receptor agonist CL (0.1 mg/kg) were determined at the times indicated. F, MABP of conscious CnAβ−/− and CnAβ+/+ mice was determined by radiotelemetry upon injection of β-adrenergic receptor agonist Iso (2 mg/kg). The time required for MABP return to resting level (dashed line) is indicated. The data presented are representative of three independent experiments with two mice per group. *, p < 0.05; #, p < 0.005.
To address the possibility of potentiated GPCR signaling pathways, we asked whether CnAβ−/− mice exhibit defects in lipolysis upon activation of the β-adrenergic receptor signaling using the specific β3 adrenergic receptor agonist CL316,243 (Fig. 4, D and E). Analogous to the effect of endogenous catecholamines released upon fasting, administration of CL316,243 increased the accumulation of triglyceride and free fatty acids (Fig. 4, D and E). CnAβ−/− mice, however, exhibited further triglyceride and free fatty acids accumulation as compared with the CnAβ+/+ mice (Fig. 4, D and E), indicating a potentiated response to β-adrenergic receptor signaling.
We further ascertained the enhanced β-adrenergic receptor signaling in CnAβ−/− mice using the general β-adrenergic receptor agonist isoproterenol and measured MABP through an implanted radiotelemetry transmitter (Fig. 4F). Injection of isoproterenol caused vasodilation and depression of MABP in conscious CnAβ−/− and CnAβ+/+ mice (Fig. 4F). In the CnAβ+/+ mice, we noted that MABP was returned to the resting level after 22 min (Fig. 4F). In CnAβ−/− mice, however, hypotension persisted for 54 min before rebounding (Fig. 4F). Together, these data indicate that CnAβ−/− mice exhibit increased responsiveness to stimulation by the β-adrenergic receptor agonist.
Sustained PKA Activation in CnAβ−/− Mice
Upon stimulation of β-adrenergic G-protein-coupled receptors, activation of adenylyl cyclases increases intracellular concentration of the second messenger cAMP, which binds to regulatory subunit of PKA holoenzyme and promotes the release of the PKA catalytic subunit for subsequent phosphorylation of its downstream targets (48). Turnover of cAMP, and hence reduction of PKA activity, is regulated by phosphodiesterases (49, 50), which are inhibited by IBMX. Dysregulation of phosphodiesterases thus would lead to aberrant intracellular levels of cAMP and subsequent malfunctions of downstream effectors regulated by PKA.
Given the potentiated response to β-adrenergic receptor agonists (Fig. 4), which channels through the cAMP/PKA signaling, we therefore determined the levels of PKA substrates phosphorylation in white and brown adipose tissues of CnAβ−/− and CnAβ+/+ mice upon stimulation with CL316,243 in vivo (Fig. 5). Administration of CL316,243 in vivo led to an increase in phosphorylation of PKA substrates in both white (Fig. 5A) and brown adipose tissues (Fig. 5B). However, ablation of CnAβ led to a much greater and sustained phosphorylation of PKA substrates (Fig. 5, A and B). These data support a role for calcineurin in the β-adrenergic G-protein-coupled, cAMP/PKA signaling pathway
FIGURE 5.

Sustained PKA activation in CnAβ−/− mice. A and B, white (WAT; A) and brown adipose tissues (BAT; B) of CnAβ−/− and CnAβ+/+ mice upon injection of β3-adrenergic receptor agonist CL (0.1 mg/kg) were determined at the times indicated. The data presented are representative of three independent experiments with two mice/group. C–E, white adipose tissues isolated from CnAβ−/− and CnAβ+/+ mice (n = 3) were challenged with β-adrenergic receptor agonist CL (1 μm) (C) or Iso (1 μm) (D and E) for the times indicated. The tissue extracts prepared were used to determine activation of PKA by immunoblotting analysis using phospho-PKA motif antibodies (phospho-PKA substrates) (C and D). E, effect of isoproterenol stimulation on phosphorylation of transcription factor CREB and HSL is also shown. Expression of tubulin is shown as a control.
Sustained activation of the cAMP/PKA signaling pathway in response to β3 adrenergic receptor agonist CL316,243 is likely adipose-specific because β3 adrenergic receptor is mainly expressed in adipocytes in mice (51). Furthermore, the rapid kinetic of potentiation of PKA substrate phosphorylation suggests the effect of calcineurin is cell autonomous. To ascertain the cell autonomous regulation by calcineurin in adipocytes, we examined activation of the β-adrenergic receptor/cAMP/PKA signaling pathway in isolated epididymal fat depots (Fig. 5, C and D). Ex vivo analysis of the isolated fat depots indicated that β-adrenergic receptor agonists, including CL316,243 (Fig. 5C) and isoproterenol (Fig. 5D), led to an increase in phosphorylation of PKA substrates. However, adipose depots isolated from CnAβ−/− mice exhibited a much greater and sustained phosphorylation of PKA substrates upon challenge with β-adrenergic receptor agonists (Fig. 5, C and D). We also determined phosphorylation of transcription factor CREB and HSL, known PKA substrates, using phospho-specific antibodies (Fig. 5E). Similar to the phosphorylation of PKA substrates, sustained phosphorylation of CREB and HSL was also found (Fig. 5E). Thus, the rapid kinetic of potentiation of PKA substrate phosphorylation indicate that a cell autonomous regulation of the β-adrenergic receptor/cAMP/PKA signaling pathway by calcineurin in adipose tissues.
Sustained PKA Activation Found in CnAβ−/− MEFs or upon CsA Treatment Is Sensitive to General Phosphodiesterase Inhibitor
Next, we determined the molecular basis of sustained phosphorylation of PKA substrates upon ablation of CnAβ. The apparent increase in PKA substrate phosphorylation could be due to the lack of dephosphorylation in the absence of CnAβ. Hence, CnAβ directly contributes to dephosphorylation and regulates the extent of PKA substrate phosphorylation. Alternatively, changes in PKA activity could account for the increased levels of phosphorylation of PKA substrates. The latter hypothesis would suggest that calcineurin indirectly affects PKA activation, which then led to changes in phosphorylation of PKA substrates.
To determine whether calcineurin directly or indirectly affects phosphorylation of PKA substrates, we examined the effect of phosphodiesterase inhibitor IBMX upon stimulation with β-adrenergic receptor agonist. In the presence of IBMX, turnover of cAMP is inhibited. If calcineurin directly affects phosphorylation of PKA substrates, potentiation of PKA substrate phosphorylation found in CnAβ−/− mice would be unaffected in the presence of IBMX. Conversely, if the levels of phosphorylation of PKA substrates in CnAβ−/− and CnAβ+/+ mice were attenuated in the presence of IBMX, these data would suggest that calcineurin indirectly regulates PKA activation.
Primary MEFs isolated from CnAβ+/+ and CnAβ−/− mice were stimulated with general β-adrenergic receptor agonist isoproterenol, which led to an increase in phosphorylation of PKA substrates (Fig. 6A). CnAβ−/− MEFs, however, exhibited a much greater and sustained phosphorylation of PKA substrates upon challenge with isoproterenol (Fig. 6A). The rapid kinetic of potentiation of PKA substrate phosphorylation supports a cell autonomous model on PKA substrate phosphorylation mediated by CnAβ. In the presence of phosphodiesterase inhibitor IBMX, the levels of phosphorylation of PKA substrates were comparable in CnAβ+/+ and CnAβ−/− MEFs (Fig. 6B), which suggests that calcineurin indirectly contributes to the sustained activation of PKA upon stimulation with β-adrenergic receptor agonist.
FIGURE 6.

Potentiation of PKA activation in CnAβ−/− mice or upon CsA treatment is sensitive to general phosphodiesterase inhibitor. MEFs (n = 3) isolated from CnAβ−/− (A and B) and CnAβ+/+ (A–D) mice were challenged with β-adrenergic receptor agonist Iso (10 μm), in the absence (A and C) or presence (B and D) of the phosphodiesterase inhibitor IBMX (0.5 mm). C–F, MEFs isolated from CnAβ+/+ mice (n = 3) (C and D) or differentiated 3T3/L1 adipocytes (E and F) were pretreated (CsA) or not (Control) with calcineurin inhibitor CsA (5 μm) for 1 h before stimulation with adrenergic receptor agonists, in the absence (−) or presence (+) of IBMX. The cell extracts prepared were used to determine activation of PKA by immunoblotting analysis using phospho-PKA motif antibodies (phospho-PKA substrates). Expression of tubulin is shown as a control.
We further ascertained the indirect regulation of calcineurin on PKA activation by using calcineurin inhibitor CsA (Fig. 6C). Inhibiting calcineurin with CsA mimics the loss of calcineurin function in CnAβ−/− MEFs. Pretreatment with CsA led to a much greater and sustained phosphorylation of PKA substrates upon challenge with isoproterenol (Fig. 6C). In the presence of IBMX, the levels of phosphorylation of PKA substrates were indistinguishable in control and CsA-treated MEFs (Fig. 6D). In addition, similar potentiation of phosphorylation of PKA substrates were found in 3T3/L1 differentiated adipocytes upon CsA treatment (Fig. 6, E and F). The increased phosphorylation of PKA substrates in CsA-treated differentiated adipocytes was also attenuated in the presence of IBMX (Fig. 6, E and F). Together, these data indicate that calcineurin inhibition by CsA recapitulates the sustained β-adrenergic receptor/PKA activation found in CnAβ−/− MEFs, which is sensitive to phosphodiesterase inhibitor IBMX.
Increased cAMP Accumulation and Reduced Phosphodiesterase Activity in CnAβ−/− MEFs
PKA activation is elicited by increase in intracellular second messenger cAMP. Next, we determined the levels of cAMP in CnAβ−/− and CnAβ+/+ MEFs (Fig. 7A). Administration of isoproterenol led to a transient accumulation of cAMP (Fig. 7A). However, ablation of CnAβ led to a much greater accumulation and prolonged elevation in the levels of cAMP (Fig. 7A). In the presence of IBMX, the levels of cAMP in CnAβ−/− and CnAβ+/+ MEFs were indistinguishable (Fig. 7A), suggesting that reduced cAMP hydrolysis by phosphodiesterases might account for the increased cAMP accumulation in the absence of CnAβ. Indeed, induction of phosphodiesterases activity by isoproterenol was attenuated in CnAβ−/− MEFs (Fig. 7B). Ablation of CnAβ, however, did not affect the transcript levels of several PDE members (Fig. 7C). Together, these data indicate that impaired cAMP hydrolysis resulting from impaired regulation of phosphodiesterases accounts for the sustained elevation of cAMP in the absence of calcineurin.
FIGURE 7.
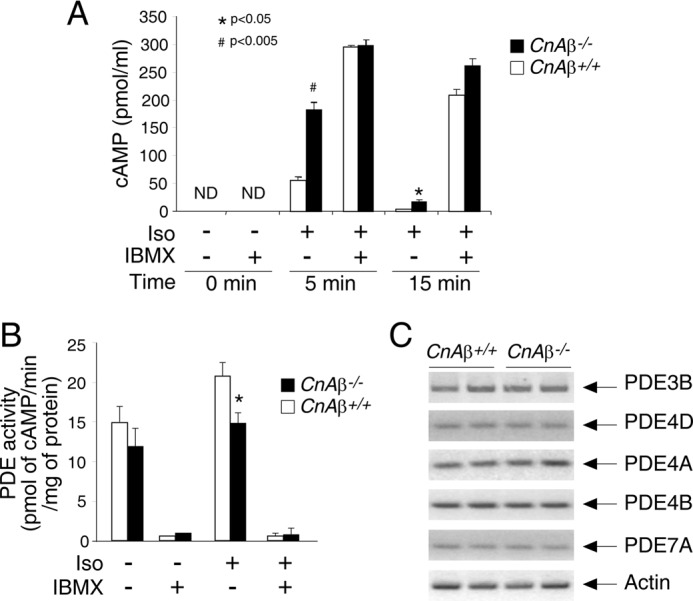
Increased cAMP accumulation and reduced phosphodiesterase activation in CnAβ−/− MEFs. A, the levels of cAMP in CnAβ−/− and CnAβ+/+ MEFs (n = 4) upon stimulation (+) or not (−) with Iso (10 μm for 5 and 15 min) were determined. The effect of the phosphodiesterase (PDE) inhibitor IBMX (0.5 mm) was also determined. B, PDE activity in CnAβ−/− and CnAβ+/+ MEFs (n = 3) upon stimulation (+) or not (−) with Iso (10 μm for 5 min) were determined. The effect of general PDE inhibitor IBMX (0.5 mm) was also determined. *, p < 0.05; #, p < 0.005. C, transcript levels of several PDE members in CnAβ−/− and CnAβ+/+ fat depots were determined by semiquantitative RT-PCR.
Calcineurin Phosphatase Regulates Expression of Phosphodiesterase 3B
Previously, we demonstrated that calcineurin regulates phosphodegron-dependent degradation of PDE4D (32). Reduced expression of PDE4D is likely accounted for by the potentiated lipolysis and hemodynamic response found in CnAβ−/− mice (Fig. 4). Glucose-induced insulin release, however, is mainly controlled by PDE3B rather than PDE4D (49, 52). Similar to PDE4D, membrane-bound PDE3B hydrolyzes cAMP and contributes to lipid homeostasis and cardiac regulation (49, 52). In addition, degradation of PDE3B, analogous to PDE4D, is sensitive to proteosome inhibitor MG132 (Fig. 8A). Thus, we hypothesized that calcineurin might also regulate expression of PDE3B and contribute to the elevated glucose-induced insulin release in CnAβ+/+ mice (Fig. 1, C and F).
FIGURE 8.
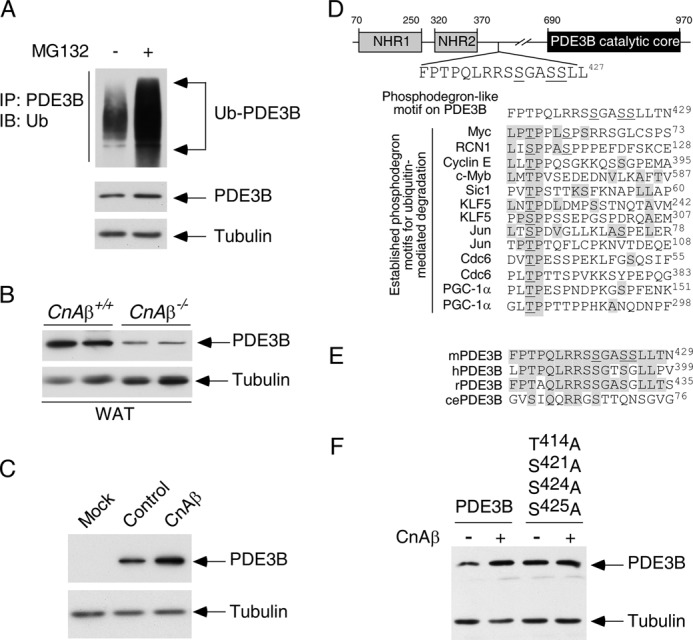
Calcineurin phosphatase regulates expression of phosphodiesterase 3B. A, COS cells were transiently transfected with PDE3B and ubiquitin. Ubiquitin conjugation of PDE3B was determined by co-immunoprecipitation assays. The presence of ubiquitin (Ub) in PDE3B immunoprecipitates (IP) was analyzed by immunoblot (IB) analysis. The effect of proteosome inhibitor MG132 (10 μm) was also shown. B, extracts prepared from CnAβ−/− and CnAβ+/+ white adipose tissues (WAT) were subjected to immunoblotting analysis to determine endogenous expression of PDE3B. The expression levels of tubulin were used as controls. C, PDE3B was co-expressed with CnAβ in COS cells. The expression levels of PDE3B were determined by immunoblot analysis. The expression levels of tubulin were used as controls. D, schematic illustration of the location of the FPTPQLRRSSGASSLLT phosphodegron on PDE3B. A potential phosphorylation site (Thr414) on PDE3B is shown. Adjacent Ser421, Ser424, and Ser425 of PDE3B, which were found phosphorylated previously (55), are also indicated. Sequences of known phosphodegrons on other proteins are also illustrated. Conserved amino acid residues and previously identified phosphorylated Thr/Ser are shaded and underlined, respectively. E, sequence alignment of the potential phosphodegron FPTPQLRRSSGASSLLT from mouse, human, rat, and C. elegans is shown. F, wild type or Ala414,421,424,425 PDE3B was co-expressed with CnAβ in COS cells. The expression levels of PDE3B were determined by immunoblot analysis. The expression levels of tubulin were used as controls.
To test this hypothesis, we examined the endogenous level of PDE3B in CnAβ−/− mice. Similar to PDE4D, immunoblotting analysis indicated reduced expression of PDE3B upon ablation of CnAβ (Fig. 8B). Conversely, the levels of PDE3B expression were potentiated upon co-expression with CnAβ (Fig. 8C). These data indicate that CnAβ regulates expression of at least two different cAMP phosphodiesterases.
How does calcineurin regulate PDE3B expression? Calcineurin regulates PDE4D expression via a phosphodegron-dependent mechanism (32). Potentiated PDE3B expression might also be regulated similarly by calcineurin. Indeed, sequence analysis identified a potential phosphodegron (FPTPQLRRSSGASSLLT428) located after the conserved NH2 terminus regulatory regions but before the PDE3B catalytic core (Fig. 8D). The putative site surrounding Thr414 of PDE3B exhibited high sequence similarity to previously established phosphodegrons in controlling protein degradation, including Myc, Jun, and cyclin E (53, 54). In addition, adjacent Ser421, Ser424, and Ser425 of PDE3B were found phosphorylated previously (55). This potential phosphodegron motif (FPTPQLRRSSGASSLLT428) on mouse PDE3B was also found across different species, including human, rat, and plausibly Caenorhabditis elegans (Fig. 8E). Given that phosphorylation of multiple sites at or nearby a phosphodegron facilitates recruitment of F-box adaptors to mediate protein degradation (54, 56), Thr414, Ser421, Ser424, and Ser425 of PDE3B might function as a phosphodegron to regulate PDE3B degradation.
Next, we determined the role of Thr414, Ser421, Ser424, and Ser425 in calcineurin regulation of PDE3B. Ala replacement of all four residues (Thr414, Ser421, Ser424, and Ser425) increased PDE3B expression (Fig. 8F). Notably, expression of Ala414,421,424,425 PDE3B was no longer potentiated by co-expression with CnAβ (Fig. 8F). These data indicate that CnAβ regulates PDE3B expression by targeting Thr414, Ser421, Ser424, and Ser425.
DISCUSSION
In this report, we demonstrate that CnAβ contributes to lipid homeostasis via lipolysis, a process mediated by the β-adrenergic G-protein-coupled receptor signaling pathway. Furthermore, we show that CnAβ participates in glucose-induced insulin release and hemodynamic response, processes also mediated by the GPCR signaling. Indeed, accumulation of cAMP caused by reduced PDE expression, including PDE3B and PDE4D (this report and Ref. 32), is found upon ablation of CnAβ. Given the importance of immunosuppressant CsA, a specific calcineurin inhibitor (6), our data provide new insights on the molecular pathology of metabolic complications found in post-transplant patients. We propose that reduced expression of PDEs upon calcineurin inhibition upsets the feedback balance of GPCR signaling and leads to sustained elevation of cAMP, a critical second messenger in controlling a diverse portfolio of downstream effectors. Thus, excess cAMP, in addition to pancreatic dysfunction and loss of insulin sensitivity (19, 25), may contribute to diabetes, cardiac dysregulations, and nephro- and neurotoxicity found in post-transplant patients treated with CsA.
Current models indicate that calcineurin plays an important role in regulating gene transcription (9, 22). One example is the calcineurin → NFAT axis for its key role in cytokine gene transcription in immune cells and in adipocytes (21, 24). The calcineurin → NFAT axis is also critical for pancreatic β-cell growth via regulating transcription of insulin and insulin receptor substrates (19, 25, 57, 58). Dephosphorylation switch mediated by calcineurin also significantly contributes to post-transcriptional regulation. For example, calcineurin modulates the gating properties of membrane channels upon dephosphorylation (59). We previously showed that calcineurin also contributes to additional post-transcriptional regulation by affecting protein degradation (32). Dephosphorylation at the phosphodegrons extends the stability of target proteins. Hence, calcineurin offers short and long term regulation to control gene expression and function. Further studies to elucidate calcineurin substrates will provide new insight for this important signaling molecule in pathophysiological conditions.
Previously, we showed that calcineurin regulates the expression of PDE4D via a phosphodegron (32). Reduced PDE4D expression could, in part, account for the potentiated GPCR signaling found in CnAβ−/− mice in response to adrenergic stimuli to trigger lipolysis in adipocytes and hemodynamic response in heart. Given that cAMP augments glucose-induced insulin release (47), it is not unexpected for the increased levels of insulin in CnAβ−/− mice. PDE3B, rather than PDE4D, is the key cAMP hydrolyase expressed in pancreatic β cells to control glucose-induced insulin release in pancreas (49, 52). Indeed, we find that the expression of PDE3B is reduced upon ablation of CnAβ (Fig. 8). Conversely, expression of CnAβ potentiates the levels of PDE3B (Fig. 8). Similar metabolic phenotypes, including dysregulations in lipid homeostasis and cardiac regulations among PDE3B null mice (49, 52) and CnAβ−/− mice, further support our model that calcineurin participates in cross-talk with cAMP-mediated signaling.
The role of calcineurin in regulating glucose-induced insulin release is further corroborated by the reduced insulin release in transgenic mice expressing constitutive active calcineurin in the pancreas (39). It is tempting to speculate that the expression levels of PDE3B are elevated in the pancreas of transgenic mice expressing constitutive active calcineurin. Elevated levels of PDE3B would then limit the accumulation of cAMP and subsequent insulin release as reported previously (60). Perhaps PDE3B inhibitor could correct the dysregulated insulin homeostasis in mice expressing constitutive active calcineurin in the pancreas.
PDE3B is phosphorylated at multiple sites by Akt and PKA upon stimulation with insulin and βAR agonist, respectively (61). PDE3B expression is increased in the presence of insulin, whereas βAR agonist reduces PDE3B levels (61). How Akt and PKA regulate the expression of PDE3B, however, remains elusive. In light of our findings that PDE3B expression could be regulated upon phosphorylation (Fig. 8), it is tempting to speculate that downstream effector protein kinases of the insulin and βAR signaling pathways play a role in modulating PDE3B expression, which is antagonized by calcineurin. Indeed, we found that GSK3β promotes degradation of PDE3B (data not shown). Perhaps induction of PDE3B expression by the insulin → Akt pathway is, in part, due to inhibition of GSK3β, which is phosphorylated and inhibited by Akt. Stabilized PDE3B, thereby, will provide a feedback mechanism to hydrolyze cAMP and turn off glucose-induced insulin release.
In addition to the insulin → Akt axis, the βAR → PKA pathway may also participate in regulating PDE3B expression. Interestingly, Ser421 of PDE3B displays consensus sequence for PKA phosphorylation and, along with Ser424 and Ser425, was found to be phosphorylated previously (55). In addition, Ser421 of PDE3B is found among different species including mouse, human, rat, and C. elegans (Fig. 8E). Phosphorylation of multiple sites at or nearby a phosphodegron would facilitate phospho-dependent protein degradation as suggested previously (54, 56). We also showed that phosphomimetic amino acid residues, such as Asp and Glu, located near the phosphodegron also contribute significantly to phospho-dependent degradation of target proteins (32). Thus, it is tempting to speculate that multiple phosphorylations, such as Ser421, Ser424, and Ser425 of PDE3B, may provide priming actions (62–65) for subsequent GSK3β-mediated phosphorylation at Thr414 and degradation of PDE3B via a phospho-dependent mechanism.
Although we have identified that Thr414, Ser421, Ser424, and Ser425 contribute significantly in calcineurin-mediated regulation of PDE3B expression (Fig. 8E), Ala replacement of these phosphoacceptor sites on PDE3B did not affect degradation facilitated by GSK3β (data not shown). In addition to GSK3β, casein kinase 1 also promotes degradation of wild type and Ala414,421,424,425 PDE3B (data not shown). Perhaps expression of GSK3β and casein kinase 1 phosphorylates additional sites and facilitates PDE3B degradation. Indeed, at least two different E3 ubiquitin ligases promote degradation of PDE3B (data not shown), suggesting that additional sites may be involved. Alternatively, the overall phosphorylation level may dictate degradation of PDE3B. If so, replacement of Thr414, Ser421, Ser424, and Ser425 of PDE3B with phosphomimetic amino acid residues should be sufficient to alter PDE3B stability.
In conclusion, we have uncovered a cell autonomous role of calcineurin in metabolic regulation. Calcineurin impinges on the cAMP signaling pathway and indirectly affects PKA activation via phosphodiesterases regulation. Our data provide new insights on the role of calcineurin in metabolic regulation. Our study also illuminates an alternative molecular pathogenesis for the metabolic complications elicited by CsA in transplant patients.
Acknowledgments
We thank Dr. Vincent Manganiello for PDE3B plasmid. We also thank members of our laboratories for their suggestions and critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health grants (to C.-W. C., J. J., D. J. L., M. P. L., J. D. M., and P. E. S.). This work was also supported by a Howard Hughes Medical Institute grant (to J. D. M.).
- used are: CsA
- cyclosporine A
- βAR
- β-adrenergic receptor
- CL
- β3 adrenergic receptor agonist CL316,243
- CnAβ
- calcineurin Aβ
- GSK3β
- glycogen synthase kinase 3β
- IBMX
- phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine
- Iso
- isoproterenol
- MEF
- primary mouse embryonic fibroblasts
- PDE
- phosphodiesterase
- CnB
- calcineurin B regulatory subunit
- CREB
- cAMP response element-binding protein
- HSL
- hormone-sensitive lipase
- MABP
- mean arterial blood pressure
- SREBP
- sterol-regulatory element-binding protein
- GPCR
- G-protein-coupled receptor.
REFERENCES
- 1. Marchetti P., Navalesi R. (2000) The metabolic effects of cyclosporin and tacrolimus. J. Endocrinol. Invest. 23, 482–490 [DOI] [PubMed] [Google Scholar]
- 2. Montori V. M., Basu A., Erwin P. J., Velosa J. A., Gabriel S. E., Kudva Y. C. (2002) Posttransplantation diabetes. A systematic review of the literature. Diabetes Care 25, 583–592 [DOI] [PubMed] [Google Scholar]
- 3. Vaziri N. D., Liang K., Azad H. (2000) Effect of cyclosporine on HMG-CoA reductase, cholesterol 7α-hydroxylase, LDL receptor, HDL receptor, VLDL receptor, and lipoprotein lipase expressions. J. Pharmacol. Exp. Ther. 294, 778–783 [PubMed] [Google Scholar]
- 4. Ballantyne C. M., Podet E. J., Patsch W. P., Harati Y., Appel V., Gotto A. M., Jr., Young J. B. (1989) Effects of cyclosporine therapy on plasma lipoprotein levels. JAMA 262, 53–56 [PubMed] [Google Scholar]
- 5. Düfer M., Krippeit-Drews P., Lembert N., Idahl L. A., Drews G. (2001) Diabetogenic effect of cyclosporin A is mediated by interference with mitochondrial function of pancreatic B-cells. Mol. Pharmacol. 60, 873–879 [PubMed] [Google Scholar]
- 6. Schreiber S. L., Crabtree G. R. (1992) The mechanism of action of cyclosporin A and FK506. Immunol. Today 13, 136–142 [DOI] [PubMed] [Google Scholar]
- 7. Rusnak F., Mertz P. (2000) Calcineurin. Form and function. Physiol. Rev. 80, 1483–1521 [DOI] [PubMed] [Google Scholar]
- 8. Guerini D. (1997) Calcineurin. Not just a simple protein phosphatase. Biochem. Biophys. Res. Commun. 235, 271–275 [DOI] [PubMed] [Google Scholar]
- 9. Crabtree G. R. (2001) Calcium, calcineurin, and the control of transcription. J. Biol. Chem. 276, 2313–2316 [DOI] [PubMed] [Google Scholar]
- 10. Aramburu J., Rao A., Klee C. B. (2000) Calcineurin. From structure to function. Curr. Top. Cell Regul. 36, 237–295 [DOI] [PubMed] [Google Scholar]
- 11. Usuda N., Arai H., Sasaki H., Hanai T., Nagata T., Muramatsu T., Kincaid R. L., Higuchi S. (1996) Differential subcellular localization of neural isoforms of the catalytic subunit of calmodulin-dependent protein phosphatase (calcineurin) in central nervous system neurons. Immunohistochemistry on formalin-fixed paraffin sections employing antigen retrieval by microwave irradiation. J. Histochem. Cytochem. 44, 13–18 [DOI] [PubMed] [Google Scholar]
- 12. Nakazawa A., Usuda N., Matsui T., Hanai T., Matsushita S., Arai H., Sasaki H., Higuchi S. (2001) Localization of calcineurin in the mature and developing retina. J. Histochem. Cytochem. 49, 187–195 [DOI] [PubMed] [Google Scholar]
- 13. Bueno O. F., Brandt E. B., Rothenberg M. E., Molkentin J. D. (2002) Defective T cell development and function in calcineurin Aβ-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 99, 9398–9403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bueno O. F., Wilkins B. J., Tymitz K. M., Glascock B. J., Kimball T. F., Lorenz J. N., Molkentin J. D. (2002) Impaired cardiac hypertrophic response in calcineurin Aβ-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 99, 4586–4591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gooch J. L., Toro J. J., Guler R. L., Barnes J. L. (2004) Calcineurin Aα but not Aβ is required for normal kidney development and function. Am. J. Pathol. 165, 1755–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang B. W., Zimmer G., Chen J., Ladd D., Li E., Alt F. W., Wiederrecht G., Cryan J., O'Neill E. A., Seidman C. E., Abbas A. K., Seidman J. G. (1996) T cell responses in calcineurin Aα-deficient mice. J. Exp. Med. 183, 413–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhuo M., Zhang W., Son H., Mansuy I., Sobel R. A., Seidman J., Kandel E. R. (1999) A selective role of calcineurin aalpha in synaptic depotentiation in hippocampus. Proc. Natl. Acad. Sci. U.S.A. 96, 4650–4655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Graef I. A., Chen F., Chen L., Kuo A., Crabtree G. R. (2001) Signals transduced by Ca2+/calcineurin and NFATc3/c4 pattern the developing vasculature. Cell 105, 863–875 [DOI] [PubMed] [Google Scholar]
- 19. Heit J. J., Apelqvist A. A., Gu X., Winslow M. M., Neilson J. R., Crabtree G. R., Kim S. K. (2006) Calcineurin/NFAT signalling regulates pancreatic β-cell growth and function. Nature 443, 345–349 [DOI] [PubMed] [Google Scholar]
- 20. Jain J., McCaffrey P. G., Miner Z., Kerppola T. K., Lambert J. N., Verdine G. L., Curran T., Rao A. (1993) The T-cell transcription factor NFATp is a substrate for calcineurin and interacts with Fos and Jun. Nature 365, 352–355 [DOI] [PubMed] [Google Scholar]
- 21. Clipstone N. A., Crabtree G. R. (1992) Identification of calcineurin as a key signalling enzyme in T-lymphocyte activation. Nature 357, 695–697 [DOI] [PubMed] [Google Scholar]
- 22. Hogan P. G., Chen L., Nardone J., Rao A. (2003) Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 17, 2205–2232 [DOI] [PubMed] [Google Scholar]
- 23. Winslow M. M., Neilson J. R., Crabtree G. R. (2003) Calcium signalling in lymphocytes. Curr. Opin. Immunol. 15, 299–307 [DOI] [PubMed] [Google Scholar]
- 24. Yang T. T., Suk H. Y., Yang X., Olabisi O., Yu R. Y., Durand J., Jelicks L. A., Kim J. Y., Scherer P. E., Wang Y., Feng Y., Rossetti L., Graef I. A., Crabtree G. R., Chow C. W. (2006) Role of transcription factor NFAT in glucose and insulin homeostasis. Mol. Cell Biol. 26, 7372–7387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goodyer W. R., Gu X., Liu Y., Bottino R., Crabtree G. R., Kim S. K. (2012) Neonatal β cell development in mice and humans is regulated by calcineurin/NFAT. Dev. Cell 23, 21–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schaffer J. E. (2003) Lipotoxicity. When tissues overeat. Curr. Opin. Lipidol. 14, 281–287 [DOI] [PubMed] [Google Scholar]
- 27. Unger R. H. (2003) The physiology of cellular liporegulation. Annu. Rev. Physiol. 65, 333–347 [DOI] [PubMed] [Google Scholar]
- 28. Shulman G. I. (2000) Cellular mechanisms of insulin resistance. J. Clin. Invest. 106, 171–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Poitout V. (2008) Glucolipotoxicity of the pancreatic β-cell. Myth or reality? Biochem. Soc. Trans. 36, 901–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Garg A., Misra A. (2002) Hepatic steatosis, insulin resistance, and adipose tissue disorders. J. Clin. Endocrinol. Metab. 87, 3019–3022 [DOI] [PubMed] [Google Scholar]
- 31. Zhou Y. T., Grayburn P., Karim A., Shimabukuro M., Higa M., Baetens D., Orci L., Unger R. H. (2000) Lipotoxic heart disease in obese rats. Implications for human obesity. Proc. Natl. Acad. Sci. U.S.A. 97, 1784–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhu H., Suk H. Y., Yu R. Y., Brancho D., Olabisi O., Yang T. T., Yang X., Zhang J., Moussaif M., Durand J. L., Jelicks L. A., Kim J. Y., Scherer P. E., Frank P. G., Lisanti M. P., Calvert J. W., Duranski M. R., Lefer D. J., Huston E., Baillie G. S., Houslay M. D., Molkentin J. D., Jin J., Chow C. W. (2010) Evolutionarily conserved role of calcineurin in phosphodegron-dependent degradation of phosphodiesterase 4D. Mol. Cell Biol. 30, 4379–4390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Miki T., Taira M., Hockman S., Shimada F., Lieman J., Napolitano M., Ward D., Taira M., Makino H., Manganiello V. C. (1996) Characterization of the cDNA and gene encoding human PDE3B, the cGIP1 isoform of the human cyclic GMP-inhibited cyclic nucleotide phosphodiesterase family. Genomics 36, 476–485 [DOI] [PubMed] [Google Scholar]
- 34. Perino A., Ghigo A., Ferrero E., Morello F., Santulli G., Baillie G. S., Damilano F., Dunlop A. J., Pawson C., Walser R., Levi R., Altruda F., Silengo L., Langeberg L. K., Neubauer G., Heymans S., Lembo G., Wymann M. P., Wetzker R., Houslay M. D., Iaccarino G., Scott J. D., Hirsch E. (2011) Integrating cardiac PIP3 and cAMP signaling through a PKA anchoring function of p110γ. Mol. Cell 42, 84–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Frank P. G., Lee H., Park D. S., Tandon N. N., Scherer P. E., Lisanti M. P. (2004) Genetic ablation of caveolin-1 confers protection against atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 24, 98–105 [DOI] [PubMed] [Google Scholar]
- 36. Duranski M. R., Elrod J. W., Calvert J. W., Bryan N. S., Feelisch M., Lefer D. J. (2006) Genetic overexpression of eNOS attenuates hepatic ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 291, H2980–H2986 [DOI] [PubMed] [Google Scholar]
- 37. Marchmont R. J., Houslay M. D. (1980) Insulin controls the cyclic AMP-dependent phosphorylation of integral and peripheral proteins associated with the rat liver plasma membrane. FEBS Lett. 118, 18–24 [DOI] [PubMed] [Google Scholar]
- 38. Arron J. R., Winslow M. M., Polleri A., Chang C. P., Wu H., Gao X., Neilson J. R., Chen L., Heit J. J., Kim S. K., Yamasaki N., Miyakawa T., Francke U., Graef I. A., Crabtree G. R. (2006) NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 441, 595–600 [DOI] [PubMed] [Google Scholar]
- 39. Bernal-Mizrachi E., Cras-Méneur C., Ye B. R., Johnson J. D., Permutt M. A. (2010) Transgenic overexpression of active calcineurin in β-cells results in decreased β-cell mass and hyperglycemia. PLoS ONE 5, e11969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Raghow R., Yellaturu C., Deng X., Park E. A., Elam M. B. (2008) SREBPs. The crossroads of physiological and pathological lipid homeostasis. Trends Endocrinol. Metab. 19, 65–73 [DOI] [PubMed] [Google Scholar]
- 41. Prentki M., Madiraju S. R. (2008) Glycerolipid metabolism and signaling in health and disease. Endocr. Rev. 29, 647–676 [DOI] [PubMed] [Google Scholar]
- 42. Robidoux J., Martin T. L., Collins S. (2004) β-Adrenergic receptors and regulation of energy expenditure. A family affair. Annu. Rev. Pharmacol. Toxicol. 44, 297–323 [DOI] [PubMed] [Google Scholar]
- 43. Duncan R. E., Ahmadian M., Jaworski K., Sarkadi-Nagy E., Sul H. S. (2007) Regulation of lipolysis in adipocytes. Annu. Rev. Nutr. 27, 79–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tentolouris N., Liatis S., Katsilambros N. (2006) Sympathetic system activity in obesity and metabolic syndrome. Ann. N.Y. Acad. Sci. 1083, 129–152 [DOI] [PubMed] [Google Scholar]
- 45. Seals D. R., Bell C. (2004) Chronic sympathetic activation. Consequence and cause of age-associated obesity? Diabetes 53, 276–284 [DOI] [PubMed] [Google Scholar]
- 46. Elia M., Zed C., Neale G., Livesey G. (1987) The energy cost of triglyceride-fatty acid recycling in nonobese subjects after an overnight fast and four days of starvation. Metabolism 36, 251–255 [DOI] [PubMed] [Google Scholar]
- 47. Shakur Y., Holst L. S., Landstrom T. R., Movsesian M., Degerman E., Manganiello V. (2001) Regulation and function of the cyclic nucleotide phosphodiesterase (PDE3) gene family. Prog. Nucleic Acid Res. Mol. Biol. 66, 241–277 [DOI] [PubMed] [Google Scholar]
- 48. Taylor S. S., Kim C., Cheng C. Y., Brown S. H., Wu J., Kannan N. (2008) Signaling through cAMP and cAMP-dependent protein kinase. Diverse strategies for drug design. Biochim. Biophys. Acta 1784, 16–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Pyne N. J., Furman B. L. (2003) Cyclic nucleotide phosphodiesterases in pancreatic islets. Diabetologia 46, 1179–1189 [DOI] [PubMed] [Google Scholar]
- 50. Houslay M. D., Adams D. R. (2003) PDE4 cAMP phosphodiesterases. Modular enzymes that orchestrate signalling cross-talk, desensitization and compartmentalization. Biochem. J. 370, 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Strosberg A. D. (1995) Structure, function, and regulation of the three β-adrenergic receptors. Obes. Res. 3, (Suppl. 4) 501S–505S [DOI] [PubMed] [Google Scholar]
- 52. Degerman E., Ahmad F., Chung Y. W., Guirguis E., Omar B., Stenson L., Manganiello V. (2011) From PDE3B to the regulation of energy homeostasis. Curr. Opin. Pharmacol. 11, 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jin J., Shirogane T., Xu L., Nalepa G., Qin J., Elledge S. J., Harper J. W. (2003) SCFβ-TRCP links Chk1 signaling to degradation of the Cdc25A protein phosphatase. Genes Dev. 17, 3062–3074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hunter T. (2007) The age of crosstalk. Phosphorylation, ubiquitination, and beyond. Mol. Cell 28, 730–738 [DOI] [PubMed] [Google Scholar]
- 55. Lindh R., Ahmad F., Resjö S., James P., Yang J. S., Fales H. M., Manganiello V., Degerman E. (2007) Multisite phosphorylation of adipocyte and hepatocyte phosphodiesterase 3B. Biochim. Biophys. Acta 1773, 584–592 [DOI] [PubMed] [Google Scholar]
- 56. Ardley H. C., Robinson P. A. (2005) E3 ubiquitin ligases. Essays Biochem. 41, 15–30 [DOI] [PubMed] [Google Scholar]
- 57. Demozay D., Tsunekawa S., Briaud I., Shah R., Rhodes C. J. (2011) Specific glucose-induced control of insulin receptor substrate-2 expression is mediated via Ca2+-dependent calcineurin/NFAT signaling in primary pancreatic islet β-cells. Diabetes 60, 2892–2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Soleimanpour S. A., Crutchlow M. F., Ferrari A. M., Raum J. C., Groff D. N., Rankin M. M., Liu C., De León D. D., Naji A., Kushner J. A., Stoffers D. A. (2010) Calcineurin signaling regulates human islet β-cell survival. J. Biol. Chem. 285, 40050–40059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Mohapatra D. P., Park K. S., Trimmer J. S. (2007) Dynamic regulation of the voltage-gated Kv2.1 potassium channel by multisite phosphorylation. Biochem. Soc. Trans. 35, 1064–1068 [DOI] [PubMed] [Google Scholar]
- 60. Härndahl L., Wierup N., Enerbäck S., Mulder H., Manganiello V. C., Sundler F., Degerman E., Ahrén B., Holst L. S. (2004) β-Cell-targeted overexpression of phosphodiesterase 3B in mice causes impaired insulin secretion, glucose intolerance, and deranged islet morphology. J. Biol. Chem. 279, 15214–15222 [DOI] [PubMed] [Google Scholar]
- 61. Oknianska A., Zmuda-Trzebiatowska E., Manganiello V., Degerman E. (2007) Long-term regulation of cyclic nucleotide phosphodiesterase type 3B and 4 in 3T3-L1 adipocytes. Biochem. Biophys. Res. Commun. 353, 1080–1085 [DOI] [PubMed] [Google Scholar]
- 62. Yang T. T., Yu R. Y., Agadir A., Gao G. J., Campos-Gonzalez R., Tournier C., Chow C. W. (2008) Integration of protein kinases mTOR and extracellular signal-regulated kinase 5 in regulating nucleocytoplasmic localization of NFATc4. Mol. Cell Biol. 28, 3489–3501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Morel C., Carlson S. M., White F. M., Davis R. J. (2009) Mcl-1 integrates the opposing actions of signaling pathways that mediate survival and apoptosis. Mol. Cell Biol. 29, 3845–3852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cole A. R., Causeret F., Yadirgi G., Hastie C. J., McLauchlan H., McManus E. J., Hernández F., Eickholt B. J., Nikolic M., Sutherland C. (2006) Distinct priming kinases contribute to differential regulation of collapsin response mediator proteins by glycogen synthase kinase-3 in vivo. J. Biol. Chem. 281, 16591–16598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Hergovich A., Lisztwan J., Thoma C. R., Wirbelauer C., Barry R. E., Krek W. (2006) Priming-dependent phosphorylation and regulation of the tumor suppressor pVHL by glycogen synthase kinase 3. Mol. Cell Biol. 26, 5784–5796 [DOI] [PMC free article] [PubMed] [Google Scholar]