

Background: Toxoplasma gondii expresses several cathepsin proteases, but little is known about how they are activated.
Results: Toxoplasma cathepsin B protease localizes to lysosome-like compartment, and its maturation requires a parasite cathepsin L protease.
Conclusion: Maturation of Toxoplasma cathepsin B is unconventionally nonself-dependent.
Significance: The unusual maturation of Toxoplasma cathepsin B suggests a novel mechanism of protease activation within the endolysosomal system.
Keywords: Cysteine Protease, Electron Microscopy (EM), Enzyme Processing, Lysosomes, Parasite, Cathepsin, Protease, Rhoptry, Toxoplasma gondii, Vacuolar Compartment
Abstract
Proteases regulate key events during infection by the pervasive intracellular parasite Toxoplasma gondii. Understanding how parasite proteases mature from an inactive zymogen to an active enzyme is expected to inform new strategies for blocking their actions. Herein, we show that T. gondii cathepsin B protease (TgCPB) does not undergo self-maturation but instead requires the expression of a second papain-family cathepsin protease, TgCPL. Using recombinant enzymes we also show that TgCPL is capable of partially maturing TgCPB in vitro. Consistent with this interrelationship, antibodies with validated specificity detected TgCPB in the lysosome-like vacuolar compartment along with TgCPL. Our findings also establish that TgCPB does not localize to the rhoptries as previously reported. Accordingly, rhoptry morphology and rhoptry protein maturation are normal in TgCPB knock-out parasites. Finally, we show that although maturation of TgCPL is independent of TgCPB, it may involve an additional protease(s) in conjunction with self-maturation.
Introduction
Cysteine proteases contribute to several key processes of pathogenic microbes including host entry, nutrient acquisition and modulation of host immune responses (1–3). Most cathepsins are members of the papain family Clan CA cysteine proteases. In many eukaryotes including protozoan parasites, papain family cathepsins function classically as globular lysosomal proteases that degrade cellular proteins or proteins endocytosed from the environment (4). For example, the human malaria parasite Plasmodium falciparum expresses three cathepsin L-like proteases (falcipain 2a, 2b, and 3) in the food vacuole to digest hemoglobin endocytosed from the host erythrocyte (1, 5–10). A fourth cathepsin, falcipain 1, resides in intracellular vesicles and is dispensable for the intraerythrocytic stages (11) but plays an important yet undefined role in parasite development mosquitoes (12). African trypanosome cathepsin B is important for parasite survival probably due to its role in degrading endocytosed host transferrin for iron acquisition (13).
Toxoplasma gondii is an obligatory intracellular protozoan parasite that can efficiently invade and replicate in many vertebrate hosts leading to a life-long infection (14). Although the chronic infection is typically contained by the host immune system, it can reactivate in patients with immune dysfunction, causing severe, even fatal, disease (15, 16). The parasite harbors three specialized secretory organelles named micronemes, rhoptries, and dense granules, which discharge their contents sequentially during and after invasion (17). Adhesive proteins secreted from micronemes facilitate parasite attachment and entry (17). Reminiscent of prohormone maturation and secretion, more than half of the known microneme proteins are synthesized as preproproteins that are maturated by endoproteolytic removal of a propeptide before storage and secretion (17–19). Initial inhibitor studies implied a role for cysteine proteases in the microneme pathway (20).
T. gondii has five cathepsin-like proteases including one cathepsin L (TgCPL),2 one cathepsin B (TgCPB), and three cathepsin Cs (TgCPC1, -2, and -3). Consistent with it contributing to the microneme pathway, genetic disruption revealed TgCPL as a maturase for at least two microneme proteins, MIC2-associated protein (M2AP) and MIC3 (21). Notably, antibodies to TgCPL identified a novel, acidic vacuolar compartment (VAC; also known as a plant-like vacuole or PLV) (21, 22). Although the VAC was proposed to function in protein degradation, TgCPL is the only protease reported to occupy this organelle. Similar to other cathepsins, the three-dimensional structure of TgCPL indicates that its propeptide occupies the active site as an autoinhibitor until maturation (21, 23). TgCPL can self-maturate by removing its own propeptide in vitro, but the extent to which it undergoes self-maturation in vivo has not been reported. Similarly, although TgCPB also exhibits a propeptide and recombinant thioredoxin-proTgCPB can self-maturate in vitro, previous studies suggesting a key role for TgCPB in rhoptry biogenesis and infection did not address its proteolytic maturation in vivo (24, 25).
Here, we show that the intracellular conversion of TgCPB from a zymogen to a mature protease is dependent on TgCPL activity rather than its own proteolytic activity, further expanding the maturase function of TgCPL. We also establish that TgCPB does not reside in the rhoptries as previously reported, and instead it co-localizes with TgCPL in the VAC. Accordingly, genetic ablation of TgCPB showed that its expression is not required for rhoptry protein maturation or rhoptry biogenesis. We also suggest based on expression of a catalytically inert mutant of TgCPL that an additional protease(s) contributes to the in vivo maturation of TgCPL beside itself. Collectively, our findings indicate that TgCPL activates and coexists with at least one additional cathepsin protease in the VAC, further underscoring the putative digestive function of this endolysosomal organelle.
EXPERIMENTAL PROCEDURES
Parasite Culture
Toxoplasma gondii tachyzoites (RHΔku80, RHΔku80Δcpl, RHΔku80ΔcplCPLWT, RHΔku80ΔcplCPLC31A, and RHΔku80Δcpb, see also Table 1) were continuously cultured in human foreskin fibroblast cells and harvested by membrane filtration as described previously (19).
TABLE 1.
Strains used in this study
| Strain | Genotype | Common name | Source |
|---|---|---|---|
| RH | Wild type | RH | (41 and 42) |
| RHΔku80::HXGPRT | Δku80::HXGPRT | RHΔku80 | (43 and 44) |
| RHΔku80::HXGPRTΔcpl::DHFR | Δku80::HXGPRTΔcpl::DHFR | RHΔku80Δcpl | This study |
| RHΔku80::HXGPRTΔcpb::CAT | Δku80::HXGPRTΔcpb::CAT | RHΔku80Δcpb | This study |
| RHΔku80::HXGPRTΔcpl::CPLWTCAT/DHFR | Δku80::HXGPRTΔcpl::CPLWTCAT/DHFR | RHΔku80ΔcplCPLWT | This study |
| RHΔku80::HXGPRTΔcpl::CPLC31ACAT/DHFR | Δku80::HXGPRTΔcpl::CPLC31ACAT/DHFR | RHΔku80ΔcplCPLC31A | This study |
| RHΔku80::HXGPRTΔcpb::CPBWTBLE/CAT | Δku80::HXGPRTΔcpb::CPBWTBLE/CAT | RHΔku80ΔcpbCPBWT | This study |
| RHΔku80::HXGPRTΔcpb::CPBC30ABLE/CAT | Δku80::HXGPRTΔcpb::CPBC30ABLE/CAT | RHΔku80ΔcpbCPBC30A | This study |
Chemicals and Reagents
Morpholinurea-leucyl-homophenyl-vinyl sulfone phenyl (LHVS), boron-dipyrromethene (BODIPY)-labeled LHVS (BODIPY-LHVS), and BODIPY-DCG-04 were kindly provided by Dr. Matthew Bogyo at Stanford University (26). Other chemicals used in this work are analytical grade.
Cloning and Expression of Recombinant TgCPB
A 1.6-kb DNA fragment encoding recombinant proform TgCPB (rproTgCPB) was PCR-amplified from a T. gondii RH cDNA library with primers CPBstreptagNdeIFwd and CPBpropepXhoIRev (supplemental Table S1) using Phusion High-Fidelity DNA polymerase (New England Biolabs). The forward and reverse primers encode strep-tactin and hexahistidine tags, respectively. The product was gel-extracted, purified, and ligated into pET22b(+). The resulting pET22b(+)-Strep-Tactin-proTgCPB-His6 expression vector was transformed into Escherichia coli strain Origami(DE3). Clones were identified by NdeI and XhoI digestion and verified by DNA sequencing. Bacteria were grown in 1 liter of LB at 37 °C to an A600 of 0.6–0.7, induced with 1 mm isopropyl 1-thio-β-d-galactopyranoside for 3 h and harvested by centrifugation at 10,000 × g for 10 min. The pellet was lysed in CelLyticTM B cell lysis buffer (Sigma) containing 0.2 mg/ml lysozyme. Bacterial DNA was fragmented by ultrasonication, and cell debris was removed by centrifugation at 20,000 × g for 30 min. The supernatant was transferred to a new tube and mixed with 2 ml of nickel-nitrilotriacetic acid beads (Qiagen) equilibrated with lysis buffer, incubated for at least 2 h at 4 °C, washed with 10 bed volumes of wash buffer (20 mm Tris-HCl, pH 7.9, 500 mm NaCl, 60 mm imidazole, 1 mm PMSF), and recovered in 10 2-ml fractions with elution buffer (20 mm Tris-HCl, pH 7.9, 500 mm NaCl, 1 m imidazole, 1 mm PMSF). SDS-PAGE analysis revealed that ∼50% of the protein had undergone self-cleavage on the column, creating mature rTgCPB. Fractions were pooled, and buffer was exchanged with PBS buffer using Amicon® Ultra 30-kDa cutoff concentrators (Millipore). Mature rTgCPB was further purified by absorbing the contaminating immature rTgCPB onto a 0.5-ml column of Strep· TactinTM Superflow-agarose beads (Novagen) for 2 h at 4 °C. Mature rTgCPB was recovered in the flow-through after adding 5 bed volumes of wash buffer (100 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1 mm EDTA, 1 mm PMSF). The purified product (∼90% homogeneous) was PBS buffer exchanged and concentrated as above and stored at −20 °C.
A 745-bp product encoding the propeptide of TgCPB was PCR-amplified using primers CPB-GST_103_BamHI_F and CPB-GST_819_XhoI_R and cloned into pGEX-4T3, creating an N-terminal fusion with glutathione S-transferase (GST). The GST-TgCPB propeptide construct was transformed in E. coli strain BL21(DE3) for growth, induction, and cell lysis under native conditions as above. The cleared supernatant was mixed with 3 ml of GST·BindTM resin (Novagen), incubated for 30 min at room temperature (RT), washed with 10 bed volumes of wash buffer (4.3 mm Na2HPO4, 1.47 mm KH2PO4, 137 mm NaCl, 2.7 mm KCl, pH 7.3) and eluted in 6 1.5-ml fractions of wash buffer containing 10 mm reduced glutathione. Fractions containing proTgCPB propeptide were pooled, and PBS buffer was exchanged, concentrated, and stored at −20 °C.
Cloning and Expression of recombinant TgCPLWT and TgCPLC31A
A QuikChangeTM II site-directed mutagenesis kit (Stratagene) was used to create a C31A mutation in the TgCPL expression plasmid, pQE30/100TgCPL (23), with primers CPLCys31DelF and CPLCys31DelR. The WT and C31A mutant constructs were introduced into competent E. coli M15[pRep4] (Qiagen), and the respective recombinant proteins were expressed, purified, and refolded as described previously (23).
Antibody Preparation
Purified mature rTgCPB or the propeptide of TgCPB were used to immunize mice to generate polyclonal antibodies. Mice were injected with 50 μg of purified recombinant protein in Freund's complete adjuvant and boosted 3 times at 2-week intervals with 50 μg of recombinant protein in Freund's incomplete adjuvant. The specificity of polyclonal antibodies was assessed by comparing their reactivity in RHΔku80 and RHΔku80Δcpb parasites by immunoblotting and immunofluorescence assay.
Generation of RHΔku80Δcpb, RHΔku80ΔcplCPLWT, RHΔku80ΔcplCPLC31A, RHΔku80ΔcpbCPBWT, and RHΔku80ΔcpbCPBC30A Strains
Genomic flanking sequences of TgCPL, TgCPB, and TgM2AP were obtained from the ToxoDB database (27). Primers CPB.-3031(KpnI).F, CPB.-27(HindIII).R, CPB.5125(BamHI).F, and CPB.8138(XbaI).R were used to amplify ∼3 kb of 5′ and 3′ DNA sequences flanking TgCPB gene. The DNA fragments were digested with restriction endonucleases and cloned upstream and downstream of the chloramphenicol resistance gene (CAT) in pTubCATSag1. The resulting TgCPB knock-out construct was electroporated into RHΔku80 tachyzoites using previously described parameters (28). Clones were screened by PCR to confirm correct integration of the knock-out construct at the TgCPB locus using the oligonucleotides shown in Fig. 2A: CPB.5Flank.-3573.F (P1), pTubpromBglII.R (P2), CAT.500.F (P3), and CPB.3Flank.+3128.R (P4).
FIGURE 2.

Targeted disruption of TgCPB does not affect the maturation of TgCPL in vivo. A, shown is a schematic illustration of the TgCPB knock-out strategy. A knock-out plasmid encompassing a CAT drug resistance cassette flanked by TgCPB targeting regions was transfected into RHΔku80 parasites for double crossover replacement of TgCPB. B, primer pairs illustrated in panel A were used to verify the replacement of TgCPB with CAT by PCR and agarose gel electrophoresis. A negative image of the gel is shown. C and D, cell lysates of parental and TgCPB deletion strains were immunoblotted with antibodies to the TgCPB catalytic domain and propeptide, respectively, to confirm the absence of TgCPB expression in the TgCPB deletion mutant. Samples were immunoblotted for actin as a loading control. E, the absence of TgCPB did not affect the expression or maturation of TgCPL, which was still processed into the mature form in TgCPB deficient parasites. F, the TgCPB-deficient strain was complemented with wild type or catalytically inert TgCPB and assessed by immunoblotting. Catalytically inert TgCPBC30A was still cleaved into the mature form, indicating that TgCPB proteolytic activity is not crucial for its in vivo maturation. G, recombinant proform TgCPB was incubated with or without in vitro activated recombinant mature TgCPL under acidic, reducing conditions. H, mature TgCPB is a single chain enzyme based on unaltered migration under non-reducing conditions and the failure to detect a light chain by immunoblotting after 16.5% Tris-Tricine peptide gel electrophoresis under the reducing conditions. Analysis by SDS-PAGE and Coomassie staining showed that TgCPB did not auto-process when incubated alone, whereas incubation with TgCPL resulted in several proteolytic products. The two dominant bands migrating ∼30 kDa (hollow arrowhead) and ∼37 kDa (solid arrowhead) were excised for N-terminal sequencing. The determined cleavage sites of each band were labeled with the same symbols within the TgCPB polypeptide sequence. The predicted sites for the mature N terminus of TgCPB and cleavage between the light and heavy chains were labeled with solid and hollow arrows, respectively.
Three pairs of primers (CPLcompSacIF and CPLcompSacIIR, CPLcompSacIIF and CPLcompNotIR, and CPLcompNotIF and CPLcompXbaIR) were used to amplify ∼1 kb of 5′ flanking sequence of TgCPL, ∼1.3 kb of TgCPL cDNA, and ∼1.4 kb of 3′ flanking region of TgM2AP, respectively. These PCR products were digested with enzymes depicted in the primer names and ligated into pTubCATSag1. The catalytically inert version of the TgCPL complementation construct was created by a single point mutation within the codon encoding the key cysteine residue within the active site to alanine using primers CPLCys31DelF and CPLCys31DelR and a QuikChangeTM II site-directed mutagenesis kit (Stratagene). The TgCPL WT and catalytically inert complementation constructs were introduced into RHΔku80Δcpl parasites by electroporation. Clones were screened for the presence of TgCPL expression by immunoblotting and immunofluorescence assay.
TgCPB genetic complementation plasmids were created using a combination of fusion PCR and conventional cloning. Briefly, 1 kb of TgCPB genomic 5′-flanking region was PCR-amplified from RH genomic DNA using primers CPB_F1 and CPB_R3_FusPcDNA. The TgCPB ORF was PCR-amplified from an RH cDNA library3 using primers CPB_F3_Fus_PrcDNA and CPB_R1. The two PCR products were gel-purified, mixed, and amplified as a fusion PCR product using primers CPB_F2_ApaI and CPB_R2_NdeI. This fusion product was gel-purified and cloned into pET15b digested with ApaI and NdeI. The resulting plasmid was digested with NdeI and ClaI to receive the TgCPB genomic 3′-flanking region amplified from RH genomic DNA using primers 3UTR_CPB_Fwd_NdeI and 3UTR_CPB_Rev_ClaI. The phleomycin resistance cassette was further introduced into the ApaI and HpaI restriction sites. The catalytically inert TgCPB complementation construct was created by site-directed mutagenesis of the catalytic cysteine (Cys30, numbered from the predicted mature N terminus; see also Fig. 2G) to alanine using primers TgCPB_cysDel_F and TgCPB_cysDel_R. Constructs were electroporated into RHΔku80Δcpb parasites. Stable populations were obtained after two rounds of selection with phleomycin. TgCPB expression was verified by immunoblotting, and the WT sequence and C30A mutation were confirmed by PCR amplification and DNA sequencing.
SDS-PAGE and Immunoblotting
Samples were prepared in 100 °C SDS-PAGE loading buffer containing 2% β-mercaptoethanol and boiled for 5 min before resolving on 10% or 12.5% SDS-PAGE mini-gels and semidry-transferred to PVDF membranes. Blots were blocked with skim milk and incubated with primary antibodies. Goat anti-mouse or anti-rabbit IgGs conjugated with horseradish peroxidase was used as the secondary antibody. Immunoblots were developed with SuperSignalTM West Pico chemiluminescent substrates (Pierce) and exposed to x-ray film.
Immunofluorescence Assay
Immunofluorescence staining was performed as described previously (19). Images were observed and digitally captured with an AxioCAM MRm camera equipped Zeiss Axiovert Observer Z1 inverted fluorescence microscope at RT with a 100×, 1.3NA oil objective lens and processed using Zeiss Axiovision 4.3 software. Mowiol was used as the mounting medium. Adobe Photoshop and Illustrator CS5 programs were used to assemble the final images.
Immunoelectron Microscopy
Monolayers of human foreskin fibroblast were infected with either RHΔku80 or RHΔku80Δcpb parasites for 30 min (pulse invasion) or 24 h (replication) before fixation, first in 4% paraformaldehyde (Electron Microscopy Sciences) in 0.25 m HEPES, pH 7.4, for 1 h at RT, then in 8% paraformaldehyde in the same buffer overnight at 4 °C. Samples were infiltrated, frozen, and sectioned as previously described (29). Sections were immunolabeled with MαTgCPB antibody at 1/5 dilution and rabbit anti-TgCPL antibody (23) at a dilution in PBS, 1% fish skin gelatin, then with IgG antibodies followed directly by 5 nm (for TgCPB) and 10 nm (for TgCPL) protein A-gold particles before examination with a Philips CM120 Electron Microscope (Eindhoven, The Netherlands) under 80 kV.
Metabolic Labeling and Immunoprecipitation
Fresh lysed tachyzoites were harvested and resuspended in ice-cold Met/Cys-free DMEM medium containing 10 mm HEPES, pH 7.0, 1% FBS and 2 mm l-glutamine. Parasites were prewarmed at 37 °C for 3 min before pulsed-labeling with 500 μCi of [35S]Met/Cys for 15 min and chasing in unlabeled medium with 5 mm methionine, 5 mm cysteine at 37 °C at 20-min intervals for 1 h. Parasites were centrifuged, washed with ice-cold Met/Cys-free medium, resuspended in 600 μl of radioimmune precipitation assay buffer (50 mm Tris-HCl, pH 7.5, 100 mm NaCl, 5 mm EDTA, 1% Triton X-100, 0.5% sodium deoxycholate, 0.2% SDS, 10 μg/ml RNase A, 20 μg/ml DNase I, and complete Mini protease inhibitor mixture (Roche Applied Science), and agitated for 30 min at RT. Cell debris was removed by centrifugation at 16,000 × g for 15 min at RT. Polyclonal antibodies against TgROP2–4 were incubated with cell lysate for 1 h at RT with gentle shaking. Twenty microliters of 25% (v/v) slurry of protein G-Sepharose beads were added to precipitate antigen-antibody complexes. The protein complexes were washed 4 times with 1 ml of radioimmune precipitation assay buffer before boiling in SDS-PAGE loading buffer and resolved on denatured 12.5% SDS-PAGE gels. Gels were incubated with fluorographic enhancer (GE Healthcare), dried in cellophane, and exposed to storage phosphor screen. The screen was scanned with TyphoonTM gel and blot imager, and intensities of the pro- and mature form TgROP2–4 were quantified with ImageQuant TL software (GE Healthcare).
Proteolytic Activity-based Profiling
Parasites grown in human foreskin fibroblast cells were harvested and resuspended in DMEM medium supplemented with 10 mm HEPES and 2 mm glutamine at a density of 5 × 108 tachyzoites/ml. Fifty microliters of parasite suspension was incubated with 0.5 μl of 20 μm BODIPY-LHVS or 200 μm BODIPY-DCG-04 or DMSO at 37 °C for 30 min. Parasite lysates were separated by SDS-PAGE, and the images were captured by TyphoonTM gel and blot imager, and processed with ImageQuant TL software.
Mapping of rTgCPL Cleavage Sites in rproTgCPB
Approximately 30 μg of rproTgCPB was mixed with 30 ng of in vitro activated rTgCPL protease and incubated at 37 °C for 20 min. The partially digested polypeptides were precipitated with trichloroacetic acid, reconstituted in SDS-PAGE sample buffer, separated by 12.5% SDS-PAGE, and transferred to the PVDF-P membrane. The blot was stained with 0.1% (w/v) Coomassie Blue R-250 in 40% methanol, 1% acetic acid solution for 5 min and destained in 50% methanol until the background was clear. After rinsing in water, bands were excised for N-terminal sequencing by Midwest Analytical (St. Louis, MO).
Testing the Structural Stability of rproTgCPLC31A
Approximately 10 μg of rproTgCPL was mixed and incubated with 10 ng of trypsin at 37 °C. Ten percent of the total reaction was sampled at 0.5, 5, 10, 15, 20, 40, and 60 min incubation. Samples were separated by 12.5% SDS-PAGE, stained with 0.1% Coomassie Blue R-250 in 50% methanol, 10% acetic acid solution for 60 min, and destained in 50% methanol, 10% acetic acid solution until the background was clear. The gels were imaged using a LI-COR Odyssey CLx instrument. Intensities of the proTgCPL bands were quantified with LI-COR image studio software and plotted against the incubation time using Prism 5 software to determine the kinetics of degradation.
RESULTS
TgCPL Expression Is Required for the Maturation of TgCPB
The loss of TgCPL causes delayed maturation of some microneme proteins including TgM2AP and TgMIC3, but not TgMIC6 or TgAMA1 (21), indicating that another maturase(s) in the parasite endocytic pathway exists. To identify additional proteases besides TgCPL, we incubated RHΔku80 (parental) and RHΔku80Δcpl (TgCPL deficient) parasites with BODIPY-DCG-04, a fluorescently conjugated inhibitor that broadly reacts with cathepsin cysteine proteases (30). This activity-based labeling revealed a 63-kDa polypeptide that became more abundant in TgCPL-deficient parasites compared with the parental strain, although another band migrating at 30 kDa disappeared in TgCPL-deficient parasites (Fig. 1A). We predicted that the 63-kDa protein is the proform of TgCPB based on the calculated molecular weight of the protease. To test this, we expressed constructs encoding recombinant TgCPB or its propeptide in E. coli for mouse polyclonal antibody production. The resulting antibodies to the TgCPB catalytic domain (MαTgCPB) or propeptide (MαTgCPBpp) were used to probe immunoblots of RHΔku80, RHΔku80Δcpl, and RHΔku80ΔcplCPLWT cell lysates. MαTgCPB recognized proteins of 63 and 30 kDa in RHΔku80 and RHΔku80ΔcplCPLWT lysates, reflecting the proteolytic maturation of the proform TgCPB to the catalytic form in vivo. Probing with MαTgCPBpp confirmed the 63-kDa band as the proform TgCPB. However, immunoblotting RHΔku80Δcpl parasite lysates revealed the absence of TgCPB maturation with consequent accumulation of proform TgCPB (Fig. 1, C and D). This finding suggests that TgCPL expression is required for TgCPB maturation.
FIGURE 1.

TgCPL is required for maturation of TgCPB. A, BODIPY-DCG-04 labeling of TgCPL knock-out strains is shown. Live parasites were treated with BODIPY-DCG-04, lysed, and assessed with SDS-PAGE and laser scanning fluorometry. A 63-kDa band labels more intensely in TgCPL-deficient parasites coincident with the loss of a 30-kDa band. B, shown is a schematic illustration of the TgCPL regions recognized by two mouse polyclonal antibodies, MαTgCPBpp and MαTgCPB. aa, amino acids. C and D, expression of TgCPB in TgCPL-deficient strains was assessed by immunoblotting using antibodies against the catalytic domain (MαTgCPB) and propeptide of TgCPB (MαTgCPBpp), respectively. TgCPB remained as a zymogenic form in TgCPL deficient strains. E, TgCPB maturation relies on TgCPL activity. Live parasites were treated with 1 μm (lane 1) or 10 μm (lane 2) LHVS, a selective inhibitor of TgCPL. Parasite lysates were immunoblotted with MαTgCPB. Proform TgCPB accumulated in LHVS-treated parasites. Samples were immunoblotted for actin as a loading control. In all panels solid arrowheads indicate proform TgCPB, and hollow arrowheads denote mature TgCPB.
To test if TgCPB maturation relies on TgCPL activity, we treated parasites with a selective TgCPL protease inhibitor, LHVS (23). LHVS treatment prevented conversion of proform TgCPB to mature TgCPB (Fig. 1E), confirming that TgCPL activity is required for TgCPB maturation. Collectively, our findings indicate that TgCPL plays an indispensible role in TgCPB maturation. This is distinct from canonical cathepsin B proteases that typically undergo automaturation in vivo (31–33).
TgCPB Activity Is Not Necessary for TgCPL Maturation or Its Own Maturation in Vivo
Although the above findings indicate that TgCPB maturation is dependent on TgCPL activity, they do not address the reciprocal, i.e. whether TgCPL maturation is dependent on TgCPB activity. To test this we created a TgCPB-deficient strain. Approximately 3 kb of the 5′- and 3′-genomic flanking regions of TgCPB were amplified by PCR and engineered to flank a chloramphenicol (CAT) resistance gene for gene replacement via double homologous recombination (Fig. 2A). After drug selection and isolation of single clones, successful double crossover integration was verified by PCR amplifying 5′- and 3′-flanking regions of TgCPB using two pairs of primers indicated in Fig. 2, A and B. The TgCPB deletion mutant was also verified by immunoblotting with MαTgCPB and MαTgCPBpp antibodies to confirm the absence of TgCPB expression (Fig. 2, C and D). This also validated the specificity of the antibodies. TgCPL expression as a ∼30-kDa mature protein was unchanged in TgCPB null parasites, indicating that TgCPL does not increase in abundance to compensate for TgCPB deficiency. That a normal amount of mature TgCPL is seen in the absence of TgCPB also demonstrates that TgCPB does not significantly contribute to the maturation of TgCPL (Fig. 2E).
Next, to investigate whether TgCPB maturation is dependent on its own activity, we genetically complemented the TgCPB-deficient strain with constructs expressing wild type (TgCPBWT) or catalytically inert (TgCPBC30A) enzyme. The TgCPB alleles were PCR-amplified from the strains and verified by DNA sequencing. Immunoblotting with MαTgCPB showed that the proform TgCPBC30A was still converted into the 30-kDa mature form in a manner identical to wild type (Fig. 2F). This finding confirms that TgCPB activity is not required for its in vivo maturation, suggesting that another protease(s) is responsible for processing TgCPB.
TgCPL Cleaves TgCPB in Vitro
Having established that TgCPL activity is required for TgCPB maturation, we next tested whether TgCPL can directly process TgCPB. E. coli derived rproTgCPB was purified, refolded, and incubated with or without active rTgCPL under acidic, reducing conditions suitable for activity. Whereas no self-digestion products were observed for TgCPB alone, incubation with TgCPL resulted in two main proteolytic products of 30 and 37 kDa (Fig. 2G). N-terminal sequencing revealed that the 30-kDa product began at AQHTT, which corresponds to a site close to where other cathepsin B proteases are split into a heavy and light chain that are held together by a disulfide bond (34). However, TgCPB SDS-PAGE migration in parasite lysates was identical under reducing and non-reducing conditions (Fig. 2H), indicating that the AQHTT cleavage site is not physiologically relevant. This conclusion is further supported by the failure to detect a TgCPB light chain (expected ∼5.5 kDa) under reducing conditions. On the other hand, the 37-kDa product sequence MPLPA begins 14 residues upstream of the hypothetical mature N terminus of TgCPB and hence could be a site used for maturation of TgCPB. We attempted to obtain N-terminal sequence from antibody affinity-purified native TgCPB isolated from parasite extracts, but the yields were insufficient for analysis. Thus, although we could not validate the in vivo physiological relevance of the MPLPA cut site, our findings raise the possibility that TgCPL initiates TgCPB maturation at this site before subsequent proteolysis to generate the final product.
TgCPB Resides in the VAC
TgCPB was reported to exist predominantly in rhoptry secretory organelles and also within an electron lucent vacuole by staining with a monoclonal antibody to amebic cysteine protease 1 (25). However, a subsequent proteomics study failed to detect TgCPB in isolated rhoptries (35). To address this apparent discrepancy, we stained parental (RHΔku80) parasites with MαTgCPB recognizing the catalytic domain of TgCPB to reassess the subcellular localization of TgCPB (21, 23). The specificity of the MαTgCPB antibody was confirmed by the absence of reactivity with TgCPB-deficient parasites (see Fig. 4A; additional examples shown in supplemental Fig. S1). Newly invaded parental parasites showed TgCPB staining in a single discrete punctum, which co-localized with TgCPL in the VAC (Fig. 3A). During intracellular replication the VAC fragments into smaller vesicles in a cell-cycle dependent manner (21, 22). Replicating parasites showed TgCPB colocalization with TgCPL in tachyzoites with a single VAC, but TgCPB often resided within vesicles that appear to be distinct from those harboring TgCPL in tachyzoites with a fragmented VAC (Fig. 3B). These findings are consistent with the cell cycle-dependent dynamic nature of the VAC, and they also suggest the existence of subpopulations of VAC-derived vesicles that are occupied by TgCPL or TgCPB.
FIGURE 4.
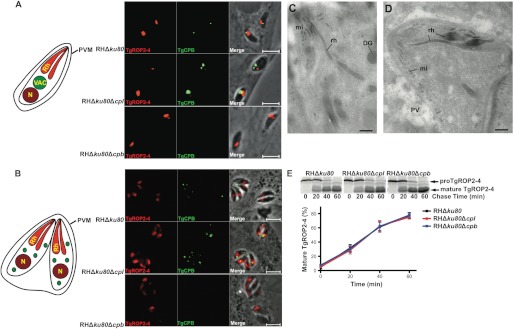
TgCPB does not contribute to TgROP2–4 protein maturation or rhoptry biogenesis. A, RHΔku80, RHΔku80Δcpl, and RHΔku80Δcpb strains were dual-stained with anti-TgROP2–4 and anti-TgCPB catalytic domain antibodies. TgCPB localization is close to but distinct from rhoptries. PVM, parasitophorous vasuole membrane. B, replicating parasites show fragmentation of TgCPB labeled vesicles that are proximal to but usually distinct from apical rhoptries. Panels are in the same order as in A. C and D, electron microscopy of RHΔku80Δcpb parasites revealed normal rhoptry morphology in either newly invaded (C) or replicating T. gondii (D). Bars, 500 nm. DG, dense granules; mi, microneme; PV, parasitophorous vacuole; rh, rhoptry; VAC, vacuolar compartment. E, maturation of TgROP2–4 was compared among RHΔku80, RHΔku80Δcpl, and RHΔku80Δcpb parasites by pulse-chase metabolic labeling and immunoprecipitation. TgROP2–4 maturation showed normal kinetics in TgCPL or TgCPB deletion mutants based on phosphorimage analysis (bars represent S.E. of three independent experiments).
FIGURE 3.
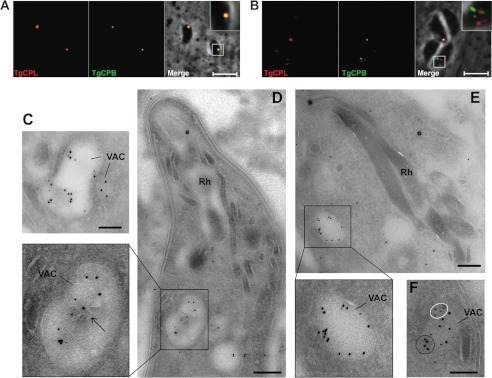
TgCPB resides in the VAC. A and B, newly invaded or overnight replicated RHΔku80 strain parasites were dual-stained with anti-TgCPL and anti-TgCPB catalytic domain antibodies, respectively. TgCPB co-localized with TgCPL in a single VAC of newly invaded parasites (A), whereas it resided in distinct puncta in replicated parasites with a fragmented VAC (B). Bars, 5 μm. TgCPL and TgCPB co-localized in the VAC of an ultrathin sectioned newly invaded parasite (C and D) or replicating parasites (E and F) immunostained with anti-TgCPL (10-nm gold particles) and anti-TgCPB catalytic domain (5-nm gold particles) antibodies. The arrow pinpoints internal membranous structures in the VAC. TgCPB (white circles), and TgCPL (black circle) also showed unique labeling of distinct clusters within the VAC upon fragmentation of this organelle. Bars, 500 nm. Rh, rhoptry.
We performed immunoelectron microscopy (IEM) analyses to verify the subcellular distribution of TgCPB in intracellular Toxoplasma. Parasites were also immunolabeled with anti-TgCPL antibodies to identify the VAC at the ultrastructural level. IEM data confirmed the co-localization of TgCPB and TgCPL in the VAC, which is a large electron-lucent organelle characterized by internal membranous structures (21, 22), both in newly invaded parasites (Fig. 3C) and replicating parasites 24 h post-infection (Figs. 3, D and E). Extensive viewing of multiple parasite sections could not detect TgCPB in rhoptries. In most sections, TgCPB, like TgCPL, was distributed on the luminal structures of the VAC (Fig. 3, C and D, arrow) or on the VAC limiting membrane (Fig. 3E). The two proteins were seen either close to each other or more distantly within the VAC. Some VAC-derived vesicles were solely labeled for TgCPL, consistent with the dynamic nature of the VAC.
TgCPB Expression Is Not Required for TgROP2–4 Protein Maturation or Rhoptry Biogenesis
Que et al. (25) observed small distorted rhoptries and swollen Golgi structures in cathepsin B inhibitor treated parasites, which also showed delayed maturation of newly synthesized TgROP2–4. We found that the discrete punctum of TgCPB staining of the VAC is adjacent to, but largely distinct from rhoptries stained for TgROP2–4 in newly invaded parental strain parasites (Fig. 4A, upper panels). TgCPL-deficient parasites exhibit an enlarged VAC, which partially overlapped with TgROP2–4 staining, confirming that the VAC is proximal to the rhoptries (Fig. 4A, middle panels). Because IEM results showed no staining of rhoptries with TgCPB antibodies, the apparent overlap is likely due to the close proximity of the VAC and rhoptries as noted previously (21). Replicating parasites showed puncta of TgCPB staining that was often proximal to, but usually discernable from rhoptries (Fig. 4B). Immunofluorescence staining suggested that the rhoptries of TgCPB-deficient parasites are indistinguishable from those of the parental strain (Fig. 4, A and B, lower panels).
Electron microscopy observations of TgCPB-deficient parasites confirmed the absence of abnormalities in the morphology and ultrastructure of rhoptries, organelles characterized by a bulbous portion with a honeycombed appearance and a tapered neck oriented toward the conoid (Fig. 4, C and D).
To assess rhoptry protein maturation, we performed pulse-chase metabolic labeling and immunoprecipitation of TgROP2–4 in parental, TgCPL-deficient, and TgCPB-deficient parasites. TgCPB-deficient parasites showed TgROP2–4 maturation kinetics that were indistinguishable from parental or TgCPL-deficient parasites (Fig. 4E). Therefore, it is unlikely that TgCPL or TgCPB are rhoptry protein maturases. Collectively, we show that TgCPB does not appear to exist in rhoptries and that it does not measurably contribute to rhoptry morphology or rhoptry protein maturation, suggesting that it is not involved in rhoptry biogenesis.
TgCPL Maturation Is Partially Dependent on Its Proteolytic Activity in Vivo
Previous studies showed that purified rproTgCPL can undergo autocatalytic maturation in vitro under acidic and reducing conditions (21, 23). These studies also established that TgCPL almost exclusively exists in a mature form in parasites that is distinct from the inefficient maturation of TgCPB (Fig. 1B). It remained unclear, however, whether TgCPL maturation is entirely self-directed in vivo or if it involves an additional parasite protease(s). To preclude TgCPL self-cleavage, we mutated the catalytic cysteine (residue 31; numbering from the N terminus of recombinant mature TgCPL according to Larson et al. (23)) to alanine to abolish proteolytic activity, and we introduced this mutant into TgCPL-deficient parasites. BODIPY-LHVS failed to label the catalytically inert TgCPLC31A mutant, consistent with the absence of activity (Fig. 5A). TgCPB maturation did not to occur in the TgCPLC31A strain, confirming the loss of activity (Fig. 5B). Interestingly, fully mature TgCPL was not observed in the catalytically inert mutant (Fig. 5B). Instead, inactive TgCPL was processed into at least three intermediate fragments migrating at ∼30–35 kDa and a species that migrated slightly slower than mature TgCPL. A minor population of TgCPLC31A remained as the immature precursor. These finding suggest that other proteases are capable of processing TgCPL in the absence of autocatalytic maturation.
FIGURE 5.

TgCPL maturation is partially dependent on its proteolytic activity in vivo. A, a catalytically inert version of the TgCPL complementation strain (RHΔku80ΔcplCPLC31A) was generated by electroporating the construct replacing catalytic cysteine to alanine into RHΔku80Δcpl strain. SDS-PAGE and fluorography of BODIPY-LHVS-labeled parasites confirmed that the RHΔku80ΔcplCPLC31A does not express active TgCPL based on the absence of labeling. B, the catalytically inert version of TgCPL was processed into at least three intermediates and one species with a similar but slightly higher molecular weight as mature TgCPL, indicating that another protease is capable of partially maturing TgCPL. C, purified rproTgCPLWT and rproTgCPLC31A were incubated with trypsin at a ratio of 1000:1 (w/w). Samples taken at different time points were separated by SDS-PAGE and stained with Coomassie. TgCPLC31A was degraded into small peptides more extensively than proform TgCPLWT. D, the intensities of proform TgCPLWT and TgCPLC31A were quantified by fluorography. Bars represent S.E. of three independent experiments. Approximately 70% of rproTgCPLWT and 30% of rproTgCPLC31A resisted the proteolytic digestion by trypsin, suggesting that TgCPLC31A was not refolded as well as TgCPLWT.
It is possible that replacing the TgCPL catalytic cysteine with alanine altered the enzyme conformation, rendering it more susceptible to proteolysis. To investigate this we expressed rTgCPLC31A in E. coli and tested it for trypsin sensitivity. Similar to WT rproTgCPL, rproTgCPLC31A was expressed in inclusion bodies; hence, we purified it under denaturing conditions and refolded in a neutral pH buffer. As expected, both proteins were stable when incubated alone in neutral pH buffer as TgCPL is inactive under these conditions (Fig. 5, C and D). Trypsin degraded ∼30% of WT proTgCPL within 20 min, but minimal degradation was seen thereafter, implying that the remaining 70% population of WT TgCPL was correctly folded and refractory to further digestion. Trypsin digested ∼70% of TgCPLC31A with a similar kinetic pattern as WT TgCPL, leaving ∼30% of the starting material refractory to digestion. These results suggest that TgCPLC31A has a greater propensity to misfold than WT TgCPL, potentially rendering it more susceptible to proteolysis within the parasite endolysosomal system. Nonetheless, because TgCPLC31A is processed within the parasite to an abundant species close to the same size as WT TgCPL, it remains possible that this pseudo-mature form would be active as a result of processing by other proteases.
DISCUSSION
Although cathepsin B proteases are typically self-maturing (31–33), we showed herein that maturation of Toxoplasma TgCPB is dependent on the expression of another papain family endoprotease, TgCPL. Moreover, TgCPL activity is required for TgCPB maturation as genetic complementation of TgCPL-deficient parasites with active TgCPL restored TgCPB maturation, whereas complementation with a catalytically inert mutant of TgCPL failed to reinstate maturation. Inhibiting TgCPL activity with LHVS also prevented TgCPB maturation. We also showed that the mature TgCPB is composed of a single chain and that its conversion from a zymogen is exclusively independent of its own catalytic activity, indicating that another protease(s) is required for its maturation. Recombinant mature TgCPL can process immature TgCPB in vitro to a pseudo-mature species 14 residues upstream of the predicted N terminus of TgCPB. Interestingly, immature human cathepsin B auto-processes in vivo and in vitro at a site six residues upstream of mature cathepsin B (36, 37). In vitro experiments showed that this pseudo-mature species is further processed by dipeptidylpeptidase I, an exopeptidase that removes the remaining six amino acids in three steps of two residues each (36). The highly conserved proline residue two positions downstream from the mature N terminus prevents further processing because dipeptidylpeptidase I cannot process a site with proline in the P1 or P1′ position (38). Based on this two-step model, we propose that TgCPL or another parasite endolysosomal protease whose activity is dependent upon TgCPL activity cleaves proTgCPB at a susceptible site in the proximal region of the propeptide followed by exopeptidase trimming to the final mature N terminus. If TgCPL initiates proTgCPB maturation at the same site in vivo as it does in vitro, then it is unlikely that a dipeptidylpeptidase finalizes the maturation because the multiple proline residues within the remaining propeptide sequence would render it refractory to processing by this enzyme.
Precisely where within the endolysosomal system the cathepsin proteases undergo maturation remains to be determined. Maturation requires acidic conditions in vitro and in vivo (21, 23, 25). Accordingly, conditions within late endosomes and the VAC are favorable for maturation because these compartments contain a vacuolar H+ pyrophosphatase and a vacuolar H+-ATPase (22). Regardless, our findings further underscore the maturase function of TgCPL for proteins that reside within or transit through the Toxoplasma endolysosomal system.
The discussion section of Larson et al. (23) indicates that expression of TgCPB is apparently increased upon deletion of TgCPL. This conclusion was based on the immunoblots using two TgCPB anti-peptide antibodies, one recognizing an epitope in the propeptide domain and the other recognizing an epitope in the catalytic domain. For reasons that remain unclear, the anti-catalytic domain antibody did not recognize the 30-kDa mature protein (supplemental Fig. S2), thus leading us to conclude that TgCPB exists in an immature form that is up-regulated in the absence of TgCPL. However, our results with anti-recombinant TgCPB antibodies strongly suggest that mature TgCPB exists in parasites and that ablation of TgCPL precludes TgCPB maturation, leading to the accumulation of proform TgCPB. These findings underscore the value of testing multiple antibodies for the analysis of proteins.
Que et al. (25) reported that TgCPB localizes to rhoptries and is responsible for rhoptry protein maturation and rhoptry biogenesis based on cathepsin B-specific inhibitor treatment. They also noted labeling of an electron lucent vacuole with the heterologous anti-cathepsin B antibodies used in the study. We show that TgCPB principally resides within the VAC, which is likely the electron lucent structure seen in the previous study. That TgCPB remains in an immature form in TgCPL-deficient parasites created a potential opportunity to readdress the function of TgCPB. The immature form of TgCPB is expected to have little or no activity on protein substrates because of active site occlusion by the TgCPB propeptide. However, this notion was somewhat clouded by the observation that immature TgCPB is susceptible to labeling with the activity-based probe BODIPY-DCG-04, a finding that is similar to that of human immature cathepsin B (39). To better understand the role of cathepsin B-like protease in Toxoplasma parasites, we created a TgCPB knock-out strain. This mutant shows normal rhoptry protein processing and rhoptry biogenesis, which is consistent with the absence of TgCPB in rhoptries but is in contrast to the pronounced effects of the cathepsin B-specific inhibitor on these events (25). The rhoptry effects observed in the previous study could be due to off target reactivity of the cathepsin B inhibitor. Alternatively, we cannot rule out that TgCPB-deficient parasites have adjusted to the absence of TgCPB by altering the expression of other proteins. Nonetheless, our finding that TgCPB does not reside in the rhoptries reduces the likelihood that it plays a key role in rhoptry biogenesis.
We and others have previously reported that the VAC is typically a single entity in extracellular parasites and newly invaded parasites in G1 phase. The VAC fragments into smaller vesicles during S phase and mitosis before apparently reforming during cytokinesis of daughter parasites (21, 22). This phenomenon resembles the fission and fusion events governing vacuolar inheritance in the budding yeast Saccharomyces cerevisiae (40). The yeast mother cell vacuole enlarges and fragments into vesicles that are shuttled into the budding cell for recreation of the vacuole by homotypic fusion. We found herein by immunofluorescence assay that T. gondii fragments its VAC into at least two distinct subpopulations of vesicles marked with TgCPL or TgCPB. IEM showed TgCPL alone in distinct vesicles, but TgCPB was not observed alone without TgCPL, perhaps reflecting a difference in sensitivity between immunofluorescence assay and IEM. Although the significance of these findings remains unclear, this heterologous fragmentation reflects distinct sorting of the proteases and possibly other luminal proteins into the subpopulations. It is possible that the different subpopulations of VAC-derived vesicles perform distinct functions during S phase and mitosis. The identification of additional markers for these subpopulations should provide clues to their prospective functions.
Catalytically inert TgCPL expressed in TgCPL null parasites is processed to several intermediate species and a final pseudo-mature product that migrates slightly slower than native mature TgCPL. These findings suggest that TgCPL activity is necessary for normal maturation. Although it is not possible to assess whether the pseudo-mature species would be active if its catalytic cysteine was intact, its close migration to wild type mature TgCPL favors the possibility of it being active. Testing the structural stability of wild type and catalytically inert TgCPL by trypsin digestion in vitro revealed that inert TgCPL is more susceptible to proteolysis. Its structural instability could result in greater susceptibility to degradation within parasite endolysosomal system. However, it remains possible that the pseudo-mature TgCPL we observed in RHΔku80ΔcplCPLC31A would have activity. In this case, the proteases acting on catalytically inert TgCPL might normally contribute to TgCPL maturation; however, our findings suggest that the final processing step requires TgCPL activity and is, therefore, autocatalytic. Regardless, the processing of catalytically inert TgCPL suggests the presence of additional proteases within the Toxoplasma endolysosomal system. Although we herein show that TgCPB also resides in the endolysosomal system, it does not appear to contribute to TgCPL maturation as TgCPL processing is normal in TgCPB-deficient parasites. Some of the additional endolysosomal proteases may contribute to the maturation of microneme proteins that are not substrates of TgCPL including TgMIC5, TgMIC6, TgMIC11, and TgAMA1, among others.
Five cathepsin proteases are encoded within the Toxoplasma genome including TgCPL and TgCPB (3). Although TgCPL has been shown to act as a maturase for some microneme proteins (21), the function of TgCPB remains unclear. The kin of Toxoplasma, the malaria parasite, encodes four cathepsin L-like proteases termed falcipain 1, 2a, 2b, and 3 but no cathepsin B-like proteases. The latter three falcipains reside in the food vacuole and are responsible for host hemoglobin protein degradation as a source of amino acids for parasite growth (1, 5–10). Evolutionary lineage analysis indicates that only Toxoplasma and Eimeria species possess cathepsin B orthologs among the sequenced organisms within the phylum Apicomplexa (3). Interestingly, Perkinsus marinus, a parasite of oysters that is taxonomically positioned near the base of the apicomplexan lineage, encodes 10 cathepsin B-like proteases, suggesting that the endopeptidase and exopeptidase roles of TgCPB have been replaced or superseded by cathepsin L and cathepsin C(s) in several apicomplexans (3). These shifts in cathepsin usage are likely adaptations to the specific needs of these parasites during infection of different cell types within their varied hosts.
Acknowledgments
We thank Jonathon Giebel, Marsha Thomas, Sarah S. Morgan, Julie Laliberté, and Tracey Schultz for assistance with some of the experiments. We also thank Kimberley Zichichi (Microscopy Facility at Yale University) for technical expertise, Con Becker (University of North Carolina, Chapel Hill) for the anti-TgROP2–4 antibody, Antonio Barragan (Karolinska Institute) and L. David Sibley (Washington University) for the TgCPB anti-peptide antibodies, and Matthew Bogyo (Stanford University) and My-Hang Huynh (University of Michigan) for critically reading the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant AI063263 (to V. B. C.). This work was also supported by an American Heart Association Postdoctoral Fellowship (to Z. D.).

This article contains supplemental Table 1 and Figs. 1 and 2.
V. B. Carruthers, unpublished information.
- TgCPL
- Toxoplasma cathepsin L protease
- CAT
- chloramphenicol acetyltransferase
- IEM
- immunoelectron microscopy
- LHVS
- morpholinurea-leucyl-homophenyl-vinyl sulfone phenyl
- RT
- room temperature
- TgCPB
- Toxoplasma cathepsin B protease
- rTgCPB
- recombinant TgCPB
- TgMIC
- Toxoplasma microneme protein
- rproTgCPB
- proform rTgCPB
- TgROP
- Toxoplasma rhoptry protein
- VAC
- vacuolar compartment
- Tricine
- N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]glycine.
REFERENCES
- 1. Rosenthal P. J. (2011) Falcipains and other cysteine proteases of malaria parasites. Adv. Exp. Med. Biol. 712, 30–48 [DOI] [PubMed] [Google Scholar]
- 2. Donnelly S., Dalton J. P., Robinson M. W. (2011) How pathogen-derived cysteine proteases modulate host immune responses. Adv. Exp. Med. Biol. 712, 192–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dou Z., Carruthers V. B. (2011) Cathepsin proteases in Toxoplasma gondii. Adv. Exp. Med. Biol. 712, 49–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hsing L. C., Rudensky A. Y. (2005) The lysosomal cysteine proteases in MHC class II antigen presentation. Immunol. Rev. 207, 229–241 [DOI] [PubMed] [Google Scholar]
- 5. Dluzewski A. R., Rangachari K., Wilson R. J., Gratzer W. B. (1986) Plasmodium falciparum. Protease inhibitors and inhibition of erythrocyte invasion. Exp. Parasitol. 62, 416–422 [DOI] [PubMed] [Google Scholar]
- 6. Rosenthal P. J., McKerrow J. H., Aikawa M., Nagasawa H., Leech J. H. (1988) A malarial cysteine proteinase is necessary for hemoglobin degradation by Plasmodium falciparum. J. Clin. Invest. 82, 1560–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rosenthal P. J., McKerrow J. H., Rasnick D., Leech J. H. (1989) Plasmodium falciparum. Inhibitors of lysosomal cysteine proteinases inhibit a trophozoite proteinase and block parasite development. Mol. Biochem. Parasitol. 35, 177–183 [DOI] [PubMed] [Google Scholar]
- 8. Rosenthal P. J., Wollish W. S., Palmer J. T., Rasnick D. (1991) Antimalarial effects of peptide inhibitors of a Plasmodium falciparum cysteine proteinase. J. Clin. Invest. 88, 1467–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rosenthal P. J. (1995) Plasmodium falciparum. Effects of proteinase inhibitors on globin hydrolysis by cultured malaria parasites. Exp. Parasitol. 80, 272–281 [DOI] [PubMed] [Google Scholar]
- 10. Rosenthal P. J., Olson J. E., Lee G. K., Palmer J. T., Klaus J. L., Rasnick D. (1996) Antimalarial effects of vinyl sulfone cysteine proteinase inhibitors. Antimicrob. Agents Chemother. 40, 1600–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eksi S., Czesny B., Greenbaum D. C., Bogyo M., Williamson K. C. (2004) Targeted disruption of Plasmodium falciparum cysteine protease, falcipain 1, reduces oocyst production, not erythrocytic stage growth. Mol. Microbiol. 53, 243–250 [DOI] [PubMed] [Google Scholar]
- 12. Sijwali P. S., Kato K., Seydel K. B., Gut J., Lehman J., Klemba M., Goldberg D. E., Miller L. H., Rosenthal P. J. (2004) Plasmodium falciparum cysteine protease falcipain-1 is not essential in erythrocytic stage malaria parasites. Proc. Natl. Acad. Sci. 101, 8721–8726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. O'Brien T. C., Mackey Z. B., Fetter R. D., Choe Y., O'Donoghue A. J., Zhou M., Craik C. S., Caffrey C. R., McKerrow J. H. (2008) A parasite cysteine protease is key to host protein degradation and iron acquisition. J. Biol. Chem. 283, 28934–28943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Montoya J. G., Liesenfeld O. (2004) Toxoplasmosis. Lancet 363, 1965–1976 [DOI] [PubMed] [Google Scholar]
- 15. Fachado A., Fonseca L., Fonte L., Alberti E., Cox R., Bandera F. (1997) Toxoplasma gondii antigenuria in patients with acquired immune deficiency syndrome. Mem. Inst. Oswaldo. Cruz. 92, 589–593 [DOI] [PubMed] [Google Scholar]
- 16. Kasper L. H., Buzoni-Gatel D. (1998) Some opportunistic parasitic infections in AIDS. Candidiasis, pneumocystosis, cryptosporidiosis, toxoplasmosis. Parasitol. Today 14, 150–156 [DOI] [PubMed] [Google Scholar]
- 17. Carruthers V. B. (2002) Host cell invasion by the opportunistic pathogen Toxoplasma gondii. Acta Trop. 81, 111–122 [DOI] [PubMed] [Google Scholar]
- 18. Zhou X. W., Blackman M. J., Howell S. A., Carruthers V. B. (2004) Proteomic analysis of cleavage events reveals a dynamic two-step mechanism for proteolysis of a key parasite adhesive complex. Mol. Cell Proteomics 3, 565–576 [DOI] [PubMed] [Google Scholar]
- 19. Harper J. M., Huynh M. H., Coppens I., Parussini F., Moreno S., Carruthers V. B. (2006) A cleavable propeptide influences Toxoplasma infection by facilitating the trafficking and secretion of the TgMIC2-M2AP invasion complex. Mol. Biol. Cell 17, 4551–4563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Teo C. F., Zhou X. W., Bogyo M., Carruthers V. B. (2007) Cysteine protease inhibitors block Toxoplasma gondii microneme secretion and cell invasion. Antimicrob. Agents Chemother. 51, 679–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Parussini F., Coppens I., Shah P. P., Diamond S. L., Carruthers V. B. (2010) Cathepsin L occupies a vacuolar compartment and is a protein maturase within the endo/exocytic system of Toxoplasma gondii. Mol. Microbiol. 76, 1340–1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Miranda K., Pace D. A., Cintron R., Rodrigues J. C., Fang J., Smith A., Rohloff P., Coelho E., de Haas F., de Souza W., Coppens I., Sibley L. D., Moreno S. N. (2010) Characterization of a novel organelle in Toxoplasma gondii with similar composition and function to the plant vacuole. Mol. Microbiol. 76, 1358–1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Larson E. T., Parussini F., Huynh M. H., Giebel J. D., Kelley A. M., Zhang L., Bogyo M., Merritt E. A., Carruthers V. B. (2009) Toxoplasma gondii cathepsin L is the primary target of the invasion-inhibitory compound morpholinurea-leucyl-homophenyl-vinyl sulfone phenyl. J. Biol. Chem. 284, 26839–26850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Que X., Wunderlich A., Joiner K. A., Reed S. L. (2004) Toxopain-1 is critical for infection in a novel chicken embryo model of congenital toxoplasmosis. Infect. Immun. 72, 2915–2921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Que X., Ngo H., Lawton J., Gray M., Liu Q., Engel J., Brinen L., Ghosh P., Joiner K. A., Reed S. L. (2002) The cathepsin B of Toxoplasma gondii, toxopain-1, is critical for parasite invasion and rhoptry protein processing. J. Biol. Chem. 277, 25791–25797 [DOI] [PubMed] [Google Scholar]
- 26. Greenbaum D., Medzihradszky K. F., Burlingame A., Bogyo M. (2000) Epoxide electrophiles as activity-dependent cysteine protease profiling and discovery tools. Chem. Biol. 7, 569–581 [DOI] [PubMed] [Google Scholar]
- 27. Gajria B., Bahl A., Brestelli J., Dommer J., Fischer S., Gao X., Heiges M., Iodice J., Kissinger J. C., Mackey A. J., Pinney D. F., Roos D. S., Stoeckert C. J., Jr, Wang H., Brunk B. P. (2008) ToxoDB: An integrated Toxoplasma gondii database resource. Nucleic Acids Res. 36, D553–D556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brydges S. D., Harper J. M., Parussini F., Coppens I., Carruthers V. B. (2008) A transient forward-targeting element for microneme-regulated secretion in Toxoplasma gondii. Biol. Cell 100, 253–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fölsch H., Pypaert M., Schu P., Mellman I. (2001) Distribution and function of AP-1 clathrin adaptor complexes in polarized epithelial cells. J. Cell Biol. 152, 595–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Greenbaum D., Baruch A., Hayrapetian L., Darula Z., Burlingame A., Medzihradszky K. F., Bogyo M. (2002) Chemical approaches for functionally probing the proteome. Mol. Cell Proteomics 1, 60–68 [DOI] [PubMed] [Google Scholar]
- 31. Mort J. S., Buttle D. J. (1997) Cathepsin B. Int. J. Biochem. Cell Biol. 29, 715–720 [DOI] [PubMed] [Google Scholar]
- 32. Barrett A. J., Kirschke H. (1981) Cathepsin B, cathepsin H, and cathepsin L. Methods Enzymol. 80, 535–561 [DOI] [PubMed] [Google Scholar]
- 33. Kirschke H., Barrett A. J., Rawlings N. D. (1998) Lysosomal Cysteine Proteases, 2nd Ed., pp. 131, Oxford University Press, Oxford [Google Scholar]
- 34. McGrath M. E. (1999) The lysosomal cysteine proteases. Annu. Rev. Biophys. Biomol. Struct. 28, 181–204 [DOI] [PubMed] [Google Scholar]
- 35. Bradley P. J., Ward C., Cheng S. J., Alexander D. L., Coller S., Coombs G. H., Dunn J. D., Ferguson D. J., Sanderson S. J., Wastling J. M., Boothroyd J. C. (2005) Proteomic analysis of rhoptry organelles reveals many novel constituents for host-parasite interactions in Toxoplasma gondii. J. Biol. Chem. 280, 34245–34258 [DOI] [PubMed] [Google Scholar]
- 36. Rowan A. D., Mason P., Mach L., Mort J. S. (1992) Rat procathepsin B. Proteolytic processing to the mature form in vitro. J. Biol. Chem. 267, 15993–15999 [PubMed] [Google Scholar]
- 37. Mach L., Schwihla H., Stüwe K., Rowan A. D., Mort J. S., Glössl J. (1993) Activation of procathepsin B in human hepatoma cells. The conversion into the mature enzyme relies on the action of cathepsin B itself. Biochem. J. 293, 437–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Turk D., Janjić V., Stern I., Podobnik M., Lamba D., Dahl S. W., Lauritzen C., Pedersen J., Turk V., Turk B. (2001) Structure of human dipeptidyl peptidase I (cathepsin C). Exclusion domain added to an endopeptidase framework creates the machine for activation of granular serine proteases. EMBO J. 20, 6570–6582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pungercar J. R., Caglic D., Sajid M., Dolinar M., Vasiljeva O., Pozgan U., Turk D., Bogyo M., Turk V., Turk B. (2009) Autocatalytic processing of procathepsin B is triggered by proenzyme activity. FEBS J. 276, 660–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fagarasanu A., Rachubinski R. A. (2007) Orchestrating organelle inheritance in Saccharomyces cerevisiae. Curr. Opin. Microbiol. 10, 528–538 [DOI] [PubMed] [Google Scholar]
- 41. Pfefferkorn E. R., Pfefferkorn L. C. (1976) Toxoplasma gondii. Isolation and preliminary characterization of temperature-sensitive mutants. Exp. Parasitol. 39, 365–376 [DOI] [PubMed] [Google Scholar]
- 42. Sabin A. B. (1941) Toxoplasmic encephalitis in children. J. Am. Med. Assoc. 116, 801–807 [Google Scholar]
- 43. Fox B. A., Ristuccia J. G., Gigley J. P., Bzik D. J. (2009) Efficient gene replacements in Toxoplasma gondii strains deficient for nonhomologous end-joining. Eukaryot. Cell 8, 520–529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Huynh M. H., Carruthers V. B. (2009) Tagging of endogenous genes in a Toxoplasma gondii strain lacking Ku80. Eukaryot. Cell 8, 530–539 [DOI] [PMC free article] [PubMed] [Google Scholar]