

Background: Cyanophages are dominant viruses in the ocean while little has been known on their nucleic acid metabolism.
Results: The RNA polymerase of cyanophage Syn5 has been purified and characterized and the Syn5 promoters identified.
Conclusion: The Syn5 RNA polymerase and promoters have unique and ocean-adapted features.
Significance: The first characterized single-subunit RNA polymerase from marine organisms.
Keywords: Cyanobacteria, DNA Viruses, Promoters, RNA Polymerase, Transcription, Cyanophage, Marine Virus
Abstract
A single subunit DNA-dependent RNA polymerase was identified and purified to apparent homogeneity from cyanophage Syn5 that infects the marine cyanobacteria Synechococcus. Syn5 is homologous to bacteriophage T7 that infects Escherichia coli. Using the purified enzyme its promoter has been identified by examining transcription of segments of Syn5 DNA and sequencing the 5′-termini of the transcripts. Only two Syn5 RNAP promoters, having the sequence 5′-ATTGGGCACCCGTAA-3′, are found within the Syn5 genome. One promoter is located within the Syn5 RNA polymerase gene and the other is located close to the right genetic end of the genome. The purified enzyme and its promoter have enabled a determination of the requirements for transcription. Unlike the salt-sensitive bacteriophage T7 RNA polymerase, this marine RNA polymerase requires 160 mm potassium for maximal activity. The optimal temperature for Syn5 RNA polymerase is 24 °C, much lower than that for T7 RNA polymerase. Magnesium is required as a cofactor although some activity is observed with ferrous ions. Syn5 RNA polymerase is more efficient in utilizing low concentrations of ribonucleotides than T7 RNA polymerase.
Introduction
Viruses are the most abundant and genetically diverse biologic entities in the ocean (1). Marine cyanophages are viruses that infect the dominant photoautotrophs, cyanobacteria. By killing 20% of marine biomass per day, cyanophages play a major role in the maintenance of the marine environment and in the cycling of marine energy (1, 2). Gene transfer between cyanophages and cyanobacteria represents the largest scale of genetic communication on earth and is believed to have played a significant role in the evolution of the biosphere (1–4). Because 60 to 80% of the sequences in cyanophage genomes are not homologous to those in the existing database, cyanophages constitute a tremendous reservoir of unexplored genetic diversity (1). Thus the establishment of laboratory cyanophage/cyanobacterium systems and the study of their interactions is an important endeavor. Recent advances in genome sequencing and bioinformatics have greatly improved our understanding of cyanophages (5–16), and studies of their gene products have provided intriguing insights into their physiology (3, 4, 17–19). However, there has been little characterization of the proteins involved in nucleic acid metabolism, one of the most critical aspects in the life cycle of cyanophage.
Syn5 is a cyanophage with a short tail isolated from the Sargasso Sea; it is homologous to bacteriophage T7 that infects Escherichia coli (20). The laboratory host, cyanobacterial strain Synechococcus sp. WH8109 (20, 21) belongs to marine cluster A of Synechococcus, clade II, one of the most widely distributed clades in the oceans (22). The genome of Syn5 has 46,214 bp containing 61 predicted open reading frames (13). The gene organization is typical of double-stranded DNA (dsDNA)3 phages with its DNA replication genes clustered in the left region of the genome, and the genes encoding its structural proteins in the right region. The gene order shares strong similarity with bacteriophage T7 as well as several other cyanophages, notably Synechococcus phage P60 and Prochlorococcus phage P-SSP7 (13).
DNA-dependent RNA polymerases are responsible for transcription, the synthesis of messenger RNAs, from a double-stranded DNA template. A homologous family of single-subunit RNAP transcribes most T7-like bacteriophage genes. These single-subunit enzymes share many of the biochemical characteristics of the larger multienzyme RNAP of their hosts; their relative simplicity has made them attractive for biochemical and structural analysis (23). One of the most extensively studied RNAP is that encoded by bacteriophage T7 (23, 24). T7 RNAP and its promoters are widely used for overexpression of recombinant genes, and in vitro transcription by T7 RNA polymerase is useful in many molecular biology studies. The RNAP of Syn5 is homologous to T7 RNAP based on DNA sequence, although it is somewhat smaller in size. Characterization of the Syn5 RNAP is particularly interesting since its host, cyanobacteria Synechococcus, is one of the most ancient bacteria and therefore may have primitive features that provide insight into the evolution of transcription systems. An important first step in understanding the transcription of the Syn5 genome is the establishment of a transcription system using purified proteins from Syn5. Furthermore, the Syn5 RNAP should possess properties that distinguish it from T7 RNAP since it is adapted to the ocean environment. In the present study we have purified the RNAP of Syn5 to homogeneity, identified its promoters biochemically, and established an in vitro Syn5 transcription system.
EXPERIMENTAL PROCEDURES
Materials
Oligonucleotides were obtained from Integrated DNA Technology. DNA purification kits and Ni-NTA resin were from Qiagen. Cellulose phosphate resin and DE81 filter disks were from Whatman. Preparative Superdex S200 for gel filtration and ion exchange column Mono Q were from GE Healthcare. Restriction endonucleases, Deep Vent® polymerase, Phusion® High-Fidelity DNA polymerase, T4 DNA ligase, and T7 RNA polymerase were from New England Biolabs. Radiolabeled nucleotides were from Perkin Elmer. FeCl2·4H2O (99.0%) and other chemicals were from Sigma-Aldrich.
Protein Purification
Syn5 genomic DNA was isolated from Syn5 particles purified by CsCl centrifugation (17). DNA fragments encoding Syn5 RNAP were amplified from the Syn5 genome using the primers listed in supplemental Table S1 and inserted into plasmid pET24a between the NdeI and NotI sites. Plasmids were used to transform E. coli BL21(DE3). The bacteria were cultured in LB medium containing 50 μg/ml kanamycin at 37 °C until they reached an A600 of ∼1.2. The gene for Syn5 RNAP was induced by the addition of 0.5 mm IPTG at 28 °C and incubation continued for 3 h. Cells were harvested, resuspended in 50 mm sodium phosphate, pH 8.0, and 100 mm NaCl, and lysed by three cycles of freeze-thaw in the presence of 0.5 mg/ml lysozyme. Cleared lysate was collected by centrifugation. His-tagged Syn5 RNAP was isolated from the lysate using Ni-NTA-agarose chromatography according to the standard Qiagen His-tagged protein purification procedure. Ammonium sulfate (40% w/v) was added to a pool of the fractions containing predominately RNAP to precipitate the protein. The pellet was then dissolved in 1 ml of 20 mm Tris-HCl pH 7.5, 50 mm NaCl, 0.5 mm DTT, and 0.5 mm EDTA and the RNAP was further purified by gel filtration chromatography on a 200 ml preparative Superdex S200 column. Fractions eluting from this column were analyzed on SDS-PAGE gels, and those containing the RNAP were pooled. This pool was then loaded onto a cellulose phosphate column. The column was washed extensively with 20 mm potassium phosphate pH 7.5, 1 mm DTT, 1 mm EDTA, 10% glycerol, and 20 mm KCl and eluted with the same buffer containing a 0.02 to 1 m KCl gradient. Syn5 RNAP eluted at ∼0.7 m KCl; fractions containing the protein were pooled and dialyzed against 20 mm potassium phosphate pH 7.5, 0.1 mm DTT, 0.1 mm EDTA, and 50% glycerol. The fractions at each step containing Syn5 RNAP with the least amount of contaminating proteins are shown in Fig. 1. For the purification of Syn5 RNAP lacking a histidine tag, the order of purification steps was adjusted to cellulose phosphate chromatography, ammonium sulfate precipitation, gel filtration chromatography, and an additional step consisting of Mono Q ion-exchange column chromatography. Syn5 RNAP eluted at ∼0.3 m NaCl from the Mono Q column. The yield of His-tagged Syn5 RNAP was 500 μg per gram of wet cells while the yield of non-tagged Syn5 RNAP was 5 μg per gram of wet cells.
FIGURE 1.
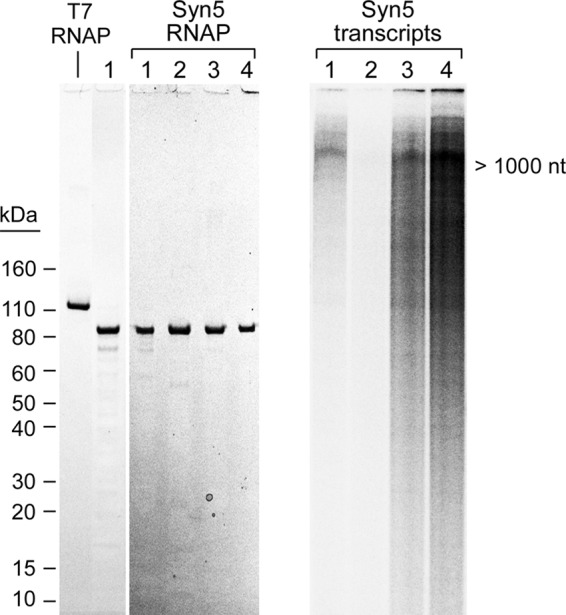
Purified Syn5 RNAP and transcription on Syn5 DNA. A, SDS-PAGE gel of purified non-tagged Syn5 RNAP (lane 1) and N-terminal His-tagged Syn5 RNAP obtained after a Ni-NTA-agarose column (lane 2), gel filtration column (lane 3), and cellulose phosphate column (lane 4). Purified T7 RNAP is shown for comparison. Proteins were stained with Coomassie Blue. B, 10% denaturing TBE-urea gel of radioactive labeled transcription products generated by Syn5 RNAPs on Syn5 genomic DNA. The lanes in B correspond to transcription by the proteins purified in A.
DNA Templates
Syn5 genomic DNA was prepared according to a previous report (13, 17). Primers used to amplify the Syn5 RNAP gene and the transcription templates are listed in supplemental Table S1. PCR reactions for DNA shorter than 3 kb were carried out using Deep Vent® DNA polymerase while reactions for DNA longer than 3 kb were carried out using Phusion® High-Fidelity DNA polymerase (New England Biolabs). PCR products were purified using Qiagen Gel Extraction Kits and DNA concentrations were determined by measuring the A260. Short transcription templates were prepared by annealing complementary synthetic DNA oligonucleotides whose sequences are shown in supplemental Table S1.
Transcription Assays
For the gel assay results shown in Figs. 1, 2, 3, and supplemental Fig. S1, reaction mixtures (20 μl) contained 40 mm Tris-HCl (pH 7.9), 6 mm MgCl2, 2 mm spermidine, 10 mm DTT, 200 μM ATP, GTP, and UTP, 10 μm [α-32P]CTP, 1.5 units/μl RNaseOUTTM recombinant ribonuclease inhibitor (Invitrogen), 100 nm Syn5 or T7 RNAP, and DNA templates as described in the figure legends (either 2 nm phage genomic DNA, 4 nm purified PCR products, or 1 μm synthetic dsDNA fragments). Reaction mixtures were incubated at 24 °C for 30 min. The reaction mixtures described in Fig. 6 contained 40 mm Tris-HCl (pH 8.0), 200 μM ATP, GTP, and UTP, 10 μM [α-32P]CTP, 1.5 units/μl RNaseOUTTM, 50 nm Syn5 RNAP, DNA templates, and various amounts of MgCl2, FeCl2, MnCl2, and KCl as described in the figure legends. After 30-min incubations at 24 °C, one unit of RQ1 RNase-free DNase (Promega) was added to each reaction mixture and incubated for an additional 20 min at 37 °C to remove the DNA templates. Reactions were then terminated by the addition of 8 μl of loading dye containing 95% formamide and 40 mm EDTA. Samples were then heated at 90 °C for 1 min and loaded onto either 10% or 25% TBE-urea denaturing gels. After electrophoresis, gels were dried and analyzed using a Fuji BAS 1000 Bioimaging Analyzer.
FIGURE 2.

Identification of the Syn5 promoter. A, entire 12 kb DNA metabolism region (beginning of the RNAP gene to the end of ribonucleotide reductase gene) of the Syn5 genome is shown in the schematic. Overlapping PCR fragments covering this region were examined for the presence of an active promoter for transcription by the purified Syn5 RNAP. Only the fragment containing the RNAP gene (template 1) was active for transcription. The following putative proteins derived from bioinformatics prediction (13) are shown: Int, integrase; SSB, ssDNA-binding protein; Endo, endonuclease; Pri/Helicase, primase/helicase; Trx, thioredoxin; DNAP, DNA polymerase; Exo, exonuclease; and Nrt for ribonucleotide reductase. B, narrowing the location of the Syn5 promoter using DNA templates with truncated 5′-ends. Based on the results obtained with templates 2 and 3, the promoter starts in the region highlighted in black background in template 2. C, determination of the 5′-end of Syn5 promoter using DNA templates with truncated 5′-ends. A series of templates each with one more nucleotide removed from the 5′-terminus were screened as effective transcription templates for Syn5 RNAP. Based on the results obtained with templates 3 and 4 the promoter starts from the A highlighted in black background in template 3.
FIGURE 3.

Characterization of the Syn5 promoter. A, determination of the 3′-end of Syn5 promoter. 3′-dATP, 3′-dGTP, 3′-dUTP replaced ATP, GTP, and UTP, respectively, as chain terminator to sequence the 5′-end of Syn5 RNAP transcript on a template same as template 1 in Fig. 2B. Once the 5′-terminus of the transcript was determined, the sequence preceding it should be the promoter and the 3′-end of the promoter can thus be defined. A 25% TBE-urea gel was used for this assay. B, cross recognition between Syn5 and T7 transcription systems. Syn5 RNAP synthesizes runoff products on a fragment of T7 DNA provided the T7 promoter is replaced by a Syn5 promoter (lane a). Syn5 RNAP fails to produce transcripts if the first nucleotide downstream of the promoter is changed from G to T (lane b). Syn5 and T7 RNAP do not recognize the heterologous promoter (lanes c and d). T7 RNAP produces runoff and “N+1” products upon its own promoter (lane e). Sequences of templates can be found in supplemental Table S1. C, schematic showing the identity of the two promoters based on the above data and previous bioinformatics analysis on the Syn5 and P-SSP7 genomes, respectively. Syn5 RNAP transcribes DNA fragments containing a Syn5 promoter to produce transcripts (>1500 for template 1, lane 1; and >200 nt for template 3, lane 3). Syn5 RNAP does not transcribe the DNA fragment covering the region between the DNA metabolism and structural genes in the middle of Syn5 genome (lane 2).
FIGURE 6.

Comparison of Syn5 RNAP transcription products in the presence of MgCl2, FeCl2, or MnCl2 on denaturing gels. Reactions contained 200 μm ATP, GTP, UTP, and 10 μm [α-32P]CTP, 50 nm Syn5 RNAP, DNA templates (1 μm template as shown in Fig. 3A for 71 nt runoff transcript, 4 nm template 3 as shown in Fig. 3C for 225 nt product, or 1 μm template 1 as shown in Fig. 3B for 37 nt runoff transcript). Amounts of MgCl2, FeCl2, MnCl2, and KCl in the reactions are shown in the figure.
Filter binding assays were used for the results shown in Figs. 4, 5, and 7. Basic reaction mixtures contained 40 mm Tris-HCl (pH 8.0 unless otherwise specified), 2 mm spermidine, 10 mm DTT, 200 μM GTP, CTP, UTP, and [3H]ATP (20 cpm/pmol), 1.5 units/μl RNaseOUTTM, 50 nm Syn5 or T7 RNAP and 4 nm plasmid pET24 DNA harboring the Syn5 RNAP gene (pET24-S5RNAP) that contains a single Syn5 promoter in the RNAP gene and a single T7 promoter preceding the gene. Reaction mixtures were incubated at 24 °C unless otherwise specified. Various salts and cofactors were added as indicated in the figure legends. For the assays described in Fig. 7, reaction mixtures contained 40 mm Tris-HCl (pH 8.0), 6 mm MgCl2, 2 mm spermidine, 10 mm DTT, 20 nm Syn5, or T7 RNAP, 20 nm pET24-S5RNAP DNA, 1 mm [3H]ATP (20 cpm/pmol), CTP, UTP (or GTP), and varying amounts of GTP (or UTP). 160 mm KCl was added for the Syn5 RNAP reactions. After various times the reactions were stopped by the addition of 20 mm EDTA. 4 μl of the mixtures were then loaded onto Whatman DE81 filter paper disks and the disks were washed to remove the unincorporated [3H]ATP. The amount of insoluble [3H]AMP, corresponding to nucleotides incorporated into newly synthesized RNA, was measured using a scintillation counter. The data were analyzed using Prism software.
FIGURE 4.
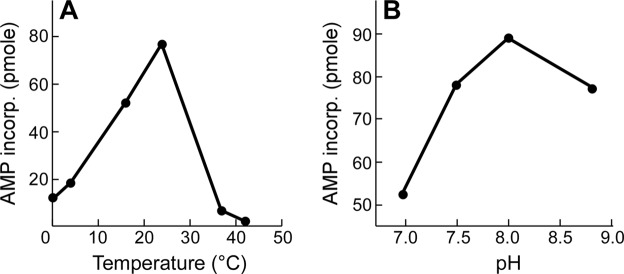
Effect of temperature and pH on Syn5 RNAP activity. A, temperature. B, pH. RNA polymerase activity was measured as described under “Experimental Procedures.” The reactions contained 6 mm MgCl2, 200 μm 4 rNTPs ([3H]ATP), 50 nm Syn5 RNAP, and 4 nm template (pET24-S5RNAP). The pH was 8.0 for assays in A, and the temperature was 24 °C for B; reactions were terminated, and the amount of AMP incorporated was measured at 3, 10, and 30 min. The AMP incorporation was linear in this time range, and the data measured at 10 min are presented.
FIGURE 5.
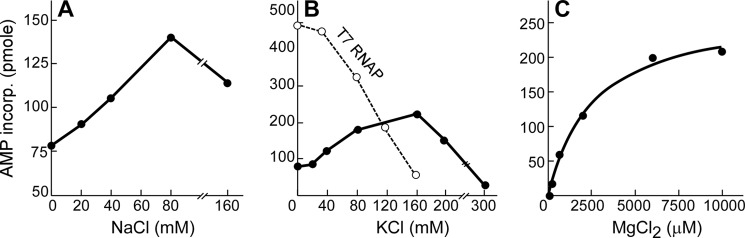
Reaction requirements for Syn5 RNAP. A, effect of NaCl on Syn5 RNAP activity. B, effect of KCl on Syn5 and T7 RNAP (dashed line) activity. C, MgCl2 concentration on Syn5 RNAP activity. Reactions contained 200 μm 4 rNTPs ([3H]ATP), 50 nm Syn5 RNAP, and 4 nm template (pET24-S5RNAP). For A and B, reactions also contained 6 mm MgCl2. For C, 160 mm KCl was present. Reactions were terminated and the amount of AMP incorporated was measured at 2.5, 5, and 10 min. AMP incorporation was linear in this time range, and the data measured at 10 min are presented.
FIGURE 7.
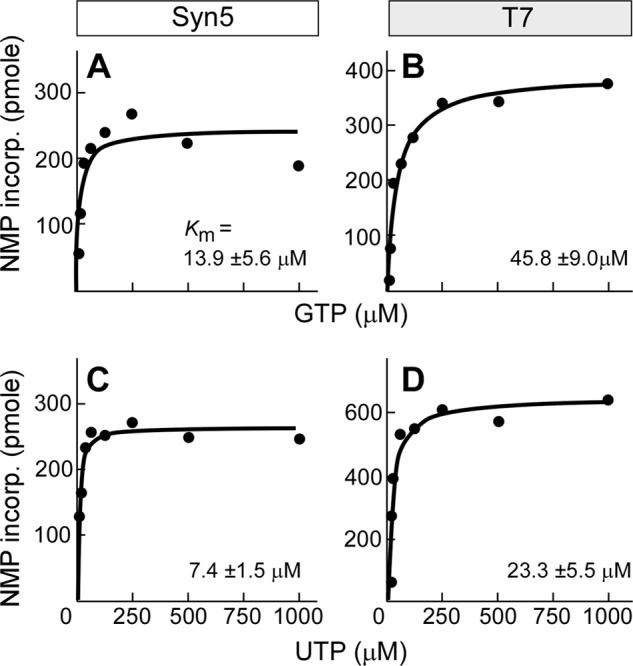
Comparison of transcription efficiency of Syn5 and T7 RNAP at various ribonucleotide concentrations. A, incorporation of NMP by Syn5 RNAP at various GTP concentrations. B, incorporation of NMP by T7 RNAP at various GTP concentrations. C, incorporation of NMP by Syn5 RNAP at various UTP concentrations. D, incorporation of NMP by T7 RNAP at various UTP concentrations. Varied ribonucleotide concentrations were 7.8, 15.6, 31.3, 62.5, 125, 250, 500, 1000 μm while other NTPs were all at 1 mm, RNAP and template were both 20 nm, and the reaction time was 5 min at 24 °C. 160 mm KCl was added for Syn5 RNAP reactions.
RESULTS AND DISCUSSION
Overproduction and Purification of Syn5 RNAP
A T7 RNAP-like single-subunit RNAP was previously predicted from the sequence of the Syn5 genome (13). The predicted protein is significantly smaller (779 residues) than the two well-characterized homologous phage RNAPs from T7 (883 residues) and SP6 (874 residues). We have cloned the Syn5 RNAP gene into a plasmid under the control of a T7 promoter and overproduced the Syn5 RNAP in E. coli. The target protein is soluble and has been purified to greater than 80% purity using cellulose phosphate chromatography, ammonium sulfate precipitation, gel filtration chromatography, and anion-exchange chromatography (Fig. 1A, lane 1). The Syn5 RNAP binds tightly (eluting at 0.7 m KCl) to cellulose phosphate resulting in the greatest purification. We were unable to further purify the protein despite using chromatography on Sepharose-Blue, ATP-agarose, and DEAE cellulose. Therefore we constructed a hybrid gene fusion that attaches a His-Tag on the N terminus of Syn5 RNAP. This tagged-protein was purified to apparent homogeneity by Ni-NTA-agarose chromatography, gel filtration chromatography, and cellulose phosphate chromatography (Fig. 1A, lane 4). Fractions from each step of this procedure were collected and their purity established by gel electrophoresis and staining with Coomassie Blue (Fig. 1A, lanes 2 and 3). During purification, fractions from each step were assayed for RNA polymerase activity by transcription on Syn5 genomic DNA (Fig. 1B). The increased protein purity significantly increased the yield of RNA transcripts, most likely due to the removal of contaminating ribonuclease activity (Fig. 1B).
The non-tagged Syn5 RNAP had biochemical properties similar to the His-tagged RNAP although it synthesized fewer transcripts compared with the tagged protein (Fig. 1B, lane 1 versus lane 4). Again, the apparent decrease in RNA polymerase activity probably reflects the lower purity of the non-tagged protein, and thus the presence of contaminating ribonuclease activity. The results shown in the following sections were all carried out using the His-tagged Syn5 RNAP.
Identification of the Syn5 RNAP Promoter
Syn5 shares many features in common with other T7 phage groups including morphology, genome size, and the presence of a terminal redundancy at the two ends of its genome (13). Another common feature is their transcription system. The phages encode their own RNAP that initiate transcription at conserved promoters distributed along the genome (25, 26). Bioinformatics identified a 12 bp sequence 5′-CCTTAATTAACT-3′ in the middle/late portion of the Syn5 genome, the only sequence that appears several times within the genome (13). This sequence was considered a potential promoter at the outset, however DNA fragments containing this sequence do not serve as templates for transcription by Syn5 RNAP.
A number of early Syn5 genes appear to have sigma70-like promoters, suggesting that the host RNAP is responsible for their transcription (13). By analogy to other T7-like phages, the genes downstream of the RNAP are most likely to be transcribed from Syn5 promoters. Therefore, in order to identify the Syn5 promoters, we prepared overlapping DNA fragments covering the entire region that encodes the predicted DNA metabolism proteins downstream of the RNAP gene. These fragments were then screened for promoters using an in vitro transcription assay with the purified Syn5 RNAP (Fig. 2A). To our surprise, Syn5 RNAP was only active on one of the templates, the template containing the RNAP gene itself. T7 phage has a promoter immediately after its RNAP gene, and thus it is reasonable that Syn5 has a promoter to transcribe its downstream genes. However, in contrast to the single promoter found in Syn5 over this region, T7 has 10 promoters distributed over the analogous region.
Using two sets of PCR fragments as transcription templates, we further narrowed the location of the Syn5 promoter to the region between 760 and 1000 nt in the RNAP gene; templates lacking this region were not transcribed by the Syn5 RNAP (supplemental Fig. S1A). A similar strategy was used to further narrow the promoter to between 797 and 842 nt in the Syn5 RNAP gene (supplemental Fig. S1B). To precisely identify the 5′ nucleotide of the promoter in this 46 nt region, we screened every 10 nt (Fig. 2B) and then every 1 nt (Fig. 2C). DNA fragments are effective transcription templates for Syn5 RNAP as long as they contain the A highlighted in black background in Fig. 2C; thus this A is designated the 5′-end of the Syn5 promoter. We determined the 3′-end of the promoter using 3′-dNTPs as chain terminators (Fig. 3A). In this experiment we used [α-32P]CTP and replaced either GTP, ATP, or UTP with their 3′-dNTP analog. 3′-dGTP blocked any transcription, suggesting that there is a G before the first C. 3′-dATP resulted in the production of a trinucleotide; thus the first three nucleotides are predicted to be pppGCA. A dinucleotide, presumably pppGC, is also present. 3′-dUTP resulted in the production of a 12 nt transcript (Fig. 3A). Combining these results we designate the sequence of the Syn5 promoter as 5′-ATTGGGCACCCGTAA-3′ (sequence in blue background in Fig. 3). The Syn5 RNAP also produces significant amounts of abortive transcripts ranging from 2–11 nt together with the runoff product (Fig. 3A). These abortive transcripts are observed with all RNAPs and occur during the transition between the initiation and elongation of transcription (23).
Syn5 RNAP does not recognize a T7 promoter, since it is unable to initiate transcription from a T7 DNA fragment containing the T7 Φ1.1 B promoter (Fig. 3B, lane c). In contrast, when we replaced the T7 promoter with the Syn5 promoter, Syn5 RNAP transcribed the template to produce a run-off product of the expected size (Fig. 3B, lane a). With T7 RNAP the first nucleotide following the promoter is important in determining transcription efficiency (27). Syn5 RNAP is unable to incorporate UTP as the first ribonucleotide (Fig. 3B, lane b). At a T7 promoter, T7 RNAP initiates transcription and produces a runoff transcript (Fig. 3B, lane e). However, at a Syn5 promoter it does not initiate transcription but does catalyze some nonspecific RNA synthesis (Fig. 3B, lane d).
Surprisingly, on the Syn5 genome there is only one other sequence identical to the Syn5 promoter sequence identified here. The second sequence is located near the right end of Syn5 genome, after the terminase gene (Fig. 3C). Transcription on a PCR fragment covering this region confirms that this sequence is indeed an active promoter (Fig. 3C, lane 3). Several sequences within the Syn5 genome were identified by alignment to be similar to the promoter (up to 73% identity) but none of them served as effective Syn5 promoters in our in vitro transcription reactions. Most T7-like phages have several strong promoters in the middle of their genomes to control the expression of their structural genes. However, Syn5 RNAP does not initiate specific transcription on a 6-kb fragment of the Syn5 genome encompassing the region from the end of the DNA metabolism genes through the end of the gene encoding the major capsid protein (Fig. 3C, lane 2).
We find support for the presence of only two cyanophage promoters from previous bioinformatics analysis. Chen and Schneider extensively analyzed the promoter systems of T7-like phages (25). For P-SSP7, the only cyanophage analyzed, no T7-like promoters were identified. However, when they aligned every 1 kb fragment of the P-SSP7 genome against the rest of the genome they found two identical sequences, 5′-AACCCCTACGTATACA-3′, one located within the RNAP gene and the other after the terminase gene (Fig. 3C). Although these two cyanophages infect different groups of host cyanobacteria, their similar distribution of promoters indicates a common transcription regulation mechanism among cyanophages, which differs from that found in other T7-like phage. Despite this similarity, there is no obvious sequence similarity between the putative promoters from these two cyanophages.
Characterization of Syn5 RNAP Transcription: Optimal Temperature and pH
We optimized the in vitro Syn5 transcription system using the purified RNAP and a plasmid containing a single Syn5 promoter. The optimum temperature for Syn5 RNAP is 24 °C (Fig. 4A). The activity decreases with lower temperatures but retains 15% of its maximum activity at 0 °C. Only 8% of the maximum activity is observed at 37 °C and 2% at 42 °C. In contrast, the maximum activity of T7 RNAP is observed at 37 °C and decreases dramatically below 20 °C (28). The lower temperature optimum for Syn5 RNAP probably reflects the temperature of the ocean environment of the host Synechococcus (22).
The pH of seawater is in the range of 7.5 to 8.4. The activity of Syn5 RNAP is highest at pH 8.0 and does not vary significantly in the range from pH 7.5 to 8.8 (Fig. 4B).
Salt and Metal Cofactors
T7 RNAP is highly sensitive to the ionic strength of the reaction (Ref. 29, Fig. 5B). Since the environment of Syn5 is the ocean, we were interested whether it would be less sensitive to salt concentration. In fact, both NaCl and KCl significantly stimulate Syn5 RNAP activity. A 2-fold increase is observed in the presence of 80 mm NaCl (Fig. 5A) and a 3-fold stimulation in the presence of 160 mm KCl (Fig. 5B). The activity decreases above 160 mm KCl, with only 10% remaining at 300 mm (Fig. 5B). MgCl2 is required as a cofactor for Syn5 RNAP activity, with a Km of about 2.5 mm in the presence of 160 mm KCl (Fig. 5C).
We tested other metal ions in place of Mg2+ as cofactors for the Syn5 RNAP using the filter-binding assay. At concentrations of 10 mm, Ca2+, Co2+, Cu2+, or Ni2+ cannot replace Mg2+ in the Syn5 RNAP reaction. A small amount of activity is observed with 10 mm Zn2+ or Mn2+. Ferrous and manganese ions are of particular interests since they are abundant in cyanobacteria (30, 31). We used denaturing gels to characterize the products by Syn5 RNAP with ferrous as a cofactor (Fig. 6), since ferrous caused nonspecific NTP precipitation in the filter binding assay. We find that FeCl2 concentrations higher than 4 mm result in retardation of radioactive NTP in the gel and bands that are not distinguishable. 2 mm FeCl2 clearly enables the Syn5 RNAP to produce a 71 nt runoff transcript identical to that produced in the presence of MgCl2 (Fig. 6, lane 4 versus lane 1). KCl stimulates the ferrous-catalyzed reaction (Fig. 6, lane 9 versus lane 10). Iron is the metal used at the active site of many important redox enzymes dealing with cellular respiration, oxidation and reduction in plants and animals. In addition, iron-sulfur clusters have been found in many nucleic acid processing enzymes including a RNAP (32). However, iron as a cofactor in a polymerase reaction has not been reported. The efficiency of iron as a cofactor, however, is several times less than that of magnesium (Fig. 6, lane 9 versus lane 7). Furthermore, in the presence of iron Syn5 RNAP does not produce longer transcripts (e.g. 225 nt, Fig. 6, lanes 11–15). Manganese, at lower concentration than that of magnesium, is also an active cofactor for Syn5 RNAP. The maximum yield of short runoff products synthesized by Syn5 RNAP is higher in the presence of manganese (0.25 to 1 mm) than that with magnesium (Fig. 6, lanes 16–20). Higher concentration of manganese inhibits the activity. It is noteworthy that the “N+1” runoff product is significantly higher with manganese than that with magnesium.
Nucleotides
We compared the catalytic efficiencies between Syn5 and T7 RNAPs at various ribonucleotide concentrations. For Syn5 RNAP, the apparent Km for GTP is 13.9 ± 5.6 μm, and the kcat is 2.5 s−1 (Fig. 7A). Under the same conditions (except that KCl was omitted) the KmGTP for T7 RNAP is 45.8 ± 9.0 μm and the kcat is 4.1 s−1 (Fig. 7B). Although the maximal efficiency of T7 RNAP is higher than that of Syn5 RNAP, the latter shows greater activity with lower GTP concentrations. The kcat/KmGTP is higher for Syn5 RNAP (0.18) than for T7 RNAP (0.09). Similar results were obtained with UTP. The KmUTP for Syn5 RNAP is 7.4 ± 1.5 μm and the kcatUTP is 2.8 s−1 (Fig. 7C) while for T7 RNAP the KmUTP is 23.3 ± 5.5 μm and the kcatUTP is 6.8 s−1 (Fig. 7D). Syn5 RNAP consistently shows higher efficiency than T7 RNAP when comparing the kcat/KmUTP (0.38 versus 0.29). The higher efficiency of ribonucleotides utilization at low concentration by Syn5 RNAP may benefit the cyanophage in the open ocean environment where nutrition is usually stringent.
Acknowledgments
We thank Steven Moskowitz (Advanced Medical Graphics) for illustrations and Drs. Seung-Joo Lee, Barak Akabayov, Huidong Zhang, and Jacqueline M. Piret for helpful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants GM54397 (to C. C. R.) and GM17980 (to J. K.).

This article contains supplemental Table S1 and Fig. S1.
- dsDNA
- double-stranded DNA
- RNAP
- RNA polymerase.
REFERENCES
- 1. Suttle C. A. (2005) Viruses in the sea. Nature 437, 356–361 [DOI] [PubMed] [Google Scholar]
- 2. Suttle C. A. (2007) Marine viruses–major players in the global ecosystem. Nat. Rev. Microbiol. 5, 801–812 [DOI] [PubMed] [Google Scholar]
- 3. Lindell D., Jaffe J. D., Johnson Z. I., Church G. M., Chisholm S. W. (2005) Photosynthesis genes in marine viruses yield proteins during host infection. Nature 438, 86–89 [DOI] [PubMed] [Google Scholar]
- 4. Lindell D., Jaffe J. D., Coleman M. L., Futschik M. E., Axmann I. M., Rector T., Kettler G., Sullivan M. B., Steen R., Hess W. R., Church G. M., Chisholm S. W. (2007) Genome-wide expression dynamics of a marine virus and host reveal features of co-evolution. Nature 449, 83–86 [DOI] [PubMed] [Google Scholar]
- 5. Chen F., Lu J. (2002) Genomic sequence and evolution of marine cyanophage P60: a new insight on lytic and lysogenic phages. Appl. Environ. Microbiol. 68, 2589–2594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Liu X., Kong S., Shi M., Fu L., Gao Y., An C. (2008) Genomic analysis of freshwater cyanophage Pf-WMP3 infecting cyanobacterium Phormidium foveolarum: the conserved elements for a phage. Microbiol. Ecol. 56, 671–680 [DOI] [PubMed] [Google Scholar]
- 7. Mann N. H., Clokie M. R., Millard A., Cook A., Wilson W. H., Wheatley P. J., Letarov A., Krisch H. M. (2005) The genome of S-PM2, a “photosynthetic” T4-type bacteriophage that infects marine Synechococcus strains. J. Bacteriol. 187, 3188–3200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu X., Shi M., Kong S., Gao Y., An C. (2007) Cyanophage Pf-WMP4, a T7-like phage infecting the freshwater cyanobacterium Phormidium foveolarum: complete genome sequence and DNA translocation. Virology 366, 28–39 [DOI] [PubMed] [Google Scholar]
- 9. Millard A. D., Zwirglmaier K., Downey M. J., Mann N. H., Scanlan D. J. (2009) Comparative genomics of marine cyanomyoviruses reveals the widespread occurrence of Synechococcus host genes localized to a hyperplastic region: implications for mechanisms of cyanophage evolution. Environ. Microbiol. 11, 2370–2387 [DOI] [PubMed] [Google Scholar]
- 10. Sullivan M. B., Coleman M. L., Weigele P., Rohwer F., Chisholm S. W. (2005) Three Prochlorococcus cyanophage genomes: signature features and ecological interpretations. PLoS Biol., 3, e144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sullivan M. B., Krastins B., Hughes J. L., Kelly L., Chase M., Sarracino D., Chisholm S. W. (2009) The genome and structural proteome of an ocean siphovirus: a new window into the cyanobacterial 'mobilome'. Environ. Microbiol. 11, 2935–2951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weigele P. R., Pope W. H., Pedulla M. L., Houtz J. M., Smith A. L., Conway J. F., King J., Hatfull G. F., Lawrence J. G., Hendrix R. W. (2007) Genomic and structural analysis of Syn9, a cyanophage infecting marine Prochlorococcus and Synechococcus. Environ. Microbiol. 9, 1675–1695 [DOI] [PubMed] [Google Scholar]
- 13. Pope W. H., Weigele P. R., Chang J., Pedulla M. L., Ford M. E., Houtz J. M., Jiang W., Chiu W., Hatfull G. F., Hendrix R. W., King J. (2007) Genome sequence, structural proteins, and capsid organization of the cyanophage Syn5: a “horned” bacteriophage of marine synechococcus. J. Mol. Biol. 368, 966–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lindell D., Sullivan M. B., Johnson Z. I., Tolonen A. C., Rohwer F., Chisholm S. W. (2004) Transfer of photosynthesis genes to and from Prochlorococcus viruses. Proc. Natl. Acad. Sci. U.S.A., 101, 11013–11018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sharon I., Alperovitch A., Rohwer F., Haynes M., Glaser F., Atamna-Ismaeel N., Pinter R. Y., Partensky F., Koonin E. V., Wolf Y. I., Nelson N., Béjà O. (2009) Photosystem I gene cassettes are present in marine virus genomes. Nature 461, 258–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sabehi G., Shaulov L., Silver D.H., Yanai I., Harel A., Lindell D. (2012) A novel lineage of myoviruses infecting cyanobacteria is widespread in the oceans. Proc. Natl. Acad. Sci. U.S.A. 109, 2037–2042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Raytcheva D. A., Haase-Pettingell C., Piret J. M., King J. A. (2011) Intracellular assembly of cyanophage Syn5 proceeds through a scaffold-containing procapsid. J. Virol., 85, 2406–2415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gao E. B., Gui J. F., Zhang Q. Y. (2012) A novel cyanophage with a cyanobacterial nonbleaching protein A gene in the genome. J. Virol. 86, 236–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thompson L. R., Zeng Q., Kelly L., Huang K. H., Singer A. U., Stubbe J., Chisholm S. W. (2011) Phage auxiliary metabolic genes and the redirection of cyanobacterial host carbon metabolism. Proc. Natl. Acad. Sci. U.S.A. 108, E757–E764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Waterbury J. B., Valois F. W. (1993) Resistance to co-occurring phages enables marine synechococcus communities to coexist with cyanophages abundant in seawater. Appl. Environ. Microbiol. 59, 3393–3399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sullivan M. B., Waterbury J. B., Chisholm S. W. (2003) Cyanophages infecting the oceanic cyanobacterium Prochlorococcus. Nature 424, 1047–1051 [DOI] [PubMed] [Google Scholar]
- 22. Zwirglmaier K., Jardillier L., Ostrowski M., Mazard S., Garczarek L., Vaulot D., Not F., Massana R., Ulloa O., Scanlan D. J. (2008) Global phylogeography of marine Synechococcus and Prochlorococcus reveals a distinct partitioning of lineages among oceanic biomes. Environ. Microbiol. 10, 147–161 [DOI] [PubMed] [Google Scholar]
- 23. Cheetham G. M., Steitz T. A. (2000) Insights into transcription: structure and function of single-subunit DNA-dependent RNA polymerases. Curr. Opin Struct. Biol. 10, 117–123 [DOI] [PubMed] [Google Scholar]
- 24. Davanloo P., Rosenberg A. H., Dunn J. J., Studier F. W. (1984) Cloning and expression of the gene for bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. U.S.A. 81, 2035–2039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen Z., Schneider T. D. (2005) Information theory based T7-like promoter models: classification of bacteriophages and differential evolution of promoters and their polymerases. Nucleic Acids Res. 33, 6172–6187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dunn J. J., Studier F. W. (1983) Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J. Mol. Biol. 166, 477–535 [DOI] [PubMed] [Google Scholar]
- 27. Milligan J. F., Groebe D. R., Witherell G. W., Uhlenbeck O. C. (1987) Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res. 15, 8783–8798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chamberlin M., Ring J. (1973) Characterization of T7-specific ribonucleic acid polymerase. 1. General properties of the enzymatic reaction and the template specificity of the enzyme. J. Biol. Chem. 248, 2235–2244 [PubMed] [Google Scholar]
- 29. Chamberlin M., Ring J. (1973) Characterization of T7-specific ribonucleic acid polymerase. II. Inhibitors of the enzyme and their application to the study of the enzymatic reaction. J. Biol. Chem. 248, 2245–2250 [PubMed] [Google Scholar]
- 30. Shcolnick S., Keren N. (2006) Metal homeostasis in cyanobacteria and chloroplasts. Balancing benefits and risks to the photosynthetic apparatus. Plant Physiol. 141, 805–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Imashimizu M., Tanaka K., Shimamoto N. (2011) Comparative Study of Cyanobacterial and E. coli RNA Polymerases: Misincorporation, Abortive Transcription, and Dependence on Divalent Cations. Genet. Res. Int. 2011, 572689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. White M. F., Dillingham M. S. (2012) Iron-sulphur clusters in nucleic acid processing enzymes. Curr. Opin. Struct. Biol. 22, 94–100 [DOI] [PubMed] [Google Scholar]