

Background: Smad7 may have biological roles other than inhibition of TGF-β signaling.
Results: Smad7 activates caspase 8 expression by recruiting IRF1, thereby inducing TRAIL-mediated apoptosis.
Conclusion: Smad7 functions as a transcriptional coactivator of IRF1 to regulate the expression of target genes.
Significance: Smad7-inducing reagents can be used as anti-cancer drugs for tumors that have defects in caspase 8 expression.
Keywords: Apoptosis, Caspase, TRAIL, Transcription Coactivators, Tumor Therapy, Apoptosis, Caspase 8, IRF1, Smad7
Abstract
Smad7 has been known as a negative regulator for the transforming growth factor-β (TGF-β) signaling pathway through feedback regulation. However, Smad7 has been suspected to have other biological roles through the regulation of gene transcription. By screening differentially regulated genes, we found that the caspase 8 gene was highly up-regulated in Smad7-expressing cells. Smad7 was able to activate the caspase 8 promoter through recruitment of the interferon regulatory factor 1 (IRF1) transcription factor to the interferon-stimulated response element (ISRE) site. Interaction of Smad7 on the caspase 8 promoter was confirmed with electrophoretic mobility shift assay and chromatin immunoprecipitation experiment. Interestingly, Smad7 did not directly interact with the ISRE site, but it increased the binding activity of IRF1 with ISRE. These results support that Smad7 recruits IRF1 protein on the caspase 8 promoter and functions as a transcriptional coactivator. To confirm the biological significance of caspase 8 up-regulation, we tested tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-mediated cell death assay in breast cancer cells. Smad7 in apoptosis-resistant MCF7 cells markedly sensitized the cells to TRAIL-induced cell death by restoring the caspase cascade. Furthermore, restoration of caspase 8-mediated apoptosis pathway repressed the tumor growth in the xenograft model. In conclusion, we suggest a novel role for Smad7 as a transcriptional coactivator for caspase 8 through the interaction with IRF1 in regulation of the cell death pathway.
Introduction
Smad7 has been described as an inhibitory Smad, which blocks transforming growth factor-β (TGF-β) signaling through feedback mechanisms (1). Smad7 is induced by TGF-β itself or other stimuli and then translocates to the cytoplasm and interacts with the activated TGF-β type I receptor to induce its ubiquitination by Smurf1 and Smurf2. The ubiquitination of the receptor leads to proteasomal degradation that terminates the transient TGF-β signal. Smad7 also associates with the kinase domain of receptors through its C-terminal region. Consequently, Smad7 suppresses the recruitment and phosphorylation of other Smad proteins and finally turns off the TGF-β signaling (2).
In addition to these conventional roles, Smad7 was shown to have tumor suppressing activity in many systems. Smad7 sensitizes tumor cells to tumor necrosis factor-α (TNF-α) and other death signals by modulating the NF-κB survival pathway (3–5). Also, overexpression of Smad7 in melanoma and mammary carcinoma cells can impair tumor growth and bone metastasis (6, 7). Smad6 or Smad7 is not just a regulator; they also serve as an important cross-talk mediator of the TGF-β signaling pathway (8, 9). Furthermore, there are some reports showing the unexpected role of Smad7 in transcriptional regulation of target genes at the nucleus. Smad7 can interact at the Smad-binding element of PAI-1 through its MH2 domain to inhibit TGF-β signaling (10). Smad7 also regulates gene transcription through interactions with histone deacetylases, such as HDAC1 and SIRT1, and acetyltransferase, p300 (11, 12).
Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)4 or apo2 ligand is a member of the TNF ligand family of type II transmembrane proteins (13). When these ligands bind to their specific receptors, adaptor molecules, such as FADD, are recruited and send different signals to the cell. Until now, there have been five human TRAIL receptors identified as follows: DR4 (TRAIL-R1), DR5 (TRAIL-R2), DcR1 (TRAIL-R3), DcR2 (TRAIL-R4), and osteoprotegerin. Both DR4 and DR5 are pro-apoptotic receptors, and interaction of TRAIL with these receptors leads to activation of caspase cascades (14). Unlike death receptors, DcR1 and DcR2 do not have the cytoplasmic functional death domain and act as decoy receptors by competing for ligand binding with the death receptors DR4 and DR5 (15). Osteoprotegerin is a soluble protein, but its role in TRAIL signaling upon binding to osteoprotegerin is not well understood.
Because TRAIL selectively kills cancer cells without causing any harmful effects on normal cells, it is a promising new agent for the treatment of cancer (16). However, many tumor cells, including breast cancer, are still resistant to TRAIL-mediated death signaling (17, 18). Many reports have shown that resistance to TRAIL-mediated apoptosis can be a consequence of various alterations in these pathways. First, the expression level of decoy receptors was correlated with sensitivity of the tumor cell to TRAIL-induced apoptosis. Highly expressed decoy receptors can block the death signal by sequestering TRAIL (19, 20). A second mechanism of resistance to TRAIL is the enhanced activity of survival factor NF-κB in the tumor cell. This transcription factor has an important role in the regulation of the expression of many anti-apoptotic genes such as inhibitor of apoptosis protein 1 (IAP1), IAP2, X-linked inhibitor of apoptosis, and FLICE-inhibitory protein (21). Another important resistance mechanism to TRAIL-induced apoptosis is the defect of caspase 8 expression in some tumor cells (22–24). Absence or suppression of caspase 8 results in resistance to apoptosis and is a major therapeutic target in childhood medulloblastoma and neuroblastoma.
In this report, we show that caspase 8 is up-regulated in Smad7-expressing MCF7 cells. Transcriptional regulation of the caspase 8 gene by Smad7 is initiated by the recruitment of interferon regulatory factor 1 (IRF1) to the interferon-stimulated response element (ISRE) sites. In this process, Smad7 enhanced the binding activity of IRF1 to the caspase 8 promoter. As a result, overexpression of Smad7 can sensitize the breast cancer cell to TRAIL-mediated apoptosis. Furthermore, reconstitution of the caspase 8-mediated cell death pathway suppressed the tumor growth in mouse xenograft model. In conclusion, our data strongly suggest that induction of Smad7 expression can be used as a therapeutic approach for some cancers that have defects in caspase 8 expression.
EXPERIMENTAL PROCEDURES
Cell Culture and Reagents
MCF7 human breast cancer cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin at 37 °C in a 5% CO2 atmosphere. HEK293 cells were obtained from the American Type Culture Collection and cultured under conditions recommended by the supplier. FLAG-tagged Smad7 or caspase 8 stably expressing cells were generated by retroviral infection with the LPCX construct, as described previously (25), and maintained with 400 ng/ml puromycin. Anti-Myc (9E10), Smad7 (N19), IRF1 (C20), caspase 8 p20 (H134), and IκBα (FL) antibodies were purchased from Santa Cruz Biotechnology. FLAG (M2) and β-actin (AC-15)-specific antibodies were obtained from Sigma. An anti-poly(ADP-ribose) polymerase (PARP) antibody was purchased from Millipore. Activated caspase 3 antibody (catalog no. 9664) was acquired from Cell Signaling. For flow cytometric analysis, phycoerythrin-conjugated anti-TRAIL-R1 or anti-TRAIL-R2 antibodies were obtained from R&D Systems.
Microarray Analysis
Total RNA was extracted from MCF7 cells using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Each total RNA sample (10 μg) was labeled with Cy3 or Cy5 dyes during cDNA synthesis using Superscript II reverse transcriptase (Invitrogen) and hybridized onto microarray slides as described previously (26). Hybridized slides were scanned on a GenePix 4000A scanner (Axon Instrument). Gene expression data were transformed and normalized, and we then calculated the average ratios of each gene from replicated experiments. Fold change filters included the requirement that the genes be present in at least 200% of controls (for up-regulated genes) and lower than 50% of controls (for down-regulated genes).
Transient Transfection and Luciferase Assays
MCF7 cells were seeded in 12-well plates at 1 × 105 cells/well and transiently transfected with pGL3B control, pGL3B-NF-κB, ISRE, or caspase 8 promoter plus Smad7-expression plasmids. β-Galactosidase was used as an internal control. Lipofectamine Plus was used as the transfection agent according to the manufacturer's instructions (Invitrogen). After a 6-h or overnight incubation, the media were changed to serum-containing media and treated with 10 ng/ml TNF-α or 100 ng/ml TRAIL (PeproTech) for 18 h. All assays were performed in triplicate using a Victor2 luminometer (PerkinElmer Life Sciences), and the data shown represent the mean of at least three independent experiments.
Immunoblot Analysis
MCF7 cells were treated with TNF-α or TRAIL for the indicated time and were lysed in a buffer containing 25 mm HEPES (pH 7.5), 150 mm NaCl, 1% Triton X-100, 10% glycerol, 5 mm EDTA, and a protease inhibitor mixture (Roche Applied Science). For immunoblotting, cell debris was removed by centrifugation, and proteins were separated by SDS-PAGE, followed by transfer to polyvinylidene difluoride membranes. After incubating with the appropriate primary and secondary antibodies, proteins were visualized by chemiluminescence, according to the manufacturer's instructions (Pierce).
RT-PCR
Total RNA from MCF7-LPCX or Smad7 cells was isolated using TRIzol reagent (Invitrogen) according to the manufacturer's protocols. For semi-quantitative RT-PCR, we used the One-step RT-PCR kit (Qiagen) with the primer set listed in supplemental Table S1. Samples were normalized to GAPDH mRNA.
Colorimetric Cell Viability Assay
MCF7 cells were grown in 96-well plates in a final volume of 100 μl of media for 12 h and treated with various concentrations of TRAIL for 24 h. The next day, 10 μl of MTT labeling solution was added to the culture media and incubated for 4 h in a 37 °C humidified CO2 incubator. 100 μl of solubilization solution (10% SDS in 0.01 m HCl) was added to each well, and the plate was placed overnight in an incubator in a humidified atmosphere. MTT activity was measured colorimetrically using an ELISA reader at 550 nm. Each value was normalized by subtracting the reference value measured at 650 nm.
DNA Fragmentation Assay
MCF7 cells were plated into 100-mm plates and treated with 100 ng/ml TRAIL in the presence of 10 μm caspase inhibitor (Z-VAD-fmk or Z-IETD-fmk) for 24 h. Cells were then treated with lysis buffer (10 mm Tris-HCl (pH 7.4), 10 mm NaCl, 10 mm EDTA, 0.5% SDS, and 0.1 mg/ml proteinase K) and were incubated at 50 °C for 2 h. The cell lysate was extracted successively with phenol, phenol/chloroform (1:1), and chloroform and was then precipitated with a 2.5× volume of ice-cold ethanol. The DNA was resuspended in Tris/EDTA buffer supplemented with 100 μg/ml RNase A. The genomic DNA bands were electrophoretically separated on a 2% agarose gel for 1 h at 50 V.
Site-directed Mutagenesis
The ISRE sequence in the caspase 8 promoter, at positions −1478 to −1479, was mutated by replacing the TTC sequences with TGA sequences. The mutants were generated using an EZchangeTM site-directed mutagenesis kit according to the manufacturer's instructions (Enzynomics, Korea).
Chromatin Immunoprecipitation (ChIP) Assay
ChIP assay was performed using ChIP assay kit according to the manufacturer's instructions (Millipore). MCF7-Smad7 cell was cross-linked by incubation in 1% formaldehyde-containing medium for 15 min at 37 °C. The reaction was stopped by the addition of glycine to a final concentration of 0.125 m. A soluble chromatin fraction, containing fragmented DNA of 500–1,000 bp, was obtained after cell lysis and sonication. Immunoprecipitation was performed by incubating the lysate with 2 μg of anti-Smad7 or IRF1 antibodies. The protein-DNA complex was collected with protein G Excellose (Bioprogen, Korea), eluted, and reverse cross-linked. Following treatment with protease K (Sigma), the samples were extracted with phenol/chloroform and precipitated with ethanol. The recovered DNA was resuspended in distilled water and used for amplification by PCR, using the primers listed in supplemental Table S1. β-Actin was use as a negative control as described previously (10).
Knockdown of Genes by Small Interfering RNA (siRNA)
To knock down the endogenous IRF1 and Smad7, we used the Stealth RNAi knockdown system (Invitrogen). As a scramble siRNA, we used Stealth RNAi negative control duplexes (catalog no. 12935). The siRNAs were transfected into cells using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's protocol. After 48 h, cells were harvested, and expression levels of targets were detected with Western blotting. For knockdown of caspase 8, we used pLKO.1-puro lentiviral short hairpin RNA (shRNA) system according to the manufacturer's instructions (Sigma). The sequences for caspase 8 shRNA are listed in supplemental Table S2.
Electrophoretic Mobility Shift Assay (EMSA) and Supershift
For EMSA analysis, MCF7 cells were gently lysed in a Nonidet P-40-containing buffer. After removing cell debris, nuclear proteins were extracted with high salt buffer. The 5 μg of nuclear proteins were incubated with 32P-labeled double-stranded ISRE probe, containing the IRF1 recognition site, at room temperature for 30 min and electrophoresed onto 6% TBE gels. The gels were dried and developed by autoradiography. Competition assay, using a 50-fold molar excess of unlabeled probe, confirmed the specificity of the band. For supershift experiments, nuclear extracts were preincubated with 1 μg of IRF1 or Smad7 antibodies. For the in vitro binding assay, recombinant Smad7 or IRF1 was isolated as GST-tagged proteins after isopropyl 1-thio-β-d-galactopyranoside induction and incubated with radiolabeled ISRE probe. After running the gel, shifted bands were detected with autoradiography. The sequences of EMSA probes are listed in supplemental Table S3.
Xenograft Experiment
For testing the effect of Smad7 on tumor formation, we used 40 of the 6-week-old female BALB/c nu/nu mice (The Jackson Laboratory) for xenograft assay. Mice were injected subcutaneously with 1 × 106 MCF7 cells in Matrigel (BD Biosciences) on the mammary fat pad after inoculating 17β-estradiol pellets (1.7 mg/pellet) 2 days earlier. Isolation and purification of TRAIL were prepared as described previously (27). After reaching the volume of 100 mm3, mice were randomly divided into four groups, and TRAIL (150 μg/mouse/day, intraperitoneally) was treated for 10 days to test the suppressive effect. Tumor volumes were continuously measured with calipers and calculated using longitudinal (L) and transverse (W) tumor diameters with formula V = (L × W2)/2. After sacrificing the mice, tumor tissues were collected, fixed with 10% formalin, and embedded in paraffin. Remaining tissues were kept at −80 °C in a deep freezer for isolating protein and RNA. Mice were housed under pathogen-free conditions and maintained on a 12-h light/dark cycle, with food and water supplied ad libitum. Animal protocol was reviewed and approved by the Institutional Animal Care and Use Committee at Gachon University.
Statistical Analysis
Statistical significance between two groups was determined using the Student's t test. Error bars for all graphs represent the standard deviation. p values <0.05 were considered statistically significant.
RESULTS
Caspase 8 Is a Target Gene of Smad7
To identify the novel biological role of Smad7 in vivo, we performed cDNA microarray analysis using MCF7 control and Smad7-stable cells (supplemental Table S4). From the screening, we found that several genes, including Smad7, are differentially regulated by Smad7 expression. Among those genes, caspase 8 gene is the most up-regulated in Smad7-stable MCF7 cells. Expression of caspase 8 was confirmed by semi-quantitative RT-PCR and Western blotting (Fig. 1, A and B). The pro-caspase 8 bands appeared at around 55 and 52 kDa in Smad7-expressing cells as expected.
FIGURE 1.
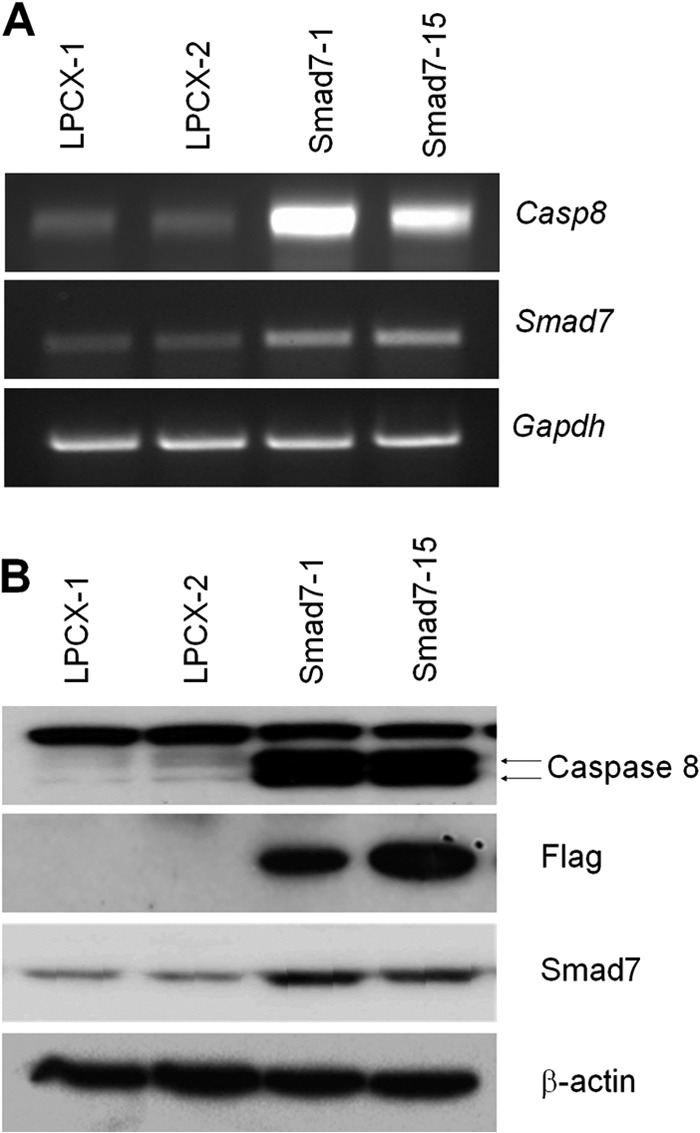
Caspase 8 is up-regulated in Smad7-expressing MCF7 cells. A, total RNA was isolated from control or FLAG-Smad7-expressing MCF7 cells, and semi-quantitative RT-PCR was performed using caspase 8 (Casp8)- or Smad7-specific primers. GAPDH was used as internal control. B, to confirm the microarray and RT-PCR results, protein lysates were prepared from MCF7 cells, and the expression of caspase 8 or FLAG-Smad7 was detected using specific antibodies. The loading amount was normalized with β-actin antibody.
To confirm whether Smad7 shows cell type specificity for caspase 8 expression, we checked other cell lines overexpressing Smad7. As shown in supplemental Fig. S1, expression of Smad7 induced the transcription of caspase 8 in human lung A549 and gastric SNU638 cancer cell lines. These data suggest that caspase 8 may be a general target of Smad7 independent of the cell context.
The down-regulation of caspase 8 has been observed in children's neuroblastoma and Ewing sarcoma cell lines, and expression of caspase 8 in these cells is suppressed through methylation of the promoter (28). So, we first checked the hypermethylation status of the caspase 8 promoter using methylation specific PCR. However, expression of Smad7 did not change the methylation status of the caspase 8 promoter (data not shown). There are some reports showing that demethylating agent or interferon-γ (IFN-γ) induced the expression of caspase 8 to enhance the apoptosis pathway (29). It has been known that IFN-γ up-regulated Smad7 expression to suppress the anti-inflammatory function of TGF-β signaling (30–32). No effect of 5-aza-2′-deoxycytidine (5-AdC) means that hypermethylation of promoter is not involved for caspase 8 regulation in MCF7 cells (data not shown). In contrast, treatment of IFN-γ induced the expression of caspase 8 in the MCF7 cell. Further experiments were performed to show the effect of Smad7 on caspase 8 expression in physiological conditions using siRNA. IFN-γ-mediated induction of caspase 8 was markedly suppressed by transfection of Smad7 siRNA with compared with scrambled siRNA-transfected cells (Fig. 2). These results suggest that transcriptional regulation may suppress the expression of caspase 8 in the MCF7 cell, and Smad7 may be an important mediator for IFN-γ-induced caspase 8 expression.
FIGURE 2.
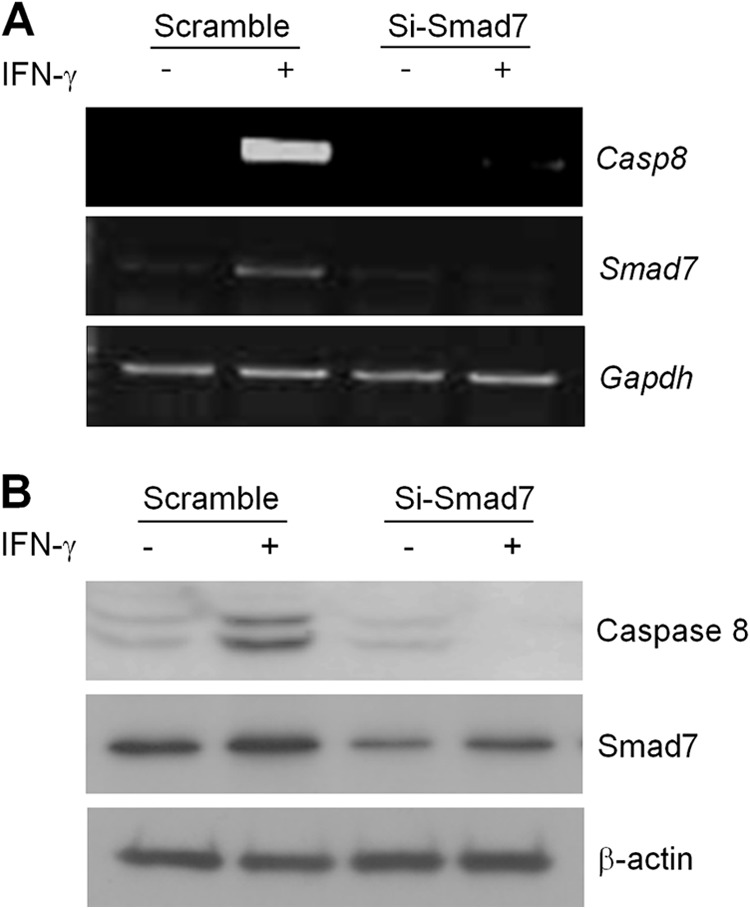
Involvement of Smad7 in IFN-γ-induced caspase 8 expression. A, total RNA was isolated in scrambled or Smad7 siRNA-transfected cells after treatment with 50 ng/ml IFN-γ for 48 h. Expression of caspase 8 (Casp8) or Smad7 was detected by semi-quantitative RT-PCR. The Gapdh gene was used as internal control. B, level of caspase 8 or Smad7 protein was confirmed with Western blotting using specific antibodies. β-Actin was used as normalization control.
Smad7 Positively Regulates the Transcription of Caspase 8 Gene
To understand the mechanism of up-regulation of caspase 8 in Smad7 cells, we generated a caspase 8 promoter-luciferase construct containing the −1615 to +16 region as reported previously and performed luciferase assays in MCF7 cells (33). The luciferase activity was markedly increased in Smad7 stable cells as compared with control cells (Fig. 3A). Additionally, transient Smad7 expression enhanced caspase 8 luciferase activity in a dose-dependent manner (Fig. 3B). These results indicate that Smad7 induces the expression of caspase 8 at the transcriptional level. To identify the regulatory region of the promoter, deletion reporter constructs were generated to perform the luciferase activity (Fig. 3C). Even the shortest construct showed a response to Smad7 in a dose-dependent manner (Fig. 3D). Interestingly, this region (−150 to +16) does not contain the Smad-binding element site but the ISRE site.
FIGURE 3.

Smad7 regulates transcription of caspase 8. A, to understand the mechanism of induced expression of caspase 8 in MCF7-Smad7 cells, we performed the luciferase assays using caspase 8 promoter-luciferase containing the promoter region (−1615/+16). Each cell was transfected with luciferase DNA using Lipofectamine, and relative luciferase activity was measured using Victor2 luminometer. The relative activity was normalized using β-galactosidase activity. B, caspase 8 luciferase construct was cotransfected with increasing amounts of Smad7 expression construct in MCF7 cells. The luciferase activity was measured with a luminometer after 36 h, and the expression level of Smad7 was detected with Western blotting. C, to identify the regulatory region of caspase 8 promoter, the deletion constructs were generated as shown in D. With deletion constructs of the caspase 8 promoter, the caspase 8 reporter assay was performed to minimize the potential regulatory region. After transfection of reporter constructs with increasing amounts of Smad7, luciferase activity was measured after 36 h and normalized with β-galactosidase activity. (n = 3 independent experiments; error bars represent S.D.)
Smad7 Cooperates with IRF1 to Activate the Caspase 8 Promoter
To validate the mechanism of Smad7 in transcriptional regulation of caspase 8, we first checked the effect of Smad7 on ISRE reporter activity. As shown in Fig. 4A, ISRE reporter activity was increased by Smad7 in a dose-dependent manner. Furthermore, Smad7 synergistically activated the ISRE reporter activity with IRF1 (Fig. 4B). Also, a point mutation on the ISRE site resulted in an obvious decrease of Smad7 and IRF1-induced transcriptional activation of caspase 8 promoter, indicating the requirement of IRF1 or Smad7 binding to the corresponding sites for caspase 8 transcription (Fig. 4B). From the EMSA experiment, ISRE probes were shifted in Smad7-expressing cells (Fig. 4C). The shifted bands disappeared in the presence of cold wild-type ISRE probe but not the mutant one, indicating the specific binding of nuclear proteins with ISRE probes (supplemental Fig. S2A). By adding Smad7 or IRF1 antibodies, ISRE bands were further shifted (supplemental Fig. S2B).
FIGURE 4.

Smad7 and IRF1 cooperate to induce caspase 8 expression. A, to test the involvement of IRF1-binding site, ISRE reporter assay was performed in MCF7 cells. After transfection with ISRE reporter construct with Smad7, cells were lysed to measure the luciferase activity. The values were normalized with β-galactosidase activity. B, wild-type or mutant ISRE reporter activity was measured after transfection with Smad7 alone or IRF1 together. The luciferase activity was normalized with β-galactosidase activity. C, after extraction of nuclear fractions from MCF7 cells, lysates were incubated with radiolabeled ISRE probe. Shifted bands were detected after running the gel and developing the autoradiography. The nuclear extracts were normalized with EMSA of the NF1 probe. D, to test the interaction of Smad7 with IRF1, expression vectors for Smad7 and IRF1 were transfected into HEK293 cells. Protein lysates were prepared and incubated with anti-FLAG antibody to precipitate Smad7 and interacting proteins. Immunoprecipitated (IP) IRF1 was detected with anti-Myc antibody. IB, immunoblot. E, to confirm the involvement of Smad7 and IRF1 in the expression of caspase 8, Smad7 or IRF1 was knocked down with siRNA. Each protein was detected with specific antibody to check the expression level of protein. F, to test the direct interaction of Smad7 on ISRE site, recombinant Smad7 or IRF1 proteins were isolated using glutathione-associated bead. Proteins were incubated with radiolabeled ISRE probe, and shifted bands were detected with autoradiography. G, to validate the interaction of Smad7 and IRF1 on caspase 8 promoter, ChIP assay was performed using antibodies against Smad7 or IRF1. Levels of protein recruitment to ISRE sites of the caspase 8 gene were measured by PCR and compared with input sample. As a negative control, β-actin primer was used.
Based on a previous report, we tested the effect of Smad7 on the expression of other IRF1 target genes (34). As expected, Smad7 positively regulated the IRF1-mediated transcription of DCC, BARD1, and BRIP1 (supplemental Fig. S3). It has been also known that the C-terminal region of Smad3 directly interacts with IRF7 to regulate the transcriptional activation of β-interferon (35). Interestingly, Smad7 only interacted efficiently with IRF1, whereas other Smad proteins showed no interaction or very weak interaction compare with Smad7 (Fig. 4D and supplemental Fig. S4A). To determine which domain of Smad7 interacts with IRF1, we performed the immunoprecipitation experiment using GST-fused Smad7 deletion constructs. Interestingly, the N-terminal MH1-linker region of Smad7 (Smad7 N; amino acids 1–258) were able to interact with IRF1 but not the MH2 region of Smad7 (Smad7 C; amino acids 259–426, supplemental Fig. S4B). Also, knockdown of IRF1 suppressed the expression of caspase 8 consistently with the depletion of Smad7 (Fig. 4E). When we checked the direct interaction of proteins with the ISRE sequence, ISRE probes could interact with only the recombinant IRF1 protein but not the recombinant Smad7 protein (Fig. 4F). Interestingly, the binding activity of IRF1 on the ISRE sequence was markedly increased in the presence of Smad7. To understand the molecular mechanism for enhancement of transcriptional activity of IRF1, we tested the effect of Smad7 on transcriptional activity of IRF1 using Gal4 transactivation assay. Gal4 DNA binding domain-fused IRF1 enhanced the transcriptional activity of the luciferase assay, but overexpression of Smad7 did not affect the IRF1-mediated transactivation (supplemental Fig. S5). Based on ChIP assay, Smad7 and IRF1 can specifically interact with the ISRE site of the caspase 8 promoter (Fig. 4G). Furthermore, Smad7 did not affect the expression level of IRF1 in various Smad7-expressing cell lines (data not shown). These results suggest that Smad7 does not directly interact with the ISRE sequence but enhances the binding activity of IRF1 through protein-protein interaction with IRF1 protein.
Smad7 Sensitizes TRAIL-induced Cell Death through Caspase 8-dependent Pathway
To understand the biological meaning of caspase 8 regulation by Smad7, we tested whether Smad7 could overcome the resistance to TRAIL-mediated apoptosis. When MCF7 cells were treated with 100 ng/ml TRAIL, there was no effect on cell growth and morphology (Fig. 5A). However, TRAIL markedly induced cell death in Smad7-expressing cells after 24 h of treatment. When cell death was measured by MTT assay, it showed that cell viability decreased with increasing concentrations of TRAIL in Smad7-stable cells (Fig. 5B). DNA laddering, an indicator of inter-nucleosomal DNA cleavage, appeared in Smad7-stable cells by treatment with TRAIL (Fig. 5C). After treatment with TRAIL, PARP as another marker for apoptosis was degraded, and the cleaved form was also detected (Fig. 5D).
FIGURE 5.

Smad7 sensitizes MCF7 cells into TRAIL-mediated apoptosis. A, representative images of mock or TRAIL-treated cells. MCF7 control or Smad7-overexpressing cells were treated with 100 ng/ml TRAIL for 24 h. B, cell viability was measured using the MTT assay. After treatment with TRAIL at indicated doses for 24 h, cells were incubated with MTT for 4 h and solubilized overnight. The intensity of color was measured with an ELISA reader and displayed as percentage of nontreated cells. C, MCF7 cells were treated with 100 ng/ml TRAIL for 24 h and harvested as described under “Experimental Procedures.” The isolated genomic DNA was separated by agarose gel electrophoresis, and the fragmented DNA was shown by staining with EtBr. D, PARP and cleaved PARP were detected by Western blotting after treatment with TRAIL. The expression of Smad7 was confirmed with anti-FLAG antibody.
To check the involvement of caspase 8 in TRAIL-mediated apoptosis, degradation of pro-caspase 8 and PARP was detected. As shown in Fig. 6, A and B, pro-caspase 8 and PARP band disappeared by treatment with TRAIL in Smad7 stable cells. The degradation of these apoptotic markers was blocked by pretreatment with the pan-caspase inhibitor Z-VAD-fmk or caspase 8-specific inhibitor Z-IETD-fmk, indicating that caspase cascades are critical for TRAIL-mediated apoptosis (Fig. 6B). Also, caspase inhibitors blocked the TRAIL-mediated DNA fragmentation in Smad7 cells (Fig. 6C). To demonstrate the direct effect of caspase 8 expression in TRAIL-induced apoptosis, the lentiviral shRNA against caspase 8 was used to block cell death. Correlated with reduced expression of caspase 8, apoptosis of MCF7-Smad7 cell was markedly inhibited even by treatment with TRAIL (Fig. 6D). To exclude the possibility of positive feedback mechanism of TRAIL-induced Smad7 expression, we performed RT-PCR for Smad7 mRNA after treatment of TRAIL in MCF7 cells. TRAIL did not induce the transcription of Smad7 even after long treatment (data not shown).
FIGURE 6.

Caspase 8 is required for TRAIL-mediated apoptosis in MCF7 cells. A, MCF7 cells were treated with 100 ng/ml TRAIL for 24 h, and protein lysates were prepared as described under “Experimental Procedures.” A total of 30 μg of protein was loaded, and Western blotting was performed using anti-caspase 8 and PARP antibody. B, to block the activation of caspase cascades, MCF7-Smad7 cells were pretreated with 10 μm of the pan-caspase inhibitor (Z-VAD-fmk) or the caspase 8-specific inhibitor (Z-IETD-fmk) for 2 h. Cells were then treated with TRAIL for 24 h, and activation of caspase was measured by analyzing degradation of PARP and caspase 8. C, after treatment with the caspase inhibitor and TRAIL, DNA fragmentation was measured by preparing the genomic DNA. The DNA was run onto 2% agarose gel and visualized by staining with EtBr. D, after knockdown of caspase 8 by lentiviral shRNA, MCF7-Smad7 cells were treated with TRAIL for 24 h. PARP and caspase 8 were detected with specific endogenous antibody, and β-actin was used as normalization control.
Previous reports showed that re-expression of caspase 8 by gene transfer can sensitize the tumor cell to death receptor- or anti-cancer drug-mediated cell death (36, 37). To generate caspase 8-stable cells, MCF7 cells were infected with retrovirus containing a caspase 8 expression vector and selected with 600 ng/ml puromycin for 2 weeks. As shown in supplemental Fig. S6A, caspase 8 expression was detected in some stable clones at ∼55 kDa. This band correlates with the upper band of caspase 8 in Smad7 stable cells. Using two stable cell clones (clones 6 and 9), sensitivity of MCF7 cells was checked by treatment with TRAIL for 24 h. Compared with control, caspase 8-stable cells were more sensitive to treatment with TRAIL (supplemental Fig. S6B). As expected, caspase 8-mediated apoptosis was blocked by incubating with caspase inhibitors (supplemental Fig. S6C). These data indicate that up-regulation of caspase 8 by Smad7 is critical for TRAIL-mediated apoptosis in MCF7 cells.
Smad7 Does Not Affect the Expression of TRAIL Receptors or Cell Survival Pathway
To test other possible mechanisms of Smad7 in TRAIL-induced apoptosis, the expression level of TRAIL receptors was checked using mRNA and protein. First, the mRNA level of two death receptors and two decoy receptors was measured using semi-quantitative RT-PCR (supplemental Fig. S7A). Death receptors were highly expressed even in control MCF7 cells and overexpression of Smad7 did not affect these expressions. It has been shown that the expression of TRAIL-R4 decoy receptor in MCF7 cell is correlated with TRAIL resistance (38). However, expression of Smad7 did not much affect the expression level of decoy receptors. To examine the surface expression of death receptors, phycoerythrin-tagged TRAIL-R1 and TRAIL-R2 antibodies were incubated with MCF7 cells and analyzed with flow cytometry. TRAIL-R1 and TRAIL-R2 expression was detected in control cells, and expression levels were not changed by expression of Smad7 (supplemental Fig. S7B).
The inhibition of NF-κB activity sensitizes cancer cells to TRAIL-mediated apoptosis (39). Although TNF-α efficiently induced the degradation of IκBα, TRAIL did not induce the degradation of IκBα in MCF7 cells and Smad7 did not affect the level of IκBα (supplemental Fig. S7C). This result was confirmed by analyzing NF-κB transcriptional activity using a, NF-κB-responsive luciferase reporter (supplemental Fig. S7D); TNF-α markedly enhanced the NF-κB luciferase activity in control cells, and expression of Smad7 blocked the luciferase activity as consistent with our previous result (3). However, TRAIL did not enhance the NF-κB luciferase activity, and Smad7 did not modulate this TRAIL activity. Based on these results, we concluded that NF-κB signaling, which can protect against TNF-α-mediated apoptosis, is not involved in resistance to TRAIL-mediated cell death in MCF7 cells.
Smad7 Suppresses Tumor Growth in Xenograft Model
To confirm the restoration of tumor suppressive activity in vivo by Smad7, we performed xenograft assay using nude mice. By inoculating the MCF7 cells in the mammary fat pad of mice, cells formed a tumor mass after 1 week. Through treatment with the TRAIL for 10 days, Smad7-expressing tumor was regressed in size compared with PBS-treated groups (Fig. 7A). In contrast, control tumors did not show any defects in growth by treatment with TRAIL. Apoptotic cell death was markedly increased in TRAIL-treated Smad7 tumors based on hematoxylin and eosin (H&E) and TUNEL staining (Fig. 7, B and C). Expression patterns of apoptotic markers, such as caspase 8, PARP, and caspase 3, also correlated with other data (Fig. 7D).
FIGURE 7.
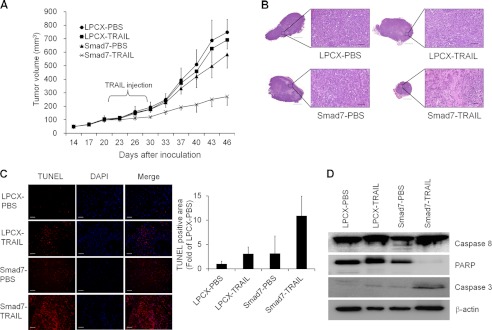
Smad7 suppresses the in vivo tumor formation. A, to check the effect of Smad7 on tumor formation, we performed xenograft experiment with 10 nude mice per group. After inoculating control and Smad7 cells onto mammary fat pads, TRAIL was loaded by intraperitoneal injection for 10 days. Tumor size was measured every 3 or 4 days, and volume was calculated as described under “Experimental Procedures.” B, after sacrificing the mice, tumor tissues were fixed and checked with H&E staining. C, to measure the induction of apoptosis in TRAIL-treated tumors, TUNEL assay was performed. Nuclei were stained with DAPI. Scale bar indicates 50 μm. D, to check expression of apoptotic markers, tumor tissues were lysed and detected with Western blotting. Caspase 8, PARP, and activated caspase 3 were detected with specific antibodies, and β-actin was used as normalization control.
DISCUSSION
In this report, we provide the evidence for a novel role of Smad7 as a transcriptional activator through recruiting IRF1 transcription factor on the ISRE site of caspase 8 promoter. Synergistic activation of caspase 8 by Smad7 and IRF1 results in restoration of the TRAIL-mediated apoptotic pathway (supplemental Fig. S8). Loss or down-regulation of caspase 8 is commonly detected in many neuroblastomas, an aggressive pediatric solid tumor, and Ewing sarcoma, an aggressive sarcoma that affects children and young adults (40, 41). Hypermethylation of caspase 8 promoter is proposed as a major suppressive mechanism in neuroblastomas. Indeed, treatment with demethylation agent, 5-AdC, activates the caspase 8 promoter in neuroblastoma cells that do not express endogenous caspase 8. However, the methylation status of caspase 8 promoter was not different between control and Smad7 cells, and treatment of 5-AdC did not induce the expression of caspase 8 in MCF7 control cells (data not shown). This result suggests that Smad7 does not modulate the expression of caspase 8 via removal of hypermethylation from the promoter.
Histone deacetylase inhibitors, such as N-butyric acid and trichostatin A, have been shown to increase caspase 8 expression and induce apoptosis in human malignant glioma cells (42). This result suggested that acetylation of histones is important for expression of caspase 8. A previous report showed that inhibitory Smads, including Smad6 and Smad7, interact with histone deacetylases, deacetylated the core histones, and repressed the target gene expression (43). In another way Smad7 can directly interact with the caspase 8 promoter and act as a transcriptional coactivator. In another study, Smad7 interacts with a specific carcinoembryonic antigen promoter region as confirmed by ChIP assay in SNU638 human gastric cancer cells. Smad7 may also recruit other transcriptional cofactors to caspase 8 promoter.5 Another inhibitory Smad protein, Smad6, recruits the transcription corepressor C-terminal binding protein to repress bone morphogenetic protein-induced Id1 transcription (44). Smad6 also interacts with homeobox c-8 as a transcriptional corepressor to inhibit Smad1-induced transcriptional activation (45). Depending on previous reports, other Smad proteins have their own specific target genes, which are identified through sequencing or microarray after ChIP experiments (46–48). To explain the transcriptional regulatory activity of Smad7, all interacting gene promoters should be identified with ChIP sequencing or ChIP-on-chip experiments. Through genome-wide analysis, the regulatory circuit between Smad7 and target genes can be understood, and the novel function of Smad7 as transcriptional regulator can be confirmed.
It has been shown that IFN-γ induced the expression of caspase 8 through IRF1 in MCF7 cells (49). In this study, we found that Smad7 cooperates with IRF1 to induce caspase 8 and functions as a transcriptional activator (Fig. 4). Without Smad7, caspase 8 was not expressed in MCF7 cells even in the presence of IRF1 (Fig. 4E). Also, Smad7 and IRF1 interacted together and were recruited at a similar region of the caspase 8 promoter (Fig. 4, D and G, and supplemental Fig. S4). It was very interesting that the MH1 region of Smad7 interacted with IRF1, unlike Smad3 that interacted with IRF7 through the MH2 domain (35). It has been known that the C-terminal region of IRF3 has similar sequences with the MH2 domain of Smad proteins and functions as a docking site for other binding partners (50, 51). However, other groups also reported that the MH1 domain of Smad proteins involves association with other proteins to regulate its functions (52, 53). As expected, Smad7 alone cannot bind to the caspase 8 promoter without the IRF1 protein (Fig. 4F). Increased binding activity of IRF1 with Smad7 means that Smad7 may induce the conformational change of IRF1 to be recruited onto the ISRE site. Smad7 may also function as a scaffold protein to recruit other transcriptional cofactors to enhance the activity of IRF1, the way Smad3 recruits the CBP/p300 to promote gene transcription (54, 55). Further studies are needed to identify the detailed mechanisms for the role of Smad7 as the transcriptional coactivator.
In this study, pro-apoptotic activity of Smad7 was confirmed using in vitro cell culture system and in vivo mouse models (Figs. 5 and 6). Also, Smad7 can induce apoptosis by inhibiting NF-κB activity or blocking TGF-β-induced metastatic gene expression (4–7). Interestingly, Smad7 expression in T cells suppressed colitis-associated tumor formation through increased expression of IFN-γ and recruitment of CD8+ and natural killer T cells (56). Consistent with previous reports, this study strongly suggests that Smad7 can be used as a chemotherapeutic agent for treatment of some human cancers.
Resistance of tumors to anti-cancer drugs is an obstacle for treatment of tumors by conventional methods. If drugs are used at high doses to show anticancer activity, this leads to serious cytotoxic activity to normal cells. To overcome the undesired side effect of drugs, combinational therapy has been tested to reduce the high doses of anti-tumor drugs (57, 58). Even subtoxic doses of chemotherapeutic drugs followed by treatment with TRAIL efficiently blocked tumor cell growth in vitro and tumor formation in nude mice in vivo. Because overexpression of Smad7 can sensitize breast cancer cells to apoptosis induced by TRAIL in this study, it will be interesting to find chemicals or natural products that increase the expression of endogenous Smad7. For example, halofuginone, a widely used alkaloid coccidiostat, can strongly induce the expression of Smad7 and block TGF-β-mediated gene regulation and fibrosis after radiation therapy (59). Interestingly, halofuginone has recently been shown to inhibit tumor growth and metastasis through the blocking of angiogenesis and matrix metalloproteinase in various tumors (60, 61). It is well known that IFN-γ induces Smad7 through the Jak1/STAT pathway to block TGF-β signaling (30–32). Because cotreatment of IFN-γ and TRAIL synergistically induced the apoptosis in neuroblastoma cells and Ewing sarcoma, it will be interesting to examine the involvement of Smad7 in this effect based on our study (28, 29, 62). Recently, Vuori and co-workers (63) developed a high throughput screening system to identify potential TRAIL-sensitizing agents that act solely in a caspase 8-dependent manner. From the screening of a library of pharmacologically active compounds, they identified several compounds that sensitize the cells specifically through caspase 8. Confirming the role of Smad7, this process has the potential to overcome the resistance of tumor cells against TRAIL treatment.
Acknowledgments
We are grateful to Jinny Choe for the critical reading of the manuscript and Dr. Ki-Baik Hahm for excellent assistance with analyzing the tissue histology. We also thank Dr. Miyazono Kohei for the generous gift for pcDNA3–6×Myc-tagged Smad constructs.

This article contains supplemental Materials and Methods, Figs. 1–8, and Tables S1–S4.
S.-J. Kim and S. Hong, unpublished data.
- TRAIL
- tumor necrosis factor-related apoptosis-inducing ligand
- IRF1
- interferon regulatory factor 1
- ISRE
- interferon stimulated response element
- 5-AdC
- 5-aza-2′-deoxycytidine
- PARP
- poly(ADP-ribose) polymerase
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- Z
- benzyloxycarbonyl
- fmk
- fluoromethyl ketone
- MH
- Mad homology.
REFERENCES
- 1. Nakao A., Afrakhte M., Morén A., Nakayama T., Christian J. L., Heuchel R., Itoh S., Kawabata M., Heldin N. E., Heldin C. H., ten Dijke P. (1997) Identification of Smad7, a TGFβ-inducible antagonist of TGF-β signalling. Nature 389, 631–635 [DOI] [PubMed] [Google Scholar]
- 2. Schmierer B., Hill C. S. (2007) TGFβ-SMAD signal transduction: molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 8, 970–982 [DOI] [PubMed] [Google Scholar]
- 3. Hong S., Lee C., Kim S. J. (2007) Smad7 sensitizes tumor necrosis factor-induced apoptosis through the inhibition of antiapoptotic gene expression by suppressing activation of the nuclear factor-κB pathway. Cancer Res. 67, 9577–9583 [DOI] [PubMed] [Google Scholar]
- 4. Schiffer M., Bitzer M., Roberts I. S., Kopp J. B., ten Dijke P., Mundel P., Böttinger E. P. (2001) Apoptosis in podocytes induced by TGF-β and Smad7. J. Clin. Invest. 108, 807–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lallemand F., Mazars A., Prunier C., Bertrand F., Kornprost M., Gallea S., Roman-Roman S., Cherqui G., Atfi A. (2001) Smad7 inhibits the survival nuclear factor κB and potentiates apoptosis in epithelial cells. Oncogene 20, 879–884 [DOI] [PubMed] [Google Scholar]
- 6. Azuma H., Ehata S., Miyazaki H., Watabe T., Maruyama O., Imamura T., Sakamoto T., Kiyama S., Kiyama Y., Ubai T., Inamoto T., Takahara S., Itoh Y., Otsuki Y., Katsuoka Y., Miyazono K., Horie S. (2005) Effect of Smad7 expression on metastasis of mouse mammary carcinoma JygMC(A) cells. J. Natl. Cancer Inst. 97, 1734–1746 [DOI] [PubMed] [Google Scholar]
- 7. Javelaud D., Mohammad K. S., McKenna C. R., Fournier P., Luciani F., Niewolna M., André J., Delmas V., Larue L., Guise T. A., Mauviel A. (2007) Stable overexpression of Smad7 in human melanoma cells impairs bone metastasis. Cancer Res. 67, 2317–2324 [DOI] [PubMed] [Google Scholar]
- 8. Choi K. C., Lee Y. S., Lim S., Choi H. K., Lee C. H., Lee E. K., Hong S., Kim I. H., Kim S. J., Park S. H. (2006) Smad6 negatively regulates interleukin 1-receptor-Toll-like receptor signaling through direct interaction with the adaptor Pellino-1. Nat. Immunol. 7, 1057–1065 [DOI] [PubMed] [Google Scholar]
- 9. Hong S., Lim S., Li A. G., Lee C., Lee Y. S., Lee E. K., Park S. H., Wang X. J., Kim S. J. (2007) Smad7 binds to the adaptors TAB2 and TAB3 to block recruitment of the kinase TAK1 to the adaptor TRAF2. Nat. Immunol. 8, 504–513 [DOI] [PubMed] [Google Scholar]
- 10. Zhang S., Fei T., Zhang L., Zhang R., Chen F., Ning Y., Han Y., Feng X. H., Meng A., Chen Y. G. (2007) Smad7 antagonizes transforming growth factor β signaling in the nucleus by interfering with functional Smad-DNA complex formation. Mol. Cell. Biol. 27, 4488–4499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kume S., Haneda M., Kanasaki K., Sugimoto T., Araki S., Isshiki K., Isono M., Uzu T., Guarente L., Kashiwagi A., Koya D. (2007) SIRT1 inhibits transforming growth factor β-induced apoptosis in glomerular mesangial cells via Smad7 deacetylation. J. Biol. Chem. 282, 151–158 [DOI] [PubMed] [Google Scholar]
- 12. Simonsson M., Heldin C. H., Ericsson J., Grönroos E. (2005) The balance between acetylation and deacetylation controls Smad7 stability. J. Biol. Chem. 280, 21797–21803 [DOI] [PubMed] [Google Scholar]
- 13. Wiley S. R., Schooley K., Smolak P. J., Din W. S., Huang C. P., Nicholl J. K., Sutherland G. R., Smith T. D., Rauch C., Smith C. A. (1995) Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 3, 673–682 [DOI] [PubMed] [Google Scholar]
- 14. Pan G., Ni J., Wei Y. F., Yu G., Gentz R., Dixit V. M. (1997) An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science 277, 815–818 [DOI] [PubMed] [Google Scholar]
- 15. Ashkenazi A., Dixit V. M. (1998) Death receptors. Signaling and modulation. Science 281, 1305–1308 [DOI] [PubMed] [Google Scholar]
- 16. Walczak H., Miller R. E., Ariail K., Gliniak B., Griffith T. S., Kubin M., Chin W., Jones J., Woodward A., Le T., Smith C., Smolak P., Goodwin R. G., Rauch C. T., Schuh J. C., Lynch D. H. (1999) Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 5, 157–163 [DOI] [PubMed] [Google Scholar]
- 17. Menke C., Bin L., Thorburn J., Behbakht K., Ford H. L., Thorburn A. (2011) Distinct TRAIL resistance mechanisms can be overcome by proteasome inhibition but not generally by synergizing agents. Cancer Res. 71, 1883–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xu J., Zhou J. Y., Wei W. Z., Wu G. S. (2010) Activation of the Akt survival pathway contributes to TRAIL resistance in cancer cells. PLoS ONE 5, e10226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lalaoui N., Morlé A., Mérino D., Jacquemin G., Iessi E., Morizot A., Shirley S., Robert B., Solary E., Garrido C., Micheau O. (2011) TRAIL-R4 promotes tumor growth and resistance to apoptosis in cervical carcinoma HeLa cells through AKT. PLoS ONE 6, e19679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Liu X., Yue P., Khuri F. R., Sun S. Y. (2005) Decoy receptor 2 (DcR2) is a p53 target gene and regulates chemosensitivity. Cancer Res. 65, 9169–9175 [DOI] [PubMed] [Google Scholar]
- 21. Irmler M., Thome M., Hahne M., Schneider P., Hofmann K., Steiner V., Bodmer J. L., Schröter M., Burns K., Mattmann C., Rimoldi D., French L. E., Tschopp J. (1997) Inhibition of death receptor signals by cellular FLIP. Nature 388, 190–195 [DOI] [PubMed] [Google Scholar]
- 22. Geiger K., Hagenbuchner J., Rupp M., Fiegl H., Sergi C., Meister B., Kiechl-Kohlendorfer U., Müller T., Ausserlechner M. J., Obexer P. (2012) FOXO3/FKHRL1 is activated by 5-aza-2-deoxycytidine and induces silenced caspase-8 in neuroblastoma. Mol. Biol. Cell 23, 2226–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Teitz T., Wei T., Valentine M. B., Vanin E. F., Grenet J., Valentine V. A., Behm F. G., Look A. T., Lahti J. M., Kidd V. J. (2000) Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat. Med. 6, 529–535 [DOI] [PubMed] [Google Scholar]
- 24. Wu Y., Alvarez M., Slamon D. J., Koeffler P., Vadgama J. V. (2010) Caspase 8 and maspin are down-regulated in breast cancer cells due to CpG site promoter methylation. BMC Cancer 10, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim B. C., Lee H. J., Park S. H., Lee S. R., Karpova T. S., McNally J. G., Felici A., Lee D. K., Kim S. J. (2004) Jab1/CSN5, a component of the COP9 signalosome, regulates transforming growth factor β signaling by binding to Smad7 and promoting its degradation. Mol. Cell. Biol. 24, 2251–2262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lee J. S., Chu I. S., Mikaelyan A., Calvisi D. F., Heo J., Reddy J. K., Thorgeirsson S. S. (2004) Application of comparative functional genomics to identify best fit mouse models to study human cancer. Nat. Genet. 36, 1306–1311 [DOI] [PubMed] [Google Scholar]
- 27. Chae S. Y., Kim T. H., Park K., Jin C. H., Son S., Lee S., Youn Y. S., Kim K., Jo D. G., Kwon I. C., Chen X., Lee K. C. (2010) Improved antitumor activity and tumor targeting of NH2-terminal-specific PEGylated tumor necrosis factor-related apoptosis-inducing ligand. Mol. Cancer Ther. 9, 1719–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lissat A., Vraetz T., Tsokos M., Klein R., Braun M., Koutelia N., Fisch P., Romero M. E., Long L., Noellke P., Mackall C. L., Niemeyer C. M., Kontny U. (2007) Interferon-γ sensitizes resistant Ewing's sarcoma cells to tumor necrosis factor apoptosis-inducing ligand-induced apoptosis by up-regulation of caspase-8 without altering chemosensitivity. Am. J. Pathol. 170, 1917–1930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fulda S., Debatin K. M. (2006) 5-Aza-2′-deoxycytidine and IFN-γ cooperate to sensitize for TRAIL-induced apoptosis by up-regulating caspase-8. Oncogene 25, 5125–5133 [DOI] [PubMed] [Google Scholar]
- 30. Monteleone G., Del Vecchio Blanco G., Palmieri G., Vavassori P., Monteleone I., Colantoni A., Battista S., Spagnoli L. G., Romano M., Borrelli M., MacDonald T. T., Pallone F. (2004) Induction and regulation of Smad7 in the gastric mucosa of patients with Helicobacter pylori infection. Gastroenterology 126, 674–682 [DOI] [PubMed] [Google Scholar]
- 31. Ulloa L., Doody J., Massagué J. (1999) Inhibition of transforming growth factor-β/SMAD signalling by the interferon-γ/STAT pathway. Nature 397, 710–713 [DOI] [PubMed] [Google Scholar]
- 32. Weng H., Mertens P. R., Gressner A. M., Dooley S. (2007) IFN-γ abrogates profibrogenic TGF-β signaling in liver by targeting expression of inhibitory and receptor Smads. J. Hepatol. 46, 295–303 [DOI] [PubMed] [Google Scholar]
- 33. De Ambrosis A., Casciano I., Croce M., Pagnan G., Radic L., Banelli B., Di Vinci A., Allemanni G., Tonini G. P., Ponzoni M., Romani M., Ferrini S. (2007) An interferon-sensitive response element is involved in constitutive caspase-8 gene expression in neuroblastoma cells. Int. J. Cancer 120, 39–47 [DOI] [PubMed] [Google Scholar]
- 34. Frontini M., Vijayakumar M., Garvin A., Clarke N. (2009) A ChIP-chip approach reveals a novel role for transcription factor IRF1 in the DNA damage response. Nucleic Acids Res. 37, 1073–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Qing J., Liu C., Choy L., Wu R. Y., Pagano J. S., Derynck R. (2004) Transforming growth factor β/Smad3 signaling regulates IRF-7 function and transcriptional activation of the β-interferon promoter. Mol. Cell. Biol. 24, 1411–1425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mühlethaler-Mottet A., Balmas K., Auderset K., Joseph J. M., Gross N. (2003) Restoration of TRAIL-induced apoptosis in a caspase-8-deficient neuroblastoma cell line by stable re-expression of caspase-8. Ann. N.Y. Acad. Sci. 1010, 195–199 [DOI] [PubMed] [Google Scholar]
- 37. Fulda S., Küfer M. U., Meyer E., van Valen F., Dockhorn-Dworniczak B., Debatin K. M. (2001) Sensitization for death receptor- or drug-induced apoptosis by re-expression of caspase-8 through demethylation or gene transfer. Oncogene 20, 5865–5877 [DOI] [PubMed] [Google Scholar]
- 38. Sanlioglu A. D., Dirice E., Aydin C., Erin N., Koksoy S., Sanlioglu S. (2005) Surface TRAIL decoy receptor-4 expression is correlated with TRAIL resistance in MCF7 breast cancer cells. BMC Cancer 5, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kim Y. S., Schwabe R. F., Qian T., Lemasters J. J., Brenner D. A. (2002) TRAIL-mediated apoptosis requires NF-κB inhibition and the mitochondrial permeability transition in human hepatoma cells. Hepatology 36, 1498–1508 [DOI] [PubMed] [Google Scholar]
- 40. Casciano I., Banelli B., Croce M., De Ambrosis A., di Vinci A., Gelvi I., Pagnan G., Brignole C., Allemanni G., Ferrini S., Ponzoni M., Romani M. (2004) Caspase-8 gene expression in neuroblastoma. Ann. N.Y. Acad. Sci. 1028, 157–167 [DOI] [PubMed] [Google Scholar]
- 41. Stupack D. G., Teitz T., Potter M. D., Mikolon D., Houghton P. J., Kidd V. J., Lahti J. M., Cheresh D. A. (2006) Potentiation of neuroblastoma metastasis by loss of caspase-8. Nature 439, 95–99 [DOI] [PubMed] [Google Scholar]
- 42. Komata T., Kanzawa T., Nashimoto T., Aoki H., Endo S., Kon T., Takahashi H., Kondo S., Tanaka R. (2005) Histone deacetylase inhibitors, N-butyric acid and trichostatin A, induce caspase-8- but not caspase-9-dependent apoptosis in human malignant glioma cells. Int. J. Oncol. 26, 1345–1352 [DOI] [PubMed] [Google Scholar]
- 43. Bai S., Cao X. (2002) A nuclear antagonistic mechanism of inhibitory Smads in transforming growth factor-β signaling. J. Biol. Chem. 277, 4176–4182 [DOI] [PubMed] [Google Scholar]
- 44. Lin X., Liang Y. Y., Sun B., Liang M., Shi Y., Brunicardi F. C., Shi Y., Feng X. H. (2003) Smad6 recruits transcription corepressor CtBP to repress bone morphogenetic protein-induced transcription. Mol. Cell. Biol. 23, 9081–9093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bai S., Shi X., Yang X., Cao X. (2000) Smad6 as a transcriptional corepressor. J. Biol. Chem. 275, 8267–8270 [DOI] [PubMed] [Google Scholar]
- 46. Kennedy B. A., Deatherage D. E., Gu F., Tang B., Chan M. W., Nephew K. P., Huang T. H., Jin V. X. (2011) ChIP-seq defined genome-wide map of TGFβ/SMAD4 targets. Implications with clinical outcome of ovarian cancer. PLoS ONE 6, e22606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Morikawa M., Koinuma D., Tsutsumi S., Vasilaki E., Kanki Y., Heldin C. H., Aburatani H., Miyazono K. (2011) ChIP-seq reveals cell type-specific binding patterns of BMP-specific Smads and a novel binding motif. Nucleic Acids Res. 39, 8712–8727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang Y., Handley D., Kaplan T., Yu H., Bais A. S., Richards T., Pandit K. V., Zeng Q., Benos P. V., Friedman N., Eickelberg O., Kaminski N. (2011) High throughput determination of TGFβ1/SMAD3 targets in A549 lung epithelial cells. PLoS ONE 6, e20319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ruiz-Ruiz C., Ruiz de Almodóvar C., Rodríguez A., Ortiz-Ferrón G., Redondo J. M., López-Rivas A. (2004) The up-regulation of human caspase-8 by interferon-γ in breast tumor cells requires the induction and action of the transcription factor interferon regulatory factor-1. J. Biol. Chem. 279, 19712–19720 [DOI] [PubMed] [Google Scholar]
- 50. Qin B. Y., Liu C., Lam S. S., Srinath H., Delston R., Correia J. J., Derynck R., Lin K. (2003) Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat. Struct. Biol. 10, 913–921 [DOI] [PubMed] [Google Scholar]
- 51. Takahasi K., Suzuki N. N., Horiuchi M., Mori M., Suhara W., Okabe Y., Fukuhara Y., Terasawa H., Akira S., Fujita T., Inagaki F. (2003) X-ray crystal structure of IRF-3 and its functional implications. Nat. Struct. Biol. 10, 922–927 [DOI] [PubMed] [Google Scholar]
- 52. Grocott T., Frost V., Maillard M., Johansen T., Wheeler G. N., Dawes L. J., Wormstone I. M., Chantry A. (2007) The MH1 domain of Smad3 interacts with Pax6 and represses autoregulation of the Pax6 P1 promoter. Nucleic Acids Res. 35, 890–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhou B., Chen L., Wu X., Wang J., Yin Y., Zhu G. (2008) MH1 domain of SMAD4 binds N-terminal residues of the homeodomain of Hoxc9. Biochim. Biophys. Acta 1784, 747–752 [DOI] [PubMed] [Google Scholar]
- 54. Feng X. H., Zhang Y., Wu R. Y., Derynck R. (1998) The tumor suppressor Smad4/DPC4 and transcriptional adaptor CBP/p300 are coactivators for Smad3 in TGF-β-induced transcriptional activation. Genes Dev. 12, 2153–2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Shen X., Hu P. P., Liberati N. T., Datto M. B., Frederick J. P., Wang X. F. (1998) TGF-β-induced phosphorylation of Smad3 regulates its interaction with coactivator p300/CREB-binding protein. Mol. Biol. Cell 9, 3309–3319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Rizzo A., Waldner M. J., Stolfi C., Sarra M., Fina D., Becker C., Neurath M. F., Macdonald T. T., Pallone F., Monteleone G., Fantini M. C. (2011) Smad7 expression in T cells prevents colitis-associated cancer. Cancer Res. 71, 7423–7432 [DOI] [PubMed] [Google Scholar]
- 57. Singh T. R., Shankar S., Chen X., Asim M., Srivastava R. K. (2003) Synergistic interactions of chemotherapeutic drugs and tumor necrosis factor-related apoptosis-inducing ligand/Apo-2 ligand on apoptosis and on regression of breast carcinoma in vivo. Cancer Res. 63, 5390–5400 [PubMed] [Google Scholar]
- 58. Hyer M. L., Croxton R., Krajewska M., Krajewski S., Kress C. L., Lu M., Suh N., Sporn M. B., Cryns V. L., Zapata J. M., Reed J. C. (2005) Synthetic triterpenoids cooperate with tumor necrosis factor-related apoptosis-inducing ligand to induce apoptosis of breast cancer cells. Cancer Res. 65, 4799–4808 [DOI] [PubMed] [Google Scholar]
- 59. Xavier S., Piek E., Fujii M., Javelaud D., Mauviel A., Flanders K. C., Samuni A. M., Felici A., Reiss M., Yarkoni S., Sowers A., Mitchell J. B., Roberts A. B., Russo A. (2004) Amelioration of radiation-induced fibrosis. Inhibition of transforming growth factor-β signaling by halofuginone. J. Biol. Chem. 279, 15167–15176 [DOI] [PubMed] [Google Scholar]
- 60. Spector I., Honig H., Kawada N., Nagler A., Genin O., Pines M. (2010) Inhibition of pancreatic stellate cell activation by halofuginone prevents pancreatic xenograft tumor development. Pancreas 39, 1008–1015 [DOI] [PubMed] [Google Scholar]
- 61. Cook J. A., Choudhuri R., Degraff W., Gamson J., Mitchell J. B. (2010) Halofuginone enhances the radiation sensitivity of human tumor cell lines. Cancer Lett. 289, 119–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Johnsen J. I., Pettersen I., Ponthan F., Sveinbjørnsson B., Flaegstad T., Kogner P. (2004) Synergistic induction of apoptosis in neuroblastoma cells using a combination of cytostatic drugs with interferon-γ and TRAIL. Int. J. Oncol. 25, 1849–1857 [PubMed] [Google Scholar]
- 63. Finlay D., Richardson R. D., Landberg L. K., Howes A. L., Vuori K. (2010) Novel HTS strategy identifies TRAIL-sensitizing compounds acting specifically through the caspase-8 apoptotic axis. PLoS ONE 5, e13375. [DOI] [PMC free article] [PubMed] [Google Scholar]