

Background: The p17 protein of avian reovirus (ARV) encoded by the S1 gene is a CRM1-independent nucleocytoplasmic shuttling protein that continuously shuttles between the nucleus and cytoplasm.
Results: ARV p17 protein triggers PTEN, AMPK, and PKR/eIF2α signaling pathways to induce autophagy.
Conclusion: ARV p17 protein functions as a positive regulator of autophagy.
Significance: This is the first evidence that ARV p17 protein triggers autophagic pathways to induce autophagy-enhancing virus replication.
Keywords: AMP-activated Kinase (AMPK), Autophagy, mTOR, Protein Kinase RNA (PKR), PTEN, Viral Replication, Avian Reovirus, p17 Protein
Abstract
Autophagy has been shown to facilitate replication or production of avian reovirus (ARV); nevertheless, how ARV induces autophagy remains largely unknown. Here, we demonstrate that the nonstructural protein p17 of ARV functions as an activator of autophagy. ARV-infected or p17-transfected cells present a fast and strong induction of autophagy, resulting in an increased level of autophagic proteins Beclin 1 and LC3-II. Although autophagy was suppressed by 3-methyladenine or shRNAs targeting autophagic proteins (Beclin 1, ATG7, and LC3) as well as by overexpression of Bcl-2, viral transcription, σC protein synthesis, and virus yield were all significantly reduced, suggesting a key role of autophagosomes in supporting ARV replication. Furthermore, we revealed for the first time that p17 positively regulates phosphatase and tensin deleted on chromosome 10 (PTEN), AMP-activated protein kinase (AMPK), and dsRNA dependent protein kinase RNA (PKR)/eIF2α signaling pathways, accompanied by down-regulation of Akt and mammalian target of rapamycin complex 1, thereby triggering autophagy. By using p53, PTEN, PKR, AMPK, and p17 short hairpin RNA (shRNA), activation of signaling pathways and LC3-II levels was significantly suppressed, suggesting that p17 triggers autophagy through activation of p53/PTEN, AMPK, and PKR signaling pathways. Furthermore, colocalization of LC3 with viral proteins (p17 and σC), p62 with LAMP2 and LC3 with Rab7 was observed under a fluorescence microscope. The expression level of p62 was increased at 18 h postinfection and then slightly decreased 24 h postinfection compared with mock infection and thapsigargin treatment. Furthermore, disruption of autophagosome-lysosome fusion by shRNAs targeting LAMP2 or Rab7a resulted in inhibition of viral protein synthesis and virus yield, suggesting that formation of autolysosome benefits virus replication. Taken together, our results suggest that ARV induces formation of autolysosome but does not induce complete autophagic flux.
Introduction
Avian reoviruses (ARVs)2 are ubiquitous among poultry flocks. ARV belongs to the Reoviridae family. They are nonenveloped viruses and consist of 10 double-stranded RNA genomes that are grouped into three groups, including large, medium, and small, depending on their electrophoretic mobility. These genomes encode at least eight structural and four nonstructural proteins. Genome segment S1 possesses three open reading frames (ORF) that are translated into p10, p17, and σC proteins, respectively. The p17 protein is a CRM1-independent nucleocytoplasmic shuttling protein that continuously shuttles between the nucleus and cytoplasm (1). It was proved that p17 plays an important role in regulating cell cycle and host cellular translation but does not induce cellular apoptosis (2–4). The σC protein is encoded by the third ORF of the S1 segment, which is responsible for host cell attachment (5) and induction of apoptosis (2, 6). Moreover, after initial binding of σC protein to receptor(s) on the cell membrane, ARV enters their host cells using a caveolin-1-mediated and dynamin-2-dependent endocytic pathway that requires activation of p38 and Src signaling pathways (7).
Autophagy is not only critical for cellular homeostasis but also participates in many physiological processes (8). Autophagy is a vacuolar degradative pathway terminating in the lysosomal compartment after forming a cytoplasmic vacuole or autophagosome that engulfs macromolecules and organelles. The autophagosome can fuse with the lysosome, and its contents and inner membrane are degraded by lysosomal enzymes (8). However, during stress, such as nutrient starvation, autophagy can also be induced as a way to create a limited amount of nutrients for the survival of cells. To date, many viruses have evolved to encode autophagy-promoting proteins to promote their survival in their host cells (9–14). Yet, it has also been proved that some viruses are able to subvert cellular autophagy to facilitate their own replication (15, 16). Thus, the role of autophagy in the host cells seems to be dependent on the viral strains.
A well-studied negative regulator of autophagy is mammalian target of rapamycin (mTOR). Previous findings suggested that autophagy is negatively regulated by mTOR through the inactivation of the Atg1·Atg13·Atg17 complex and suppression of the class III PI3K·Beclin 1/Atg6·UVRAG·Bif-1 kinase complex (17, 18). Formation of the autophagosome requires a conjugate of Atg5-Atg12, as well as a second conjugation system in which LC3-I and LC3-II are produced from the cleavage of LC3 (19–21). A compelling finding has recently shown that the Beclin 1 (Atg6), which is involved in the initial step of autophagosome formation, is directly targeted by signaling pathways and is discovered in a complex with Vps34, a class III PI3K (22). This complex may supply phosphatidylinositol 3-phosphates to preautophagosome membranes, thereby facilitating the localization of Atg proteins (18). Autophagy assays in mammalian cells frequently involve analysis of the LC3-II, an ortholog of yeast Atg8 (23). LC3-I is present as a cytosolic form, which during autophagy is conjugated to phosphatidylethanolamine. The resulting form, LC3-II, is associated with autophagosomal membranes and is essential for autophagosome formation (21). To date, various publications have also begun to address the relationship between virus infection and autophagy, a degradation pathway in which bulk amounts of cytoplasm are sequestered by double-membrane vesicles (15, 24).
Although ARV is capable of inducing autophagy (25), the protein of ARV that is responsible for induction of autophagy remains unknown. In this study, we revealed that p17 is an autophagy-promoting activator. Suppression of p53, PTEN, AMPK, and PKR signaling by shRNAs and overexpression of Bcl-2 in BHK-21 cells resulted in a significant reduction of ARV- or p17-induced formation of autophagosome as well as σC protein synthesis and virus yield, suggesting that ARV p17 triggers autophagy through activation of p53-PTEN-mTORC1, AMPK, and PKR/eIF2α signaling pathways. Furthermore, we also revealed that ARV triggers formation of autolysosome that is beneficial for its own replication. ARV, like other viruses, has evolved to encode an autophagy-promoting protein to alter the physiology of the host cells to enhance its own replication.
EXPERIMENTAL PROCEDURES
Cells
Both immortalized chicken embryo fibroblast (DF-1) cells and African green monkey kidney (Vero) cells used in this study were maintained in Dulbecco's modified Eagle's medium and minimum essential medium (MEM) supplemented with 10% fetal bovine serum (FBS), respectively. The BHK-21 cell line stably overexpressing Bcl-2 was described previously (26). DF-1 cells were seeded in 6-cm cell culture dishes with 1 × 106 cells 1 day before each experiment in a 37 °C incubator with 5% CO2. Vero cells were performed in the same conditions under a 37 °C incubator environment. Cultured cells were used in this study after refreshing the MEM containing 2% FBS overnight. The S1133 strain of ARV was used in this study.
Reagents and Antibodies
Rapamycin was purchased from Merck. Thapsigargin (TG) was purchased from Sigma. Lysosome tracker (LysoTracker® Red DND-99) was from Invitrogen. 3-Methyladenine (3-MA) was purchased from Sigma. Monoclonal antibodies against σC, σA, and p17 proteins of ARV are our laboratory stock (27, 28). Rabbit anti-p-ULK1 Ser-757, rabbit anti-ULK1, rabbit anti-Beclin 1, rabbit anti-p-PTEN 380/Thr-382/Thr-383, rabbit anti-PTEN, mouse anti-p-p38 Thr-180/Tyr-182, rabbit anti-p38, rabbit anti-p-AMPK Thr-172, rabbit anti-AMPK, rabbit anti-mTOR, rabbit anti-p-mTOR Ser-2448, rabbit anti-p-Akt Thr-308, rabbit anti-p-Akt Ser-473, rabbit anti-Akt, rabbit anti-p-eIF4E Ser-209, rabbit anti-eIF4E, mouse anti-p-p70S6k1 Thr-389, rabbit anti-p70S6k1, rabbit anti-PKR, rabbit anti-p-eIF2α Ser-51, rabbit anti-eIF2α, rabbit anti-Atg7, rabbit anti-Bcl-2, mouse anti-p53, mouse anti-IFN-α, and rabbit anti-Na/K-ATPase antibodies were from Cell Signaling Technology (Danvers, MA). Rabbit anti-p-PKR Thr-451 and mouse anti-actin antibodies were from Millipore (Billerica, MA). Mouse anti-p62 and rabbit anti-LAMP2 antibodies were purchased from Abcam Inc (Cambridge, UK). Anti-mouse IgG (H+L) and anti-rabbit IgG (H+L) antibodies were purchased from Kirkegaard & Perry Laboratories (Washington, D. C.).
Plasmid Construct and Transfection
The constructs containing the respective p17-, σC-, and σA-encoding genes of ARV have been described previously (3, 4). In the control assay, cells were transfected with the σC- and σA-encoding genes of ARV. For transfection, cells were seeded into 6-cm cell culture dishes. At about 75% confluence, cells were transfected with respective constructs by using Lipofectamine reagent according to the manufacturer's instructions (Invitrogen).
shRNA Constructs
To confirm the role of p53, PTEN, AMPK, PKR, Atg7, Beclin 1, LC3, LAMP2, and Rab7a in the regulation of autophagy, Vero or DF-1 cells at 75% confluence were transfected with gene-specific shRNAs targeting p53, PTEN (PTEN Gene ID 5728, human shRNA TG320498), AMPK (Prkaa1 Gene ID 105787, mouse shRNA TG505729), PKR (EIF2AK2 Gene ID 5610, human shRNA TG320493), ATG7 (ATG7 Gene ID 10533, human shRNA TG314609), Beclin 1 (BECN1 Gene ID 8678, human shRNA TG314484), LC3 (Map1lc3b Gene ID 67443, mouse shRNA TG503902), LAMP2 (LAMP2 Gene ID 3920, human shRNA TG311794), Rab7a (RAB7A Gene ID 7879, human shRNA TG309983), and scrambled shRNA (29-mer noneffective scrambled pGFP-V-RS vector) and pGFP-V-RS vector. All the shRNAs and scrambled shRNAs (TR30013) from OriGene Co. (Rockville, MD) were constructed in the vector pGFP-V-RS (TR30007) plasmid. Each set of shRNA containing four different shRNA sequences targeting the respective proteins was tested in DF-1 and Vero cells. Initial experiments revealed that the PTEN shRNA construct GI379210 (CTTGACCAATGGCTAAGTGAAGATGACAA), AMPK shRNA construct GI505440 (CTCCAAGACCAGGAAGTCATACAATAGAA), PKR shRNA construct GI379191 (CTGAAGGTGACTTCTCAGCAGATACATCA), Atg7 shRNA construct GI358430 (CTTGGCTGCTACTTCTGCAATGATGTGGT), Beclin 1 shRNA construct GI357930 (TGTCAGAACTACAAACGCTGTTTGGAGAT), LC3 shRNA construct GI515131 (TGGACAAGACCAAGTTCCTGGTGCCTGAC), Lamp2 shRNA construct GI347171 (GGAAGTTCTTATATGTGCAACAAAGAGCA), and Rab7a shRNA construct GI339927 (GATGACCTCTAGGAAGAAAGTGTTGCTGA) resulted in the most significant down-regulation of respective target protein expression in DF-1 cells, respectively. In Vero cells, initial experiments revealed that the PTEN shRNA construct GI379210 (CTTGACCAATGGCTAAGTGAAGATGACAA), AMPK shRNA construct GI505439 (CGCAGACTCAGTTCCTGGAGAAAGATGGC), Atg7 shRNA construct GI358429 (TGCCAGCTCGCTTAACATTGGAGTTCAGT), Beclin 1 shRNA construct GI357930 (TGTCAGAACTACAAACGCTGTTTGGAGAT), LC3 shRNA construct GI515132 (TTGCAGCTCAATGCTAACCAAGCCTTCTT), LAMP2 shRNA construct shRNA construct GI347171 (GGAAGTTCTTATATGTGCAACAAAGAGCA), and Rab7a shRNA construct GI339928 (GACTTTCTGACCAAGGAGGTGATGGTGGA) resulted in the most significant down-regulation of respective target protein expression. These shRNAs were utilized for further studies. The transfection was performed according to the manufacturer's protocol (Thermo Fisher Scientific Inc.).
To further examine whether all signaling pathways triggered by ARV can be suppressed by the p17 shRNA, cells were transfected with the p17 shRNA (29). The p17 shRNA sequence targeting the S1 gene of ARV is 1272GACATGACCGTTAACGCTCAC1292. The expression level of p17 and signaling molecules were analyzed by Western blot at 24 hpi.
Isolation of the Fractions of Cell Membrane and Cytoplasm
To check the expression level of membrane-associated PTEN in both p17-transfected DF-1 and Vero cells, the fractions of cell membrane and cytoplasm were isolated according to manufacturer's protocol (BioChain Institute, Hayward, CA).
Semi-quantitative RT-PCR
To study whether ARV infection and p17 transfection affect Beclin 1 transcription, Vero cells were transfected with p17 and infected with ARV with an m.o.i. of 10. All cultures were harvested and lysed at 24 hpi. Total RNAs from the above treatments were extracted and subjected to semi-quantitative RT-PCR according to previously published methods (30). The glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene was used as an internal control for normalization. Beclin 1 with the forward primer (5′-GAAAAGTGTGGAGAACCAGATGCGTTATGC,798–827) and the antisense primer (5′-GCTTTTGTCCATTGCTCCTCAGAGTTAAA-3′, 1312–1340) was designed based on GenBankTM accession number NM-001006332. The expected size of the Beclin 1 gene is 543 bp in length. The mRNA level of Beclin 1 was compared with mock-treatments. To further investigate whether autophagic proteins (Beclin 1 and LC3) affect σC and p17 gene transcription, Vero cells were transfected with shRNAs targeting Beclin 1 and LC3 for 24 h and were then infected with ARV at an m.o.i. of 10 for 12 h. Total RNAs from the above treatments were extracted and subjected to semi-quantitative RT-PCR as described above. The primers designed for amplification of the σC- and p17-encoding genes of ARV were based on the published sequences of the S1 gene of the ARV S1133 strain (31, 32). The primers for amplification of the σC-encoding gene of ARV are as follows: forward primer (5′-ATGGCGGGTCTCAATCCA, 1–18) and reverse primer (5′-TTCGGTGTCGATGCCGGTACG-3′, 981–961). The primers for amplification of the p17-encoding gene of ARV are as follows: forward primer (5′-ATGCAATGGCTCCGCCATACGA-3′, 1–22) and the antisense primer (5′-TCATAGATCGGCGTCAAATCGC-3′, 441–420). The expected sizes of the σC- and p17-encoding genes of ARV are 981 and 441 bp in length, respectively. The internal control, GAPDH with the forward primer (5′-CACCACCATGGAGAAGGCTGGGGCTCA-3′, 357–383) and the antisense primer (5′-GGCAGGTTTCTCCAGACGGCAGGTCAG-3′, 810–784), was designed based on the published sequences of GenBankTM accession number NM-204305. The expected size is 454 bp in length.
Coimmunoprecipitation of p17, σC, and LC3
The 6-cm cell culture dishes were seeded with 1 × 106 Vero cells, and then the cells were cultured in media containing 2% FBS overnight. Cells were infected with ARV at an m.o.i. of 10 and collected 24 hpi. The cells were collected and washed twice in phosphate-buffered saline and scraped in 200 μl of lysis buffer. About 500 μg of cellular proteins was incubated with 4 μg of anti-p17 or σC antibodies at 4 °C overnight. The immunoprecipitated proteins were separated by SDS-PAGE followed by Western blotting, and then proteins were detected with indicated antibodies.
Colocalization of ARV Proteins with LC3, p62 with LAMP2, and LC3 with Rab7 by Immunostaining
To study whether ARV promotes formation of autolysosome, TG was used to induce autophagy arrest (33). As previously demonstrated, TG blocks recruitment of Rab7 to autophagosome and blocks complete autophagy flux (33). In this study, Vero cells in 24-well culture plates were pretreated with TG (3 nm) for 2 h and then infected with ARV at an m.o.i. of 10 for 12 h. All treated and untreated cells were then fixed by methanol and stained with ARV proteins (p17 and σC), LC3, Rab7a, and LAMP2 antibodies. Colocalization of ARV proteins (p17 and σC) with LC3, p62 with LAMP2, and LC3 with Rab7a was observed under a fluorescence microscope.
Colocalization of GFP-LC3 and Lysosome
GFP-LC3-transfected and ARV-infected Vero cells were incubated with LysoTracker solution (50 nm) for 2 h and then were observed under a fluorescence microscope. GFP-LC3 plasmid was kindly provided by Dr. Shieh at the Institute of Biomedical Sciences, National Chung Hsing University, Taiwan.
Virus Titration
ARV-infected cell supernatant was collected at 24 hpi. Virus titer was determined by an agar overlay plaque assay performed in triplicate. Cells in 6-cm cell culture dishes were incubated for 1 h with diluted virus in 100 μl of serum-free MEM. The cells were then washed twice with MEM to remove unabsorbed viruses and overlaid with 2 ml of 1% agarose in MEM containing 2% FBS and antibiotics. Plaques were examined after an incubation period of 2 days at 37 °C by staining with neutral red for 3 h.
Electrophoresis and Western Blot
After each experimental procedure as described, culture medium was removed. Cells in 6-cm cell culture dishes were washed twice with PBS and lysed with 2.5× Laemmli loading dye. Cells were harvested by scraping and boiled for 10 min. Equal amount of samples was run in a 10% SDS-polyacrylamide gel and then transferred to a PVDF membrane. Expression of the individual proteins was examined by respective antibodies, followed by secondary antibody conjugated with horseradish peroxidase (HRP). After incubation with enhanced chemiluminescence (ECL Plus) (Amersham Biosciences), the result was exposed to x-ray films (Eastman Kodak). The intensity of each protein was calculated using the program Photocapt (Vilber Lourmat, France).
RESULTS
Induction of Autophagy Promotes Viral Growth
To determine the role of autophagy in ARV replication, we investigated the effect of rapamycin on autophagosome induction. Autophagy can be pharmacologically induced by inhibiting negative regulators such as mTOR with rapamycin (34). In this study, our results show that induction of autophagy through rapamycin treatment increased viral protein synthesis and viral yield (Fig. 1A). These results further suggest a critical role of autophagy in the virus life cycle, although we cannot rule out other effects on viral replication, independent of autophagy.
FIGURE 1.
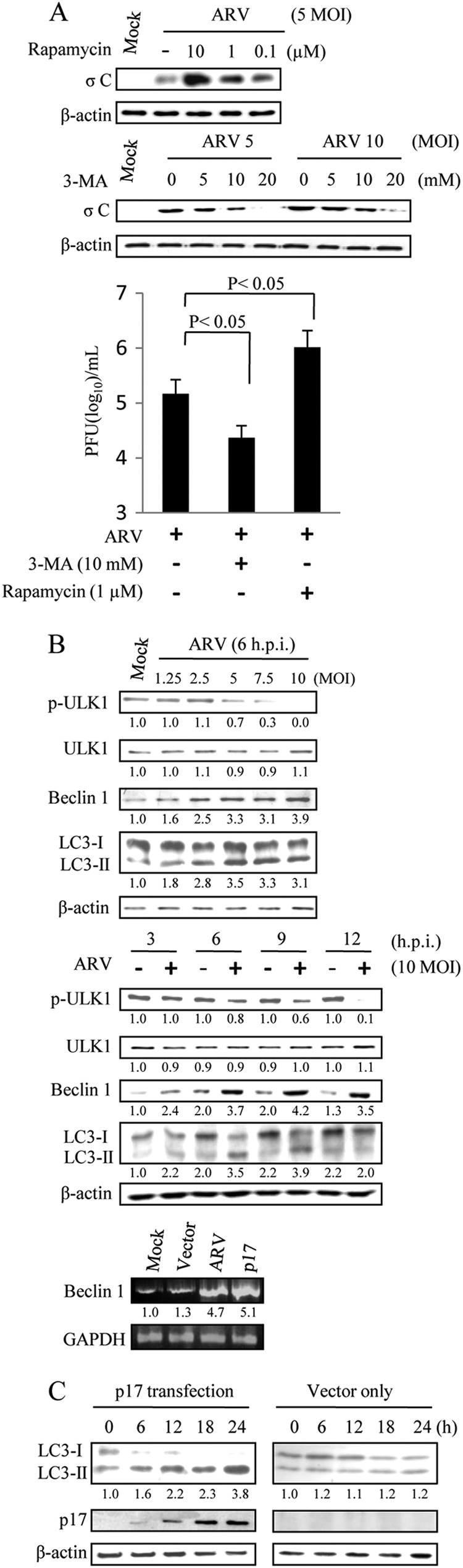
ARV induces autophagy. A, Vero cells were pretreated with different concentrations of 3-MA or rapamycin for 2 h and then infected with ARV at m.o.i. of 10. Cells lysates were separated by SDS-PAGE and immunoblotted with antibodies against ARV σC and β-actin, as indicated. The supernatants of ARV-infected cells in each well were harvested at 24 hpi for viral titration. Each value represents the mean of three independent experiments ± S.D. B, Vero cells were infected with different m.o.i. or time points as indicated. Cells lysates were separated by SDS-PAGE and immunoblotted with antibodies directed against ULK1, LC3-I/II, Beclin 1, and β-actin, as indicated. In semi-quantitative RT-PCR amplification of the Beclin 1 gene, Vero cells were transfected with p17 or infected with ARV at an m.o.i. of 10. In the bottom panel, the p17-transfected or ARV-infected cells were collected at 24 hpi, and total RNAs were extracted for semi-quantitative RT-PCR. After electrophoretic separation in an agarose gel and staining with ethidium bromide, the expression rate of the target gene is assessed by measuring the intensity of the band corresponding to a generated amplicon. The Beclin 1 mRNA levels in ARV-infected and p17-transfected cells were compared with those in mock-treated cells. The mRNA levels were normalized to that for GAPDH. Numbers below each lane are percentages of the control level of a specific protein in the mock treatment. C, Vero cells were transfected with p17-pcDNA3.1 and pCDN3.1, respectively. Cell lysates were harvested at 6, 12, 18, and 24 hpi and immunoblotted with respective antibodies against p17, LC3, and β-actin. Similar results were obtained from three independent experiments.
Autophagy can be pharmacologically inhibited with a widely used selective inhibitor of autophagy, 3-MA, by targeting the class III PI3K, which involves autophagosome formation (35). It inhibits intracellular protein degradation without affecting protein synthesis (35). To determine whether the autophagosome induction during ARV infection was a host antiviral response or a viral replication mechanism, we further tested the effect of 3-MA on ARV replication. As shown in Fig. 1A, treatment with 3-MA resulted in a dose-dependent reduction in σC protein synthesis and viral yield. This finding suggests that autophagic machinery may play an important role in the replication of ARV.
p17 Functions as a Positive Regulator of Autophagy
The correlation between the accumulation of ARV p17 protein and the appearance of autophagic markers raised the question whether a viral protein encoded by ARV constitutes an autophagic stimulus in infected cells. Therefore, experiments were designed to analyze the level of accumulation of lipid-conjugated LC3-II protein. The conversion of LC3-I to LC3-II and its translocation from the cytosol to the autophagosome is a reliable marker for autophagy. Furthermore, immunoblotting analysis also indicated that the levels of autophagic proteins Beclin 1 and LC3-II were elevated in a dose- and time-dependent manner in ARV-infected cells (Fig. 1B, upper and middle panels). It is important to note that the accumulation of lipid-conjugated LC3-II protein induced by ARV becomes evident at 3 hpi and then slightly decreased at 12 hpi. The decrease in ULK 1 phosphorylation level was found in a dose- and time-dependent manner (Fig. 1B, upper and middle panels). As shown in Fig. 1B (bottom panel), the mRNA level of Beclin 1 was elevated in ARV-infected and p17-transfected cells. Furthermore, the accumulation of lipid-conjugated LC3-II protein was also seen in p17-transfected cells in a time-dependent manner (Fig. 1C).
To determine the potential mechanism of induction of the autophagosome, we examined the phosphorylation status of mTOR and eIF2α, two key regulators that control autophagosome formation. It has been shown that eIF2α phosphorylation positively regulates the autophagic pathway (36), whereas mTOR is a negative regulator of autophagosome formation (37). In this study, we found that the increase in PKR and eIF2α phosphorylation is evident at 3 hpi (Fig. 2, upper panel), corresponding to the increased level of Beclin 1 and LC3-II in ARV-infected cells. The regulation of autophagy operated by mTOR occurs primarily at the initiation step and involves the phosphorylation and inhibition of ULK kinase and its associated protein ATG13 (38, 39). Here, we illustrated that the decreased level of mTOR and ULK1 phosphorylation occurs at 3 hpi (Fig. 1B, lower panel and Fig. 2, upper panel). ARV infection can suppress mTORC1 and activate ULK1 (Figs. 1B, upper and middle panels, and 2, upper panel). Following the ULK1 kinase induction, the Beclin 1·class III PI3K complex was activated to produce phosphatidylinositol 3-phosphate-enriched membrane domains that act as platforms capable of recruiting factors required for autophagosome formation. Immunoblotting analysis indicates that the levels of autophagic proteins Beclin 1 and LC3-II were evident at 3 hpi (Fig. 2, upper panel). Furthermore, the remarkable increase of phosphorylation level of p38, AMPK, PKR, and eIF2α proteins was found in ARV-infected cell in a time-dependent manner, accompanied by the decreased level of p-Akt, p-mTOR, p-p70S6K, and p-eIF4E in ARV-infected Vero cells (Fig. 2, upper panel). In a control assay, we found that bovine ephemeral fever virus activated neither p38 nor AMPK signaling (Fig. 2, lower panel). Our previous study and this work indicated that at the middle to late stages of ARV infection, activation of Akt and mTOR was inhibited in ARV-infected cells. Previously, we have found that Akt is activated by ARV during the early stages of infection (from 30 min to 2 h), and then declined at 3 hpi, resulting in delayed apoptosis in Vero cells (40). The kinetics of Akt and mTOR inhibition correlated with conversion of LC3-I to LC3-II, suggesting a role for Akt and mTOR as negative mediators of ARV-induced autophagy.
FIGURE 2.

ARV activates PTEN, p38, AMPK, and PKR signaling pathways in a time-dependent manner. DF-1 cells were infected with an m.o.i. of 5. The results of Western blot analysis of the total cellular protein isolated from ARV-infected DF-1 cells (upper panel) and from bovine ephemeral fever virus (BEFV)-infected Vero cells (lower panel) at the indicated time points are shown. A nonrelated virus, bovine ephemeral fever virus, was used as a control to examine whether activation of p38 and AMPK by ARV is specific. Phosphorylation and protein levels were determined by Western blot assay with the respective antibodies. The β-actin was included as an internal control for normalization. Similar results were obtained in three independent experiments.
Furthermore, the remarkable increase of phosphorylation level of PTEN, p38, AMPK, PKR, and eIF2α proteins was found in a time-dependent manner, accompanied by the decreased level of p-Akt, p-mTOR, p-p70S6K, p-eIF4E in p17-transfected DF-1 and Vero cells (Fig. 3A). Interestingly, we observed that p17 not only enhances PTEN stability by elevating phosphorylation of cytoplasmic PTEN to prevent it from NEDD4-1-mediated proteasome degradation (41) but also increases the level of membrane-associated PTEN to conduct its biological function (Fig. 3A). p17 also increases the phosphorylation level of PKR, eIF2α, p38, and AMPK in a time-dependent manner. In our earlier study, we have demonstrated that ARV triggers the p38-AMPK pathway enhancing virus replication (42). This study further illustrates that p17 is the viral protein responsible for activation of p38 and AMPK signaling. The active form of AMPK was detected 6 h after transfection, coincident with the decrease in p-mTOR and its downstream target p70S6K (Fig. 3A). Furthermore, Western blot assay indicated that the increased level of autophagic proteins Beclin 1 and LC3-II was seen in p17-transfected cells at 6 h after transfection (Figs. 1D and 3A). It is interesting to note that p17 protein can activate PKR but does not increase the level of IFN (Fig. 3A). In control assays, we found that only σA can reduce the level of p-PKR and LC3-II but not σC (Fig. 3B). The levels of p-PTEN and p-AMPK were not changed in both σC- and σA-transfected cells as compared with vector only (Fig. 3B).
FIGURE 3.

ARV p17 protein regulates PTEN, Akt, mTOR, S6K1, p38, AMPK, PKR, and eIF2α, and Beclin 1 in a time-dependent manner. DF-1 and Vero cells were transfected with p17 (A) as well as σC and σA (B) for 24 h. To ensure that p17 is the viral protein to activate PTEN, PKR, and AMPK, cells transfected with the σC- and σA-encoding genes of ARV were used as additional controls. The results of Western blot analysis of the total cellular protein isolated from p17-transfected DF-1 and Vero cells at the indicated time points are shown. Phosphorylation and protein levels were determined by Western blot assay with the indicated antibodies. The β-actin was included as an internal control for normalization. Similar results were obtained in three independent experiments.
Suppression of PTEN, PKR-eIF2α, and AMPK Reduced Autophagosome Formation and ARV Replication
As mentioned above, ARV triggers PTEN, AMPK, and PKR signaling; we next used shRNAs to specifically knock down these signaling proteins in ARV-infected cells. At 24 hpi, we found that depletion of PTEN, AMPK, or PKR by respective shRNAs reduced the level of LC3-II, σC protein synthesis and viral yield as compared with negative controls (Fig. 4, A–D). Depletion of PKR also resulted in the decrease of p-eIF2α, Beclin 1, and LC3-II (Fig. 4C), suggesting that PKR is an upstream inducer of eIF2α and autophagic proteins (Beclin 1 and LC3). Taken together, our results illustrate the critical role of PTEN, AMPK, and PKR-mTOR signaling pathways triggering autophagy that enhances virus replication.
FIGURE 4.

Knockdown of PTEN, AMPK, and PKR gene expression reduced ARV σC protein synthesis and viral yield. DF-1 and Vero cells transfected with respective shRNA or scramble shRNA were mocked or infected with ARV at m.o.i. of 10 for 24 h. Cell lysates were analyzed by Western blotting with antibodies against PTEN (A), AMPK (B), and PKR (C), as well as ARV σC, LC3-II or β-actin, as indicated. D, supernatants of ARV-infected and shRNA-transfected cells in each well were harvested at 24 hpi for viral titration. Each value represents the mean of three independent experiments ± S.D.
Knockdown of the Genes Critical for Autophagosome Formation Reduced ARV Replication
To expand the studies with pharmacological agents, we employed shRNAs targeting the autophagic genes (ATG7, Beclin 1, and LC3) required for different steps of autophagosome formation to assess the effect of autophagosome disruption on viral replication. Cells were transfected with shRNAs to deplete the genes required for different steps of autophagosome formation. In this study, depletion of ATG7 blocked viral protein synthesis and viral yield (Fig. 5, A and E). The autophagy protein ATG7 is an enzyme essential for activation of autophagosome formation and maturation machinery (43, 44). It was also demonstrated that deletion of ATG7 in mice has been shown to result in reduced autophagosome formation and impaired degradation of proteins and organelles induced by starvation (45). Furthermore, disruption of the class III PI3K signaling complex required for autophagosome formation by Beclin 1 shRNA resulted in significant reduction in σC protein synthesis and viral yield (Fig. 5, B and E). It is noteworthy that Beclin1-mediated induction of autophagy is blocked by overexpression of Bcl-2, as revealed by the decreased level of LC3-II (Fig. 5C). Depletion of LC3 through the use of shRNA reduced viral protein synthesis and viral yield (Fig. 5, D and E).
FIGURE 5.

Knockdown of ATG7, Beclin 1, and LC3 reduced ARV σC protein synthesis and viral yield. DF-1 and Vero cells transfected with respective shRNA or scrambled shRNA were mock-infected or infected with ARV at m.o.i. of 10 for 24 h. Cell lysates were analyzed by Western blotting with antibodies against ATG7 (A), Beclin 1 (B), LC3 (D), ARV σC or β-actin, as indicated. C, BHK-21 cells stably expressing Bcl-2 were infected with ARV at an m.o.i. of 10 for 24 h. Cells lysates were analyzed by Western blot with antibodies against LC3-II, σC, or β-actin, respectively. E, supernatants of ARV-infected and shRNA-transfected cells in each well were harvested at 24 hpi for viral titration. Each value represents the mean of three independent experiments ± S.D. F, in semi-quantitative RT-PCR amplification of the σC and p17-encoding gene of ARV, DF-1 cells were transfected with specific Beclin 1 and LC3 shRNAs for 24 h and then infected with ARV at an m.o.i. of 10. The ARV-infected cells were collected at 24 hpi, and total RNAs were extracted for semi-quantitative RT-PCR. The quantitated assay was carried out as described in Fig. 1B. The σC and p17 mRNA levels in LC3- and Beclin 1-depleted cells were compared with those in ARV-infected and mock-treated cells. The mRNA levels were normalized to that for GAPDH. Numbers below each lane are percentages of the control level of a specific protein in shRNA-treated cells. The shRNAs used are indicated on the top of the gel. Similar results were obtained from three independent experiments.
Blockade of Beclin 1 and LC3 Reduced the mRNA Level of σC- and p17-encoding Gene of ARV
To study whether the autophagic proteins Beclin 1 and LC3 affect viral transcription, knockdown of Beclin 1 or LC3 was carried out to examine viral transcription. In the semi-quantitative RT-PCR, transcription of the σC- and p17-encoding genes of ARV was inhibited (up to 79–88% inhibition) in the presence of Beclin 1 or LC3 shRNAs (Fig. 5F). Our results indicated that suppression of Beclin 1 and LC3, two proteins necessary for autophagosome formation, also led to a reduced viral transcription and/or ARV RNA replication.
ARV p17 Protein Triggered Autophagosome Formation through Activation of p53-PTEN-mTOR, AMPK, and PKR-eIF2α Signaling Pathways
The above-described results promoted us to investigate whether p17 triggers the formation of autophagosome through activation of p53-PTEN-mTOR, AMPK, and PKR-eIF2α signaling pathways. Therefore, we next utilized shRNAs to specifically targeting p53, PTEN, AMPK, and PKR or autophagic proteins (Beclin 1 and ATG 7) in p17-transfected cells. Previous studies have shown that transcriptional activation of the PTEN gene is regulated by p53 protein (46). Therefore, we next examined whether p17 regulates the transcription of the PTEN gene in a p53-dependent pathway. As shown in Fig. 6 (3rd and 4th lanes), p53 knockdown led to a reduction in PTEN level in mock-transfected and p17-transfected Vero and DF-1 cells. Immunoblotting analysis indicated that p17 not only decreases the level of p-Akt and p-mTOR but also increases the level of PTEN, Beclin 1, and LC3-II in Vero and DF-1 cells (Fig. 6, 5th lane), further suggesting that p53 is the upstream inducer of PTEN and that p17 induces autophagosome formation, at least in part, via the p53-PTEN-mTOR pathway.
FIGURE 6.

Blockade of p53 by p53 shRNA-inhibited p17 up-regulated PTEN, Beclin 1, and LC3-II. Vero and DF-1 cells were transfected with p53 shRNA, pcDNA3.1, and p17 for 24 h followed by Western blot analysis. The Western blot results of different combinations of treatments in Vero and DF-1 cells are shown. Phosphorylation and protein levels were determined by immunoblotting with the appropriate antibodies, as indicated. Similar results were obtained in three independent experiments.
As demonstrated above, PTEN, AMPK, and PKR/eIF2α are activated by ARV 17. As shown in Fig. 7, A–C, PTEN, AMPK, and PKR knockdown led to a significant reduction in the level of LC3-II protein as compared with negative controls. Simultaneous knockdown of PKR and AMPK or PKR and Beclin 1 in p17-transfected cells significantly reduced the level of LC3-II protein as compared with negative controls (Fig. 7, D and E). Interestingly, the reduction in Beclin 1 level was also seen in PKR shRNA-transfected DF-1 cells (Fig. 7C), suggesting that PKR may mediate Beclin 1 expression. Furthermore, depletion of autophagic protein Atg7 significantly reduced the level of LC3-II as compared with negative controls (Fig. 7F). In this study, PTEN, AMPK, and PKR signaling were suppressed in p17 shRNA-treated cells (Fig. 7G). Altogether, these data revealed that ARV p17-triggered autophagy needs simultaneous activation of multiple signaling pathways, including p53-PTEN-mTOR, PKR-eIF2α, and AMPK signaling pathways.
FIGURE 7.

Knockdown of PTEN, AMPK, PKR, and ATG7 reduced the level of LC3-II protein in p17-transfected cells. DF-1 and Vero cells were transfected with the respective shRNA, scrambled shRNAs, and mock transfection, respectively, for 24 h. Cell lysates were analyzed by Western blotting with antibodies against PTEN (A), AMPK (B), PKR (C), PKR and AMPK (D), PKR and Beclin 1 (E), ATG7 (F), and β-actin, as indicated. G, PTEN, AMPK, and PKR signalings were suppressed in p17 shRNA-treated cells. Similar results were obtained in three independent experiments.
p17 and σC Proteins of ARV Colocalized with Autophagy Marker LC3
Formation of the autophagosomes can be detected by immunofluorescence analysis, as the staining of LC3-I is diffusely cytoplasmic, whereas the staining of LC3-II is punctate (23). Although the steady-state levels of LC3-I and LC3-II, as detected by immunoblotting, may vary among different cell lines or independent cultures of a cell line over long periods of time, the amount of LC3-II among samples within an individual experiment correlates with the number of autophagosomes (47). Immunofluorescence results indicated that viral proteins (p17 and σC) colocalize with LC3 (Fig. 8A). The numbers of LC3 puncta were significantly higher in ARV-infected and p17-transfected cells as compared with negative controls (Fig. 8B). As shown in Fig. 8C, p17 and σC proteins did not coimmunoprecipitate with LC3. This suggests that ARV proteins and LC3 can exist in the same subcellular compartment but do not interact each other.
FIGURE 8.

ARV p17 promotes autophagosome and autolysosome formation that benefits virus replication. A, Vero cells were infected with ARV for 12 h and then fixed and processed for immunofluorescence staining of p17, σC, and LC3. The colocalization of ARV proteins (p17 and σC) was observed under a fluorescence microscope. Scale bar, 20 μm. B, quantitation results from Fig. 1 represents mean LC3 puncta per cell. n = 13. C, coimmunoprecipitation of p17, σC, and LC3 was carried out. Cells were infected with ARV at an m.o.i. of 10 and collected 24 h postinfection. About 500 μg of cellular proteins was incubated with 4 μg of anti-p17 or σC antibodies at 4 °C overnight. The immunoprecipitated proteins were separated by SDS-PAGE followed by Western blot, and then proteins were detected with indicated antibodies. Similar results were obtained in three independent experiments. IB, immunoblot. D, uninfected Vero cells were pretreated with TG for 2 h or without TG treatment (mock) in complete media as well as without TG treatment but in amino acid-free media for 2 h (starvation). In parallel experiments, two sets of Vero cells were infected with ARV at m.o.i. of 10 or transfected with p17-pCDNA3.1 plasmid, respectively. Cells lysates were collected at 24 hpi or post-transfection and then analyzed by Western blot with antibodies against p62. E, Vero cells were infected with ARV at m.o.i. of 10 for 12 h and then fixed and processed for immunofluorescence staining of p62 and LAMP2. The procedures for the TG and starvation treatments are described in D. Colocalization of p62 and LAMP2 was observed under a fluorescence microscope. Scale bar, 20 μm. F, p62- and LAMP2-positive cells were quantified by counting the numbers of positive cells in an individual field under a fluorescence microscope. Each value represents the mean of 20 fields ± S.D. G, Vero cells were transfected with LC3-GFP plasmid for 24 h and then infected with ARV at m.o.i. of 10 for 12 h. The cells were then fixed and processed for immunofluorescence staining of LC3-GFP and Rab7a. Colocalization of LC3-GFP and Rab7a was observed under a fluorescence microscope. Scale bar, 20 μm. H, Vero cells were transfected with LC3-GFP plasmid for 24 h and then infected with ARV at m.o.i. of 10 for 12 h. The cells were then fixed and processed for immunofluorescence staining of LC3-GFP and lysosome. In the p17 transfection assay, cells were transfected with p17-pCDNA3.1 plasmid. The treatment conditions for TG and starvation are described as in D. I, Vero cells were transfected with shRNAs targeting LAMP2 or Rab7a as well as scrambled shRNA for 24 h and then infected with ARV at an m.o.i. of 10 for 24 h. Cells lysates were analyzed by Western blot with antibodies against LAMP2, σC, and actin, respectively. The supernatants of LAMP2 and Rab7a knockdown in ARV-infected cells were harvested at 24 hpi for viral titration. Each value represents the mean of three independent experiments ± S.D.
Expression Level of p62 Was Slightly Reduced in ARV-infected Cells
The purpose of this set of experiments was to further investigate autolysosome formation in ARV infection and specifically to determine whether the pathway could proceed to completion or whether it could be blocked at later stages by ARV. p62 is a multifunctional protein that associates with protein aggregates, interacts with LC3-II, and is selectively degraded by the autophage-lysosome pathway (48). The protein p62 has also been considered a marker for autophagy-mediated protein degradation or autophagic flux. A commonly used method to detect autophagic flux involves monitoring the levels of polyubiquitin-binding protein p62. The expression level of p62 was increased at 18 hpi and then slightly decreased at 24 hpi as compared with mock infection or TG treatments (Fig. 8D). This suggests that ARV may prevent p62 from degradation in an unknown mechanism before completion of virus replication. In contrast, during starvation-induced autophagy in Vero cells, autophagosomes fuse with lysosomes, leading to the degradation of intra-autophagosomal LC3-II and p62 by lysosomal proteases (Fig. 8D). It is worth noting that ARV promotes the fusion of p62 with LAMP2 as well as LC3 and Rab7 to form autolysosome (Fig. 8, E and G). The p62/LAMP2-positive cells were also significantly higher than that of the mock control (Fig. 8F). Furthermore, GFP-LC3 colocalized with lysosome was more evident in ARV-infected and p17-transfected Vero cells as compared with mock infection and TG treatment (Fig. 8H). Taken together, our results suggest that ARV induces autolysosome formation without enhancing autophagic protein degradation before 18 hpi.
Depletion of LAMP2 or Rab7a Reduced ARV Replication
As mentioned above, ARV promotes autophagosome-lysosome fusion. Therefore, we next examined whether prevention of autophagosome-lysosome fusion affects viral replication. Both Rab7a and LAMP2 are critical for autophagosome-lysosome fusion (49–52). In this study, we revealed that depletion of LAMP2 or Rab7a by shRNAs resulted in the decrease of viral protein synthesis and virus yield (Fig. 8I). As blocking the fusion between autophagosomes and lysosomes can reduce viral yield, these results indicated that autophagosome maturation likely plays a critical role in ARV replication.
DISCUSSION
Autophagy is a dynamic process of subcellular degradation, which has recently sparked great interest as it is now recognized to be involved in regulation of virus replication. Successful invading pathogens have evolved various strategies to evade host autophagy. For instance, some bacterial or viral proteins, such as IcsB secreted by Shigella flexneri (53) and ICP34.5 encoded by herpes simplex virus type 1 (HSV-1) (54) have been documented to block or evade cellular autophagy. Other pathogens adopt different approaches in battling host autophagic defense by subverting it to facilitate their own procreation. In fact, several viruses have been reported to induce autophagy to facilitate their own replication (9–14). A previous study and this work demonstrated that the induction of autophagy by rapamycin enhanced ARV replication (25). In this work, we revealed that inhibition of autophagy by 3-MA or shRNA knockdown of cellular genes, including ATG7, Beclin 1, and LC3, which are important for the formation of autophagosomes, significantly reduced ARV replication, supporting that autophagy positively regulates ARV replication. The cross-talk between autophagy and apoptosis has become evident (55–57). Induction of autophagy has often connected to inhibition of apoptosis. Here, we found that ARV-induced autophagosome at 3 hpi correlates with our previous findings that ARV triggers Akt at 30 min to 2 hpi, resulting in delayed apoptosis in Vero cells (40). It was proved that ARV induces cell death in the middle to late stages of infection (2, 40). ARV seems to trigger survival signaling in the early stage of infection that protects the host cells from caspase-dependent apoptotic cell death.
Accumulating evidence has suggested that viral proteins can induce autophagy, such as Epstein-Barr virus LMP-1, human papillomavirus 16 (HPV 16) E7, simian virus 40 (SV40) small T antigen, influenza virus M2 protein, poliovirus 2BC and 3A proteins, hepatitis C virus NS4B, and Rotavirus NSP4 (9–14), whereas some viral proteins suppress autophagy, such as HSV-1 protein IVP34.5 and bovine herpesvirus type 1 (BHV-1) bICP0 (15, 16). In this work, we found that p17 protein exerts a novel role responsible for ARV-induced autophagy. Based on our findings, we propose that p17-triggering autophagy relies on activation of p53-PTEN-mTORC1, AMPK, and PKR-eIF2α signaling pathways. Interestingly, ARV, like other viruses, has evolved to encode a nonstructural protein that alters the physiology of the host cells to enhance its own replication.
Proteins that inhibit autophagy include oncogenes, such as PI3K, Akt, mTORC1, and Bcl-2 (55, 58). Regulators that induce autophagy include tumor suppressors, such as PTEN, TSC1·TSC2 complexes, stress-activated signaling molecules, such as JNK1, and those that respond to low energy or endoplasmic reticulum stress, and molecules involved in innate immune signaling (13, 36, 59, 60). As alluded to earlier, the process of autophagosome formation is tightly controlled. Central in the regulation of autophagy are two proteins, mTOR and Beclin 1 (22, 37). The mTOR is one of the key regulators of autophagy and shuts off autophagy (37). Downstream of mTORC1 and Beclin 1 is at the heart of a regulatory complex for the class III PI3K/hVps34 whose activity is required for pre-autophagosome formation (22). In this study, depletion of Beclin 1 by shRNAs or overexpression of Bcl-2 reduced the expression level of LC3-II, leading to the significant reduction of viral transcription and virus yield. These results may explain why Bcl-2 overexpression in ARV-infected cells reduces virus yield (26).
Cap-dependent translation is initiated through assembly of several proteins called translation initiation factors (61). It has been reported that several viruses influence cellular protein synthesis impeding cap-dependent translation that potentially facilitates translation of viral transcripts or slow down production of cellular proteins (62, 63). The major substrate of PKR is eIF2α, resulting in inhibition of protein synthesis under various stress conditions (64). Both PKR and phosphorylated eIF2α have been proved to promote autophagy (36). In addition to regulation of eIF2α, p17 also promotes phosphorylation of eukaryotic translation elongation factor (eEF2), thereby inhibiting cellular protein synthesis (65). The present results and our earlier findings provide evidence to support our conclusion that the ability of p17 to block mTORC1 and to mediate the phosphorylation of eIF2α and eEF2 results in induction of autophagy and enhancement of virus replication. This finding is consistent with our previous study suggesting that p17 causes translation shutoff but promotes virus replication (4).
The σA protein of ARV has been suggested to be an inhibitor of PKR (66). Conversely, we revealed that the p17 protein of ARV functions as an activator of PKR. A previous study suggested that autophagy is antagonized by HSV-1 γ34.5 or ICP34.5 to mediate dephosphorylation of eIF2α (54). During nutrient starvation and viral infection, the synchronous regulation of translation and autophagy by the eIF2α kinase signaling pathway may be a fundamental mechanism that permits eukaryotic cells to successfully adapt to environmental stress. As part of their virulence strategy, ARV may subvert this host adaptive mechanism. Although PKR can be activated without viral dsRNA or viral infection by different stresses or through protein-protein interactions (67), a still unanswered question is how PKR can be activated by p17.
The regulation of autophagy by signaling pathways overlaps the control of cell growth, proliferation, cell survival, and death. The evolutionarily conserved mTORC1 plays an important role in the control of autophagy by growth factors and nutrient signaling and in response to stress situations. An important function of AMPK is sensing energy shortage. We have demonstrated previously that ARV triggers the p38-AMPK signaling pathway enhancing its own replication (42). However, the involved viral proteins and the underlying mechanism of AMPK promoting virus replication remain unclear. This study further identifies a novel role of p17 in triggering AMPK that enables activation of autophagy and enhances virus replication. Studies by Kumar and Rangarajan (13) suggested that SV40 small T antigen inactivates PP2A to activate AMPK that enables survival of cancer cells under glucose deprivation via inhibition of protein synthesis and activation of autophagy as an alternative energy source. Conversely, ARV triggers AMPK under normal levels of glucose concentration in host cells (42). Hence, ARV likely exploits different strategies to activate AMPK, inducing autophagy.
Colocalization of autophagosome marker LC3 with viral proteins reveals that autophagy occurs in cells with ARV. This study provide evidence indicating that autophagy machinery proteins are required for viral transcription and/or viral RNA replication as well as virus replication, as suppression of autophagy with shRNAs targeting Beclin 1 or LC3 leads to inhibition of viral transcription or virus replication. It is very likely that autophagosomes provide the membranous support for viral transcription and virus replication. Moreover, a compelling observation is that knockdown of either LAMP2 or Rab7a, two critical proteins responsible for the fusion of autophagosome with lysosome (49–52), significantly reduces virus replication. ARV seems to evolve a strategy to promote formation and maturation of the autophagosome to support its own replication. Conversely, several other viruses, such as coxsackieviruses, hepatitis C virus, influenza virus A, human immunodeficiency virus type 1 (HIV-1), and rotaviruses (68–71) have also evolved mechanisms to subvert autophagy by suppressing the fusion between autophagosomes and lysosomes. The molecular mechanisms for this suppression of membrane fusion are largely unknown, although both influenza virus A and HIV-1 apparently target Beclin 1, a protein factor that is important for the fusion between autophagosomes and lysosomes. In this study, the new insights into the underlying mechanisms of p17-induced autophagosome and autolysosome formation broaden our understanding of ARV-induced autophagy enhancing virus replication and may facilitate new strategies for combating ARV-caused diseases. A model depicting the ARV p17 triggers signaling pathways to facilitate autophagy is shown in Fig. 9.
FIGURE 9.
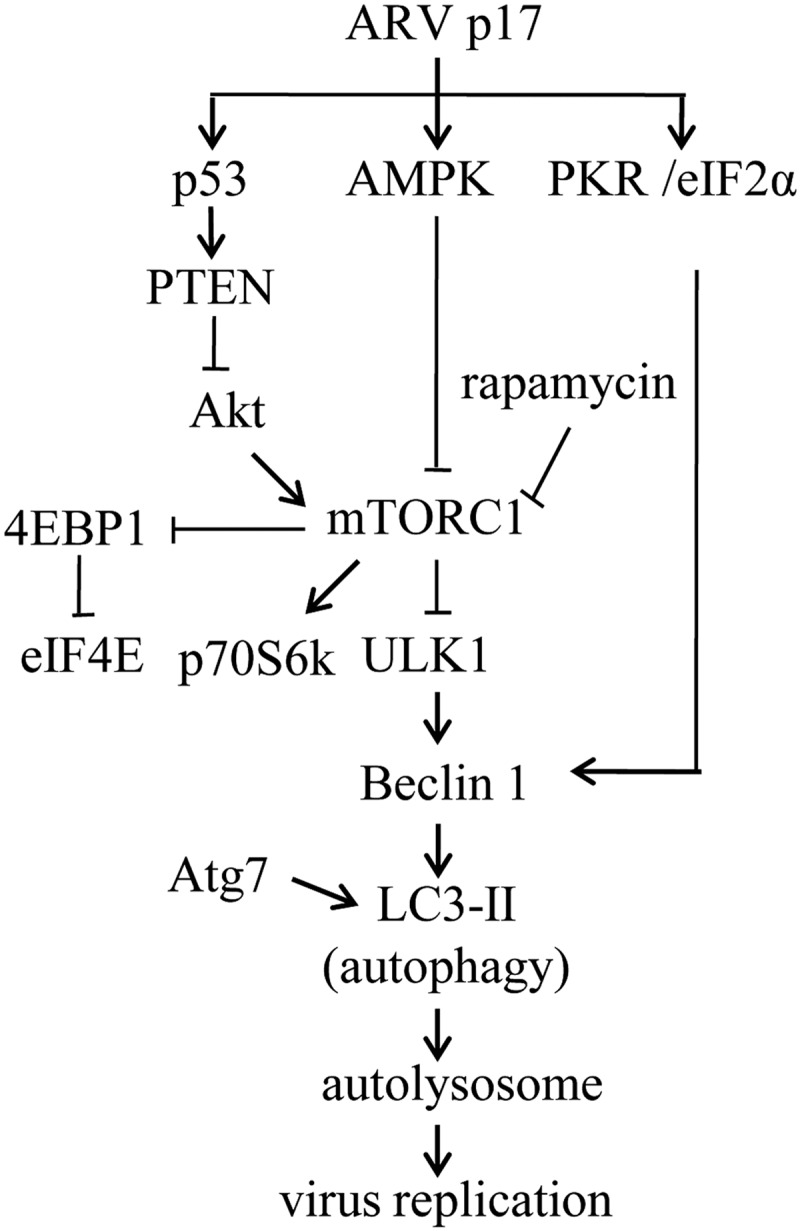
Model depicting the mechanism of ARV p17 triggers autophagy. This study established a new regulatory network of p17 linking p53/PTEN/mTOR, AMPK, and PKR/eIF2α pathways, which up-regulate ULK1, Beclin 1, and LC3. p17 increases the levels of phosphorylated p53, PTEN, and AMPK, which in turn down-regulate Akt and mTORC1. p17 also activates PKR to phosphorylate eIF2α. Taken together, PTEN, AMPK, and PKR signaling pathways triggered by p17 that induce autophagy enhance virus replication.
This work was supported by National Science Council, Taiwan, Grant NSC 99-2321-B-005-015-MY3 (to H.-J. L.) and by the Ministry of Education, Taiwan under the Aiming for Top University plan.
- ARV
- avian reovirus
- PKR
- dsRNA-dependent protein kinase
- AMPK
- AMP-activated protein kinase
- m.o.i.
- multiplicity of infection
- TG
- thapsigargin
- MEM
- minimum essential medium
- 3-MA
- 3-methyladenine
- PTEN
- phosphatase and tensin deleted on chromosome 10
- hpi
- hours post-infection.
REFERENCES
- 1. Costas C., Martínez-Costas J., Bodelón G., Benavente J. (2005) The second open reading frame of the avian reovirus S1 gene encodes a transcription-dependent and CRM1-independent nucleocytoplasmic shuttling protein. J. Virol. 79, 2141–2150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Shih W. L., Hsu H. W., Liao M. H., Lee L. H., Liu H. J. (2004) Avian reovirus σC protein induces apoptosis in cultured cells. Virology 321, 65–74 [DOI] [PubMed] [Google Scholar]
- 3. Liu H. J., Lin P. Y., Lee J. W., Hsu H. Y., Shih W. L. (2005) Retardation of cell growth by avian reovirus p17 through the activation of p53 pathway. Biochem. Biophys. Res. Commun. 336, 709–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chulu J. L., Huang W. R., Wang L., Shih W. L., Liu H. J. (2010) Avian reovirus nonstructural protein p17-induced G2/M cell cycle arrest and host cellular protein translation shutoff involve activation of p53-dependent pathways. J. Virol. 84, 7683–7694 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5. Grande A., Rodriguez E., Costas C., Everitt E., Benavente J. (2000) Oligomerization and cell-binding properties of the avian reovirus cell-attachment protein σC. Virology 274, 367–377 [DOI] [PubMed] [Google Scholar]
- 6. Chen Y. T., Lin C. H., Ji W. T., Li S. K., Liu H. J. (2008) Proteasome inhibition reduces avian reovirus replication and apoptosis induction in cultured cells. J. Virol. Methods 151, 95–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Huang W. R., Wang Y. C., Chi P. I., Wang L., Wang C. Y., Lin C. H., Liu H. J. (2011) Cell entry of avian reovirus follows a caveolin-1-mediated and dynamin-2-dependent endocytic pathway that requires activation of p38 mitogen-activated protein kinase (MAPK) and Src signaling pathways as well as microtubules and small GTPase Rab5 protein. J. Biol. Chem. 286, 30780–30794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Levine B., Kroemer G. (2008) Autophagy in the pathogenesis of disease. Cell 132, 27–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Berkova Z., Crawford S. E., Trugnan G., Yoshimori T., Morris A. P., Estes M. K. (2006) Rotavirus NSP4 induces a novel vesicular compartment regulated by calcium and associated with viroplasms. J. Virol. 80, 6061–6071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Taylor M. P., Kirkegaard K. (2007) Modification of cellular autophagy protein LC3 by poliovirus. J. Virol. 81, 12543–12553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lee D. Y., Sugden B. (2008) The LMP1 oncogene of EBV activates PERK and the unfolded protein response to drive its own synthesis. Blood 111, 2280–2289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhou X., Münger K. (2009) Expression of the human papillomavirus type 16 E7 oncoprotein induces an autophagy-related process and sensitizes normal human keratinocytes to cell death in response to growth factor deprivation. Virology 385, 192–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kumar S. H., Rangarajan A. (2009) Simian virus 40 small T antigen activates AMPK and triggers autophagy to protect cancer cells from nutrient deprivation. J. Virol. 83, 8565–8574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Su W. C., Chao T. C., Huang Y. L., Weng S. C., Jeng K. S., Lai M. M. (2011) Rab5 and class III phosphoinositide 3-kinase Vps34 are involved in hepatitis C virus NS4B-induced autophagy. J. Virol. 85, 10561–10571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Orvedahl A., Alexander D., Tallóczy Z., Sun Q., Wei Y., Zhang W., Burns D., Leib D.A., Levine B. (2007) HSV-1 ICP34.5 confers neurovirulence by targeting the Beclin 1 autophagy protein. Cell Host Microbe 1, 23–35 [DOI] [PubMed] [Google Scholar]
- 16. Geiser V., Rose S., Jones C. (2008) Bovine herpesvirus type 1 induces cell death by a cell-type-dependent fashion. Microb. Pathog. 44, 459–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mizushima N., Yoshimori T. (2007) How to interpret LC3 immunoblotting. Autophagy 3, 542–545 [DOI] [PubMed] [Google Scholar]
- 18. Suzuki K., Kirisako T., Kamada Y., Mizushima N., Noda T., Ohsumi Y. (2001) The pre-autophagosomal structure organized by concerted functions of APG genes is essential for autophagosome formation. EMBO J. 20, 5971–5981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mizushima N., Sugita H., Yoshimori T., Ohsumi Y. (1998) A new protein conjugation system in human. The counterpart of the yeast Apg12p conjugation system essential for autophagy. J. Biol. Chem. 273, 33889–33892 [DOI] [PubMed] [Google Scholar]
- 20. Mizushima N., Yoshimori T., Ohsumi Y. (2003) Role of the Apg12 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 35, 553–561 [DOI] [PubMed] [Google Scholar]
- 21. Tanida I., Ueno T., Kominami E. (2004) LC3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 36, 2503–2518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kihara A., Kabeya Y., Ohsumi Y., Yoshimori T. (2001) Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep. 2, 330–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 19, 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shelly S., Lukinova N., Bambina S., Berman A., Cherry S. (2009) Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity 30, 588–598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Meng S., Jiang K., Zhang X., Zhang M., Zhou Z., Hu M., Yang R., Sun C., Wu Y. (2012) Avian reovirus triggers autophagy in primary chicken fibroblast cells and Vero cells to promote virus production. Arch. Virol. 157, 661–668 [DOI] [PubMed] [Google Scholar]
- 26. Chulu J. L., Lee L. H., Lee Y. C., Liao S. H., Lin F. L., Shih W. L., Liu H. J. (2007) Apoptosis induction by avian reovirus through p53 and mitochondria-mediated pathway. Biochem. Biophys. Res. Commun. 356, 529–535 [DOI] [PubMed] [Google Scholar]
- 27. Hsu C. J., Wang C. Y., Lee L. H., Shih W. L., Chang C. I., Cheng H. L., Chulu J. L., Ji W. T., Liu H. J. (2006) Development and characterization of monoclonal antibodies against avian reovirus σC protein and their application in detection of avian reovirus isolates. Avian Pathol. 35, 320–326 [DOI] [PubMed] [Google Scholar]
- 28. Huang P. H., Li Y. J., Su Y. P., Lee L. H., Liu H. J. (2005) Epitope mapping and functional analysis of σA and σNS proteins of avian reovirus. Virology 332, 584–595 [DOI] [PubMed] [Google Scholar]
- 29. Ji W. T., Chulu J. L., Lin F. L., Li S. K., Lee L. H., Liu H. J. (2008) Suppression of protein expression of three avian reovirus S-class genome segments by RNA interference. Vet. Microbiol. 129, 252–261 [DOI] [PubMed] [Google Scholar]
- 30. Guénin S., Mauriat M., Pelloux J., Van Wuytswinkel O., Bellini C., Gutierrez L. (2009) Normalization of qRT-PCR data: the necessity of adopting a systematic, experimental condition-specific, validation of references. J. Exp. Bot. 60, 487–493 [DOI] [PubMed] [Google Scholar]
- 31. Liu H. J., Lee L. H., Hsu H. W., Kuo L. C., Liao M. H. (2003) Molecular evolution of avian reovirus: evidence for genetic diversity and reassortment of the S-class genome segments and multiple cocirculating lineages. Virology 314, 336–349 [DOI] [PubMed] [Google Scholar]
- 32. Hsu H. W., Su H. Y., Huang P. H., Lee B. L., Liu H. J. (2005) Sequence and phylogenetic analysis of P10- and P17-encoding genes of avian reovirus. Avian Dis. 49, 36–42 [DOI] [PubMed] [Google Scholar]
- 33. Ganley I. G., Wong P. M., Gammoh N., Jiang X. (2011) Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol. Cell 42, 731–743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rubinsztein D. C., Gestwicki J. E., Murphy L. O., Klionsky D. J. (2007) Potential therapeutic applications of autophagy. Nat. Rev. Drug Discov. 6, 304–312 [DOI] [PubMed] [Google Scholar]
- 35. Seglen P. O., Gordon P. B. (1982) 3-Methyladenine. Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. U.S.A. 79, 1889–1892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tallóczy Z., Jiang W., Virgin H. W., 4th, Leib D. A., Scheuner D., Kaufman R. J., Eskelinen E. L., Levine B. (2002) Regulation of starvation- and virus-induced autophagy by the eIF2α kinase signaling pathway. Proc. Natl. Acad. Sci. U.S.A. 99, 190–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Codogno P., Meijer A. J. (2005) Autophagy and signaling. Their role in cell survival and cell death. Cell Death Differ. 12, 1509–1518 [DOI] [PubMed] [Google Scholar]
- 38. Hara T., Takamura A., Kishi C., Iemura S., Natsume T., Guan J. L., Mizushima N. (2008) FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J. Cell Biol. 181, 497–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chan E. Y., Longatti A., McKnight N. C., Tooze S. A. (2009) Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol. Cell. Biol. 29, 157–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lin P. Y., Liu H. J., Liao M. H., Chang C. D., Chang C. I., Cheng H. L., Lee J. W., Shih W. L. (2010) Activation of PI 3-kinase/Akt/NF-κB and Stat3 signaling by avian reovirus S1133 in the early stages of infection results in an inflammatory response and delayed apoptosis. Virology 400, 104–114 [DOI] [PubMed] [Google Scholar]
- 41. Wang X., Trotman L. C., Koppie T., Alimonti A., Chen Z., Gao Z., Wang J., Erdjument-Bromage H., Tempst P., Cordon-Cardo C., Pandolfi P. P., Jiang X. (2007) NEDD4–1 is a proto-oncogenic ubiquitin ligase for PTEN. Cell 128, 129–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ji W. T., Lee L. H., Lin F. L., Wang L., Liu H. J. (2009) AMP-activated protein kinase facilitates avian reovirus to induce mitogen-activated protein kinase (MAPK) p38 and MAPK kinase 3/6 signalling that is beneficial for virus replication. J. Gen. Virol. 90, 3002–3009 [DOI] [PubMed] [Google Scholar]
- 43. Klionsky D. J., Emr S. D. (2000) Autophagy as a regulated pathway of cellular degradation. Science 290, 1717–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Klionsky D. J. (2007) Autophagy. From phenomenology to molecular understanding in less than a decade. Nat. Rev. Mol. Cell Biol. 8, 931–937 [DOI] [PubMed] [Google Scholar]
- 45. Komatsu M., Waguri S., Ueno T., Iwata J., Murata S., Tanida I., Ezaki J., Mizushima N., Ohsumi Y., Uchiyama Y., Kominami E., Tanaka K., Chiba T. (2005) Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 169, 425–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stambolic V., MacPherson D., Sas D., Lin Y., Snow B., Jang Y., Benchimol S., Mak T. W. (2001) Regulation of PTEN transcription by p53. Mol. Cell 8, 317–325 [DOI] [PubMed] [Google Scholar]
- 47. Tanida I., Minematsu-Ikeguchi N., Ueno T., Kominami E. (2005) Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 1, 84–91 [DOI] [PubMed] [Google Scholar]
- 48. Seibenhener M. L., Babu J. R., Geetha T., Wong H. C., Krishna N. R., Wooten M. W. (2004) Sequestosome 1/p62 is a polyubiquitin chain-binding protein involved in ubiquitin proteasome degradation. Mol. Cell. Biol. 24, 8055–8068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kimura S., Noda T., Yoshimori T. (2007) Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3, 452–460 [DOI] [PubMed] [Google Scholar]
- 50. Gutierrez M. G., Munafó D. B., Berón W., Colombo M. I. (2004) Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J. Cell Sci. 117, 2687–2697 [DOI] [PubMed] [Google Scholar]
- 51. Jäger S., Bucci C., Tanida I., Ueno T., Kominami E., Saftig P., Eskelinen E. L. (2004) Role for Rab7 in maturation of late autophagic vacuoles. J. Cell Sci. 117, 4837–4848 [DOI] [PubMed] [Google Scholar]
- 52. Saftig P., Beertsen W., Eskelinen E. L. (2008) LAMP-2. A control step for phagosome and autophagosome maturation. Autophagy 4, 510–512 [DOI] [PubMed] [Google Scholar]
- 53. Ogawa M., Yoshimori T., Suzuki T., Sagara H., Mizushima N., Sasakawa C. (2005) Escape of intracellular Shigella from autophagy. Science 307, 727–731 [DOI] [PubMed] [Google Scholar]
- 54. Tallóczy Z., Virgin H. W., 4th, Levine B. (2006) PKR-dependent autophagic degradation of herpes simplex virus type 1. Autophagy 2, 24–29 [DOI] [PubMed] [Google Scholar]
- 55. Maiuri M. C., Zalckvar E., Kimchi A., Kroemer G. (2007) Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 8, 741–752 [DOI] [PubMed] [Google Scholar]
- 56. Levine B., Sinha S., Kroemer G. (2008) Bcl-2 family members. Dual regulators of apoptosis and autophagy. Autophagy 4, 600–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Djavaheri-Mergny M., Maiuri M. C., Kroemer G. (2010) Cross talk between apoptosis and autophagy by caspase-mediated cleavage of Beclin 1. Oncogene 29, 1717–1719 [DOI] [PubMed] [Google Scholar]
- 58. Pattingre S., Levine B. (2006) Bcl-2 inhibition of autophagy. A new route to cancer? Cancer Res. 66, 2885–2888 [DOI] [PubMed] [Google Scholar]
- 59. Wei Y., Sinha S., Levine B. (2008) Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy 4, 949–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sinha S., Levine B. (2008) The autophagy effector Beclin 1. A novel BH3-only protein. Oncogene 27, S137–S148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Merrick W. C. (2004) Cap-dependent and cap-independent translation in eukaryotic systems. Gene 332, 1–11 [DOI] [PubMed] [Google Scholar]
- 62. Kleijn M., Vrins C. L., Voorma H. O., Thomas A. A. (1996) Phosphorylation state of the cap-binding protein eIF4E during viral infection. Virology 217, 486–494 [DOI] [PubMed] [Google Scholar]
- 63. Perales C., Carrasco L., Ventoso I. (2003) Cleavage of eIF4G by HIV-1 protease. Effects on translation. FEBS Lett. 533, 89–94 [DOI] [PubMed] [Google Scholar]
- 64. Williams B. R. (1999) PKR. A sentinel kinase for cellular stress. Oncogene 18, 6112–6120 [DOI] [PubMed] [Google Scholar]
- 65. Ji W. T., Wang L., Lin R. C., Huang W. R., Liu H. J. (2009) Avian reovirus influences phosphorylation of several factors involved in host protein translation including eukaryotic translation elongation factor 2 (eEF2) in Vero cells. Biochem. Biophys. Res. Commun. 384, 301–305 [DOI] [PubMed] [Google Scholar]
- 66. González-López C., Martínez-Costas J., Esteban M., Benavente J. (2003) Evidence that avian reovirus σA protein is an inhibitor of the double-stranded RNA-dependent protein kinase. J. Gen. Virol. 84, 1629–1639 [DOI] [PubMed] [Google Scholar]
- 67. Li S., Min J. Y., Krug R. M., Sen G. C. (2006) Binding of the influenza A virus NS1 protein to PKR mediates the inhibition of its activation by either PACT or double-stranded RNA. Virology 349, 13–21 [DOI] [PubMed] [Google Scholar]
- 68. Jackson W. T., Giddings T. H., Jr., Taylor M. P., Mulinyawe S., Rabinovitch M., Kopito R. R., Kirkegaard K. (2005) Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 3, e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Wong J., Zhang J., Si X., Gao G., Mao I., McManus B. M., Luo H. (2008) Autophagosome supports coxsackievirus B3 replication in host cells. J. Virol. 82, 9143–9153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sir D., Chen W. L., Choi J., Wakita T., Yen T. S., Ou J. H. (2008) Induction of incomplete autophagic response by hepatitis C virus via the unfolded protein response. Hepatology 48, 1054–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gannagé M., Dormann D., Albrecht R., Dengjel J., Torossi T., Rämer P.C., Lee M., Strowig T., Arrey F., Conenello G., Pypaert M., Andersen J., García-Sastre A., Münz C. (2009) Matrix protein 2 of influenza A virus blocks autophagosome fusion with lysosomes. Cell Host Microbe 6, 367–380 [DOI] [PMC free article] [PubMed] [Google Scholar]