

Background: Arg75 in connexins is highly conserved.
Results: Disease-causing mutations at this position cause loss of function; for Cx32, loss of a positive charge appears to be critical.
Conclusion: Positive charge at position 75 is required for normal Cx32 function.
Significance: Better understanding of the effects of mutations of this position in Cx32 may have relevance to pathogenesis of several human diseases.
Keywords: Connexin, Gap Junctions, Gating, Ion Channels, Nerve, Charcot Marie Tooth Disease
Abstract
Charcot Marie Tooth disease (CMT) is a group of inherited disorders characterized clinically by exclusively or predominantly peripheral nerve dysfunction. CMT1X, the most common form of X-linked CMT is caused by mutations in connexin 32 (Cx32). In this work, we used dual whole cell patch clamp recording to examine the functional effects of mutations at the Arg75 position. This residue is highly conserved among members of the connexin family, and disease-causing mutations have been identified at this (or the corresponding) position in Cx26, Cx43, and Cx46. Thus, a better understanding of the effects of mutations of this position in Cx32 may have relevance to pathogenesis of a number of different human diseases. All three mutants associated with CMT1X (R75P, R75Q, and R75W) showed very low levels of coupling similar to those of the cells transfected with vector alone. Heterotypic pairing with Cx32 WT showed that the absence of coupling for these mutants in the homotypic configuration could be explained by shifts in their hemichannel Gj-Vj relations. Examination of the expression levels and gating characteristics of seven additional mutants (R75A, R75D, R75E, R75H, R75K, R75L, and R75V) at this position suggest that the positive charge at position 75 in Cx32 is required for normal channel function but not for gap junction assembly. Our studies also suggest that disease treatment strategies for CMT1X, which correct trafficking abnormalities in Cx32, may be ineffective for the group of mutations also conferring changes in gating properties of Cx32 channels.
Introduction
Connexins comprise a family of >20 homologous integral membrane proteins that form gap junction channels (1), typically providing a pathway for the diffusion of small molecules and ions between neighboring cells (2, 3). Data suggest that functional intercellular channels do not form in isolation (4); rather, they are organized into two dimensional arrays visible at the light microscopic level (using direct fluorescent tagging (4, 5) or immunocytochemistry (6)) as intercellular puncta. In myelinating Schwann cells, immunohistochemical studies show Cx32 expression in the noncompact myelin of the paranodal loops and Schmidt-Lantermann incisures (7–12). Freeze-fracture replica immunogold labeling (FRIL) (13) confirmed these localizations but also showed connexin 32 (Cx32)2 gap junctions in internodes, between the outermost non-compact layer of myelin and the second outermost, partially compact layer. These findings suggest that Cx32 forms an intracellular reflexive pathway between adjacent layers of non-compacted myelin and thus can provide a radial diffusion pathway for signaling molecules, metabolites, or ions from the inner (adaxonal) to the outer (abaxonal) cytoplasm of a myelinating Schwann cell. This pathway is calculated to be 300 to 1000 times shorter than the circumferential pathway (2, 7) and is likely to play an important role in normal Schwann cell function and axon-Schwann cell interactions.
Balice-Gordon and colleagues (9) functionally demonstrated this pathway by injecting the junction-permeable fluorescent dye 5,6-carboxyfluorescein near the Schwann cell nucleus and tracking its transfer to the adaxonal region via radial paths thought to correspond to Schmidt-Lantermann incisures. However, they were also able to demonstrate similar dye transfer in nerve fibers from Cx32 KO mice. Thus, it remains to be shown whether Cx32 plays a role in the Schwann cell reflexive pathway.
Charcot Marie Tooth disease (CMT) is a group of inherited disorders characterized clinically by exclusively or predominantly peripheral nerve dysfunction (14). CMT1X, the most common form of X-linked CMT is caused by mutations in connexin 32 (Cx32) (15), a gap junction protein expressed in Schwann cells and oligodendrocytes as well as in several non-neural cell types (16). Hundreds of different mutations in Cx32 have been associated with this disorder (see The Mutation Database of Inherited Peripheral Neuropathies for a partial list). Scherer and colleagues, (17–19), we, (20), and others (21, 22) have shown that a number of Cx32 mutants show abnormal trafficking when expressed in transfected mammalian cell lines. Thus, a reasonable approach to treatment of CMT1X would be targeted toward correcting the trafficking defects. However, such an approach is only feasible if the mutations do not induce other significant changes in functional properties of the protein. Furthermore, the study of the functional properties of mutations in Cx32 should lead to a better understanding of structure-function relationships in this protein. Such studies, in conjunction with structural data such as that already available for Cx26 (23), may lead to strategies to correct the functional alterations demonstrated by these mutants.
In this work, we examine the functional effects of mutations at the Arg75 position of Cx32. This residue is highly conserved among members of the connexin family, and disease-causing mutations have been identified at this (or the corresponding) position in Cx26 (24), Cx43 (25), and Cx46 (26) (see supplemental Table 1 for additional references.) Thus, a better understanding of the effects of mutations of this position in Cx32 may have relevance to pathogenesis of a number of different human diseases.
MATERIALS AND METHODS
Generation of Mutants
The gene for human Cx32 was amplified by polymerase chain reaction (PCR) from human chromosomal DNA. Some mutants were then produced by oligonucleotide-directed PCR as described previously (27), screened by restriction digestion, and sequenced using primers encompassing the entire cloned PCR product and flanking restriction sites. Other mutants were produced using the Stratagene QuikChange Lightening site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA) followed by sequencing of the full coding sequence.
Transfection of Neuro2a Cell Lines
Confluent Neuro2A cells were transiently transfected with the DNA for Cx32WT or mutant subcloned into pIRES2-EGFP (Clontech, Palo Alto, CA) or pIRES2-DsRed (28). These IRES elements allow for the independent expression of the fluorescent proteins, which then act as cytoplasmic as markers of transfection. One day later, transfected cells were washed, and cells expressing a connexin-pIRES2-EGFP construct were either plated directly onto poly-d-lysine-coated 10-mm coverslips for homotypic pairing, or first mixed with cells expressing a connexin-pIRES2-DsRed construct for heterotypic pairing.
Immunofluorescence Microscopy
Neuro2a or HeLa cells were transiently transfected with 500 ng of WT or mutant Cx32 constructs using LTX and Plus Reagent (Invitrogen) per the manufacturer's instructions. The cultures were incubated for 24 h before being processed for immunofluorescence staining, and immunostained for Cx32 using mouse anti Cx32 (7C6.7C, gift of Dr. E. Hertzberg, 1:1000) and Alexa Fluor 594 goat anti mouse secondary antibodies and counterstained with DAPI as described previously (28). Slides were imaged at 100× using an Olympus Bx61 microscope outfitted for epifluorescence in conjunction with a Retiga digital CCD camera using a uniform exposure time (Q-imaging, Surrey, BC Canada). Individual images were montaged and adjusted for brightness and contrast in Adobe Photoshop.
Dual Whole-cell Recording
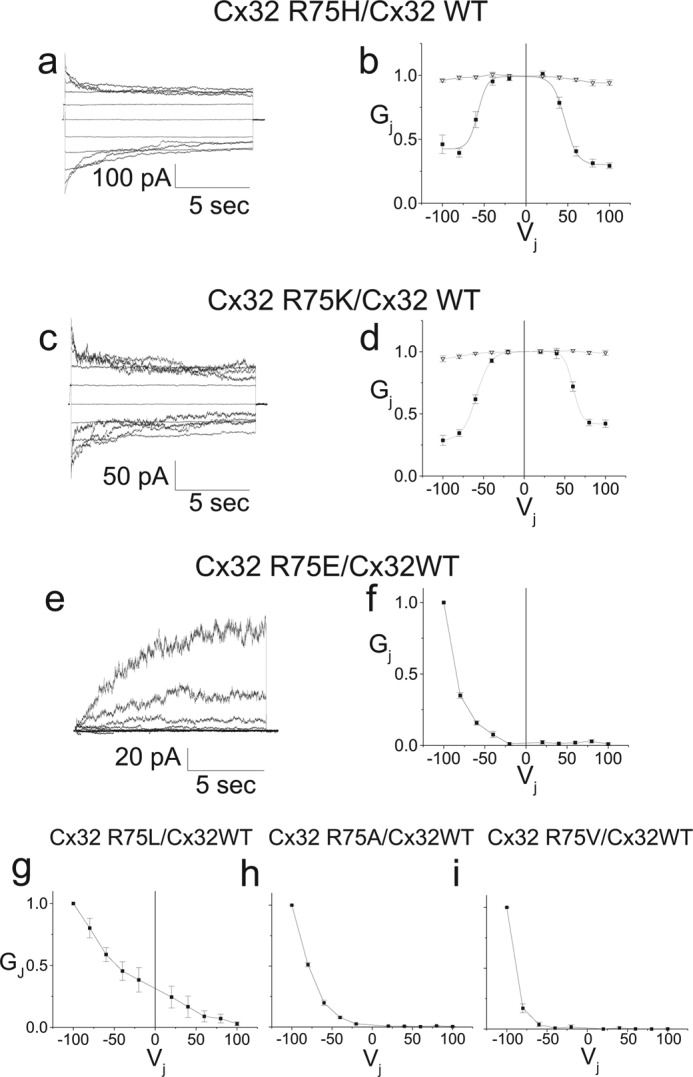
Voltage clamping of pairs of cells and analyses were performed as described previously (20). Heterotypic pairings are described as “connexin expressed in cell 2/connexin expressed in cell 1” with cell 1 being the cell pulsed. Both cells of a pair were voltage clamped at an identical potential (usually 0 mV) to yield a transjunctional voltage gradient (Vj) of zero. Conductance measurements were made by pulsing from Vj = 0 to ± 40 mV. Cytoplasmic bridges were excluded by demonstrating the sensitivity of junctional conductance to application of bath solution containing octanol. Values for different pairing configurations were compared using the Kruskal-Wallis test with Dunn's multiple comparison test. For determination of normalized steady-state junctional conductance-junctional voltage (Gj-Vj) relations, cells were held between each pulse at Vj = 0 for 22.5 s, and cell 1 was pulsed in 20-mV increments from −100 to +100 mV for 12.5 s. Any resulting change in current in cell 2, the apposed cell, is attributable to junctional current. Currents are displayed with positive up for both cells; thus, a positive Vj step in cell 1 causes a downward deflection in cell 2. For WT and mutants with measurable conductance at Vj = 0 mV, voltage pulses were preceded by a short (∼200 mS) pulse to −20 mV for normalization. Data were collected as described (27). Steady-state (t = ∞) conductances were determined by fitting each current trace to a sum of exponentials. Dividing the current at t = ∞ by the applied voltage gives the steady-state junctional conductance. For determination of Boltzmann parameters, steady-state plots were fit to the product of two Boltzmann distributions of the form,
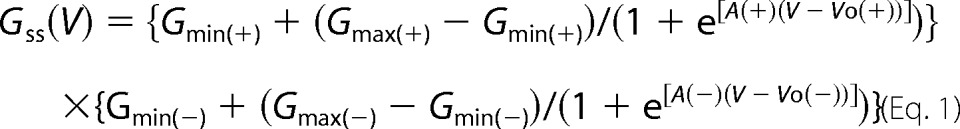 |
where Gss is the steady-state junctional conductance normalized to Vj = 0, Gmax is the maximal normalized conductance, and Gmin is the normalized residual conductance, which in macroscopic recordings is approached as the absolute value of Vj is increased, V0 is the voltage at which the conductance is one-half of the difference between Gmin and Gmax and roughly corresponds to the voltage at which a single connexin hemichannel has an open probability of 50%, and A is a parameter that reflects the slope of the Gj-Vj plot and is a measure of voltage sensitivity. A = nq/kT where n is the effective gating charge and q, k, and T have their usual meanings. The + and − designations indicate that the parameters are fits to the positive or negative limbs of the Gj-Vj relation. The model makes two assumptions that may not always be met for connexin gating: 1) it assumes a two state gating process where the energy difference between the states is proportional to the applied voltage, and 2) the steady-state Gj-Vj arises from the gating of two independent apposed hemichannels. However, the parameters generated provide a useful basis for comparison among channels produced by various pairing configurations of mutant and WT connexins. Homotypic pairs of Cx32WT-IRES2-EGFP served as a positive control and pairs of empty vector expressing cells served as negative controls for homotypically paired mutants. For heterotypic cell pair controls, an additional “heterotypic” pairing of Cx32WT-IRES2-EGFP with Cx32WT-IRES2-DSRed expressing cells was performed. The pipette solution consisted of the following: 145 mm CsCl, 5 mm EGTA, 1.4 mm CaCl2, 5.0 mm HEPES, pH 7.2; and bath solution consisted of the following: 150 mm NaCl, 4 mm KCl, 1 mm MgCl2, 2 mm CaCl2, 5 mm dextrose, 2 mm pyruvate, 5 mm HEPES; pH 7.4.
Predictions from X-ray Crystal Structure of Cx26
Salt bridges were predicted using ESBRI (29). A homology model of Cx32 WT and each of the mutants was created based on the average equilibrated structure of Cx26 (30) using MODELLER. For each mutant, 500 conformers were examined and the lowest statistical potential energy conformer (discrete optimized potential energy) was selected for further analysis. For each pair of residues examined, electrostatic energies were calculated in CHARMM by solution of Coulombs law using the dielectric constant of a vacuum, and van der Waals (vdW) energies were calculated by solution of Lennard-Jones potentials. All side chain atoms of the relevant residues were considered in each calculation.
RESULTS
CMT1X Mutants at Arg75 Show Reduced Levels of Functional Expression in Neuro2a Cells
To explore the role of mutations in codon 75 of GJB1 (Cx32) on the pathogenesis of CMT1X, we used dual whole cell patch clamping to examine coupling between isolated pairs of Neuro2a cells homotypically expressing the R75P, R75Q, or R75W mutants.
As shown in Table 1, homotypic cell pairs transiently transfected with DNA for Cx32WT produce average junctional conductances ∼42 μs, whereas the three CMT1X mutants, R75P, R75Q, and R75W, produced very low levels of coupling similar to those for pairs of cells transfected with empty vector (pIRES2-EGFP). This result might have been predicted on the basis of prior studies; As noted above, gap junction mediated coupling is thought to occur only between cells connected by gap junction plaques (4), and Yum and colleagues (19) have shown that stably transfected cells expressing the R75P, R75Q, and R75W mutants show an abnormal localization of connexin protein in a pattern suggestive of predominant accumulation in the Golgi, with few, if any, gap junction plaques. Our results in transiently transfected HeLa cells, shown in Fig. 1 are similar to those of Yum et al. (19) However, transiently transfected Neuro2a cells expressing the R75P and R75Q mutant show a somewhat different pattern (see Fig. 2 and the discussion below).
TABLE 1.
Junctional coupling for Cx32WT, mutants, and controls expressed in N2a cells in the homotypic and heterotypic configuration
| Pairing | n | Mean (ps) | S.D. | S.E. | p vs. WT | P vs. ie |
|---|---|---|---|---|---|---|
| 32WT/32WT | 7 | 41,900 | 41,900 | 13,100 | *** | |
| R75P/R75P | 7 | 7.71 | 2.40 | 7.71 | *** | ns |
| R75Q/R75Q | 7 | 22.1 | 43.6 | 16.5 | ** | ns |
| R75W/R75W | 9 | 10.4 | 21.7 | 7.24 | *** | ns |
| R75D/R75D | 7 | 35.3 | 74.7 | 28.2 | ** | ns |
| R75E/R75E | 5 | 49.2 | 58.8 | 19.3 | ** | ns |
| R75H/R75H | 31 | 827 | 2930 | 526 | *** | ns |
| R75K/R75K | 14 | 6720 | 10,600 | 2840 | ns | ** |
| R75A/R75A | 22 | 2750 | 9950 | 2120 | *** | ns |
| R75L/R75L | 9 | 13,700 | 18,900 | 6310 | ns | ** |
| R75V/R75V | 15 | 18.3 | 48.6 | 12.5 | *** | ns |
| E47R*R75E/E47R*R75E | 8 | 16.3 | 30.2 | 11.7 | *** | ns |
| R75E*E186R/R75E*E186R | 17 | 172 | 275 | 72.5 | ** | ns |
| 32WTie/32WTir | 15 | 20,600 | 21,800 | 5830 | *** | |
| R75Pie/32WTir | 16 | 29.4 | 63.3 | 15.8 | *** | ns |
| R75Qie/32WTir | 11 | 205 | 222 | 67.2 | * | ns |
| R75Wie/32WTir | 11 | 53.6 | 81.3 | 24.5 | *** | ns |
| R75Die/32WTir | 10 | 62.0 | 140 | 44.5 | *** | ns |
| R75Eie/32WTir | 7 | 27 | 37.63 | 14.22 | ** | ns |
| R75Hie/32WTir | 28 | 11,900 | 19,100 | 3620 | ns | *** |
| R75Kie/32WTir | 19 | 12,400 | 19,200 | 4400 | ns | *** |
| R75Aie/32WTir | 6 | 0 | 0 | 0 | *** | ns |
| R75Lie/32WTir | 12 | 5280 | 7280 | 2100 | ns | ** |
| R75Vie/32WTir | 6 | 0 | 0 | 0 | *** | ns |
| E47R*R75E/32WTir | 7 | 0 | 0 | 0 | *** | ns |
| R75E*E186R/32WTir | 5 | 56 | 125 | 56 | ** | ns |
| ie (empty vector) | 10 | 0 | 0 | 0 | *** |
Homotypic expressing cells were all transfected using pIRES2-EGFP vectors. ie indicates pIRES2-EGFP; ir indicates pIRES2-dsRed; ns indicates not significant.
*, p < 0.05;
**, p < 0.01;
***, p < 0.001.
FIGURE 1.

Localization of Cx32WT and selected mutants at the Arg75 position expressed in HeLa cells. HeLa cells were transiently transfected with DNA coding for the noted variants of Cx32 and stained with mouse anti-connexin antibodies in conjunction with an Alexa Fluor 594 secondary. As shown, Cx32WT and the R75L mutant show elongated puncta at intercellular appositions (arrows), consistent with gap junction plaques. R75P, R75Q, and R75L all exhibit a pattern of staining consistent with localization to the Golgi compartment. Note that the expression level for the R75W mutant is very low. Scale bar, 20 μm.
FIGURE 2.

Plaque formation by Cx32WT and mutants at the Arg75 position. Neuro2a cells were transfected with DNA coding for the noted forms of Cx32 and stained with mouse anti-connexin antibodies in conjunction with an Alexa Fluor 594 secondary. As shown, Cx32WT and all studied mutants except R75W show elongated puncta at intercellular appositions (arrows), consistent with gap junction plaques. Note that the expression level for the R75W mutant is very low. Scale bar, 15 μm.
The findings for all three mutations are consistent with a mechanism of disease of loss of function of Cx32. Mutations in connexins may lead to reduced levels of coupling either because they inhibit formation of junctions, for example, by causing defects in trafficking or assembly of connexins or hexameric connexons, or because the junctions form but have altered functional properties. As shown in Fig. 2, transiently transfected Neuro2a cells expressing the R75P and R75Q mutants each express gap junction plaques, making the lack of plaque formation a less likely explanation for the observed loss of function of these mutants in our studies. In contrast, no plaques are seen between R75W expressing cells, but the expression levels for this mutant are generally extremely low.
Alterations in the Voltage Dependence of Gating Can Account for Reduced Functional Expression of the R75P, R75Q, and R75W Mutants
Alterations in the voltage dependence of gating from that seen for the wild-type can lead to loss of gap junction mediated intercellular communication. Cx32WT hemichannels close when the transjunctional voltage is more negative than −40 with respect to the cell in which that hemichannel is expressed; thus, Cx32 is said to have negative gating polarity.3 Homotypic gap junction channels are composed of two identical apposed hemichannels. When two such hemichannels are apposed the resulting symmetric homotypic cell-cell channel will typically have a characteristic symmetric bell-shaped steady-state junctional conductance-junctional voltage (Gj-Vj) relation (see for example, the Gj-Vj relation for Cx32WT, Fig. 3b), where each limb of the curve is due to gating of one of the two hemichannels. (See below for a discussion of exceptions to this rule.)
FIGURE 3.
Representative current traces (a, c, and e) and Gj-Vj relations (b, d, and f) for R75P, R75Q, and R75W expressed in Neuro2a cells and paired heterotypically with Cx32 WT. The steady-state Gj-Vj relations are markedly shifted to the left with steady-state conductances at Vj = 0 that are markedly reduced compared with the conductance at −100 mV, consistent with substantially reduced open probabilities at Vj = 0. Steady-state Gj-Vj relations for each of these parings show very small Gmin, suggesting that these channels gate to a nonconducting (or very low conductance) state. Because the Cx32 hemichannel is predicted to be fully open at Vj > −40 mV, the low open probabilities at large positive Vj values are likely ascribable to the mutant hemichannel.
We (27) and others (2) have shown that for a number of mutants of Cx32 associated with CMT1X, the voltage-open probability relation is shifted so that even at junctional voltages much more positive than −40 mV, the hemichannels will still have a very low open probability. If this shift is of large enough magnitude, there will be no voltage at which both apposed hemichannels of a homotypic channel have a significant open probability. Such a situation would lead to non-conducting junctions. For the reflexive Schwann cell pathway, the relevant parameter is the open probability at Vj = 0, as significant voltage gradients are unlikely to develop across reflexive gap junctions (2).
The ideal way to demonstrate a shift in hemichannel voltage dependence of gating for a mutant would be to pair the mutant hemichannel heterotypically with a hemichannel that remains open throughout the voltage range being probed. In that case, all observed gating would be attributable to the mutant hemichannel. This situation is approximated by pairing the mutants with Cx32WT, which has a hemichannel open probability close to 1.0 for voltages more positive than −40 and has a residual conductance ∼20% of the maximum at more negative potentials. As shown in Fig. 3, when each of the CMT1X mutants is paired with Cx32WT, the resulting currents and normalized conductances show strong and asymmetric dependence on the applied trans-junctional voltage. These asymmetries reflect the very low open probability of these channels at Vj = 0 and are sufficient to explain the failure to observe coupling for these mutants when paired homotypically. In addition, the ability of these mutants to participate in formation of heterotypic currents, albeit with abnormal voltage dependences, suggests that in Neuro2a cells, these mutants are capable of trafficking to the plasma membrane and of docking with the Cx32WT hemichannels in the apposed cell. The apparent discrepancy between the measurable heterotypic R75W/Cx32WT currents (Fig. 3, e and f) and the lack of homotypic plaques by immunocytochemistry (Fig. 2) for the R75W mutant likely reflects the greater sensitivity of the electrophysiological studies to detect coupling between cell pairs with very low expression levels. Alternatively heterotypic pairing of R75W with the WT may induce or stabilize gap junction channel formation by the R75W mutant.
Normal Gating of Cx32 Requires a Positive Charge at Position 75
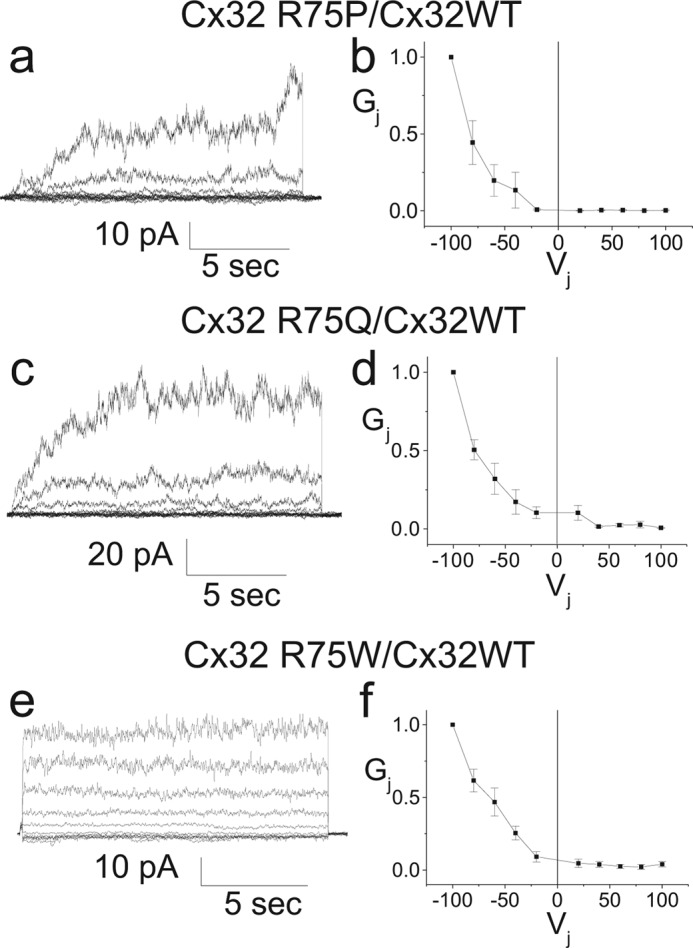
As noted above, the arginine at position 75 in Cx32 is highly conserved across all connexins and all species. In addition, mutations in this residue (or its homologous residue at position 76) have been associated with at least four different human disorders. Furthermore, the crystal structure of Cx26, a connexin closely related to Cx32, shows that the Arg75 residue to be a key component of an electrostatic cluster having a central role in interprotomeric interactions of the Cx26 hemichannel (23, 31). These facts suggest that the arginine at position 75 is critical for the normal functioning of Cx32. To further clarify the role(s) of this residue in the function of Cx32, we examined the effects of substitution of additional amino acids at this position. As shown in Table 1, cells expressing mutants in which the positively charged arginine has been replaced with a negatively charged aspartate or glutamate (R75D or R75E) failed to show functional coupling in the homotypic configuration above that of the control cells, whereas homotypically paired cells expressing mutants expressing a positively charged lysine or histidine at position 75 (R75K or R75H) both produced cell pairs with connexin mediated coupling. However, for the R75H mutant, the level of coupling was neither consistent enough, nor of great enough magnitude to achieve statistical significance. Fig. 2 shows that all tested mutants (except R75W, as noted above) formed clearly identifiable gap junction plaques when expressed in transiently transfected Neuro2a cells, making the failure of plaque formation an unlikely explanation for the absence of homotypic coupling.
As shown in Fig. 4, c and d, current traces and Gj-Vj relations for the homotypic R75K channels exhibit a symmetric bell shape with conductances falling to ∼20% percent of maximum at ± 100 mV and differing only subtly from those of Cx32WT, shown in Fig. 4, a and b. Boltzmann parameters for fits to the data are shown in Table 2. The current traces and steady-state Gj-Vj relation for the R75H mutant, shown in Fig. 4, e and f, show some asymmetry. In this case, small positive voltages reduce conductance, whereas small negative voltages increase it. As noted above, homotypic channels are composed of two identical apposed hemichannels, predicting a symmetric response to junctional voltage. The most straightforward explanation for the asymmetry is Vm (also called Vin-out) dependence (32), wherein gap junction channels show sensitivity to transmembrane voltage; in this case, positive transmembrane voltage tends to close the channel, whereas negative transmembrane voltage tends to open it. This asymmetry is reflected in the Boltzmann parameters shown in Table 2.
FIGURE 4.
Representative current traces (a, c, e, and g) and Gj-Vj relations (b, d, f, and h) for Cx32WT, R75K, R75H, and R75L expressed in Neuro2a cells and paired homotypically. The R75K and R75H mutants show steady-state conductance voltage relations similar to those for WT, whereas the R75L mutant shows a markedly increased Gmin.
TABLE 2.
Boltzmann parameters for homotypic pairings of Cx32 WT and mutant forms of Cx32
Steady-state Gj-Vj relations were determined for each type of junction, as described under “Materials and Methods” and fit to Boltzmann distributions. Data for negative (−) and positive (+) Vi are given separately. Gmin, Gmax, V0, and A have their usual meanings and are defined under “Materials and Methods.”
| 32WT/32WT | R75K/R75K | R75H/R75H | R75K/32WT | R75H/32WT | |
|---|---|---|---|---|---|
| Gmax (+V) | 1.02 | 1.07 | 1.00 | 0.99 | 1.00 |
| Gmin (+V) | 0.22 | 0.15 | 0.27 | 0.41 | 0.31 |
| +V0 | 49.37 | 45.18 | 47.75 | 60.34 | 46.76 |
| K (+V) | 0.098 | 0.054 | 0.091 | 0.18 | 0.14 |
| Gmax (−V) | 1.04 | 1.09 | 1.00 | 1.01 | 0.99 |
| Gmin (−V) | 0.21 | 0.19 | 0.27 | 0.28 | 0.42 |
| −V0 | −48.06 | −48.04 | −62.38 | −58.84 | −57.60 |
| K (−V) | −0.094 | −0.072 | −0.13 | −0.11 | −0.18 |
In contrast to the R75H and R75K mutants, the R75L mutant produces currents and a steady-state Gj-Vj relation dramatically different than those of WT, with a residual state of ∼0.8 instead of 0.2 (see Fig. 4, g and h). The most straightforward explanation for this finding is that the hemichannel open probability at Vj = 0 is low; thus, most of the conductance seen at Vj = 0 represents substate conductance, and the reduction in conductance seen with increases in the absolute value of Vj represent closure of the remaining open channels to a substate or closure of a fraction of the channels from substate to the fully closed state (loop gating). We also examined the R75A and R75V mutants and found that neither mutant consistently induced currents above those of the negative control. The small amount of conductance noted for cells expressing the R75A mutant, resulting from the contribution of two well coupled pairs, is of unclear significance.
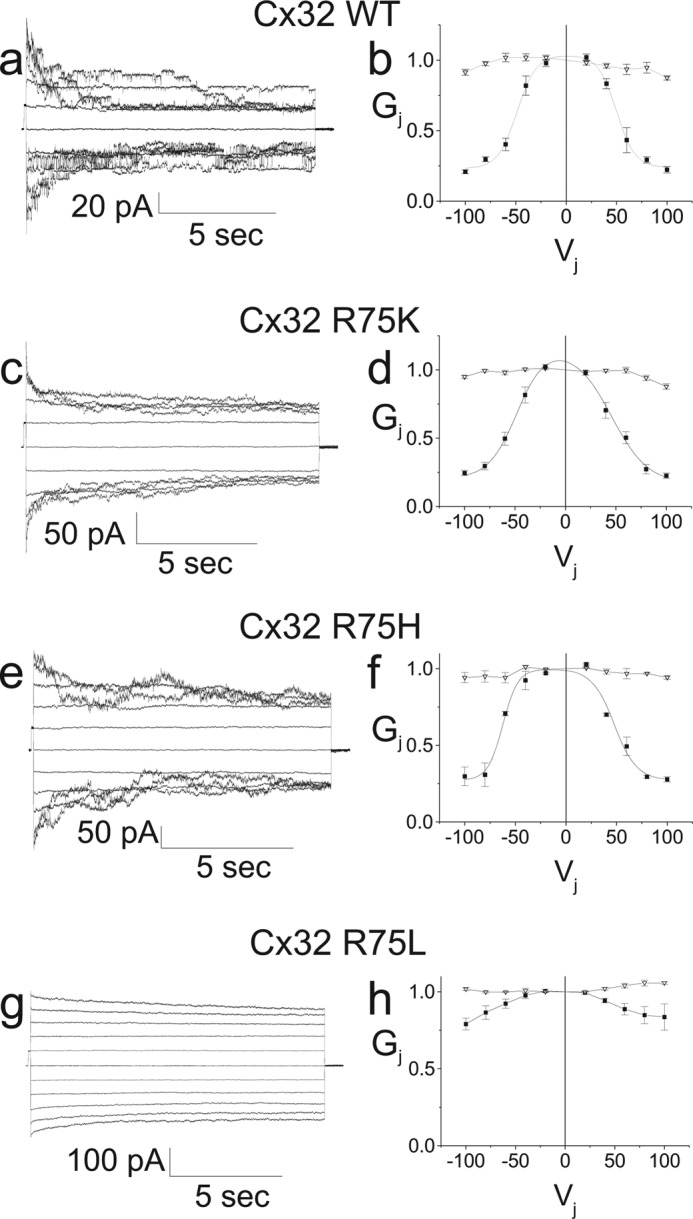
Loss of Positive Charge at Position 75 of Cx32 Shifts the Relationship between Hemichannel Open Probability and Junctional Voltage
To further investigate the reasons for our failure to observe currents induced by the homotypically paired R75D, R75E, R75V, and R75A mutants and to further examine the properties of the R75H, R75K, and R75L mutants, we recorded from each of these mutants in the heterotypic configuration with Cx32 WT. Fig. 5, a and b, show representative current traces and average Gj-Vj relation for the R75H/32WT junctional currents. The currents and Gj-Vj relation are similar to those for the homotypic Cx32WT pairs, however as seen in Fig. 5, a and b, and quantitated in the Boltzmann parameters summarized in Table 2, the V0 and Gmin values are somewhat higher for the limb corresponding to gating of the Cx32 WT hemichannel than for the limb corresponding to the R75H hemichannel. The Gj-Vj relation for R75K/32WT differs only subtly from that of the Cx32 WT homotypic channels, as shown in Fig. 5, c and d, and quantified in the Boltzamnn parameters in Table 2. On the other hand, as shown in Fig. 5g, the Gj-Vj relation for R75L/32WT differs very significantly from that of the Cx32 WT homotypic channels and is consistent with a substantial shift in hemichannel open probability-voltage relation, though not as great as those for R75P, R75Q, R75W, R75E, R75A, or R75V. As shown in Fig. 5, e and f, when paired with Cx32 WT, the R75E mutant induces currents that increase as Vj is made more negative than −20 with respect to the cell expressing Cx32 WT. As is the case for the R75P, R75Q, and R75W mutants, the failure to detect junctional coupling could be entirely explained by this shift in the hemichannel Gj-Vj relation. A similar argument can also be made to explain the failure to detect currents in homotypically paired R75A (see Fig. 5h) and R75V (see Fig. 5i) expressing cells. Only the R75D mutant failed to consistently induce junctional currents when paired heterotypically with Cx32 WT; we saw small amounts of current activation at −100 mV with respect to Cx32WT in a few cell pairs (data not shown). This finding suggests that the R75D mutation may induce shifts in voltage dependence that are even greater than those for R75E or the other examined mutations, leading to very little R75D hemichannel activation even at large negative voltages with respect to Cx32 WT. The well expressed plaques seen in by immunocytochemistry for the R75D mutant (Fig. 2) are consistent with this notion. Overall, the observed degree of shift in hemichannel open probability for the Arg75 mutants at Vj = 0 is WT ≈ Lys ≈ His < Leu < Trp ≈ Gln < Glu ≈ Ala ≈ Pro < Val.
FIGURE 5.
Representative current traces (a, c, and e) and Gj-Vj relations (b, d, and f) for R75H, R75K, and R75E expressed in Neuro2a cells and paired with Cx32 WT, and Gj-Vj relations (g, h, and i) for R75L, R75A, and R75V expressed in Neuro2a cells and paired heterotypically with Cx32 WT. The R75H and R75K mutants show relatively symmetric steady-state conductance voltage relations. The conductance voltage relation for the R75E, R75L, R75A, and R75V mutants show marked shifts in their Gj-Vj relations, although the shift for R75L appears to be less than those for R75E, R75A, and R75V.
In Silico Mutagenesis Provides Insight into the Effects of Amino Acid Substitutions at Position 75 in Cx32
To further explore structural changes that may result from substitution of Arg75 by other charged and uncharged amino acids, we performed in silico mutagenesis of a Cx32 homology model created with MODELLER as described under “Materials and Methods.” As a prelude to a complete characterization by molecular dynamics simulation, we determined the extent to which mutations of Arg75 analyzed in this study altered non-bonded interactions (van der Waals and Columbic forces) in the unequilibrated Cx32 homology model. The analysis in effect estimates changes in the potential energy of the structure and thus provides a preliminary measure of the relative perturbation to the homology model caused by any given substitution. We reasoned that this approach, although preliminary, is preferable to inference of structural defects based on the Cx26 crystal structure, which is characterized by several energetically unfavorable van der Waals interactions in the region of Arg75. The crystal structure also failed to identify the intersubunit interaction between Arg75 and Glu42, the intrasubunit interaction of Arg184 with Glu42, and the intrasubunit interaction between Lys188 and Asp46 that are evident in the atomic structure following all-atom molecular dynamics simulations. Consequently, the electrostatic network that includes Arg75 is less extensive in the crystal structure than in the equilibrated structure.
There are two important caveats to the interpretation of these data. First, these values of non-bonded energy reflect the perturbations to the unequilibrated homology model of Cx32 that is based on an equilibrated model of the open state of Cx26 (30). Thus, they can only provide an indication of the degree of perturbation arising from substitution of each of the residues noted and cannot predict the final structure that would be obtained by MD simulation. Second, these values are based solely on perturbations to the open structure. Because the open probability of a voltage-dependent channel at any given voltage is a function of the difference in free energies of the open and closed state at that voltage, these calculated values provide a necessarily incomplete picture of the effects of the mutations. Ultimately, a detailed understanding of structural perturbations caused by these mutations requires atomistic models of both open and closed states.
In Cx26, Arg75 is a component of a large inter- and intrasubunit electrostatic network that links all six connexin subunits and stabilizes the loop-gate permeability barrier when the channel resides in the open state. The loop-gate permeability barrier is formed primarily by a segment of the channel pore termed the parahelix (residues 42–51) (31, 33, 34). In the equilibrated open state Cx26 model, Arg75 forms intrasubunit salt bridges with Glu47 and intersubunit salt bridges with Glu42 and Glu187. The open state is also stabilized by an extensive intrasubunit vdW network that we do not consider in this analysis, although it is likely that Arg75 substitutions will also change the composition and dynamics of this network, as the two are linked (31).
The analyses of MD trajectories show that fluctuations within the electrostatic network are directly coupled to structural fluctuations of the permeability barrier in Cx26 hemichannels. This finding, and the position of the electrostatic network in or near the channel pore, strongly suggested that Arg75 forms part of the connexin loop-gate voltage sensor. Thus, amino acid substitutions at this position are expected to produce shifts in voltage gating that could arise as a consequence of changes in both gating charge and the relative free energy of open and closed states.
The computed changes in Columbic and vdW energies are shown in Table 3. We consider both electrostatic and vdW interactions of all side chain atoms in wild type and mutant Cx32. The electrostatic values are estimates of the Columbic energies of Arg75 or the given mutated residue at position 75 in the open channel with the Glu at position 47 or 186 and the Ser at position 42. (Ser42 in Cx32 is included because in Cx26 both side chains and backbone carbonyl atoms of Glu42 have significant interactions with Arg75. Glu186 in Cx32 is the homolog of Glu187 in Cx26.) It should be noted that although neutral residues have a net charge of 0, partial charges on atoms exist in CHARMM force fields and these were used to calculate changes in electrostatic energy. For example, atom Cβ of alanine carries a partial charge of −0.27. Also important are the positive charges found directly above the centers of the aromatic rings of tryptophan. This distribution of charge allows π-cation interactions.
TABLE 3.
Electrostatic (elec) and Van der Waals (vdW) interaction energies between the noted mutant and Glu47, Glu186, or Ser42 in a Cx32 homology model
Also given are the total electrostatic and vdW interaction energies for the three interactions.
| Mutant | Glu47-Arg75 |
Arg75-Glu186 interaction |
Ser42-Arg75 interaction |
Total |
||||
|---|---|---|---|---|---|---|---|---|
| elec | vdW | elec | vdW | elec | vdW | elec | vdW | |
| R75R (WT) | 211.29 | −0.210 | −223.22 | −0.390 | −46.11 | 0.280 | −480.63 | −0.321 |
| R75K | −86.06 | −0.089 | −109.19 | −0.263 | −16.15 | −0.144 | −211.40 | −0.495 |
| R75H_1+ | −39.85 | −0.024 | −61.30 | −0.152 | −14.50 | −0.254 | −115.66 | −0.429 |
| R75H_0 | −19.48 | −0.024 | −40.13 | −0.157 | −22.58 | −0.232 | −82.19 | −0.413 |
| R75E | −10.09 | −0.015 | −33.48 | −0.236 | −40.12 | −0.124 | −83.68 | −0.375 |
| R75D | −5.67 | −0.007 | −21.31 | −0.121 | −34.53 | −0.048 | −61.51 | −0.176 |
| R75Q | −19.75 | −0.023 | −40.65 | −0.224 | −37.20 | −0.169 | −97.59 | −0.416 |
| R75W | −39.28 | 0.743 | −47.54 | 0.500 | −39.84 | 2.206 | −126.66 | 3.450 |
| R75L | −17.75 | −0.042 | −50.27 | 0.173 | −18.81 | −0.186 | −86.83 | −0.055 |
| R75V | −12.63 | −0.025 | −35.08 | −0.253 | −10.38 | −0.059 | −58.09 | −0.337 |
| R75P | −13.70 | −0.031 | −24.06 | −0.133 | −6.49 | −0.041 | −44.25 | −0.205 |
| R75A | −5.12 | −0.007 | −15.22 | −0.103 | −3.16 | −0.013 | −23.50 | −0.123 |
The most conservative mutation, R75K had the smallest effect on the total interaction energies between R75K and residues 42, 47, and 186 (Table 3). The decrease in electrostatic energies almost certainly reflects the reduced length of the lysine side chain relative to that of the arginine residue. Similarly, the increase in vdW interaction energies determined by solution of Lennard-Jones potential energy function (6–12 potential) for Glu47-R75K and R75K-Glu186 is consistent with an increase in distance among atoms. Notably, in the wild-type homology model the calculated vdW energy between residue Arg75 and Ser42 is slightly positive, 0.28 kcal/mol. This result most likely reflects a decrease in distance between Ser42 and Arg75 from that between Glu42 and Arg75 in the equilibrated Cx26 structure. Note that the Cx32 structure is based on the validated open state model of Cx26 that resulted from MD equilibration (Kwon et al. (31)). The slight positive value obtained for the Cx32 Ser42-Arg75 interaction persisted following energy minimizations that optimized the position of the serine side chain in the homology model. The decrease in vdW interaction energy calculated for Ser42-R75K is again consistent with the reduced length of the lysine side chain. The smaller side chain, would increase the distance between all pairs of atoms in Ser42 and Lys75, and consequently reduce the repulsive force, which in Lennard-Jones potential function increases by the 12th power. Thus, the short range energies calculated by Lennard-Jones equations are very sensitive to the distance of separation, with repulsive forces increasing dramatically as atoms are moved closer together. In general, vdW energies calculated by LJ are minimized when atoms are separated by ∼2 Å.
The changes in the electrostatic and vdW energies are smallest for R75K substitution relative to all other mutations. This suggests that R75K would perturb structure less than other substitutions following model equilibration, as suggested by our electrophysiological measurements.
The largest changes in electrostatic and vdW energies are for the R75A substitution. This most likely reflects the decrease in volume of the alanine side chain and rather small partial charge on atom Cβ that would make only small contribution to calculated Columbic energy. This result suggests that R75A open channel will undergo substantial conformational change during energy optimization by MD in that reconfiguration of both electrostatic and vdW networks will likely be required to obtain a low energy structure. If the energy of the resulting structure is greater than that of wild type, then one would reasonably predict that the open state would be destabilized and that this would result in a shift in the open-probability-voltage relation. Interestingly, R75W electrostatic energies are decreased but to a lesser extent than those of other neutral and negatively charged substitutions. This result can be explained by π-cation interactions. However, R75W causes a substantial and the largest increase in calculated vdW energies of all mutations. This increase in energy is most likely a consequence of the large volume of the tryptophan side chain. It is expected that this interaction energy, which reflects relatively short distance interactions among atoms, will likely play a major role in the optimization of R75W structure by MD.
Although these preliminary calculations of energies cannot by themselves explain the degree of shift in open hemichannel probability observed, they provide an indication of the degree to which different mutations might perturb Cx32 structure. Notably, with the exception of R75K, all mutations cause significant perturbations of the electrostatic network that are expected to cause structural changes during geometry optimization by MD. In all cases, the mutations are expected to decrease the strength of the electrostatic interactions emanating from Arg75, which plays an important role in stabilizing open channel structure. Determination of the degree to which mutations destabilize the open structure awaits equilibration of WT and mutant structures by MD simulation. A complete understanding of the role of a given Arg75 mutation also requires knowledge of the perturbation of the closed state. Computational methods such as free energy perturbation molecular dynamics can provide measures of relative changes in the free energy of open and closed states and consequently allow direct comparison with experimental results.
The electrostatic interaction energies for the protonated form of R75H are even lower, again potentially providing some basis for the observed low level of homotypic coupling seen for this mutant. With the exception of R75W, all the other studied mutants show even greater destabilization. With respect to the three aliphatic substitutions, this analysis also suggests that R75L is a less destabilizing mutation than either the R75A or R75V mutations, which may explain the relatively high expression and less shifted voltage dependence of the R75L mutant. This in turn may be due, at least in part to the reduced steric perterbations induced by an Arg to Leu as opposed to an Arg to Ala or to Val substitution. Nonetheless our analysis does not account for the apparent greatly increased stability of R75L when compared with the Trp, Gln, or Glu mutations, all of which show similar electrostatic interaction energies but more markedly shifted steady-state Gj-Vj relations. Regarding effects on vdW forces, the effect conferred by the R75W substitution is the largest seen for any mutant studied and the only one which would be predicted to be significantly destabilizing. It is likely a result of the disruption caused by the bulky hydrophobic side chain of this residue.
As noted above, the positive charge at Arg75 in Cx32 is predicted to interact with both Glu47 and Glu186. Thus we reasoned that it might be possible to restore the functional deficits induced by the R75E mutation by also placing an arginine at either position 47 or 186 to produce the Cx32-R75E*E47R or Cx32-R75E*E186R double mutants. As shown in Table 1, when examined in the homotypic configuration or heterotypic pairing with Cx32WT, neither the Cx32-E47R*R75E nor the Cx32-R75E*E186R mutants induced coupling significantly above that of control cells. (The very low levels of coupling noted in some cell pairs had the characteristics of the endogenous gap junction channels occasionally seen between control Neuro2a cells.) However, in reality, each of these charged residues is involved in an extensive electrostatic network involving multiple charged residues (30); thus, in retrospect, it was unrealistic to posit that simply inverting two of the charges within the network would normalize the gating behavior of the channel.
DISCUSSION
Implications of Our Findings for CMT1X and Other Connexin Diseases
In this study, we examined the effects of substitutions for the Arginine at position 75 on Cx32-mediated coupling and on the properties of the channels underlying the coupling. We were specifically motivated to study these effects because of the highly conserved nature of Arg75 across all connexins and because amino acid changes at this position in four human connexins are associated with disease. Because of our interest in the pathogenesis of CMT1X, associated with mutations in Cx32, we began by examining the effects of three CMT1X-associated mutations in Cx32. All three mutations (R75P, R75Q, and R75W) induced no homotypic junctional coupling above base line and showed shifts in their heterotypic Gj-Vj characteristics sufficient to explain this. This is an important finding because it highlights the complicated effects that mutations may have on Cx32. In this case, each of these three mutant proteins was previously shown to have an abnormal subcellular distribution, failing to form gap junctions and primarily co-localizing with markers for the Golgi apparatus. The reasonable conclusion is that these are trafficking mutants. Because all available data suggest that most, if not all, cases of CMT1X are due loss of connexin function, it would be tempting to conclude that treatment aimed at bypassing the trafficking defect should ameliorate the disease. However, our results clearly indicate that simply correcting the abnormal subcellular distribution caused by these and many other mutations in Cx32 may not be sufficient to ameliorate the disease state.
Implications of Our Findings for the Role of Charge at Position 75
Kwon et al. (31) have shown that Arg75 is part of an electrostatic network, which they hypothesize forms the loop-gate voltage sensor. To investigate the role of Arg75 in voltage gating, we examined the effects of substitution of the positively charged arginine at position 75 by negatively charged (Asp and Glu,) neutral (Ala, Leu and Val), and positively charged (His and Lys) residues. Based on these experiments and the steady-state Gj-Vj, characteristics of these mutants in the homotypic and heterotypic configuration with WT Cx32, we conclude that a positive charge at position 75 is required for the channel formed by Cx32 to have a WT-like gating characteristic. However, based on data for junctional coupling, we conclude that the presence of a positively charged residue (e.g. R75K) at this position does not ensure completely normal levels of junctional coupling. The relevance of results for R75H to the role of charge at this position must be interpreted with caution because the pK of the imidazole side chain of His is 6.0 in free solution, meaning that (neglecting the potential contribution of to local environment to shifts in pK), the residue would be predicted to carry only a small partial positive charge at the pH of our internal pipette recording solutions. This predicted reduced positive charge may also help to explain the finding that homotypically coupled cells expressing the R75H mutant produced coupling ∼1/40th that of WT (see Table 1). Reduced coupling would be predicted if a significant fraction of the R75H hemichannels were in a state where their open probability is shifted such that it is very low at Vj = 0, a situation that could arise if the unprotonated imidazole stabilizes the closed state. Assuming that the delivery of R75H hemichannels to gap junction plaques occurs with the same efficiency as for WT, ∼84% of the hemichannels would need to be in this stabilized closed state. (This is because both apposed hemichannels need to be in the unshifted state to produce a WT-like cell-cell channel, so that a conductance of 1/40th of WT means that the probability of an individual hemichannel being in the unshifted state is √(1/40) ≈ 0.16.) Alternatively, the R75H mutant may interfere with trafficking of hemichannels to gap junctions. Regardless of which explanation one adopts, the degree of heterotypic coupling of R75H with Cx32WT is ∼3.6-fold higher than that that predicted from the homotypic coupling. This may reflect either ability of the WT hemichannels to recruit R75H channels into the gap junction or an ability of these channels to stabilize the “unshifted” state of the R75H hemichannel or a combination of these two mechanisms.
Although the absence of a positive charge at position 75 leads to a channel with altered voltage dependence of gating in every case tested, based on our results with the R75L mutant, we conclude that the lack of positive charged residue does not in itself limit levels of junctional coupling. As shown in Table 1, the conductance of homotypically paired cells expressing the R75L mutation is ∼40% of that of WT. Assuming that the single channel conductance of the R75L channel is similar to that of WT, this reduction in coupling can be fully accounted for by the reduction in hemichannel open probability at Vj = 0 which, as determined from the data shown in Fig. 4g, is no greater than 31% of maximum.
Shifts in the Gj-Vj relation such as seen for the Ala, Glu, Leu, Pro, Gln, Val, and Trp substitutions and inferred for the Asp substitution likely reflect an increased stability of the closed state relative to that of the open state. The findings of relatively normal gating for the Lys and His substitutions make it reasonable to speculate that the open state is stabilized by electrostatic interactions involving Arg75 as one partner. In their studies of an energy minimized structure of Cx26, Kwon et al. (31) identified three residues that would be predicted to have electrostatic interactions with Arg75. One is an intrasubunit interaction with Glu47, and two are with Glu187 or Glu42 of the adjacent subunit. We used MODELLER to build an homology model of Cx32 using the structure of human C32 aligned x-ray crystal structure of Cx26. As for Cx26, intrasubunit interactions are predicted for Glu47 and with a Glu186 of the adjacent subunit (homologous to Cx26-Glu187), with no other potential electrostatic interactions within 4 Å. Glu42 in Cx26 is replaced by a Ser in Cx32; thus, interactions between Arg75 and this residue would likely be much weaker.
Implications of Our Findings in the Context Other Studies on Mutations at Position 75 in Cx26 and Cx32
Several studies have been undertaken to study the properties of pathogenic and nondisease-causing mutations of Arg75 in Cx26 and Cx32. These are summarized in columns 5 and 6 of supplemental Table 1. Most functional studies have been performed for mutant forms of Cx26. The R75W mutant showed no cell-cell coupling in one study (35), and in another study (36), no cell coupling was seen when the R75A, R75D, R75N, R75K, R75F, R75Y, or R75W mutants were expressed in the homotypic configuration in Xenopus oocytes. Based on the results of our study, one might have predicted that the R75K mutant would produce functional channels. However, these data may not be directly comparable with those in our study because the effect of the mutations in Cx26 on formation of gap junction plaques, a prerequisite for functional coupling (37), was not determined. Another study of Cx26 mutations (38) showed dye transfer between HeLa cells expressing the Cx26-R75D above that of control cells, suggesting that Cx26-R75D can form functional homotypic channels; the discrepancy between these data and those of Deng et al. (36) may relate to the reduced specificity of the dye transfer technique (for example, coupling may occur due to up-regulation of an endogenous connexin or cytoplasmic bridges (39)) or less likely to differences between the behavior of the Cx26-R75D mutant in Xenopus oocytes and HeLa cells. More puzzling is the report of Wang et al. (40) that Cx32-R75Q forms functional homotypic channels in Neuro2a cells. However, the specificity of their assay is difficult to assess, as they provide no data on voltage or chemical dependence of the coupling currents measured, which would allow for the exclusion of cytoplasmic bridges (39) as a cause of junctional coupling.
Conclusions
In this work, we have examined 10 mutations, including two that preserve the positive charge at position 75, and eight others that do not. Only the charge preserving mutations show normal gating behavior, supporting the hypothesis that a positively charged residue at position 75 is sufficient and necessary to form a channel with normal gating behavior. Conversely, expression levels of mutants with charge preserving mutations are reduced, whereas expression levels of the R75L mutant are unaffected. These findings suggest that a positively charged residue at position 75 is neither sufficient nor necessary to ensure normal trafficking and assembly, both of which would need to occur to have normal levels of coupling.
As noted above, disease-causing mutations have been identified in four connexins at position 75. In addition to Cx32, mutations have been identified in Cx26, Cx43, and Cx46. It will be interesting to see whether the charge at position 75 is as critical for channel gating in these connexins as it appears to be in Cx32.
This work was supported by National Institutes of Health Grant 1R01NS050705 and Muscular Dystrophy Association research grants (to C. K. A.).

This article contains supplemental Table 1 and additional references.
In general, two separate kinds of gates control the conducting state of each hemichannel. The Vj gate of a connexin hemichannel closes the channel to a substate, whereas the loop or slow gate closes the channel fully. These gates can be described as having a negative or positive gating polarity, depending on whether they close on negative or positive potential with respect to the cell in which they are expressed. For Cx32, both the Vj gate and the loop gate have a negative polarity (41, 42). See Bukauskas and Verselis (43) for further discussion.
- Cx32
- connexin 32
- CMT
- Charcot Marie Tooth disease
- EGFP
- enhanced GFP
- IRES
- internal ribosome entry site
- vdW
- van der Waals.
REFERENCES
- 1. Willecke K., Eiberger J., Degen J., Eckardt D., Romualdi A., Güldenagel M., Deutsch U., Söhl G. (2002) Structural and functional diversity of connexin genes in the mouse and human genome. Biol. Chem. 383, 725–737 [DOI] [PubMed] [Google Scholar]
- 2. Oh S., Ri Y., Bennett M. V., Trexler E. B., Verselis V. K., Bargiello T. A. (1997) Changes in permeability caused by connexin 32 mutations underlie X-linked Charcot-Marie-Tooth disease. Neuron 19, 927–938 [DOI] [PubMed] [Google Scholar]
- 3. Kumar N. M., Gilula N. B. (1996) The gap junction communication channel. Cell 84, 381–388 [DOI] [PubMed] [Google Scholar]
- 4. Bukauskas F. F., Jordan K., Bukauskiene A., Bennett M. V., Lampe P. D., Laird D. W., Verselis V. K. (2000) Clustering of connexin 43-enhanced green fluorescent protein gap junction channels and functional coupling in living cells. Proc. Natl. Acad. Sci. U.S.A. 97, 2556–2561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gaietta G., Deerinck T. J., Adams S. R., Bouwer J., Tour O., Laird D. W., Sosinsky G. E., Tsien R. Y., Ellisman M. H. (2002) Multicolor and electron microscopic imaging of connexin trafficking. Science 296, 503–507 [DOI] [PubMed] [Google Scholar]
- 6. Janssen-Timmen U., Dermietzel R., Frixen U., Leibstein A., Traub O., Willecke K. (1983) Immunocytochemical localization of the gap junction 26 K protein in mouse liver plasma membranes. EMBO J. 2, 295–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scherer S. S., Deschênes S. M., Xu Y. T., Grinspan J. B., Fischbeck K. H., Paul D. L. (1995) Connexin32 is a myelin-related protein in the PNS and CNS. J. Neurosci. 15, 8281–8294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Altevogt B. M., Kleopa K. A., Postma F. R., Scherer S. S., Paul D. L. (2002) Connexin29 is uniquely distributed within myelinating glial cells of the central and peripheral nervous systems. J. Neurosci. 22, 6458–6470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Balice-Gordon R. J., Bone L. J., Scherer S. S. (1998) Functional gap junctions in the schwann cell myelin sheath. J. Cell Biol. 142, 1095–1104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sutor B., Schmolke C., Teubner B., Schirmer C., Willecke K. (2000) Myelination defects and neuronal hyperexcitability in the neocortex of connexin 32-deficient mice. Cereb. Cortex 10, 684–697 [DOI] [PubMed] [Google Scholar]
- 11. Chandross K. J., Kessler J. A., Cohen R. I., Simburger E., Spray D. C., Bieri P., Dermietzel R. (1996) Altered connexin expression after peripheral nerve injury. Mol. Cell Neurosci. 7, 501–518 [DOI] [PubMed] [Google Scholar]
- 12. Mambetisaeva E. T., Gire V., Evans W. H. (1999) Multiple connexin expression in peripheral nerve, Schwann cells, and Schwannoma cells. J. Neurosci. Res. 57, 166–175 [DOI] [PubMed] [Google Scholar]
- 13. Meier C., Dermietzel R., Davidson K. G., Yasumura T., Rash J. E. (2004) Connexin32-containing gap junctions in Schwann cells at the internodal zone of partial myelin compaction and in Schmidt-Lanterman incisures. J. Neurosci. 24, 3186–3198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Siskind C. E., Shy M. E. (2011) Genetics of neuropathies. Semin Neurol. 31, 494–505 [DOI] [PubMed] [Google Scholar]
- 15. Bergoffen J., Scherer S. S., Wang S., Scott M. O., Bone L. J., Paul D. L., Chen K., Lensch M. W., Chance P. F., Fischbeck K. H. (1993) Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science 262, 2039–2042 [DOI] [PubMed] [Google Scholar]
- 16. Wilgenbus K. K., Kirkpatrick C. J., Knuechel R., Willecke K., Traub O. (1992) Expression of Cx26, Cx32 and Cx43 gap junction proteins in normal and neoplastic human tissues. Int. J. Cancer 51, 522–529 [DOI] [PubMed] [Google Scholar]
- 17. Deschênes S. M., Walcott J. L., Wexler T. L., Scherer S. S., Fischbeck K. H. (1997) Altered trafficking of mutant connexin32. J. Neurosci. 17, 9077–9084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kleopa K. A., Yum S. W., Scherer S. S. (2002) Cellular mechanisms of connexin32 mutations associated with CNS manifestations. J. Neurosci. Res. 68, 522–534 [DOI] [PubMed] [Google Scholar]
- 19. Yum S. W., Kleopa K. A., Shumas S., Scherer S. S. (2002) Diverse trafficking abnormalities of connexin32 mutants causing CMTX. Neurobiol. Dis. 11, 43–52 [DOI] [PubMed] [Google Scholar]
- 20. Abrams C. K., Freidin M., Bukauskas F., Dobrenis K., Bargiello T. A., Verselis V. K., Bennett M. V., Chen L., Sahenk Z. (2003) Pathogenesis of X-linked Charcot-Marie-Tooth disease: differential effects of two mutations in connexin 32. J. Neurosci. 23, 10548–10558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matsuyama W., Nakagawa M., Moritoyo T., Takashima H., Umehara F., Hirata K., Suehara M., Osame M. (2001) Phenotypes of X-linked Charcot-Marie-Tooth disease and altered trafficking of mutant connexin 32 (GJB1). J. Hum. Genet. 46, 307–313 [DOI] [PubMed] [Google Scholar]
- 22. Martin P. E., Mambetisaeva E. T., Archer D. A., George C. H., Evans W. H. (2000) Analysis of gap junction assembly using mutated connexins detected in Charcot-Marie-Tooth X-linked disease. J. Neurochem. 74, 711–720 [DOI] [PubMed] [Google Scholar]
- 23. Maeda S., Nakagawa S., Suga M., Yamashita E., Oshima A., Fujiyoshi Y., Tsukihara T. (2009) Structure of the connexin 26 gap junction channel at 3.5 A resolution. Nature 458, 597–602 [DOI] [PubMed] [Google Scholar]
- 24. Richard G., White T. W., Smith L. E., Bailey R. A., Compton J. G., Paul D. L., Bale S. J. (1998) Functional defects of Cx26 resulting from a heterozygous missense mutation in a family with dominant deaf-mutism and palmoplantar keratoderma. Hum. Genet. 103, 393–399 [DOI] [PubMed] [Google Scholar]
- 25. Paznekas W. A., Boyadjiev S. A., Shapiro R. E., Daniels O., Wollnik B., Keegan C. E., Innis J. W., Dinulos M. B., Christian C., Hannibal M. C., Jabs E. W. (2003) Connexin 43 (GJA1) mutations cause the pleiotropic phenotype of oculodentodigital dysplasia. Amer. J. Hum. Genet. 72, 408–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Burdon K. P., Wirth M. G., Mackey D. A., Russell-Eggitt I. M., Craig J. E., Elder J. E., Dickinson J. L., Sale M. M. (2004) A novel mutation in the Connexin 46 gene causes autosomal dominant congenital cataract with incomplete penetrance. J. Med. Genet. 41, e106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Abrams C. K., Freidin M. M., Verselis V. K., Bennett M. V., Bargiello T. A. (2001) Functional alterations in gap junction channels formed by mutant forms of connexin 32: evidence for loss of function as a pathogenic mechanism in the X-linked form of Charcot-Marie-Tooth disease. Brain Res. 900, 9–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Orthmann-Murphy J. L., Freidin M., Fischer E., Scherer S. S., Abrams C. K. (2007) Two distinct heterotypic channels mediate gap junction coupling between astrocyte and oligodendrocyte connexins. J. Neurosci. 27, 13949–13957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Costantini S., Colonna G., Facchiano A. M. (2008) ESBRI: a web server for evaluating salt bridges in proteins. Bioinformation 3, 137–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kwon T., Harris A. L., Rossi A., Bargiello T. A. (2011) Molecular dynamics simulations of the Cx26 hemichannel: evaluation of structural models with Brownian dynamics. J. Gen. Physiol. 138, 475–493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kwon T., Roux B., Jo S., Klauda J. B., Harris A. L., Bargiello T. A. (2012) Molecular dynamics simulations of the Cx26 hemichannel: insights into voltage-dependent loop-gating. Biophys. J. 102, 1341–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Verselis V. K., Bennett M. V., Bargiello T. A. (1991) A voltage-dependent gap junction in Drosophila melanogaster. Biophys. J. 59, 114–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tang Q., Dowd T. L., Verselis V. K., Bargiello T. A. (2009) Conformational changes in a pore-forming region underlie voltage-dependent “loop gating” of an unapposed connexin hemichannel. J. Gen. Physiol. 133, 555–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Verselis V. K., Trelles M. P., Rubinos C., Bargiello T. A., Srinivas M. (2009) Loop gating of connexin hemichannels involves movement of pore-lining residues in the first extracellular loop domain. J. Biol. Chem. 284, 4484–4493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen Y., Deng Y., Bao X., Reuss L., Altenberg G. A. (2005) Mechanism of the defect in gap-junctional communication by expression of a connexin 26 mutant associated with dominant deafness. Faseb J. 19, 1516–1518 [DOI] [PubMed] [Google Scholar]
- 36. Deng Y., Chen Y., Reuss L., Altenberg G. A. (2006) Mutations of connexin 26 at position 75 and dominant deafness: essential role of arginine for the generation of functional gap-junctional channels. Hear Res. 220, 87–94 [DOI] [PubMed] [Google Scholar]
- 37. Bukauskas F. F., Bukauskiene A., Bennett M. V., Verselis V. K. (2001) Gating properties of gap junction channels assembled from connexin43 and connexin43 fused with green fluorescent protein. Biophys. J. 81, 137–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Oshima A., Doi T., Mitsuoka K., Maeda S., Fujiyoshi Y. (2003) Roles of Met-34, Cys-64, and Arg75 in the assembly of human connexin 26. Implication for key amino acid residues for channel formation and function. J. Biol. Chem. 278, 1807–1816 [DOI] [PubMed] [Google Scholar]
- 39. Bukauskas F. F., Kempf C., Weingart R. (1992) Cytoplasmic bridges and gap junctions in an insect cell line (Aedes albopictus). Exp. Physiol. 77, 903–911 [DOI] [PubMed] [Google Scholar]
- 40. Wang H. L., Chang W. T., Yeh T. H., Wu T., Chen M. S., Wu C. Y. (2004) Functional analysis of connexin-32 mutants associated with X-linked dominant Charcot-Marie-Tooth disease. Neurobiol. Dis. 15, 361–370 [DOI] [PubMed] [Google Scholar]
- 41. Oh S., Rubin J. B., Bennett M. V., Verselis V. K., Bargiello T. A. (1999) Molecular determinants of electrical rectification of single channel conductance in gap junctions formed by connexins 26 and 32. J. Gen. Physiol. 114, 339–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Oh S., Abrams C. K., Verselis V. K., Bargiello T. A. (2000) Stoichiometry of transjunctional voltage-gating polarity reversal by a negative charge substitution in the amino terminus of a connexin32 chimera. J. Gen. Physiol. 116, 13–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bukauskas F. F., Verselis V. K. (2004) Gap junction channel gating. Biochim. Biophys. Acta 1662, 42–60 [DOI] [PMC free article] [PubMed] [Google Scholar]